

Abstract
Organocatalysts derived from diethylenetriamine effect the rapid isomerization of non-native protein disulfide bonds to native ones. These catalysts contain a pendant hydrophobic moiety to encourage interaction with the non-native state, and two thiol groups with low pKa values that form a disulfide bond with a high E°′ value.
The formation of native disulfide bonds is at the core of oxidative protein folding.1–4 In oxidizing environments, reduced proteins with multiple cysteine residues tend to oxidize rapidly and nonspecifically. To attain a proper three-dimensional fold, any non-native disulfide bonds must isomerize to the linkages found in the native protein.5 In eukaryotic cells, this process is mediated by the enzyme protein disulfide isomerase (PDI; EC 5.3.4.1).4,6–14
Catalysis of disulfide-bond isomerization by PDI involves thiol–disulfide interchange chemistry. A putative mechanism commences with the nucleophilic attack by a thiolate on a non-native disulfide bond, generating a mixed-disulfide and a new substrate thiolate (Fig. 1).15 This thiolate can then attack another non-native disulfide bond, inducing further rearrangements to achieve the stable native state. The ability of PDI to catalyze disulfide-bond isomerization (rather than dithiol oxidation) makes the enzyme essential to the viability of the yeast Saccharomyces cerevisiae.7,16
Fig. 1.
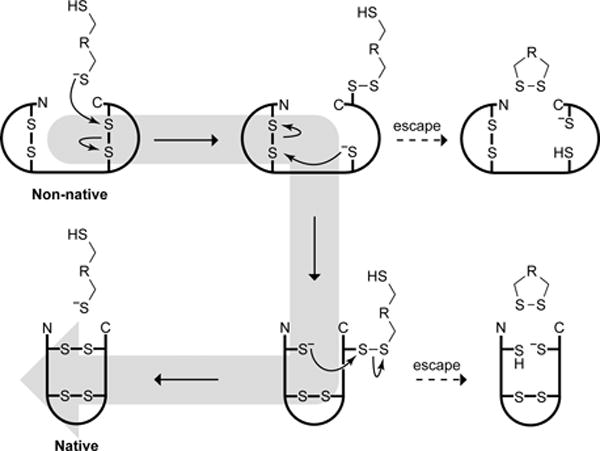
Putative mechanism for catalysis of protein-disulfide isomerization by protein disulfide isomerase (PDI) and small-molecule dithiol catalysts.
PDI is abundant in the endoplasmic reticulum (ER) of eukaryotic cells. The enzyme contains four domains: a, a′, b, and b′.12 The a and a′ domains each contain one active-site CGHC motif—a pattern analogous to that in many other oxidoreductases, whereas the b and b′ domains appear to mediate substrate binding.12,17,18 The physicochemical properties of its active-site make PDI an ideal catalyst for the reshuffling of disulfide bonds in misfolded proteins. The deprotonated thiolate of its N-terminal active-site cysteine residue (CGHC) initiates catalysis (Fig. 1).19 The amount of enzymic thiolate present is dependent on two factors.20,21 One is the pKa of the active-site cysteine residue; the other is the reduction potential (E°′) of the disulfide bond formed between the two active-site cysteine residues. In PDI, the cysteine pKa is 6.7, and the disulfide E°′ is −0.18 V.22,23 Given the properties of the ER (pH 7.0; Esolution = −0.18 V), ⅓ of PDI active sites contain a reactive thiolate.16,24 Moreover, the high (less negative) reduction potential of PDI renders the protein as a weak disulfide-reducing agent, ensuring that ample time isavailable for the catalyst to rearrange all of the disulfide bonds before reducing its protein substrate to “escape” (Fig. 1). If necessary, however, the second active-site cysteine residue can engage to rescue the enzyme from non-productive mixed-disulfide intermediates.7,25,26
Efficient oxidative protein folding requires a redox environment that supports both thiol oxidation and disulfide-bond isomerization. In vitro and in cellulo, this environment can be provided by a redox buffer consisting of reduced and oxidized glutathione. For example, the oxidative folding of a favorite model protein, bovine pancreatic ribonuclease (RNase A; EC 3.1.27.5), occurs readily in the presence of 1 mM glutathione (GSH) and 0.2 mM oxidized glutathione (GSSG).27 Adding PDI accelerates the process, but the large-scale use of PDI as a catalyst for folding proteins in vitro is impractical due to its high cost and conformational instability, and the complexity imposed by its separation from a substrate protein. Accordingly, the development of small-molecule PDI mimics has become a high priority.
