

Abstract
Although human papillomavirus (HPV) was identified as an etiological factor in cervical cancer, the key human gene drivers of this disease remain unknown. Here we apply an unbiased approach integrating gene expression and chromosomal aberration data. In an independent group of patients, we reconstruct and validate a gene regulatory meta-network, and identify cell cycle and antiviral genes that constitute two major sub-networks up-regulated in tumour samples. These genes are located within the same regions as chromosomal amplifications, most frequently on 3q. We propose a model in which selected chromosomal gains drive activation of antiviral genes contributing to episomal virus elimination, which synergizes with cell cycle dysregulation. These findings may help to explain the paradox of episomal HPV decline in women with invasive cancer who were previously unable to clear the virus.
INTRODUCTION
Although it has been known for decades that persistent infection with high-risk human papillomavirus (HPV) is the major risk factor for the development of cervical cancer 1, the exact means by which the virus induces tumor growth and the genomic changes that promote tumors have not been completely elucidated 2. On the one hand, it is known that failure to mount an effective immune response facilitates viral persistence and integration into a host genome leading to genomic aberrations and a subsequent development of cervical cancer after several decades 3. Episomal virus can cause some genomic instability during early stages of viral infection even before integration into a host genome 4–5. On the other hand, there is evidence that the loss of episomal HPV is important in disease progression after virus integration. For example, data from biopsies suggest that pre-cancerous lesions have more episomal virus than high-grade tumors 6–7, and data from cervical cell lines show that cervical keratinocytes that lose episomal virus show increased expression of viral oncogenes coded by the integrated form 8. This increase seems to be due to the loss of the regulatory viral gene, E2, which is frequently disrupted on integration, and which normally suppresses the expression of the viral oncogenes E6 and E7 2. The E6 and E7 transforming proteins have multiple mechanisms to alter cell cycle control and to cause genomic instability that lead to an increase of chromosomal aberrations and mutations in cellular genes 9. The loss of episomal virus in the in vitro cell lines coincides with an increase of antiviral immune response, and this has led to another model of carcinogenesis suggesting that an augment in antiviral activity results in the elimination of episomal virus aiding tumor progression 8.
There are thus two main questions that are currently awaiting answers: is there evidence of a similar antiviral immune response in cervical tumors in vivo and, if yes, why would women that have not been able to deal with HPV infection for a long time suddenly acquire an antiviral response strong enough to generate a dramatic decrease of viral load?
Here we sought to identify in unbiased way molecular mechanisms and key gene drivers that may be involved in the cervical carcinogenesis using an integrative systems biology approach. Although we did not originally aim to ask the above questions our results have positively answered the first question and provided the basis for solution of the paradox contained in the second one.
We started by generating gene expression data from 40 tumors and 20 normal tissue samples. Combining these data with data from 4 other independent studies, we revealed a robust set of differentially expressed genes and used it to reconstruct a gene co-expression network of cervical cancer. We found three major sub-networks, one containing genes involved in the cell cycle, another related to the antiviral activity, and a third minor network related to epithelial cell differentiation. Integrating these data with data on genomic (chromosomal) aberrations, we found that the cell cycle and antiviral sub-networks were regulated by genes located within the same regions of chromosomal amplification. We suggest that activation of antiviral genes that occurs late in women that have not otherwise made strong this response is driven by selected chromosomal changes that occur in HPV-transformed cells. We propose a model in which frequent aberrations contain regulatory genes that stimulate antiviral response and cell cycle, and consequently promote the elimination of inhibitory episomal virus and uncontrolled proliferation, allowing the dysplastic cell to become a malignant tumor.
RESULTS
Chromosomal aberrations regulating gene expression
We performed a meta-analysis of gene expression microarray data using our dataset and four others from the literature (Supplementary Table S1) to identify genes with altered expression in cervical tumor samples versus normal tissue. We identified 1268 genes with a false discovery rate (FDR) less than 1%, of which 526 genes were down-regulated in tumor samples (Down-genes) and 742 genes were up-regulated (Up-genes) (Fig. 1a, Supplementary Data 1).
Figure 1. Chromosomal aberrations regulating expression of genes from frequent chromosomal gain or loss.

(a) Heat map of the 1268 genes differentially expressed between cervical tumor and normal tissue samples in the five data sets used for the meta-analysis of gene expression microarray data. red-upregulated, green-downregulated, grey-missing value. (b) Frequency of gain (FqG, red) or loss (FqL, green) in the genome detected in the meta-analysis of comparative genomic hybridization studies using cervical cancer samples. (c) Distribution of the delta values (FqG − FqL) for the 1268 genes. Chromosomal regions with delta values > 0.32 or < −0.2 were considered to be regions of frequent gains or losses, respectively. Bar graph shows the number of up- (Up) or down-regulated (Dn) genes in each region. There is an association between gene expression and chromosomal aberrations for the genes in the regions of frequent gains and losses (p < 0.0001; Fisher exact test).
As the chromosomal imbalances may cause alterations in gene expression, we questioned whether the identified differentially expressed genes (DEGs) were located in the regions of frequent chromosomal gains or losses. To identify regions of chromosomal aberrations in cervical cancer we performed a meta-analysis of published comparative genomic hybridization studies in this cancer (Supplementary Table S2). We used a total of 269 cases from ten studies and determined the frequencies of gains and losses for the whole genome (Fig. 1b). Next, we retrieved information on chromosomal aberration frequencies for 1268 DEGs according to their gene locations (Supplementary Fig. S1a, Supplementary Data 2) and calculated the difference between frequency of Gain (FqG) and frequency of Loss (FqL) for each gene. In order to identify cut-off values for frequent losses and gains, we plotted the density distribution for these differences (Supplementary Fig. S1b) and searched for local minima on both sides of the distribution. The cut-off values were −0.2 and 0.32 of (FqG − FqL) for frequent losses and frequent gains, respectively (Supplementary Fig. S1b). The frequent loss regions were located on chromosomes 2 (2q33-q37), 3 (3p12-p26), 4 (4p12-p15.2, 4q13-q35), 11 (11q23-q25) and 13 (13q21) and the frequent gain regions on chromosome 3 (3q13.2-q29) (Supplementary Fig. S1c). In the loss regions there were 89 DEGs (66 Down- and 23 Up-genes), and in the gain regions 62 DEGs (53 Up- and 9 Down-genes) (Fig. 1c). Thus, we found a strong association between chromosomal Gains or Losses and Up or Down-regulation of gene expression (p<0.0001; Fisher exact test) indicating that the majority of differentially expressed genes located in the regions of frequent chromosomal aberrations are regulated by these aberrations.
