

Abstract
Tissue development and disease progression are multi-stage processes controlled by an evolving set of key regulatory factors, and identifying these factors necessitates a dynamic analysis spanning relevant time scales. Current omics approaches depend on incomplete biological databases to identify critical cellular processes. Herein, we present TRACER (TRanscriptional Activity CEll aRrays), which was employed to quantify the dynamic activity of numerous transcription factor (TFs) simultaneously in 3D and networks for TRACER (NTRACER), a computational algorithm that allows for cellular rewiring to establish dynamic regulatory networks based on activity of TF reporter constructs. We identified major hubs at various stages of culture associated with normal and abnormal tissue growth (i.e., ELK-1 and E2F1, respectively) and the mechanism of action for a targeted therapeutic, lapatinib, through GATA-1, which were confirmed in human ErbB2 positive breast cancer patients and human ErbB2 positive breast cancer cell lines that were either sensitive or resistant to lapatinib.
Introduction
Many biological processes are dynamic in nature, such as tissue regeneration or tumor progression. The active cellular processes that control those evolving phenotypes change with time. Time-series microarrays, RNA-seq and phosphoproteomics are common techniques to follow cellular dynamics, but they rely on incomplete biological databases to determine the most relevant active processes1,2. Meanwhile, transcription factors (TFs) are powerful regulators of cellular responses, as well as the liaison between signaling and gene expression. TF activity, which results from the integration of intracellular and extracellular signals within the signal transduction network, can lead to subsequent changes in gene expression. Here we present transcriptional activity cell arrays (TRACER), a technology that enables the monitoring of the dynamic activity of a multitude of TFs in 3D over tissue formation time scales in real time.
The complexity of intracellular signal transduction networks provides the robustness and versatility necessary for the regulation of normal cellular processes, yet this complexity also makes challenging the identification of critical signals driving dysregulated growth in cancer progression. In cancer research, biomarkers are being actively pursued to predict patient prognosis or to serve as novel therapeutic targets3, of which ErbB2 (HER2/Neu) is a leading example4. ErbB2 is overexpressed in 25–30% of breast cancers and is associated with aggressive cancer biology5. Several drugs have been developed to target ErbB2, such as trastuzumab (Herceptin; Genentech) or lapatinib (Tykerb; GlaxosmithKline), but not all ErbB2 overexpressing patients respond6, and resistance can develop in those that do7. The impact of constitutive ErbB2 signaling on dynamic activity within the intracellular signal transduction network on the time scales of tissue formation has not been well characterized. Moreover, an improved mechanistic understanding of currently available anti-ErbB2 agents could be employed to discriminate the agent or combination of agents that would be most efficacious based on the tumor biology, thereby further refining personalized therapeutic strategies.
Herein, TRACER8,9 is applied for the large-scale real-time quantification of dynamic TF activity associated with constitutive ErbB2 signaling during the development of mammary epithelial cells into pre-invasive structures. TF activity is obtained through parallel delivery of TF reporter (TFr) constructs in an array, with bioluminescence imaging employed for dynamic quantification. The array is also implemented to investigate the mechanism of action for a targeted ErbB2 therapeutic, lapatinib. We generated dynamic networks based on activity of the TFrs resulting from constitutive ErbB2 activity, which correlate with observed phenotypes, and provide insight into the mechanism of action of lapatinib, using TRACER data in combination with prior biological knowledge, inference methods and optimization techniques. The ability to define TF dynamics at a large-scale presents a strategy towards identifying key cellular processes associated with normal and dysregulated cell growth, as well as the mechanisms underlying the effects of therapeutic agents.
Results
Constitutive ErbB2 activity leads to dysregulated growth
The cell line MCF10A/ErbB210, referred to as 10A/ErbB2, exhibits ErbB2 dimerization and constitutive activity in the presence of a dimerizing agent (DA). 10A/ErbB2 cells, expressing inducible ErbB2 homodimers, formed large highly disorganized structures within basement membrane extract (BME) over a period of 10 days in the presence of DA. Untreated control and cells treated with EGF resulted in the formation of spherical structures, with EGF stimulation producing larger spheres. The phenotypic differences in structures formed by DA treated and untreated cells became more pronounced as time progressed. β-catenin, a marker for epithelial cell-cell junctions, was strongly localized to the basal lamina for all structures with some lateral localization within cells on the structure perimeters, indicating that the cells have not lost their adhesiveness (Fig. 1A–B).
Figure 1. Culture of 10A/ErbB2 cells in BME with different stimuli.

10A/ErbB2 cells grown in BME for a period of 10 d following no stimulation or stimulation with EGF or DA form multicellular structures with different morphologies over time. Cells were imaged repeatedly with phase contrast microscopy (A) and confocal microscopy at day 10 (B). (Scale bars represent 50 μm; red – nuclei, green – β-catenin) (C) Transduced cell arrays measured activities of TFrs in 10A/ErbB2 cells growing in BME following stimulation with no stimulant (blue), EGF (green), or DA (red). Normalized transcription factor reporter intensities for TFrs above the background are represented (TCF/LEF, SMAD and NC are excluded). Shaded areas around the average lines represent ± standard error. Significance TFrs under DA stimulation are highlighted with a red rectangle; under EGF stimulation under a green rectangle; purple rectangles indicate that the TF reporter is significant under both stimulations (p-value ≤0.15).
We subsequently investigated TFr activity for 10A/ErbB2 cells growing in BME using an improved viral technology based on previously established TRACER11 that enable longer experimental times. Cells were parallel transduced with 23 TF firefly luciferase (FLuc)-based lentiviral reporter constructs (Supplementary Table 1) and a control construct with a TATA-box for basal FLuc expression (TA). 10A/ErbB2 cells expressing reporter constructs were seeded in BME in parallel, with bioluminescence imaging used to quantify TFr activities at multiple time points. Light measured in wells containing transduced cells was consistently above background levels observed in control wells with blank BME, persisted throughout the experimental time course, and normalized TFr activity was highly consistent between samples and experiments (Supplementary Fig. 1). Clear differences were observed in TFr activities between the treatments. Constitutive ErbB2 dimerization induced by DA increased the activity of more constructs relative to EGF treatment (Fig. 1C, Supplementary Table 2). Interestingly, constitutive ErbB2 signaling revealed activation of multiple TFrs at the later time points, such as E2F1, SP1, SRE, STAT and YY1 reporters. In contrast, EGF stimulation induced a more transient response that largely resolved after 3 days in culture.