To date, most PDI mimics have been designed to replicate the physicochemical properties of the CGHC active site—low thiol pKa and high disulfide E°′.28 Previously, we reported on (±)-trans-1,2-bis(mercaptoacetamido)cyclohexane (1; BMC) (Fig. 2), a small molecule that catalyzes the formation of native disulfide bonds in proteins, both in vitro and in cellulo.29 In 2005, other workers screened 14 reagents for their ability to fold a variety of proteins, and concluded that BMC was the best of known small-molecule catalysts.30 Though effective, BMC has shortcomings. For example, its low disulfide E°′ renders the compound too reducing for optimal catalysis of disulfide-bond isomerization. Subsequently, various CXXC and CXC peptides, aromatic thiols, and selenium-based catalysts were developed and employed with some success.31–42 Nevertheless, these organocatalysts had non-optimal thiol pKa and disulfide E°′ values. Moreover, they did not mimic a hallmark of enzymic catalysts—binding to the substrate.43
Fig. 2.
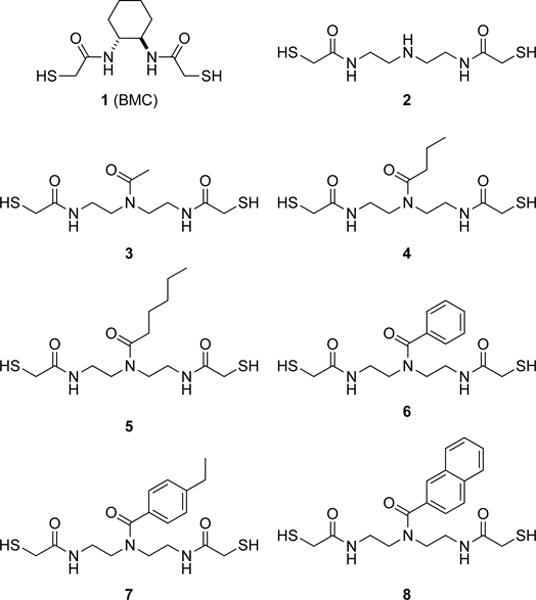
Small-molecule PDI mimics synthesized and assessed in this study.
The b and b′ domains of PDI have an exposed hydrophobic patch. The two patches unite to form a continuous hydrophobic surface between the two active sites.10,12,13,44,45 This hydrophobic surface could entice PDI to bind to unfolded or misfolded proteins, which tend to expose more hydrophobic residues than do proteins in their native state.46 Accordingly, we set out to design organocatalysts that not only have low thiol pKa and high disulfide E°′ values but also emulate substrate binding by PDI. We were inspired by the demonstrated ability of the hydrophobic effect to induce proximity in aqueous solution and thereby accelerate a variety of chemical reactions, such as O→N acyl transfer,47,48 ester hydrolysis,49,50 and dithiol oxidation.51,52 We reasoned that analogous induced proximity could enhance disulfide-bond isomerization in a misfolded protein, which is the key step in oxidative protein folding.7,16
We reasoned that dithiol 2 (Fig. 2) would provide an appropriate scaffold for the development of useful catalysts. We were drawn to dithiol 2 for three reasons. First, its mercaptoacetamido groups are known to have low thiol pKa values.29,53 Secondly, the disulfide bond of its oxidized form resides in a large, 13-membered ring containing two secondary amides, which should lead to a high reduction potential. Finally, dithiol 2 has an amino group that can be condensed with hydrophobic carboxylic acids to mimic the b and b′ domains of PDI.