Gene network reconstruction and identification of sub-networks
Although we found that chromosomal aberrations regulated the expression of about 80% of DEGs located within these regions, those genes represented only ~9% of all DEGs. It is known, however, that differentially expressed genes are not totally independent from each other but rather represent a regulatory network driven by different factors, with chromosomal aberrations being one of them. Therefore, some DEGs can be indirectly regulated by chromosomal gains or losses, if their expression is regulated by another gene directly affected by a chromosomal aberration. To uncover such regulatory relationships we employed an approach of network reconstruction that we have previously applied to disclose gene-gene interactions in immunodeficiency10. We analyzed correlations between all gene pairs formed by the DEGs and found 3161 correlated gene pairs with FDR ≤ 0.1%. The network was composed of 738 genes with only a few gene pairs not connected to the main network (Fig. 2; p < 10−300; Erdős–Rényi model, see Supplementary Methods). We analyzed this network to find subnetworks which may have specific biological functions. We found three major sub-networks that contained 112 genes (sub-network 1), 54 (sub-network 2) and 19 genes (sub-network 3), respectively (Fig. 2). Using functional enrichment analysis, we identified “cell cycle” (subnetwork 1), “response to virus” (sub-network 2, which we name “antiviral” in the manuscript) and “epithelial cell differentiation” (sub-network 3) as the main Gene Ontology terms over-represented in these sub-networks (Supplementary Table S3). The cell cycle and antiviral subnetworks consisted mostly of Up-regulated genes and the epithelial cell differentiation subnetwork of Down-regulated genes.
Figure 2. Gene regulatory network reconstructed using the differentially expressed genes in cervical cancer.

Dots are genes (red, up-regulated; green, down-regulated); lines indicate presence of correlation between genes. The three identified sub-networks indicated by circles were named after Gene Ontology terms they represent as Cell Cycle, Antiviral (equivalent to GO term “response to virus”), and Epithelial differentiation.
Hierarchy in the sub-networks
To search for key regulators of the process and to test the regulatory relationships among the genes we needed to establish a hierarchy within the sub-networks. It was previously shown for different cancers that alteration in expression of genes located within chromosomal aberration can be a critical event driving a disease11. Furthermore, we based our analyses on a mathematical/statistical concept that correlation is a result of causation unless it is found by chance. The correlations, according to “Reichenbach’s common cause principles” 12–13, should reflect regulatory relationships. Specifically, if there is a correlation between expression of gene X and Y, then there are 3 possibilities: X regulates Y, Y regulates X, or there is Z that regulates both. Thus, for the case when X correlates with Y and there is a known cause for X (i.e. chromosomal aberration) the most likely scenario will be that X regulates Y (see Supplementary Fig. S2).
We used two criteria for ranking genes within sub-networks for their potential to be key regulators: first, frequency of chromosomal aberrations in patient population concordant with a direction of the change in gene expression (i.e. gain corresponding to up-regulation; loss to down-regulation). Second criterion was the number of connections with other genes in the network, as this feature had been shown to be a distinctive characteristic of major regulators14. The top-ranked genes were considered as gene-regulators and placed on the top of the hierarchy, followed by the genes directly linked to them (targets) and then by the genes that did not have direct links to the regulators. We could establish these hierarchies for sub-networks 1 (Fig. 3a) and 2 (Fig. 3b) but not for sub-network 3 (Supplementary Fig. S3a) as there was no gene that met the first criterion of a regulator in this sub-network.
Figure 3. Hierarchy in the cell cycle and antiviral sub-networks.

(a) Sub-network 1- Cell Cycle. (b) Sub-network 2- Antiviral. The sub-networks 1 and 2 have genes located in the regions of frequent DNA gain and were organized hierarchically with the regulator genes on the top and the target genes below them. (c) Testing the hierarchy of the sub-networks by perturbagens analysis using the Connectivity Map (http://www.broadinstitute.org/cmap). Twenty acting and 20 non-acting perturbagens were tested in each group of genes (regulators, targets and other sub-network), in the sub-network 1 and sub-network 2. Enrichment score (or connectivity score) is a measure of how perturbagens influence a gene expression signature where high positive score means significant induction (designated by **p < 0.0001 and *p < 0.001; Mann-Whitney Test).
In order to test the hierarchies of the sub-networks in the experimental settings, we took an advantage of the CMAP database 15 which contains global gene expression data of in vitro responses to a big variety of perturbations. First we divided each sub-network into two groups of genes: the first mainly containing regulators and the second one targets (Supplementary Fig. S3b, c). We tested the hypothesis that perturbagens that stimulate regulators should also stimulate targets of the given sub-network.
For each sub-network we chose 20 perturbagens which induced regulators (i.e. acting perturbagens). Next we tested if these perturbagens also acted on the target genes. As controls, we also selected perturbagens which did not act on the regulator genes (non-acting perturbagens). As a specificity control, we tested these perturbagens on the genes from the other sub-network. For both sub-networks, we observed that perturbagens selected on the basis of their ability to stimulate regulators also stimulated targets of a given sub-network, but not genes of another sub-network (Fig. 3c). Perturbagens with no effect on regulators did not show any effect on the targets either. Therefore, these results give us experimental support for the two ideas emerging from network analysis: first, no or minimal cross-regulation between genes of different subnetworks; and the second, that genes defined as regulators can drive the expression of target genes from the same subnetwork. It is important, though, to interpret these results in combination with other data because in isolation they do not rule out a possibility that regulators and targets are under common regulatory mechanisms affected by perturbagens.
Validation of genomic aberrations and gene expression network
Although the meta-analyses of chromosomal aberrations and gene expression network were performed using many independent studies, we wanted to validate these results by analyzing another cohort of patients. For this, we used an independent dataset containing CGH data on 97 patients with cervical cancer 16 and obtained gene expression data for 82 of them. First, we checked for the overall concordance of the genomic aberrations of the drivers/regulators genes between this new dataset and our meta-analysis. We observed a remarkably strong correlation between the two results (r = 0.8, p < 0.0001, Pearson correlation, Fig. 4a) further supporting the idea that genomic aberrations in those genes are a general feature of invasive cervical cancers.
Figure 4. Reproducibility of the meta-analysis in a new patient dataset.