TFr activity was analyzed for consistency with available transcriptomic data from MCF10A cells grown in BME that were untreated or treated with EGF at 1.5, 3, 5, 7, 9 days12. We computationally determined the most likely active TFs that regulated the differentially expressed genes (fold change FC ≥3 and p-value ≤ 0.001) by predicting TF binding sites in the promoter regions of those genes using available position weighted matrices (PWM) from TRANSFAC13. TFr activities measured by TRACER and those predicted by transcriptomics show that TRACER had a medium high sensitivity, 0.75, indicating that TRACER correctly identified activated TFs predicted from the gene expression profiles in the microarray data. The specificity of TRACER was similar, 0.72(Supplementary Fig. 2). Specifically, predictions of TF activity from the microarray data for MCF10A cells treated with EGF confirmed the activity of ELK1, IRF1, NFAT, NFKB, STAT1 and STAT4 when 10A/ErbB2 cells are treated with EGF. While TRACER was highly consistent with microarray data, some results differed, which is not unexpected for large-scale data. The microarray data did not predict activation of CRE, HIF or SP1 in EGF stimulated MCF10A cells (Supplementary Fig. 2). The activity of those non-predicted reporters may reflect binding of alternative TFs with currently unavailable PWMs or just being the result of the false negative predictions of the computational method employed.
A dynamic TFr regulatory network was subsequently developed in order to identify potential causal relationships between the TFr that were activated or deactivated upon treatment of 10A/ErbB2 cells with EGF or DA. Network topology was determined using NTRACER, by applying multiple inference methods (partial least square regression (PLSR), mutual information and Bayesian networks) to the TFr activity data obtained from the 3D TRACER, combined with literature curated protein-protein, protein-DNA and indirect interactions between the different stimuli and the TFs obtained from TRANSFAC14, IPA (Ingenuity® Systems, www.ingenuity.com) and GeneGO (MetaCore from Thomson Reuters). The resulting network contained 675 TFs and more than 1000 connections. An initial network topology was obtained by combining the inferred network with the simplified prior knowledge network (PKN), whose nodes represent treatments given to the cells (i.e., DA or EGF) and the significant TFrs. The inferred network and PKN overlapped for 13% of the total number of the interactions, indicating that TRACER identified novel interactions that have not been captured by other experimental techniques. Of note, the interactions determined by the ensemble of inference methods (53% of the total number of connections) may not be direct, but indirect TF-TF interactions, such as phosphorylation or dephosphorylation of proteins responsible to the activation or deactivation of TFs, which are not included in the PKN. Finally, the network topology allowed for inhibitory mechanisms (InhM) that are not easily described by TF-TF interactions alone, such as receptor endocytosis or degradation, apoptosis or standard dephosphorylation of the activation site for the TF that led to a decline in TF activity.
The active edges between nodes at various time points were identified using a modification of CellNOptR15, which minimizes the difference between the experimental data and the output of the logic model using a genetic algorithm. Data were modeled as a three-level Boolean paradigm, where TFrs and treatments are the nodes and the edges or gates indicate the relationship between them, (i.e., activating or inhibiting). Consensus networks yielded distinct dynamic patterns of activity within the TFr network upon treatment of 10A/ErbB2 cells with either DA or EGF (Fig. 2) and revealed two types of key reporters. First, reporters that are directly affected by the external stimuli, in the case of DA and EGF included the nodes AP1 (Fig. 2A) and ELK-1 (Fig. 2G) for instance. These nodes represent the initial connections between the external stimulus and the intracellular activity. The second type of reporter included those that serve as hubs, which have large numbers of connections to other nodes in the network, and thus disseminate the signal throughout the network. These hubs may differ between time points, and thus they are dynamic. For stimulation with DA, AP1 is a hub between 5 to 7 days of culture indicated by 7 out-going connections (Fig. 2D). Between days 7 and 10, AP1 has 2 out-going connections, similar to SRF (Fig 2E). Taken together, the hubs identified during ErbB2 dimerization and after EGF treatment were distinct, and the timing at which these TFrs served as hubs similarly varied.
Figure 2. Dynamic transcription factor activity networks upon DA or EGF treatment of 10A.ErbB2 cells.

Dynamic transcriptional consensus networks upon DA activation between 0 to day 1 in culture (A), day 1 to day 3 (B), day 3 to day 5 (C), day 5 to day 7(D) and day 7 to day 10 (E). Panels F to J represent the corresponding dynamic transcriptional networks upon EGF activation. Treatments, TFrs and inhibitory mechanism are represented as nodes, while the connections between them are represented by directed edges. Active treatments are symbolized by green circles; inhibitory mechanism, by a red circle. TFrs that are activated with respect to the beginning of the culture based on their median experimental discretize value are purple; deactivated TFrs are orange and no change in activity is denoted by yellow. TFrs that are not modulated by the treatment at any of the explored experimental time points are represented in white. Active or present edges at a given culture time interval are colored in black. Activating edges end in an arrow, deactivating edges end in a T. Edges obtained from prior knowledge are indicated by continuous lines; identified by inferences methods are denoted by discontinuous lines. Those identified by both methods are marked by two parallel lines.
Construction of the dynamic networks made it easy to visualize that ErbB2 dimerization yielded maximal levels of TFr activities towards the end of the culture, while EGF primarily produced transient TFr activity between days 1 and 5. Cells activated through ErbB2 dimerization responded with a slow activation of TFr up to 5 days in culture. Between days 5 to 7, numerous TFrs became activated through E2F1, YY1 and STAT3 reporters, with the AP1 reporter being the hub with the most outgoing connections between those time points. A majority of the TFr constructs had increased activities toward the end of culture, while the P53 reporter was down-regulated through PTTG and AP1 reporters. In contrast, EGF stimulation induced ELK1 reporter activity by day 2, which served to control the activation of numerous other TFrs. ELK1 reporter also functioned as a hub, along with NFkB and SP1. Activated TFrs had their activities decline to basal levels (i.e., untreated 10A/ErbB2 cells) by day 5, with some TFrs having activity dropped below control by day 10. Taken together, EGF stimulation was translated as a transient activation of ELK1 reporter, whereas ErbB2 dimerization and constitutive activity were translated through the network to increase activity of E2F1, YY1 and STAT3 reporters at later times of culture.
Next, we examined patient data to investigate the translational relevance of the dynamic TF network to two breast tumor subtypes: ErbB2 positive (ErbB2+) and triple negative (TN). Analysis of transcriptional data from The Cancer Genome Atlas (TCGA) identified 143 genes that were differentially expressed between ErbB2+ and TN tumors (FC ≥1.2 and p-value≤0.01). Analysis of the promoter regions of differentially expressed genes revealed that ErbB2+ tumors had significantly activated E2F4, STAT1 and STAT5A (Fig. 3) relative to TN tumors. Our dynamic TF network identified that the E2F1 reporter had increased activity at later times in culture, and E2F4 would be expected to bind the E2F1 reporter (Supplementary File 1). Furthermore, the activation of STAT1 and STAT5A in the ErbB2+ tumors is consistent with the increased activity through the STAT1 reporter (Supplementary File 1), which had increased activity in our dynamic TF network. The differential gene expression did not support increased activity for the other TFs (AP1, SRF and YY1) in the ErbB2+ tumors.