Our experimental work commenced with the synthesis of dithiol 2 from diethylenetriamine in a few high-yielding steps (see: Supporting Information). To determine its thiol pKa values, we monitored its A238 nm as a function of pH.29,54 We found pKa values of 8.0 ± 0.2 and 9.2 ± 0.1 (Table 1). These values are slightly less than those of BMC, presumably due to the additional electronegative nitrogen atom. To determine the reduction potential of its oxidized form, we equilibrated equimolar amounts of dithiol 2 and oxidized β-mercaptoethanol, and quantified the amount of each reduced and oxidized species with analytical HPLC.29,55 We found a disulfide E°′ value of (–0.192 ± 0.003) V. This value indicates that dithiol 2 is a weaker reducing agent than is BMC, which is consistent with BMC being more preorganized for disulfide-bond formation. Finally, to probe the effect of increasing hydrophobicity on catalyzing the formation of native disulfide bonds in proteins, we synthesized dithiols 3–8. We isolated dithiols 3–6 as colorless oils, and dithiols 7 and 8 as white solids. None had a strong odor.
Table 1.
Properties of PDI and mimics 1–8.
| Catalyst | pKa | Disulfide E°′ | logPa | Folding yield (%)b |
|---|---|---|---|---|
| (None) | — | — | — | 45 ± 2 |
| PDI | 6.7c | –0.180 V | — | 87 ± 2 |
| 1 (BMC) | 8.3; 9.9d | –0.232 V | — | 42 ± 2 |
| 2 | 8.0; 9.2 | –0.192 V | 0.10 | 50 ± 2 |
| 3 | ND | ND | –0.74 | 45 ± 2 |
| 4 | ND | ND | 0.66 | 54 ± 4 |
| 5 | 8.1; 9.3 | –0.203 V | 1.67 | 57 ± 1 |
| 6 | ND | ND | 0.90 | 60 ± 2 |
| 7 | 8.1; 9.4 | –0.206 V | 1.82 | 66 ± 2 |
| 8 | ND | ND | 2.06 | ND |
Values were calculated for dimethylamine in dithiol 2 and the tertiary amide moiety in dithiols 3–8 (e.g., N,N-dimethylacetamide for dithiol 3) with software from Molinspiration (Slovenský Grob, Slovak Republic), and are similar to known experimental values.63
Values are for the unscrambling of sRNase A to give native RNase A by 1 mM catalyst in 5 h, as in Fig. 4.
Value for the N-terminal cysteine residue in the active site of PDI.24
Values are from ref. 29.
ND, not determined.
Enzymatic catalysis provides an extremely sensitive measure of native protein structure.56 RNase A contains eight cysteine residues, which could form 105 (= 7 × 5 × 3 × 1) distinct fully oxidized species, only one of which gives rises to enzymatic activity (Fig. 3).57,58 Accordingly, we tested the ability of this panel of compounds to catalyze the isomerization of “scrambled” RNase A (sRNase A), which is a random mixture of oxidized species, to its native state. The isomerization reaction was monitored by measuring the gain of catalytic activity.59 Dithiol 8 was excluded from the analysis due to its low solubility in aqueous solution.
Fig. 3.
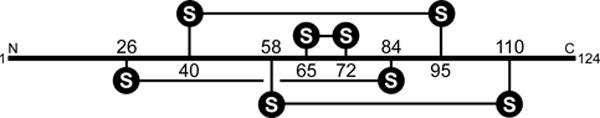
Scheme showing the connectivity of the four disulfide bonds in native RNase A. There are 104 other fully oxidized forms.
Some, but not all, of the PDI mimics led to a significant increase in the yield of oxidative protein folding (Fig. 4A). Most notably, the data with dithiols 2–7 revealed an overall trend toward higher yield with increasing hydrophobicity of the pendant carboxamide (Fig. 4B). This trend culminated with dithiol 7, which increased the yield of folded RNase A by 47% compared to that in the absence of a catalyst. These data contrast markedly with those using monothiols (e.g., glutathione), which reduce the yield of properly folded protein by favoring the accumulation of mixed-disulfide species.27
Fig. 4.