(a) Correlation between frequency of gain in the meta-analysis and in validation dataset for the 36 regulator genes from the cell cycle and antiviral sub-networks (r = 0.8; p < 0.0001; spearman rank correlation). (b) Gene expression data of 82 patients were used to build a network from the genes that comprised the meta-network. Red nodes are up-regulated genes, blue nodes are down-regulated genes, white and black edges represent positive and negative correlations, respectively. Three sub-networks are indicated. (c) Chromosomal gains increase expression of key driver genes. Expression level of each of the six genes in patients with gains, losses or no change in the corresponding chromosomal region. Each symbol represents an individual tumor. NAT13, LAMP3, p<0.01; others, p<0.001 for comparison of ‘gain’ vs. ‘none’. (d) Chromosomal gains in the key driver genes regions regulate expression of their respective target genes. Driver genes are indicated on x axis. Each dot represents ratio for individual target genes that are calculated by dividing average expression of a target gene in tumors with gains by its expression in tumors with no gains.
Next we wanted to ensure the reproducibility of the gene expression meta-network. Using gene expression data of 82 tumors, we analyzed connections of the meta-network in the new data and found that ~96% of the significant correlations (FDR <10%) had the same directions as in the original network. Next we analyzed the substructure of this network and found three major subnetworks, which corresponded to those previously revealed in the original meta-network (Fig. 4b). Remarkably, each of the three subnetworks contained about 90% of genes out of the original subnetworks and all but one of the identified major key regulators were within the subnetworks. After a detailed inspection, we found that the missing major regulator gene (GMPS) actually does not have a probe on the Illumina platform that we have used for this new experiment. Overall, these results show that the meta-network we have reconstructed is highly reproducible in a new set of patients, demonstrating the robustness of our approach to meta-network reconstruction.
Because the validation patient dataset had both genomic aberrations data and gene expression data, we could directly ask whether the chromosomal gains in the key driver genes influence their expression. We found (as was predicted by our previous analysis, Fig. 1c) that expression of the key drivers was higher in tumors that had chromosomal amplifications in regions where these genes were located than in tumors with no aberrations or with losses in these regions (Fig. 4c).
Next we asked whether the genes we predicted to be targets of the key drivers have different expression depending whether the drivers have chromosomal gains or not. We tested this question by comparing the expression signature of target genes directly connected in the meta-network to the six driver genes between tumors with and without gains for the driver genes. To avoid biases, we only tested those target genes that were not located on the same chromosome with their corresponding driver. We observed that the signatures of target genes respective to each key driver had higher expression in tumors with gains than in tumors with no gains (loss and no aberration) suggesting that majority of the targets are to be regulated by their respective driver genes (Fig. 4d). These new results give an independent experimental support for our claim that the key driver genes we identified may drive the expression of the genes connected to them in the network.
Frequent aberrations contain cell cycle and antiviral drivers
Although both the cell cycle and antiviral sub-networks were up-regulated in tumors, we observed only a few correlations between gene expression of these two sub-networks. Because regulator/driver genes for both of these sub-networks were located in the regions of frequent chromosomal gains, we hypothesized that there might be relationships between the two subnetworks at the genomic level. In this analysis, we could only include the 117 patients from the meta-analysis whose individual data on genomic aberrations were available as wells as data from 97 patients of the validation dataset 16.
We asked if aberrations in the regions with the drivers of two functional classes (antiviral and cell cycle) appear in different samples reflecting the heterogeneity of cervical cancer or if these aberrations are present in the same tumors.
In the meta-analysis and the validation dataset, we found a much higher frequency of co-presence of the aberrations in regions that contained cell cycle and antiviral drivers in the same samples than might be expected by chance (meta-analysis dataset: p<0.0001, validation dataset: p<0.002; Chi-squared test, Fig. 5a).
Figure 5. Drivers of cell cycle and antiviral sub-networks are located in the same regions of frequent gains.

(a) Frequency of patients with chromosomal gains containing the driver genes from the Cell cycle and Antiviral sub-networks together and separately in the patients from meta-analysis and from validation dataset (p<0.0001, p<0.002, Chi-squared test respectively). (b) An integrative network reconstructed with data on chromosomal aberrations and gene expression correlation using the driver genes of cell cycle and antiviral sub-networks. All correlations in this network were positive. (c) Distribution of number of frequent chromosomal aberrations (gains) present in the same tumor. The regions used for this analysis were those containing regulator genes (1q, 1p, 3q, 5p, 8q, 17q, 19q, 20q). For b and c, n=97 patients from the validation dataset.
In order to understand the nature of this co-presence we reconstructed a chromosomal aberration network between gene-regulators of the cell cycle and antiviral sub-networks. Because only the validation dataset contained quantitative values of the level of chromosomal amplification for each gene in each patient, we used this dataset to analyze correlations between these values and to re-construct a network (Fig. 5b). As expected, genes located in a physical (i.e. genomic) proximity to one another showed high correlations at the level of genomic aberrations (green edges, Fig. 5b) despite their affiliations with different gene expression sub-networks (cell cycle and antiviral). Note that genes for several other processes are located in the same chromosomal gains as cell cycle and antiviral gene drivers. Many of those genes were also among differentially expressed but they were not classified as drivers in the analysis. For example, in the frequently gained region 3q13.2 to 3q27.3, there were 46 over-expressed genes but only 7 were identified as regulators. Thus it’s only a minority of the genes located within frequent chromosomal aberrations that are identified as regulators by our analysis, but all of those represented either cell cycle or antiviral genes.
We next asked if each tumor contained aberrations from only one chromosomal region. This was not the case. The majority of tumors contained gains in four or more chromosomal loci with a total of 58 unique combinations across 97 tumors (Figure 5c; Supplementary Fig. S4). Differently from the link between cell-cycle and antiviral drivers, we did not find any particularly preferred combination pair. For example, two most frequent aberrations in 3q and 1q appear in 76% and 64% of tumors, respectively. The proportion of tumors where these two aberrations co-occur is ~ 50% which is almost the same as would be expected by chance (48%). The analysis of more complex combinations (three to seven aberrations) was precluded by the available sample size.
Thus, altogether these results suggested that combinations of frequent chromosomal aberrations that contain cell cycle and antiviral drivers seem to be necessary for the development of invasive carcinoma indicating potential synergy between the two biological processes.