Figure 3. Transcription factor targets overexpression in breast cancer human tumors.

Chi-square in log10 scale for AP1, SRF, STATs, E2Fs and YYs families during ErbB2 activation according to the dynamic network (Fig. 2) in ErbB2 positive human breast cancer tumors in comparison with triple negative human breast cancer tumors. Horizontal lines indicate level of significance (p-value≤0.01). Blue bars represent the overexpression of a given TF when considering consensus mammal promoters; red bars represent the overexpression of a given TF when considering just human promoters.
Mode of lapatinib drug action within induced 10A/ErbB2 cells
We subsequently investigated the mechanism of action for lapatinib. 10A/ErbB2 cells growing in BME were stimulated with DA and subsequently treated with lapatinib or no therapeutic. At 3 d of culture, lapatinib treatment reduced viability by approximately 80% compared with untreated cells, consistent with a similar study16. A 3D TRACER was subsequently applied to lapatinib treatment of DA-treated 10A/ErbB2 cells growing in BME. Lapatinib treatment resulted in activity of numerous TFrs at day 1 and day 2 of culture, with significant reductions in activity for all reporters (except P53) at day 3, which coincided with the significant decrease in cell viability (Fig. 4A). The TFrs with greatest activity change at day 1 were E2F1, ELK1, GATA, P53 and STAT4, which may suggest that the TFs that bind to those reporters are the downstream targets of lapatinib (Supplementary Table 3).
Figure 4. Treatment of cells with ErbB2-targeting therapeutic and dynamic transcription factor activity networks upon lapatinib treatment of DA activated 10A.ErbB2 cells.

A)Transduced cell arrays measured activities of TFrs in DA activated 10A/ErbB2 cells growing in BME following no treatment (blue), or lapatinib treatment (red). Normalized transcription factor reporter intensities for TFrs above the background are represented (TCF/LEF, SMAD and NC are excluded). Shaded areas around the average lines represent ± standard error. Significance TFrs under lapatinib treatment are highlighted with a red rectangle (fdr corrected p-value ≤ 0.05). B) Dynamic transcriptional consensus networks upon lapatinib treatment between 0 to day 1 in culture (B), day 1 to day 2 (C), and day 2 to day 3 (D). See Figure 2 legend for more details.
Lapatinib most directly increased activity of ELK1, GATA, P53, and RAR reporters (Fig. 4B–D), with SP1 and STAT4 reporters being the initial dynamic hubs that modulated the activity of many of the remaining TFrs. After 2 days of culture, NTRACER infers that E2F1 reporter activity was inhibited by factors associated with GATA, P53 and SP1 reporters. At 3 days in culture, most TFrs had activity that decreased relative to day 2 and were similar or had decreased activity relative to pre-lapatinib treatment. The P53 reporter was the sole exception, which had activity increased relative to day 0 that served to decrease activity of E2F1 and CMYC reporters. This decrease in TFr activity relative to day 2 may result from apoptosis of cells as the cell viability decreased (Supplementary Fig. 3). We subsequently investigated the hypothesis that GATA was a key factor modulating the biological effects of lapatinib. GATA was selected as its reporter construct had increased activity at the initial time point, yet the modeling predicted a down-regulation of E2F1 reporter by day 2. E2F1 has multiple cellular functions such as cell proliferation and p53 dependent and independent apoptosis17. Multiple TFs can bind to GATA reporter (Supplementary File 1), and GATA1 was investigated as literature reports connect GATA1 to E2F, specifically E2F418, and its role in regulating growth in other cell types19. To test our hypothesis, Western blotting for cells with and without lapatinib treatment revealed increased levels of phosphorylated GATA1 after 1 d of lapatinib treatment, consistent with its increased activity (Fig. 5A–B). Subsequently, we confirmed that the GATA reporter could identify changes in GATA1 activity. Overexpression of GATA1, confirmed with Western blots (Supplementary Fig. 4), led to increased activity through the GATA reporter (Fig. 5C), and produced smaller and less disorganized 3D structures after ErbB2 dimerization with DA (Fig. 5D–E). Cell viability was decreased with overexpression of GATA1, with measurement of viability similar to levels measured for 10A/ErbB2 cells treated with lapatinib (Fig. 5F). However, lapatinib treatment of GATA1 overexpressing cells led to a further decrease in viability. These phenotypic results and protein analyses are consistent with lapatinib treatment acting, in part, through activation of GATA1.10A/ErbB2 only included the intracellular domain of ErbB2 and, hence, the therapeutic antibodies trastuzumab and pertuzumab did not stimulate the key TFrs found using lapatinib (Supplementary Table 3 and Supplementary Fig. 5). Notably, these studies were performed for 3D culture, which has been proposed as essential for investigating mechanisms of drug action 20, as drug mechanisms can differ between 2D and 3D culture.
Figure 5. Overexpression of GATA1 decreased structure disorganization and cell viability.

10A/ErbB2 cells were stimulated with DA and treated with lapatinib for 1 d and lysates were probed on Western blots. A) Sample bands detected for phosphorylated GATA1 (p-GATA1), total GATA1 (GATA1), and β-actin. B) Quantification of relative p-GATA1 and GATA1 expression normalized to β-actin, represented by mean ± s.d. from five distinct samples (*** p<0.001). C) 10A/ErbB2 and 10A/ErbB2/GATA1 cells were transduced with GATA reporter gene and imaged with bioluminescence imaging to confirm increased activity. D) Sample images of 10A/ErbB2 and 10A/ErbB2/GATA1 cells growing in BME without and with DA stimulation for 10 d (scale bars represent 200 μm), with a quantification of cell coverage area (E) (solid bars – 10A/ErbB2; spotted bars – 10A/ErbB2/GATA1). F) Relative viability of cells stimulated with DA and subsequently treated with lapatinib for 3 d, with significant differences indicated by letters (a, b, c, d; α=0.05). For C, E, and F, bars represent mean ±s.d. from at least three replicates and three distinct experiments (***p<0.001).