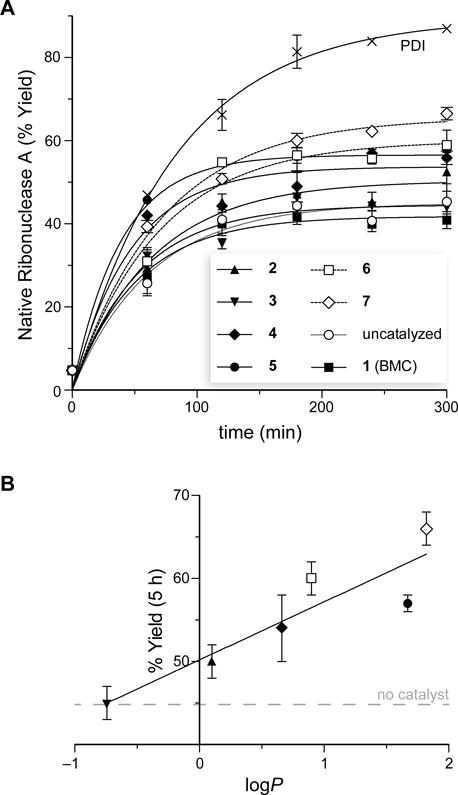
Catalysis of disulfide-bond isomerization by PDI and PDI mimics 1–7. (A) Graph of the time-course for the isomerization of sRNase A to give native RNase A. All assays were performed in triplicate at 30 °C in 50 mM Tris–HCl buffer, pH 7.6, containing GSH (1.0 mM), GSSG (0.2 mM), and PDI or dithiol 1–7 (1.0 mM). (B) Graph of the yield of native RNase A achieved by PDI mimics 2–7 after 5 h as a function of the logP value of its side chain (Table 1).
The apparent correlation of catalytic efficacy with hydrophobicity could be due to a physicochemical property other than hydrophobicity. Accordingly, we determined the thiol pKa and disulfide E°′ values of the most efficacious dithiols containing an alkyl (5) and aryl (7) carboxamide. We found dithiol 5 to have thiol pKa values of 8.1 and 9.3 and a disulfide E°′ value of −0.203 V (Table 1). We found dithiol 7 to have similar physicochemical properties, with thiol pKa values of 8.1 and 9.4 and a disulfide E°′ value of −0.206 V. Both of these compounds possess thiol acidity and disulfide stability similar to those of parent dithiol 2, affirming that hydrophobicity is indeed correlative with catalytic efficacy.
Our data are the first to indicate that adding a hydrophobic moiety to a small-molecule PDI mimic can have a profound effect on its ability to catalyze disulfide-bond isomerization. Still, none of the organocatalysts were as efficacious as PDI itself. We note, however, that the molecular mass of PDI (57 kDa) is >102-fold greater than any of its mimics, enabling optimization of substrate binding and turnover beyond that attainable with small-molecule catalysts. Also, each molecule of PDI has two active sites, and thus provides a higher concentration of dithiol than do the organocatalysts.
Like the substrate-binding domains of PDI, the hydrophobicity of dithiols 4–7 likely encourages their interaction with unfolded or misfolded proteins.10,12,13,44,45,60,61 Dithiols having moieties with higher logP values perform better, and aromatic moieties seem to be especially efficacious (Fig. 4B). We note that a more hydrophobic catalyst could also increase the rate of the underlying thiol–disulfide interchange chemistry, as nonpolar environments are known to lower the free energy of activation for this reaction.62
Conclusions
We have designed, synthesized, and characterized novel organocatalysts that enhance the efficiency of oxidative protein folding. Moreover, we have demonstrated that increasing the hydrophobicity of the catalysts has a marked effect on their catalytic efficacy. The production of proteins that contain disulfide bonds by recombinant DNA technology often leads to the aggregation of misfolded proteins.64,65 These aggregates must be reduced, denatured, and solubilized to enable proper folding. Approximately 20% of all human proteins66 and many proteins of high pharmaceutical relevance67,68 contain at least one disulfide bond between cysteine residues. For example, antibodies contain at least 12 intrachain and 4 interchain disulfide bonds,69 and there are >300 distinct antibodies in clinical development,70 including ~30 antibody–drug conjugates.71 The ability to mimic the essential function of PDI7,16 in a small molecule could have a favorable impact on the production of antibodies and other biologics, and usher in a new genre of organocatalysts for oxidative protein folding.