LAMP3 as a regulator of antiviral genes in cervical cancer
The increased activity of the cell cycle genes and decreased epithelial differentiation are well known attributes of different malignancies, including cervical cancer17. The finding of increased expression of genes related to the antiviral response was surprising, because persistent high-risk HPV infection is highly associated with malignancy 1. Among these genes there were innate immune sensors of viruses (ADAR, AIM2), molecules involved in antigen presentation (HLA, TAP, RFX5), transcription regulators (IRF1, IRF7, IRF9, and STAT1) and other genes of innate immunity (such as HERC5, MX1, OAS2, ISG15, and RSAD2 (viperin)) directly involved in the elimination of viruses 18–19.
In order to test whether the antiviral sub-network genes would be active in other infections we compared expression of this signature between cervical cancer and several viral and bacterial infections. The great majority of the genes of the antiviral subnetwork were concordantly regulated in cervical cancer and in influenza virus, rhinovirus and respiratory sincitial virus infections whereas there was no similarity to bacterial infections (Supplementary Fig. S5) indicating that cervical cancer has gene expression properties of activated antiviral response represented by genes of the corresponding subnetwork.
In order to test if genes we have predicted as antiviral from ex vivo tumor data are actually regulated in the context of a well-defined experimental HPV infection, we took advantage of the gene expression data obtained in the in vitro W12 system of cervical carcinogenesis8. In this model, the authors demonstrated that outgrowth of HPV-integrated cells was associated with the loss of episomal HPV and simultaneous activation of antiviral response. Therefore, we tested if the genes from the antiviral sub-network we found in patients were associated with the loss of episomal virus in this in vitro system. From the 54 genes of antiviral sub-network, 36 were detected in the vitro model, and of those, 18 were significantly regulated (FDR<10%), with 16 out of 18 genes having higher expression during episomal HPV elimination in vitro (Fig. 6a). Out of 18 genes that did not pass the threshold, 11 also had higher expression at the stage of episomal E2 drop. This result supports the idea that activation of antiviral genes in vivo might be involved in the elimination of episomal virus in tumors. Further, analyzing effect of interferon treatment we found that tumor-derived antiviral signature contained pathways dependent on both types of interferon (Supplementary Fig. S6).
Figure 6. Antiviral sub-network genes are regulated by LAMP3.
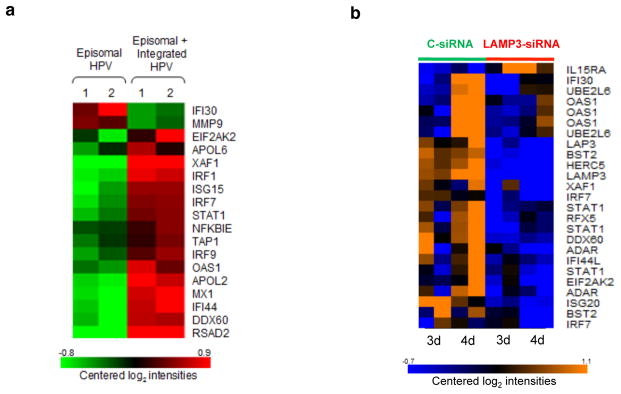
(a) Expression of genes from the antiviral sub-network in an in vitro culture W12 before and during elimination of episomal HPV in the presence of integrated HPV in the study of Pett et al. (data retrieved from GSE4289). (b) Knock down of LAMP3 by siRNA leads to down-regulation of many antiviral IFN-dependent genes. HeLa cells were pre-treated with control or LAMP3 siRNA overnight, then 1 ng/ml IFN-alpha was added for 3 or 4 days and gene expression was assessed. Log intensity values are represented by colors: orange - high expression, blue – low expression.
We demonstrated that LAMP3 was one of the major regulators of antiviral subnetwork with chromosomal amplifications of its genomic region occurring in more than 50% of patients. Although this gene is known to be regulated by interferon20 and might be involved in antigen presentation21, its precise role in immune response is uncertain. Therefore, we decided to further test the role of LAMP3 as a regulator of antiviral genes. We compared the expression signature of the genes from the antiviral subnetwork between LAMP3 siRNA and control siRNA treated HeLa cells after interferon stimulation. All 17 genes but one (IL15RA) that showed significant difference (FDR<10%) in gene expression between cells treated with control- and LAMP3-siRNA were regulated as predicted by our meta-analysis, i.e. had lower expression in cells where LAMP3 was knocked down (Fig. 6b). Several well-known IFN-dependent antiviral genes such as STAT1, IRF7, HERC5, ISG20, and OAS1 were affected by LAMP3 down-regulation. Thus, taking together the knock-down and chromosomal amplification results we conclude that LAMP3 plays a key regulatory role in the activation of interferon-dependent antiviral genes in cervical cancer.
DISCUSSION
As only a small percentage of women harboring the virus develop the cancer 22, HPV infection alone appears to be insufficient for the progression of cervical cancer 23. Thus, the interaction between the infected cells, immune system, and virus seems to be involved in the malignant progression of cervical cancer. Aiming to understand this complex process, we started by integrating global transcriptional profiling and genomic data and found that only small proportion (~9%) of differentially expressed genes can be directly regulated by frequent chromosomal aberrations. This result led us to hypothesize that the genes regulated by chromosomal aberrations might themselves regulate other genes. To substantiate this claim, we reconstructed a co-expression gene network of cervical cancer. There were multiple pieces of evidence indicating that this co-expression network reflects causative relationships between genes. First, we found that in vitro perturbation of major regulators of each individual subnetwork, located within chromosomal amplifications, resulted in a similar effect on target genes of the same sub-network, but did not affect unrelated genes (Fig. 3c). Second, using independent cohort of patients in whom we could obtain gene expression and genomic aberrations data we demonstrated that genes predicted to be targets of key drivers had higher expression in tumors with genomic gains than in tumors without gains for respective drivers (Fig. 4d). The last but not the least, we showed that in vitro knockdown of LAMP3 leads to down-regulation of many antiviral genes predicted to be stimulated by this molecule by our analysis. Altogether these findings strongly support our model that majority of transcriptional events in cervical cancer are directly and indirectly driven by chromosomal aberrations.
The identification of two sub-networks, cell cycle and epithelial cell differentiation, was not surprising, as both were extensively studied in cervical cancer17. From the cell cycle subnetwork, six genes (CEP70, GMPS, MCM2, NAT13, RFC4 and TOPBP1) located in regions of frequent chromosomal aberrations were identified as the main drivers in this sub-network. All these genes are located on chromosome 3q, where a frequent DNA gain occurs. This amplification has been previously described in cervical cancer 24. Although some of these genes have been also demonstrated as regulated by chromosomal gains, our study is the first to point to their key role in driving cell cycle transcriptional program in cervical cancer.