Finally, the translational relevance of these dynamic TF networks was investigated using human ErbB2+ cell lines (i.e., BT474 and SKBR3). Analysis of transcriptomic data for lapatinib sensitive ErbB2+ cell lines21, 22 identified a total of 592 genes that were differentially expressed upon lapatinib treatment relative to control. For this differential gene expression, a significant increase was not observed for the ELK-1, P53 or RAR targets. However, a significant enrichment of GATA1 targets was observed (Fig. 6A). Conversely, in a BT474 lapatinib insensitive cell line, 680 differentially expressed genes were identified and the GATA1 target enrichment was not observed (Fig. 6B).
Figure 6. Transcription factor targets overexpression after lapatinib treatment in two breast cancer cell lines.
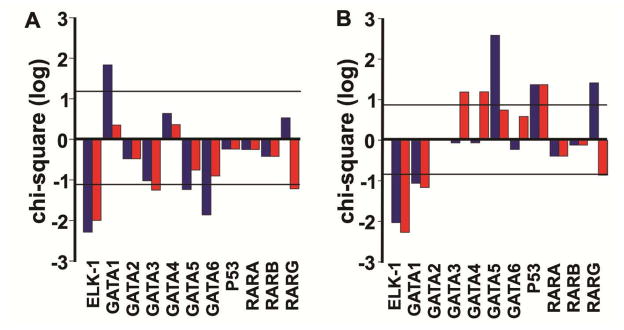
Chi-square in log10 scale for ELK-1, GATA, P53 and RAR families during lapatinib treatment according to the dynamic network (Fig. 4) in (A) BT474 and SKBR3 cell lines in comparison with no lapatinib treatment and in (B) a resistant BT474 cell line in comparison with no lapatinib treatment. Horizontal lines indicate level of significance (p-value≤0.01). Blue bars represent the overexpression of a given TF when considering consensus mammal promoters; red bars represent the overexpression of a given TF when considering just human promoters.
Discussion
We have applied 3D TRACER arrays to analyze dynamic TF activity in a model of cancer progression and in response to a targeted therapeutic. These arrays monitor intracellular signaling on a large scale with parallel delivery of TFrs8, 9. Herein, TRACER employed 23 TFrs, which has the potential to be expanded (approximately 1400 TFs in humans23). More traditional approaches for dynamic analysis of cell signaling include microscopic techniques, which can analyze few pathways24, time series transcriptomics25 and proteomics2. The latter omics methods have greater coverage than TRACER as they quantify the abundance of >10,000 cellular components (i.e., mRNA, proteins), with pathway activity inferred indirectly from expression level26. Phosphoproteomics can be a more direct measure of pathway activity, but there are constraints on the extent to which post-translational modifications can be characterized27. The activity measurements from TRACER offer the potential to identify key TFrs associated with cellular processes. The promoter region of each TF reporter construct may bind multiple TFs, and identifying the specific TFs that are active at the promoter region can be accomplished through validation with omics data (e.g., microarrays) or biological techniques such as overexpression, knockdown, or Western blotting. Interestingly, and unlike other methods, quantification is compatible with 3D culture of cells in hydrogels and over time scales that support formation of multicellular structures over multiple weeks that resemble the range of pathologies of native tissues28. Relative to growth on 2D polystyrene, cells growing in 3D matrices can exhibit differential responsiveness to chemotherapeutics that more faithfully represent patient responses29. Importantly, TFr activity is quantified through non-invasive bioluminescence imaging, and the cells were repeatedly imaged, thus the cost of the array does not scale with the number of measured time points, as cells are not lysed.
TRACER provides data with two unique aspects: firstly, the experimental determination of TF activity, and secondly, TF activity is monitored dynamically over several days. These unique aspects motivated the development of the computational approaches (Supplementary Fig. 6). Analysing dynamic TFr activity data involved generating dynamic networks from a limited amount of temporal data and employing a combination of prior knowledge and an ensemble of inference methods. Approaches have been developed to handle inference in dynamic data1; however, few focus on a limited number of available time points in the range of days and weeks30 and most assume a static network31. The connections in TF regulatory networks are not necessarily present in all cells or at all times32,33; thus, we proposed to model our system as dynamic networks in order to capture cellular re-wiring.
Currently, no single inference method can determine all network motifs present in a biological network. Combining multiple inference methods takes advantage of the method preference for certain types of motifs34. Additionally, due to the nature of the experimental data, TF activity, the TF regulatory networks have encompassed multiple processes (e.g., transcription, translation, and possibly phosphorylation) that are represented by the edges between nodes. Each process can be non-linear and thus their combination is likely to be non-linear as well. Therefore inference methods that can handle non-linear interactions are included for identifying edges in non-linear processes, such as mutual information or Bayesian network inference methods, as well as linear inference methods such as PLSR.
Moreover, prior knowledge is commonly used in reverse engineering of networks1, 35, although biological databases, from which prior knowledge is acquired, are incomplete36 and their information is not cell specific37. However, connections between nodes of the transcription factor (TF) regulatory network are indirect interactions of the two TFs (i.e., TF A binds to the promoter region of the gene that encodes TF B, or TF A binds to the promoter region of a gene whose production modulates the activity of TF B). The experimental system measures the activity of the two TFs, yet cannot distinguish between the different indirect pathways of interaction. The available prior knowledge is in the form of direct protein-DNA interactions, and does not account for all possible indirect connections. Thus, inferred connections only overlap with prior knowledge connections in the case that one TF directly regulates the transcription of a second TF, which based on our results, occurs at relatively low levels (13%). Therefore, combining prior knowledge with inference methods incorporates connections that are established in the literature, yet allows for new cell-specific interactions that are determined through inference.
NTRACER equally weighted both sources of information to accommodate novel interactions or indirect TF-TF interactions, which provided an initial network topology that contained multiple false positive edges originating from the union of diverse inference methods as well as the previously established connections obtained from the literature, as the connections were established from a variety of cell types. We employed a structure optimization methodology to identify the most likely connections present at each time point while penalizing network complexity, based on a modification of CellNOptR15 to accommodate TF-TF interactions and dynamic data, to remove false positive edges coming from prior knowledge as well as inference methods.
Dynamic networks identified TFrs associated with phenotypes that developed over 10 days resulting from ErbB stimulation. ErbB family signaling pathways have been extensively studied due to their implication in cancer38, with most signaling studies performed on short time scales (i.e., hours). We established that the E2F family was active in ErbB2+ tumors, specifically E2F4. Previous work in human breast cancer tumors has shown E2F4 nuclear expression to be associated with markers of poor prognosis39. Furthermore, increased E2F4 expression correlated to both decreased distant metastasis and overall survival from breast cancer39. Additionally, our findings that STAT1 and STAT5A were highly activated in ErbB2+ tumors relative to TN tumors confirmed prior observations40, 41.