Supplementary Material
Acknowledgments
K.A.A. was supported by a predoctoral fellowship from the PhRMA Foundation and by Molecular and Cellular Pharmacology Training Grant T32 GM008688 (NIH). This work was supported by grant R01 GM044783 (NIH). NMR spectra were obtained at NMRFAM, which is supported by grant P41 GM103399 (NIH).
Footnotes
Electronic Supplementary Information (ESI) available: Synthetic and analytical procedures. See DOI: 10.1039/c000000x/
Notes and references
- 1.Jocelyn PC, editor. Biochemistry of the SH Group: The Occurence, Chemical Properties, Metabolism and Biological Function of Thiols and Disulfides. London, U.K.: 1972. [Google Scholar]
- 2.Buchner J, Moroder L, editors. Oxidative Folding of Peptides and Proteins. The Royal Society of Chemistry; Cambridge, UK: 2009. [Google Scholar]
- 3.Lindahl M, Mata-Cabana A, Kieselbach T. Antioxid Redox Signal. 2011;14:2581–2642. doi: 10.1089/ars.2010.3551. [DOI] [PubMed] [Google Scholar]
- 4.Oka OB, Bulleid NJ. Biochim Biophys Acta. 2013;1833:2425–2429. doi: 10.1016/j.bbamcr.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 5.Anfinsen CB. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 6.Robinson AS, Hines V, Wittrup KD. Biotechnology. 1994;12:381–384. doi: 10.1038/nbt0494-381. [DOI] [PubMed] [Google Scholar]
- 7.Laboissière MCA, Sturley SL, Raines RT. J Biol Chem. 1995;270:28006–28009. doi: 10.1074/jbc.270.47.28006. [DOI] [PubMed] [Google Scholar]
- 8.Guzman NA, editor. Prolyl Hydroxylase, Protein Disulfide Isomerase, and Other Structurally Related Proteins. Marcel Dekker; New York, NY: 1998. [Google Scholar]
- 9.Woycechowsky KJ, Raines RT. Curr Opin Chem Biol. 2000;4:533–539. doi: 10.1016/s1367-5931(00)00128-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freedman RB, Klappa P, Ruddock LW. EMBO Rep. 2002;3:136–140. doi: 10.1093/embo-reports/kvf035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kersteen EA, Raines RT. Antioxid Redox Signal. 2003;5:413–424. doi: 10.1089/152308603768295159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tian G, Xiang S, Noiva R, Lennarz WJ, Schindelin H. Cell. 2006;124:61–73. doi: 10.1016/j.cell.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 13.Gruber CW, Cemazar M, Heras B, Martin JL, Craik DJ. Trends Biochem Sci. 2006;31:455–464. doi: 10.1016/j.tibs.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 14.Denisov AY, Maattanen P, Dabrowski C, Kozlov G, Thomas DY, Gehring K. FEBS J. 2009;276:1440–1449. doi: 10.1111/j.1742-4658.2009.06884.x. [DOI] [PubMed] [Google Scholar]
- 15.Kersteen EA, Barrows SR, Raines RT. Biochemistry. 2005;44:12168–12178. doi: 10.1021/bi0507985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chivers PT, Laboissière MCA, Raines RT. EMBO J. 1996;15:2659–2667. [PMC free article] [PubMed] [Google Scholar]
- 17.Holmgren A. J Biol Chem. 1979;254:9627–9632. [PubMed] [Google Scholar]
- 18.Edman JC, Ellis L, Blacher RW, Roth RA, Rutter WJ. Nature. 1985;317:267–270. doi: 10.1038/317267a0. [DOI] [PubMed] [Google Scholar]
- 19.Darby NJ, Creighton TE. Biochemistry. 1995;34:3576–3587. doi: 10.1021/bi00011a012. [DOI] [PubMed] [Google Scholar]