The increased expression of antiviral genes was surprising, because one could expect a reduction in the antiviral response to occur during persistent HPV infection as E6/E7 proteins were shown to inhibit interferon related responses 25. The natural history of infection, however, shows that in most cases antiviral response prevails over the inhibitory activity of HPV as ~90% of women eliminate the virus. Also, Pett and colleagues8 made an interesting observation in an in vitro system that activation of antiviral immune response genes was associated with the loss of episomal HPV16, over-expression of E6/E7 oncogenes and outgrowth of cervical keratinocytes containing integrated HPV. These results led them to propose a model of cervical carcinogenesis where elimination of episomal virus, by activated immune response, is a critical step in tumor progression because it removes the inhibitory component of the episomal virus (E2) and releases oncogenes expression2. Although the new model8 insightfully proposed the antiviral response as a critical step in tumor progression, the trigger of this gene activation, remained a mystery. Our results provide the first in vivo evidence that chromosomal amplifications in the antiviral genes selected during cancer development trigger and sustain the their activation in the tumor possibly providing additional help for the host immune system which was unable to eliminate the virus alone. It is remarkable that regions of frequent chromosomal gains simultaneously contained drivers not only of antiviral but also of cell cycle genes in the majority of patients (Fig. 5). This result indicates that both processes might be necessary for tumor survival and, as might be expected, it would be cost-effective to select one aberration that simultaneously affects both functions (i.e. killing two birds with one stone). Activation of the two processes may have synergistic effect on the tumor progression, because while antiviral response ultimately would lead to the block of cell cycle repressors (p53 and retinoblastoma) by permitting over-expression of E6/E7, the second group of drivers directly promotes cell cycle (Fig. 7). Although this is the most plausible model, other interpretations may exist such as that stimulation of immune response by “antiviral” drivers might be tumor-promoting independently on the episomal HPV decline.
Figure 7. A revised model of cervical carcinogenesis.

Persistent high risk HPV infection may result in the integration of virus into host genome leading to the increased genomic instability and aberrations, however, the expression of E6/E7 oncogenes is still under control of E2 if the episomal virus is around. If the chromosomal aberrations (gains) occur in the regions containing antiviral genes, they will induce the elimination of inhibitory episomal E2 8, release of E6/E7 that will block suppressors of cell cycle (p53, retinoblastoma). The same chromosomal gains contain drivers of cell cycle that directly induce cell proliferation. The two processes act synergistically allowing the dysplastic cell to become a malignant tumor.
One of the major drivers of the antiviral sub-network, LAMP3, was the only driver from this sub-network located in the same gain region (3q) as the cell cycle drivers. An over-expression of this gene has been associated with the enhanced metastatic potential and poor prognosis in cervical cancer 26. It was not clear, however, which cellular process is affected and leads to cancer progression as a result of LAMP3 elevation. Our results suggest that this gene is one of the major drivers of immune/antiviral genes activated in the process of cervical oncogenesis, a result that can hardly be obtained without the comprehensive reconstruction and analysis of a genomic/transcriptomic network. Although LAMP3 had not been shown before as a regulator of immunity, its increased expression was associated with better response to antiviral treatment in patients with Hepatitis C 27. Thus, our results in tumors (Fig. 4d) and knockdown of LAMP3 by siRNA in cell line (Fig. 6b) is the first direct demonstration that LAMP3 drives the IFN-dependent gene expression signature, which is highly enriched for antiviral genes such as STAT1, IRF7, HERC5, ISG20, OAS1 among others. Taken together with the findings that LAMP3 is involved in the cancer metastatic process 26, these results suggest that LAMP3 may have a dual role in cervical cancer promoting expression of antiviral genes and possibly contributing to elimination of episomic HPV and increasing the migratory capacity of tumor cells.
Other regulators of the antiviral sub-network were located outside of 3q in chromosomes 1 and 20 but it was recently shown that those regions also have characteristic aberrations in advanced intraepithelial lesions with a high short-term risk for progression 28. Furthermore, in vitro modulation of expression of RFX529 and AIM230, located on 1q, leads to the expected changes in expression of predicted targets (Supplementary Fig. S7). Interestingly, of the nine genes from the antiviral sub-network regulated by chromosomal aberrations and considered to be regulator genes, eight are located on these regions (3q, 1q, 1p, 20q).
Herein we show that antiviral genes we found overexpressed in cervical cancer are active in several viral but not bacterial infections (Supplementary Fig. S5) suggesting that host uses common mechanism for elimination of different viruses including HPV. These genes contain a mixed pattern of IFN type I and type II pathways, while the in vitro results of Pett et al. 8 indicated an involvement of only type I interferon. This partial discrepancy can probably be explained by the mixture of different cell types, including immune cells, that generates antiviral signature in vivo. This hypothesis is supported by the fact that expression of some genes from antiviral subnetwork is known to be limited to immune cells (e.g. CD163, FCGR1B, granzyme B; Supplementary Fig. S8). In addition, it agrees with the data showing that high expression of granzyme B is associated with poor outcome of cervical cancer patients 31.
In contrast to the cell cycle and antiviral sub-networks, we did not find any evidence that protein coding genes located within chromosomal aberrations can be regulators of the epithelial differentiation sub-network. Our bioinformatics analysis (Supplementary Methods; Supplementary Fig. S9), however, indicated that the genes from this sub-network might be down-regulated by mir-15b and mir-16-2 that are located within amplification in 3q region and had been already shown over-expressed in cervical cancer 32–34.
A recent paper using a similar strategy has discovered major drivers of a melanoma 11. Thus, the successful application of this approach in such different malignancies as cervical cancer and melanoma suggests its potential usefulness for the analysis of almost any tumor, since genomic aberrations are generic features of malignancies. Akavia and colleagues demonstrated that their algorithm discovers critical drivers of carcinogenesis in “any tumor cohort containing matched data for copy number aberrations and gene expression” 11. The critical advantage of our approach is that we have revealed key drivers of the process using non-matched genomic and transcriptomic datasets with the only connection between those being the fact that all data were collected from patients with cervical cancer.