The combination of TFr activity data and computational analysis identified novel mechanisms for lapatinib action. Lapatinib was found to exert its effects through activation of GATA1 in the 10A/ErbB2 cells, and also two more clinically relevant breast cancer cell lines (BT474 and SKBR3), which were derived from ErbB2+ breast cancer patients. Collectively, these findings support the capacity of our systems biology network assessment to identify hubs of TF activity that may translate into drug targets and also inform mechanisms of drug action and resistance. Notably, these studies were performed for 3D culture, which has been proposed as essential for investigating mechanisms of drug action20.
Materials and methods
Cell line and maintenance
10A/ErbBB2 cells10, generously provided by Dr. S. K. Muthuswamy, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, were cultured using DFCI-1 media described previously42
3D cell culture
10A/ErbB2 cells were cultured in BME (Trevigen, Gaithersburg, MD) using the cell overlay technique43 on 16-well chamber slides (Nalgene Nunc International, Rochester, NY). Cold BME solution was diluted to 12 mg/ml and 40 μl was added to wells and incubated at 37°C for 45 min to solidify. 10A/ErbB2 cells in DFCI media containing 5 ng/ml EGF and 2% (v/v) BME were subsequently seeded on top of BME underlay (2250 cells/well). After 3 d of culture, media was removed and replaced with DFCI containing 2% BME and i) no EGF and EtOH vehicle, ii) 5 ng/ml EGF and EtOH vehicle, or iii) no EGF and 500 nM dimerizing agent (DA) (AP1510, ARIAD pharmaceuticals, Cambridge, MA; or B/B homodimerizer, Clontech). Media was changed every 3 d thereafter. A Leica microscope was used to capture phase images of structures every 3 d. ImageJ was used to quantify the areas occupied by cells in images taken with a 5x objective (at least three fields of view sampled from triplicate conditions, each experiment was performed with at least two replicates).
Immunostaining and confocal microscopy
Following 10 days of culture, cells were fixed and immunostained as described by Muthuswamy et al.10 with some modifications. Cells were fixed for 30 min with 4% paraformaldehyde, washed with phosphate buffered saline (PBS) containing 100 mM glycine, permeabilized with 0.5% Triton-X in PBS for 5 min, washed with immunofluorescence buffer (IF)10, blocked with 2% bovine serum albumin for 1 h, stained with mouse anti-β-catenin (1:100 dilution; Millipore, Billerica, MA) overnight, washed with IF, stained with anti-mouse-AlexaFluor 488 (1:500 dilution, Invitrogen, Carlsbad, CO) for 1 h, washed with IF, counterstained with TOPRO-3 (5 μM, Invitrogen) for 10 min, and washed with PBS. A Leica confocal microscope fitted with a 40x immersion lens was used to image structures. A student’s t-test with false discovery rate adjustment was used to assess cell growth data.
Treatments and assessment of viability
10A/ErbB2 cells (2250 cells seeded/well) were cultured in 96-well plates (Becton Dickinson and Company, Franklin Lakes, NJ) using the cell overlay culturing technique. After 3 days of culture, 5 ng/ml EGF in media was replaced with 500 nM DA, then after another 3 d the following treatments were added in combination with DA: i) no additional treatment, or ii) 1.5 μM lapatinib (Santa Cruz Biotechnology, Santa Cruz, CA). We also used two other ErbB2 therapeutics as negative controls, i) 20 μg/ml trastuzumab (generously provided by Genentech/Roche, South San Francisco, CA), and ii) 25 μg/ml pertuzumab (Genentech/Roche), as 10A/ErbB2 cells do not contain the ErbB2 extracellular domain that is binding by these antibodies, trastuzumab and pertuzumab. After 3 d of treatment, alamarBlue reagent (Invitrogen) was used to assess viability using the manufacturer recommendation with a BioTek Synergy 4 plate reader. A student’s t-test with false discovery rate adjustment was used to assess viability.
Transfer vector constructs
An HIV-based transfer vector encoding CMV-GFP44 was modified to encode TA-FLuc (plenti-TA-FLuc) by exchanging the CMV-GFP cassette with TA-FLuc from the Panomics translucent control vector (Panomics, Madison, WI) using NheI and XbaI restriction enzymes. This construct was further modified to create a library of lentivirus-producing transfer vector constructs with TF-responsive binding elements. Sequences derived from Panomics constructs were digested out of the constructs also using NheI and XbaI and ligated into the plenti-TA-FLuc backbone. For non-Panomics constructs, such as TCF/LEF45, CMYC14, NOTCH146, and PTTG47, custom oligonucleotides were synthesized (Sigma Aldrich), annealed, and inserted into the plenti-TA-FLuc backbone using NheI and BglII. A vector encoding Gaussia luciferase (GLuc), TA-GLuc, was also constructed by transferring the GLuc gene from pCMV-GLuc (New England Biolabs, Ipswich, MA) into the Panomics vector using HindIII and XbaI, and subsequently following the same procedure as the other Panomics-derived constructs.
Lentivirus production
Lentivirus for each TFr was produced by co-transfecting HEK-293T cells with one of the transfer vector constructs and three packaging plasmids (pMDL-GagPol, pRSV-Rev, and pIVS-VSV-G)48 using techniques described previously49. Number of physical particles (PP) for each virus batch was determined using an HIV-1 p24 Antigen ELISA kit (ZeptoMetrix, Buffalo, NY). GATA1 and GFP expression lentiviral packaging plasmid pRRL-GATA1-GFP (graciously provided by JD Crispino, Northwestern University) was used to produce lentiviral particles in an identical fashion as TFr transfer vector constructs.
Formation of 3D transduced cell arrays
GLuc expression was used to normalize for cell number. 10A/ErbB2 cells were transduced with lentivirus encoding TA-GLuc (25,000 PP/cell) by centrifugation (800 g, 32°C, 45 min) and cultured continually to create a stable TA-GLuc encoding cell line, which was used in all array experiments. To form an array, cells were transduced with lentivirus encoding TA-FLuc or one of the TFr genes (10,000 PP/cell) by centrifugation, resuspended in media containing 5 ng/ml EGF and 2% BME, and seeded into wells of a black 384-well plate (Greiner BioSciences, Monroe, NC) previously containing BME (1000 cells seeded/well). Stimulations and treatments were added at times and concentrations described above.
Measuring reporter gene activities
Bioluminescence imaging was utilized to assess FLuc activity. D-luciferin (1 mM, Caliper, Hopkinton, MA) was added to wells and plates were incubated at 37°C for 30 min, followed by imaging with an IVIS 200 system (Perkin Elmer, Waltham, MA). For assessing GLuc activity, a GLuc activity kit (New England Biolabs) was used. Media (10 μl/well) was sampled and placed in a black 384-well plate. Substrate solution (20 μl/well) was added and luminescence was measured with a 1 s integration time using a Synergy 4 plate reader (BioTek, Winooski, VT). After each time point, media in wells was partially replaced with the addition of appropriate stimulants and treatments.