- 20.Gilbert HF. Adv Enzymol. 1990;63:69–172. doi: 10.1002/9780470123096.ch2. [DOI] [PubMed] [Google Scholar]
- 21.Chivers PT, Prehoda KE, Raines RT. Biochemistry. 1997;36:4061–4066. doi: 10.1021/bi9628580. [DOI] [PubMed] [Google Scholar]
- 22.Hawkins HC, Freedman RB. Biochem J. 1991;275:335–339. doi: 10.1042/bj2750335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lundstrom J, Holmgren A. Biochemistry. 1993;32:6649–6655. doi: 10.1021/bi00077a018. [DOI] [PubMed] [Google Scholar]
- 24.Hwang C, Sinskey AJ, Lodish HF. Science. 1992;257:1496–1502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- 25.Walker KW, Lyles MM, Gilbert HF. Biochemistry. 1996;35:1972–1980. doi: 10.1021/bi952157n. [DOI] [PubMed] [Google Scholar]
- 26.Walker KW, Gilbert HF. J Biol Chem. 1997;272:8845–8848. doi: 10.1074/jbc.272.14.8845. [DOI] [PubMed] [Google Scholar]
- 27.Lyles MM, Gilbert HF. Biochemistry. 1991;30:619–625. doi: 10.1021/bi00217a005. [DOI] [PubMed] [Google Scholar]
- 28.Lees WJ. Curr Opin Chem Biol. 2008;12:740–745. doi: 10.1016/j.cbpa.2008.08.032. [DOI] [PubMed] [Google Scholar]
- 29.Woycechowsky KJ, Wittrup KD, Raines RT. Chem Biol. 1999;6:871–879. doi: 10.1016/s1074-5521(00)80006-x. [DOI] [PubMed] [Google Scholar]
- 30.Willis MS, Hogan JK, Prabhakar P, Liu X, Tsai K, Wei Y, Fox T. Protein Sci. 2005;14:1818–1826. doi: 10.1110/ps.051433205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Woycechowsky KJ, Hook BA, Raines RT. Biotechnol Progr. 2003;19:1307–1314. doi: 10.1021/bp0257123. [DOI] [PubMed] [Google Scholar]
- 32.Woycechowsky KJ, Raines RT. Biochemistry. 2003;42:5387–5394. doi: 10.1021/bi026993q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gough JD, Gargano JM, Donofrio AE, Lees WJ. Biochemistry. 2003;42:11787–11797. doi: 10.1021/bi034305c. [DOI] [PubMed] [Google Scholar]
- 34.Gough JD, Lees WJ. J Biotechnol. 2005;115:279–290. doi: 10.1016/j.jbiotec.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 35.Gough JD, Lees WJ. Bioorg Med Chem. 2005;15:777–781. doi: 10.1016/j.bmcl.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 36.Gough JD, Barrett EJ, Silva Y, Lees WJ. J Biotechnol. 2006;125:39–47. doi: 10.1016/j.jbiotec.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 37.Beld J, Woycechowsky KJ, Hilvert D. Biochemistry. 2008;47:6985–6987. doi: 10.1021/bi8008906. [DOI] [PubMed] [Google Scholar]
- 38.Beld J, Woycechowsky KJ, Hilvert D. Biochemistry. 2009;48:4662–4662. [Google Scholar]
- 39.Beld J, Woycechowsky KJ, Hilvert D. J Biotechnol. 2010;150:481–489. doi: 10.1016/j.jbiotec.2010.09.956. [DOI] [PubMed] [Google Scholar]
- 40.Wang GZ, Dong XY, Sun Y. Biochem Eng J. 2011;55:169–175. [Google Scholar]
- 41.Patel AS, Lees WJ. Bioorg Med Chem. 2012;20:1020–1028. doi: 10.1016/j.bmc.2011.11.049. [DOI] [PubMed] [Google Scholar]
- 42.Lees WJ. ChemBioChem. 2012;13:1725–1727. doi: 10.1002/cbic.201200288. [DOI] [PubMed] [Google Scholar]
- 43.Jencks WR. Catalysis in Chemistry and Enzymology. McGraw-Hill; New York, NY: 1969. [Google Scholar]
- 44.Shouldice SR, Heras B, Walden PM, Totsika M, Schembri MA, Martin JL. Antioxid Redox Signal. 2011;14:1729–1760. doi: 10.1089/ars.2010.3344. [DOI] [PubMed] [Google Scholar]