In summary, by reconstructing and analyzing a gene regulatory network of cervical cancer, we infer a model of carcinogenesis in which genomic aberrations that promote cell cycle progression and elimination of episomal HPV are the major drivers of the process and orchestrate the growth advantage of the tumor cells carrying them (Fig. 7). In addition to the novel analytical approach and surprising biological insight, our study has potentially important implications for future clinical practice. For example, current World Health Organization guidelines on HPV vaccination do not contain any specific recommendations in regard to HPV status of the vaccine recipient 35. Also, anti-HPV therapy with immune-modulators inducing interferon pathways has been proposed to treat HPV-infected women36. Our results, however, suggest that antiviral therapy or anti-HPV therapeutic vaccination might be dangerous for women whose lesions already present signs of HPV integration. Furthermore, we compiled a comprehensive list of genomic and transcriptomic markers that if detected in pre-invasive cancer lesions might be indicative of poor prognosis.
METHODS
Meta-analysis of gene expression data
We searched PubMed at the NCBI database (http://www.ncbi.nlm.nih.gov/pubmed/) for studies of microarray in cervical cancer (published until 03/2009) and selected four studies 37–40 that: (i) had publicly available microarray data, (ii) used tumor and normal clinical samples, (iii) used oligonucleotide arrays and (iv) had sample size in each class >=5 (Supplementary Table S1). The data were analyzed using BRB Array-Tools v3.6.0 beta 3 developed by Drs. Richard Simon and Amy Peng Lam (http://linus.nci.nih.gov/BRB-ArrayTools.html) using the original normalization used in three studies 37–38,40 and median normalization over entire the array for 39. For all studies, we only considered genes found in at least 70% of arrays.
Besides publicly available data used in this study we analyzed gene expression in samples collected from two unrelated cohorts of patients, first one became a part of meta-analysis and the second was used for validation. The first dataset consisted of cervical cancer biopsies (n=40) and normal adjacent tissue samples (n=20) (Gene Expression Omnibus accession code GSE26342). The second dataset consisted of cervical cancer samples from 82 patients (GSE27469). See details of both cohorts in Supplementary Methods.
Differentially expressed genes (DEGs) between tumor and normal samples were identified for each of the five studies using random-variance t test and then combined in a meta-analysis using Fisher’s inverse chi-square method 41 as described in Supplementary Methods.
Meta-analysis of comparative genomic hybridization data
We selected ten publications which studied tumor clinical samples and provided either individual comparative genomic hybridization (CGH) data for their samples or graphical diagrams 42–51. A total of 269 cervical cancer cases were identified from the ten studies (Supplementary Table S2). From each study, we extracted the number of cases with chromosomal aberrations (gain and loss) in each chromosomal band to calculate the frequency of gain (FqG), frequency of loss (FqL) and the delta between them (FqG−FqL). We used the standard ISCN 1995 (an International System for human Cytogenetic Nomenclature 1995) resolution at the 400-band level 52. Chromosomal gains and losses in each study were defined according to the thresholds reported in the original papers.
Integration of gene expression and chromosomal aberrations
The chromosomal locations of the 1268 DEGs identified in the gene expression meta-analysis were determined using the NCBI database. When we did not find the location, we searched the Ensembl database. Based on the chromosomal location, the values of the frequencies (FqG, FqL and delta FqG-FqL) were identified for each gene. We used the delta values of all genes to construct a histogram that displayed their distribution (Supplementary Fig. S1b). Using this distribution, we determined the delta values which were considered to be the cut-offs that identify regions with frequent gain or loss. These were the values of local minima on both sides of the distribution. We applied the Fisher’s exact test to test if the altered expression of genes in these regions was significantly associated with chromosomal aberrations. A p value <0.05 was considered significant.
Reconstruction of the gene expression meta-network
Using gene expression data from tumors in five datasets, we reconstructed networks by performing meta-analysis of correlations for gene pairs using similar approach employed for gene expression. The networks were visualized in the Cytoscape Software 2.6.3 53. To identify sub-networks of correlated genes, we used MCODE v1.2 (Molecular Complex Detection) plug-in for Cytoscape 54. To establish a hierarchy within the sub-networks, we organized the genes as follows: first line, genes located in regions of frequent chromosomal aberrations and with 12 or more links (see Supplementary Methods for estimation of links number); second line, genes located in regions with less frequent chromosomal aberrations (delta values between −0.2 and −0.1 and between 0.1 and 0.32); third line, genes directly connected with genes from the first line; fourth line, genes connected with genes from the second line and last line, the rest of the genes. Genes in lines 3 and further did not have chromosomal aberrations (delta −0.1 to 0.1). See details in Supplementary Methods.
Perturbagens analysis
We used the Connectivity Map 02 (CMAP) 15 to perform the perturbagen analysis. The genes of each sub-network were divided into two groups: regulators and targets (Supplementary Fig. S3). The regulator genes were used to select the perturbagens and the target genes to test them. As controls, we used genes from another sub-network and we also analyzed perturbagens which did not act on the regulator genes. In total, we selected 20 perturbagens that acted on the regulator genes and 20 that did not do so. See details in Supplementary Methods.
Chromosomal aberrations network
We used data from our meta-analysis with the available individual patient results on genomic aberrations (n=117) and results from another independent dataset 16; ArrayExpress accession no. E-TABM-398 for reconstruction of a chromosomal aberrations network. We reconstructed the genomic aberrations network using the same approach employed for reconstruction of gene expression network (details in Supplementary Methods).
LAMP3 knockdown experiment
HeLa cells obtained directly from ATCC were transfected with LAMP3-specific siRNA or control siRNA (ON-TARGETplus SMARTpool), then stimulated with interferon and gene tested for genes expression as described in Supplementary Methods.
Supplementary Material
Acknowledgments
We thank Richard Simon, Ronald Germain, Yuri Kotliarov for helpful discussions and suggestions; Constantin Vasilyev for help with matching ambiguous gene locations in databases; Tim Myers and the staff of NIAID microarray facility for excellent technical support.
This research was partially supported by FAPESP (Fundação de Amparo a Pesquisa do Estado de São Paulo), Brazil; AFIP (Associação Fundo de Incentivo à Psicofarmacologia), São Paulo, Brazil; and Intramural Research Program of the NIAID, NIH; startup funds for AM from Oregon State University, OR, US.