Normalization and statistical significance
Data were analyzed using R50. The initial methodology to normalize and determine statistical significance11 was slightly modified. Data from the array was log2 transformed and filtered to eliminate all intensities below background (p<0.05). Background was defined as the measured intensity in non-infected cells subject to the same treatment at the same time and plate. Each TFrFluc intensity data point was subsequently normalized by Gluc at the initial experimental time, t=0 days, and multiplied by the ratio of initial measured activity to the average of all initial measured activities for a given TFr, such that the initial normalized value was the same across all conditions for the same TFr. Finally, data for each TFr were normalized by the control reporter, TA, and outliers were removed (p<0.01). Quality assessment also included at least a Pearson correlation of 0.75 intra- and inter-arrays. The R package limma51 was employed to determine differentially activated TFr versus initial time and experimental control (no treatment). False discovery rate (fdr) was used to correct for multiple comparisons.
Western blotting
Cells were washed with PBS and lysed with ice cold IP Lysis/Wash Buffer (Pierce) containing protease inhibitor cocktail (Sigma) and Halt phosphatase inhibitor (Pierce), followed by centrifugation and storage at −80°C. A BCA Assay (Pierce) was performed to quantify protein concentrations. Proteins were reduced with 20 mM DTT, resolved on a NuPage 4–12% Tris-Bis gel (Invitrogen), transferred to a PVDF membrane, and blocked overnight in TBS-T containing 5% BSA. Membranes were subsequently blotted on a SnapID system (Millipore) using antibodies against p-GATA1 (Assay biotech), GATA1, or β-actin (Santa Cruz Biotechnology) (all diluted 3:1000), and species appropriate HRP-conjugated secondary antibodies (3:1000). HRP was detected with ECL-Plus (GE Healthcare) using a Typhoon imaging system. Blotting was quantified with densitometry using ImageJ. Sample bands were normalized to the corresponding β-actin band and then normalized to the average appropriate control samples. A student’s t-test with fdr adjustment was used to assess Western blot data.
Determination of TFr specificity
P-match13 from Explain 3.0 was selected to identify TFs that might bind to the DNA sequence of a given reporter. High specific PWMs from vertebrates that minimized the sum of false positives and negatives were selected to explore the most likely TFs that bind to a given DNA sequence. PWMs that predict binding to a given sequence with a core score ≥0.9 and a matrix score ≥0.9 were deemed as significant (Supplementary File 1).
Living cell array consensus with publically available microarray data in MCF10A cell line
GSE18938 microarrays12 were downloaded from the Array Express library52. Briefly, MCF10A were treated with EGF or left untreated over a period of 9 days. Time-series HuGene 1.0st v1 arrays were background corrected using robust multi-array average (rma)53, quantile normalized54 and probe set summarized with the oligo package55. Inadequate experiments were previously removed (see supplemental methods). Differentially expressed probes versus the untreated MCF10A cells were identified using limma package and were deemed significant at a fold change, FC ≥3 and p-value ≤0.00151. P-match13 was exploited to identify the most likely TFs that might regulate the expression of such genes, using no significant genes (FC ≤ 1.001 and p-value ≥ 0.5) as a background set. Search was performed between −1000 to 500 base pairs with respect to the transcription starting site (TSS). The most significant PWMs associated with the most likely TFs were contrasted against the PWMs associated with each TFr (Supplementary File 1) to establish whether the activity of any TFr could be altered by any of the active TF. Sensitivity and specificity of TRACER were calculated assuming the computational results from the transcriptomic measurements as reference. Results are presented in Supplementary Figure 2 as well as the receiving operating characteristic (ROC) and precision-recall curves.
Generation of GATA1 overexpressing cells
10A/ErbB2 cells were seeded in a 6-well plate at 1.5e5 cell/cm2 and subsequently GATA1/GFP-expressing lentivirus and Polybrene (4 μg/ml; Sigma) were added. Following 3 d in culture, transduction efficiency was assessed by imaging GFP expression.
Dynamic network generation
Initial network topology was originated from an equally weighted ensemble of prior knowledge sources and inference methods. Prior knowledge information includes directed human protein-DNA interactions, either proximal or distal regulation with respect to their TSS and external stimuli and TFs, obtained from TRANSFAC14, IPA (Ingenuity® Systems, www.ingenuity.com) and GeneGO (MetaCore, Thomson Reuters). An ensemble of inference methods was employed to determine novel connections that have not been explored before experimentally: PLSR56, mutual information (MI)57–60 and Bayesian networks (BN)61. MI methods were only considered to determine the interactions between the external stimuli and TFrs at the initial time point only. Inference networks from PLSR, MI and BN were combined with equal weights and finally merged with the prior knowledge network. Measurements for the cell array were taken on the time scale of days while signaling networks have activity that occurs on much shorter time scales62. We thus assume that experimental data were under a pseudo-steady state. Data were discretized in three levels, 1, 0 and −1, and modeled in a Boolean paradigm. Present edges at each pseudo-steady state were identified using structure optimization, by minimizing the difference between the experimental data and the fit of the model. Complex structures were penalized to avoid over fitting. A total of 500 runs were performed and reported dynamic consensus networks were obtained by generating an ensemble of the top 1% networks (see Supplementary Methods for more details). The raw data and source code are available at http://www.bme.umich.edu/labs/shea/publications.php.
Testing for edge significance in the final dynamic consensus networks
The significance of edges present in the consensus networks was identified using a total of 1000 bootstrapping samples, which were randomly generated to determine the probability of an edge to be present. Finally, the probability that an edge was present was compared with the probabilities generated by a random model using 1000 samples. An edge was deemed significant if it was at least three times more probable to be observed compared with the random model for a given time and treatment (Supplementary Figs. 7 and 8). Edges that were determined to not be statistically significant were removed from the consensus graphs. P-values for each edge were calculated based on the area under the permutation curve for the same probability as the given edge according to the bootstrapping runs and are summarized in Supplementary File 2 as well as the probability of each edge (see Supplementary Methods for more details).
Dynamic network target validation in human breast cancer tumors and lapatinib treated BT474 and SKBR3 cell line
22 ErbB2 positive and 58 triple negative Agilent microarrays were downloaded from TCGA (http://cancergenome.nih.gov/). Possible TFs that could regulate those genes and are direct targets of ErbB2 overexpression based on the dynamic network (AP-1, STAT, SRF, E2F and YY families) were explored. Two additional experiments of lapatinib treated BT474, BT474-J4 and SKBR3 cell lines were employed in the validation studies (E-GEOD-16179 and E-MEXP-440). The entire set of raw microarrays are not available for E-MEXP-440, so the significant genes obtained by O’Neil et al.63 were used in that case (see the reference for details on the analysis). Possible TFs that could regulate those significant genes and are direct targets of lapatinib overexpression based on the dynamic network (ELK-1, RAR, GATA and P53 families) were explored.