- 45.Kober FX, Koelmel W, Kuper J, Drechsler J, Mais C, Hermanns HM, Schindelin H. J Biol Chem. 2013;288:2029–2039. doi: 10.1074/jbc.M112.410522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lins L, Brasseur R. FASEB J. 1995;9:535–540. doi: 10.1096/fasebj.9.7.7737462. [DOI] [PubMed] [Google Scholar]
- 47.Knowles JR, Parsons CA. Chem Commun. 1967:755–757. [Google Scholar]
- 48.Blyth CA, Knowles JR. J Am Chem Soc. 1971;93:3017–3021. [Google Scholar]
- 49.Knowles JR, Parsons CA. Nature. 1969;221:5553–5554. doi: 10.1038/221053a0. [DOI] [PubMed] [Google Scholar]
- 50.Blyth CA, Knowles JR. J Am Chem Soc. 1971;93:3021–3027. [Google Scholar]
- 51.Wang GZ, Dong XY, Sun Y. Biotechnol Progr. 2011;27:377–385. doi: 10.1002/btpr.517. [DOI] [PubMed] [Google Scholar]
- 52.Liu H, Dong XY, Sun Y. Biochem Eng J. 2013;79:29–32. [Google Scholar]
- 53.Lees WJ, Singh R, Whitesides GM. J Org Chem. 1991;56:7328–7331. [Google Scholar]
- 54.Lukesh JC, Palte MJ, Raines RT. J Am Chem Soc. 2012;134:4057–4059. doi: 10.1021/ja211931f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lamoureux GV, Whitesides GM. J Org Chem. 1993;58:633–641. [Google Scholar]
- 56.Knowles JR. Science. 1987;236:1252–1258. doi: 10.1126/science.3296192. [DOI] [PubMed] [Google Scholar]
- 57.Raines RT. Chem Rev. 1998;98:1045–1066. doi: 10.1021/cr960427h. [DOI] [PubMed] [Google Scholar]
- 58.Klink TA, Woycechowsky KJ, Taylor KM, Raines RT. Eur J Biochem. 2000;267:566–572. doi: 10.1046/j.1432-1327.2000.01037.x. [DOI] [PubMed] [Google Scholar]
- 59.Kelemen BR, Klink TA, Behlke MA, Eubanks SR, Leland PA, Raines RT. Nucleic Acids Res. 1999;27:3696–3701. doi: 10.1093/nar/27.18.3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guddat LW, Bardwell JC, Zander T, Martin JL. Protein Sci. 1997;6:1148–1156. doi: 10.1002/pro.5560060603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gilbert HF. J Biol Chem. 1997;272:29399–29402. doi: 10.1074/jbc.272.47.29399. [DOI] [PubMed] [Google Scholar]
- 62.Fernandes PA, Ramos MJ. Chem Eur J. 2004;10:257–266. doi: 10.1002/chem.200305343. [DOI] [PubMed] [Google Scholar]
- 63.Sangster J. J Phys Chem Ref Data. 1989;18:1111–1227. [Google Scholar]
- 64.Marston FAO. Biochem J. 1986;240:1–12. doi: 10.1042/bj2400001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.De Bernardez Clark E. Curr Opin Biotechnol. 1998;9:157–163. doi: 10.1016/s0958-1669(98)80109-2. [DOI] [PubMed] [Google Scholar]
- 66.Martelli PL, Fariselli P, Casadio R. Proteomics. 2004;4:1665–1671. doi: 10.1002/pmic.200300745. [DOI] [PubMed] [Google Scholar]
- 67.Fahey RC, Hunt JS, Windham GC. J Mol Evol. 1977;10:155–160. doi: 10.1007/BF01751808. [DOI] [PubMed] [Google Scholar]
- 68.Thorton JM. J Mol Biol. 1981;151:261–287. doi: 10.1016/0022-2836(81)90515-5. [DOI] [PubMed] [Google Scholar]
- 69.Liu H, May K. mAbs. 2012;4:17–23. doi: 10.4161/mabs.4.1.18347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pharmaceutical Research and Manufacturers of America report “Medicines in Development—Biologics, 2013 Report”.
- 71.Mullard A. Nat Rev Drug Discov. 2013;12:329–332. doi: 10.1038/nrd4009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.