Footnotes
AUTHOR CONTRIBUTIONS
KLM participated in study design, conducted experiments and analyses, drafted the manuscript; NS designed the study, analyzed and interpreted the data, co-proposed biological model, wrote the manuscript; AY, designed and developed statistical analyses including inference of causality model and drafted related part of the manuscript; SV and JS developed some statistical analyses and drafted related part of the manuscript; MR, TD, MB conducted siRNA experiments, GFOS conducted microarray experiments; ML and HL conducted the microarray experiments and analyses for validation dataset; NV conducted bioinformatics analysis; SMFB, CRNC, and JCLR collected tumor samples and performed clinical evaluation; PM drafted the manuscript and critical revision of the study; IDCGS supervised sample collection, participated in study design; MGL participated in study design, supervised the study; AM designed the study, analyzed and interpreted the data, co-proposed biological model, contributed to inference of causality model, wrote the manuscript; supervised and provided scientific leadership of the project. MGL and AM are co-senior authors.
COMPETING FINANCIAL INTERESTS
The authors declared no competing financial interests.
References
- 1.zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2:342–350. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 2.Woodman CB, Collins SI, Young LS. The natural history of cervical HPV infection: unresolved issues. Nat Rev Cancer. 2007;7:11–22. doi: 10.1038/nrc2050. [DOI] [PubMed] [Google Scholar]
- 3.Patel S, Chiplunkar S. Host immune responses to cervical cancer. Curr Opin Obstet Gynecol. 2009;21:54–59. doi: 10.1097/GCO.0b013e32831a9890. [DOI] [PubMed] [Google Scholar]
- 4.Duensing S, et al. Centrosome abnormalities and genomic instability by episomal expression of human papillomavirus type 16 in raft cultures of human keratinocytes. J Virol. 2001;75:7712–7716. doi: 10.1128/JVI.75.16.7712-7716.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duensing S, Munger K. Mechanisms of genomic instability in human cancer: insights from studies with human papillomavirus oncoproteins. Int J Cancer. 2004;109:157–162. doi: 10.1002/ijc.11691. [DOI] [PubMed] [Google Scholar]
- 6.Tonon SA, et al. Physical status of the E2 human papilloma virus 16 viral gene in cervical preneoplastic and neoplastic lesions. J Clin Virol. 2001;21:129–134. doi: 10.1016/s1386-6532(01)00155-x. [DOI] [PubMed] [Google Scholar]
- 7.Cricca M, et al. Molecular analysis of HPV 16 E6I/E6II spliced mRNAs and correlation with the viral physical state and the grade of the cervical lesion. J Med Virol. 2009;81:1276–1282. doi: 10.1002/jmv.21496. [DOI] [PubMed] [Google Scholar]
- 8.Pett MR, et al. Selection of cervical keratinocytes containing integrated HPV16 associates with episome loss and an endogenous antiviral response. Proc Natl Acad Sci U S A. 2006;103:3822–3827. doi: 10.1073/pnas.0600078103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2010;10:550–560. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- 10.Shulzhenko N, et al. Crosstalk between B lymphocytes, microbiota and the intestinal epithelium governs immunity versus metabolism in the gut. Nat Med. 2011;17:1585–1593. doi: 10.1038/nm.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akavia UD, et al. An integrated approach to uncover drivers of cancer. Cell. 2010;143:1005–1017. doi: 10.1016/j.cell.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reichenbach H. The Direction of Time. University of California Press; 1956. [Google Scholar]
- 13.Judea P. Causality: Models, Reasoning, and Inference. Cambridge University Press; 2000. [Google Scholar]
- 14.Luscombe NM, et al. Genomic analysis of regulatory network dynamics reveals large topological changes. Nature. 2004;431:308–312. doi: 10.1038/nature02782. [DOI] [PubMed] [Google Scholar]
- 15.Lamb J, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 16.Lando M, et al. Gene dosage, expression, and ontology analysis identifies driver genes in the carcinogenesis and chemoradioresistance of cervical cancer. PLoS Genet. 2009;5:e1000719. doi: 10.1371/journal.pgen.1000719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCance DJ. Human papillomaviruses and cell signaling. Sci STKE. 2005;2005:pe29. doi: 10.1126/stke.2882005pe29. [DOI] [PubMed] [Google Scholar]
- 18.Tang Y, et al. Herc5 attenuates influenza A virus by catalyzing ISGylation of viral NS1 protein. J Immunol. 2010;184:5777–5790. doi: 10.4049/jimmunol.0903588. [DOI] [PubMed] [Google Scholar]
- 19.Jiang D, et al. Identification of five interferon-induced cellular proteins that inhibit west nile virus and dengue virus infections. J Virol. 2010;84:8332–8341. doi: 10.1128/JVI.02199-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mohty M, et al. IFN-alpha skews monocyte differentiation into Toll-like receptor 7-expressing dendritic cells with potent functional activities. J Immunol. 2003;171:3385–3393. doi: 10.4049/jimmunol.171.7.3385. [DOI] [PubMed] [Google Scholar]
- 21.de Saint-Vis B, et al. A novel lysosome-associated membrane glycoprotein, DC-LAMP, induced upon DC maturation, is transiently expressed in MHC class II compartment. Immunity. 1998;9:325–336. doi: 10.1016/s1074-7613(00)80615-9. [DOI] [PubMed] [Google Scholar]
- 22.Tindle RW. Immune evasion in human papillomavirus-associated cervical cancer. Nat Rev Cancer. 2002;2:59–65. doi: 10.1038/nrc700. [DOI] [PubMed] [Google Scholar]
- 23.Castellsague X. Natural history and epidemiology of HPV infection and cervical cancer. Gynecol Oncol. 2008;110:S4–7. doi: 10.1016/j.ygyno.2008.07.045. [DOI] [PubMed] [Google Scholar]
- 24.Lockwood WW, Coe BP, Williams AC, MacAulay C, Lam WL. Whole genome tiling path array CGH analysis of segmental copy number alterations in cervical cancer cell lines. Int J Cancer. 2007;120:436–443. doi: 10.1002/ijc.22335. [DOI] [PubMed] [Google Scholar]
- 25.Nees M, et al. Papillomavirus type 16 oncogenes downregulate expression of interferon-responsive genes and upregulate proliferation-associated and NF-kappaB-responsive genes in cervical keratinocytes. J Virol. 2001;75:4283–4296. doi: 10.1128/JVI.75.9.4283-4296.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanao H, et al. Overexpression of LAMP3/TSC403/DC-LAMP promotes metastasis in uterine cervical cancer. Cancer Res. 2005;65:8640–8645. doi: 10.1158/0008-5472.CAN-04-4112. [DOI] [PubMed] [Google Scholar]