TF gene targets were identified in two manners. First, from experimentally validated targets obtained from GeneGO (MetaCore, Thomson Reuters), a list of more than 7000 interaction was compiled for the above TF families. Secondly, computationally predicted targets were extracted by exploring the promoter regions of the entire human genome, NCBI36/hg18, (from the Regulatory Sequence Analysis Tools, http://rsat.ulb.ac.be/) and the consensus mammalian promoter regions64 between −2000 to 2000 from TSS. Mammalian consensus and human promoter regions were investigated using MATCH65 and FIMO66 at 0.999 matrix scores and 10−6 uncorrected p-value (Supplementary Files 3 and 4).
The most likely active TFs were calculated using a hypergeometric test for both, experimentally and computationally obtained targets, and a z-score test for the computationally acquired targets26. Results from the three different methods were consolidated using a meta-analysis approach for the same type of experiment (i.e, E-MEXP-440 results and BT474 from E-GEOD-16179 were combined using the meta-analysis method). Median chi-square values were reported due to the skew of the bootstrapping results.
Conclusions
We have applied 3D TRACERs to monitor long-term dynamics of intracellular signaling that can be connected to cellular phenotype and response to therapeutics. NTRACER enabled determination of key dynamic hubs, and the temporal relationship between them, that contribute to cellular phenotype. These findings were validated in human breast cancer cell lines and tumor tissue. This identification of key signaling hubs may facilitate the development of treatment strategies or drug combinations that will further improve outcomes for patients with aggressive breast cancer subtypes, including patients with ErbB2 overexpression.
Supplementary Material
Insight box.
We present a new combination of experimental and computational technologies to quantify the dynamic activity of numerous TFs through differentiation in 3D culture, as TF activity is the integration of intracellular and extracellular signals that powerfully regulate cell fate. TRACER allows quantification of key signalling pathway activity over time scales of days to weeks that corresponds to complex cell fate decisions, while the computational approach is aimed at identifying the critical pathways that modulate cell fate. The potential of this experimental/computational combination was demonstrated through identifying TF hubs associated with normal and abnormal 3D tissue formation that correlated with clinical breast cancer samples, or critical TFs stimulated following drug treatment that identified novel mechanisms of action.
Acknowledgments
Confocal microscopy was performed at the Northwestern University Biological Imaging Facility, and bioluminescence imaging was performed at the Northwestern University Center for Advanced Molecular Imaging. Funding: Support for this work was provided by the National Institutes of Health (NIH; P50GM081892, R01GM097220) and the Chicago Biomedical Consortium with support from the Searle Funds at The Chicago Community Trust. MSW and BPB were both supported by an NIH training grant (T32GM008449).
Footnotes
The authors declare that they have no conflict of interest.
Electronic Supplementary Information (ESI) available: The raw data and source code as well as the supplementary information and methods are available at http://www.bme.umich.edu/labs/shea/publications.php.
Author Contributions: MSW, JSJ and LDS conceived the project; MSW designed the experiments, construct the viral reporters and performed GATA1 validation studies; MWS, SJD, MDM and ADB obtained transcriptional activity cell array data; SS developed the GATA1 overexpressing cells; SA designed and produced new TFrs; BPB and DB developed a pipeline to analyze TRACER data; BPB developed NTRACER and performed TF enrichment studies; MSW, BPB, JSJ, LDS wrote the manuscript; JSR helped with CellNOptR modifications; JSR and LJB critically revised the paper and; DAT, LJB, JSJ and LDS supervised and guided the research.
Notes and references
- 1.Bar-Joseph Z, Gitter A, Simon I. Nature reviews Genetics. 2012;13:552–564. doi: 10.1038/nrg3244. [DOI] [PubMed] [Google Scholar]
- 2.Ciaccio MF, Wagner JP, Chuu CP, Lauffenburger DA, Jones RB. Nat Methods. 2010;7:148–155. doi: 10.1038/nmeth.1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bernards R. Cell. 2010;141:13–17. doi: 10.1016/j.cell.2010.03.018. [DOI] [PubMed] [Google Scholar]
- 4.Baselga J, Norton L. Cancer Cell. 2002 doi: 10.1016/s1535-6108(02)00066-1. [DOI] [PubMed] [Google Scholar]
- 5.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 6.Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, Slamon DJ, Murphy M, Novotny WF, Burchmore M, Shak S, Stewart SJ, Press M. J Clin Oncol. 2002;20:719–726. doi: 10.1200/JCO.2002.20.3.719. [DOI] [PubMed] [Google Scholar]
- 7.Nahta R, Esteva FJ. Breast Cancer Res. 2006;8:215. doi: 10.1186/bcr1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bellis AD, Bernabe BP, Weiss MS, Shin S, Weng S, Broadbelt LJ, Shea LD. Biotechnol Bioeng. 2013;110:563–572. doi: 10.1002/bit.24718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weiss MS, Penalver Bernabe B, Bellis AD, Broadbelt LJ, Jeruss JS, Shea LD. Plos One. 2010;5:e14026. doi: 10.1371/journal.pone.0014026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muthuswamy S, Li D, Lelievre S, Bissell M, Brugge JS. Nature Cell Biology. 2001;3:785–792. doi: 10.1038/ncb0901-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weiss MS, Bernabé B Peñalver, Bellis AD, Broadbelt LJ, Jeruss JS, Shea LD. Plos One. 2010;5:e14026. doi: 10.1371/journal.pone.0014026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pradeep CR, Kostler WJ, Lauriola M, Granit RZ, Zhang F, Jacob-Hirsch J, Rechavi G, Nair HB, Hennessy BT, Gonzalez-Angulo AM, Tekmal RR, Ben-Porath I, Mills GB, Domany E, Yarden Y. Oncogene. 2012;31:907–917. doi: 10.1038/onc.2011.