- 27.Taylor MW, et al. Changes in gene expression during pegylated interferon and ribavirin therapy of chronic hepatitis C virus distinguish responders from nonresponders to antiviral therapy. J Virol. 2007;81:3391–3401. doi: 10.1128/JVI.02640-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilting SM, et al. Chromosomal signatures of a subset of high-grade premalignant cervical lesions closely resemble invasive carcinomas. Cancer Res. 2009;69:647–655. doi: 10.1158/0008-5472.CAN-08-2478. [DOI] [PubMed] [Google Scholar]
- 29.Amit I, et al. Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses. Science. 2009;326:257–263. doi: 10.1126/science.1179050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee J, Li L, Gretz N, Gebert J, Dihlmann S. Absent in Melanoma 2 (AIM2) is an important mediator of interferon-dependent and -independent HLA-DRA and HLA-DRB gene expression in colorectal cancers. Oncogene. 2012;31:1242–1253. doi: 10.1038/onc.2011.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guzman VB, et al. High levels of granzyme B expression in invasive cervical carcinoma correlates to poor response to treatment. Cancer Invest. 2008;26:499–503. doi: 10.1080/07357900701805678. [DOI] [PubMed] [Google Scholar]
- 32.Lui WO, Pourmand N, Patterson BK, Fire A. Patterns of known and novel small RNAs in human cervical cancer. Cancer Res. 2007;67:6031–6043. doi: 10.1158/0008-5472.CAN-06-0561. [DOI] [PubMed] [Google Scholar]
- 33.Wang XH, et al. Aberrant Expression of Oncogenic and Tumor-Suppressive MicroRNAs in Cervical Cancer Is Required for Cancer Cell Growth. Plos One. 2008;3 doi: 10.1371/journal.pone.0002557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee JW, et al. Altered MicroRNA expression in cervical carcinomas. Clin Cancer Res. 2008;14:2535–2542. doi: 10.1158/1078-0432.CCR-07-1231. [DOI] [PubMed] [Google Scholar]
- 35.WHO. Human papillomavirus vaccines-WHO position paper. WHO-Weekly epidemiological record. 2009;84:117–132. [Google Scholar]
- 36.Koromilas AE, Li S, Matlashewski G. Control of interferon signaling in human papillomavirus infection. Cytokine Growth Factor Rev. 2001;12:157–170. doi: 10.1016/s1359-6101(00)00023-x. [DOI] [PubMed] [Google Scholar]
- 37.Zhai Y, et al. Gene expression analysis of preinvasive and invasive cervical squamous cell carcinomas identifies HOXC10 as a key mediator of invasion. Cancer Res. 2007;67:10163–10172. doi: 10.1158/0008-5472.CAN-07-2056. [DOI] [PubMed] [Google Scholar]
- 38.Biewenga P, et al. Gene expression in early stage cervical cancer. Gynecol Oncol. 2008;108:520–526. doi: 10.1016/j.ygyno.2007.11.024. [DOI] [PubMed] [Google Scholar]
- 39.Scotto L, et al. Identification of copy number gain and overexpressed genes on chromosome arm 20q by an integrative genomic approach in cervical cancer: potential role in progression. Genes Chromosomes Cancer. 2008;47:755–765. doi: 10.1002/gcc.20577. [DOI] [PubMed] [Google Scholar]
- 40.Pyeon D, et al. Fundamental differences in cell cycle deregulation in human papillomavirus-positive and human papillomavirus-negative head/neck and cervical cancers. Cancer Res. 2007;67:4605–4619. doi: 10.1158/0008-5472.CAN-06-3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hedges LV, Olkin I. Statistical Methods for Meta-Analysis. Academic Press, Inc; Orlando: 1985. [Google Scholar]
- 42.Heselmeyer K, et al. Gain of chromosome 3q defines the transition from severe dysplasia to invasive carcinoma of the uterine cervix. Proc Natl Acad Sci U S A. 1996;93:479–484. doi: 10.1073/pnas.93.1.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heselmeyer K, et al. Advanced-stage cervical carcinomas are defined by a recurrent pattern of chromosomal aberrations revealing high genetic instability and a consistent gain of chromosome arm 3q. Genes Chromosomes Cancer. 1997;19:233–240. [PubMed] [Google Scholar]
- 44.Dellas A, et al. Prognostic value of genomic alterations in invasive cervical squamous cell carcinoma of clinical stage IB detected by comparative genomic hybridization. Cancer Res. 1999;59:3475–3479. [PubMed] [Google Scholar]
- 45.Hidalgo A, et al. Human papilloma virus status and chromosomal imbalances in primary cervical carcinomas and tumour cell lines. Eur J Cancer. 2000;36:542–548. doi: 10.1016/s0959-8049(99)00323-8. [DOI] [PubMed] [Google Scholar]
- 46.Jee KJ, Kim YT, Kim KR, Aalto Y, Knuutila S. Amplification at 9p in cervical carcinoma by comparative genomic hybridization. Anal Cell Pathol. 2001;22:159–163. doi: 10.1155/2001/174645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rao PH, et al. Chromosomal amplifications, 3q gain and deletions of 2q33-q37 are the frequent genetic changes in cervical carcinoma. BMC Cancer. 2004;4:5. doi: 10.1186/1471-2407-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lyng H, et al. Intratumor chromosomal heterogeneity in advanced carcinomas of the uterine cervix. Int J Cancer. 2004;111:358–366. doi: 10.1002/ijc.20258. [DOI] [PubMed] [Google Scholar]
- 49.Huang FY, et al. Genetic abnormalities and HPV status in cervical and vulvar squamous cell carcinomas. Cancer Genet Cytogenet. 2005;157:42–48. doi: 10.1016/j.cancergencyto.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 50.Huang KF, et al. Chromosomal gain of 3q and loss of 11q often associated with nodal metastasis in early stage cervical squamous cell carcinoma. J Formos Med Assoc. 2007;106:894–902. doi: 10.1016/S0929-6646(08)60059-5. [DOI] [PubMed] [Google Scholar]
- 51.Wilting SM, et al. Increased gene copy numbers at chromosome 20q are frequent in both squamous cell carcinomas and adenocarcinomas of the cervix. J Pathol. 2006;209:220–230. doi: 10.1002/path.1966. [DOI] [PubMed] [Google Scholar]
- 52.Schreck RR, Disteche CM, Adler D. ISCN standard idiograms. Curr Protoc Hum Genet. 2001 doi: 10.1002/0471142905.hga04bs18. Appendix 4, Appendix 4B. [DOI] [PubMed] [Google Scholar]
- 53.Cline MS, et al. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc. 2007;2:2366–2382. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003;4:2. doi: 10.1186/1471-2105-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.