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chekmenev DS, Haid C, Kel AE. Nucleic Acids Res. 2005;33:W432–437. doi: 10.1093/nar/gki441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matys V. Nucleic Acids Research. 2006;34:D108–D110. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Terfve C, Cokelaer T, Henriques D, MacNamara A, Goncalves E, Morris MK, van Iersel M, Lauffenburger DA, Saez-Rodriguez J. BMC systems biology. 2012;6:133. doi: 10.1186/1752-0509-6-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosh R, Narasanna A, Wang SE, Liu S, Chakrabarty A, Balko JM, González-Angulo AM, Mills GB, Penuel E, Winslow J, Sperinde J, Dua R, Pidaparthi S, Mukherjee A, Leitzel K, Kostler WJ, Lipton A, Bates M, Arteaga CL. Cancer Res. 2011;71:1871–1882. doi: 10.1158/0008-5472.CAN-10-1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Polager S, Ginsberg D. Trends Cell Biol. 2008;18:528–535. doi: 10.1016/j.tcb.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 18.Yu M, Riva L, Xie HF, Schindler Y, Moran TB, Cheng Y, Yu DN, Hardison R, Weiss MJ, Orkin SH, Bernstein BE, Fraenkel E, Cantor AB. Mol Cell. 2009;36:682–695. doi: 10.1016/j.molcel.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ezoe S, Matsumura I, Gale K, Satoh Y, Ishikawa J, Mizuki M, Takahashi S, Minegishi N, Nakajima K, Yamamoto M, Enver T, Kanakura Y. J Biol Chem. 2005;280:13163–13170. doi: 10.1074/jbc.M413461200. [DOI] [PubMed] [Google Scholar]
- 20.Weigelt B, Lo AT, Park CC, Gray JW, Bissell MJ. Breast Cancer Research and Treatment. 2010;122:35–43. doi: 10.1007/s10549-009-0502-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu L, Greger J, Shi H, Liu Y, Greshock J, Annan R, Halsey W, Sathe GM, Martin AM, Gilmer TM. Cancer Res. 2009;69:6871–6878. doi: 10.1158/0008-5472.CAN-08-4490. [DOI] [PubMed] [Google Scholar]
- 22.Hegde PS, Rusnak D, Bertiaux M, Alligood K, Strum J, Gagnon R, Gilmer TM. Mol Cancer Ther. 2007;6:1629–1640. doi: 10.1158/1535-7163.MCT-05-0399. [DOI] [PubMed] [Google Scholar]
- 23.Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM. Nature Reviews Genetics. 2009;10:252–263. doi: 10.1038/nrg2538. [DOI] [PubMed] [Google Scholar]
- 24.Li GW, Xie XS. Nature. 2011;475:308–315. doi: 10.1038/nature10315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Nature biotechnology. 2010;28:511–U174. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sui SJH, Mortimer JR, Arenillas DJ, Brumm J, Walsh CJ, Kennedy BP, Wasserman WW. Nucleic Acids Research. 2005;33:3154–3164. doi: 10.1093/nar/gki624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Terfve C, Saez-Rodriguez J. Advances in experimental medicine and biology. 2012;736:19–57. doi: 10.1007/978-1-4419-7210-1_2. [DOI] [PubMed] [Google Scholar]
- 28.Debnath J, Brugge JS. Nat Rev Cancer. 2005;5:675–688. doi: 10.1038/nrc1695. [DOI] [PubMed] [Google Scholar]
- 29.Weaver VM, Lelièvre S, Lakins JN, Chrenek MA, Jones JCR, Giancotti F, Werb Z, Bissell MJ. Cancer Cell. 2002;2:205–216. doi: 10.1016/s1535-6108(02)00125-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Honkela A, Girardot C, Gustafson EH, Liu YH, Furlong EE, Lawrence ND, Rattray M. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:7793–7798. doi: 10.1073/pnas.0914285107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Przytycka TM, Singh M, Slonim DK. Brief Bioinform. 2010;11:15–29. doi: 10.1093/bib/bbp057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neph S, Stergachis AB, Reynolds A, Sandstrom R, Borenstein E, Stamatoyannopoulos JA. Cell. 2012;150:1274–1286. doi: 10.1016/j.cell.2012.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luscombe NM, Babu MM, Yu HY, Snyder M, Teichmann SA, Gerstein M. Nature. 2004;431:308–312. doi: 10.1038/nature02782. [DOI] [PubMed] [Google Scholar]
- 34.Marbach D, Costello JC, Kuffner R, Vega NM, Prill RJ, Camacho DM, Allison KR, Kellis M, Collins JJ, Stolovitzky G, Consortium D. Nature Methods. 2012;9:796-+. doi: 10.1038/nmeth.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hecker M, Lambeck S, Toepfer S, van Someren E, Guthke R. Biosystems. 2009;96:86–103. doi: 10.1016/j.biosystems.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 36.Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, Chen Y, DeSalvo G, Epstein C, Fisher-Aylor KI, Euskirchen G, Gerstein M, Gertz J, Hartemink AJ, Hoffman MM, Iyer VR, Jung YL, Karmakar S, Kellis M, Kharchenko PV, Li Q, Liu T, Liu XS, Ma L, Milosavljevic A, Myers RM, Park PJ, Pazin MJ, Perry MD, Raha D, Reddy TE, Rozowsky J, Shoresh N, Sidow A, Slattery M, Stamatoyannopoulos JA, Tolstorukov MY, White KP, Xi S, Farnham PJ, Lieb JD, Wold BJ, Snyder M. Genome Res. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arvey A, Agius P, Noble WS, Leslie C. Genome Res. 2012;22:1723–1734. doi: 10.1101/gr.127712.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Citri A, Yarden Y. Nat Rev Mol Cell Biol. 2006;7:505–516. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- 39.Rakha EA, Pinder SE, Paish EC, Robertson JF, Ellis IO. Journal of Pathology. 2004;203:754–761. doi: 10.1002/path.1573. [DOI] [PubMed] [Google Scholar]
- 40.Han W, Carpenter RL, Cao X, Lo HW. Mol Carcinog. 2013;52:959–969. doi: 10.1002/mc.21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wagner KU, Rui H. J Mammary Gland Biol Neoplasia. 2008;13:93–103. doi: 10.1007/s10911-008-9062-z. [DOI] [PubMed] [Google Scholar]
- 42.Band V, Sager R. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:1249–1253. doi: 10.1073/pnas.86.4.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Debnath J, Muthuswamy S, Brugge J. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 44.Miyoshi H, Blömer U, Takahashi M, Gage FH, Verma IM. Journal of virology. 1998;72:8150–8157. doi: 10.1128/jvi.72.10.8150-8157.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Veeman MT, Slusarski DC, Kaykas A, Louie SH, Moon RT. Current Biology. 2003;13:680–685. doi: 10.1016/s0960-9822(03)00240-9. [DOI] [PubMed] [Google Scholar]
- 46.Samon JB, Champhekar A, Minter LM, Telfer JC, Miele L, Fauq A, Das P, Golde TE, Osborne BA. Blood. 2008;112:1813–1821. doi: 10.1182/blood-2008-03-144980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pei L. Journal of Biological Chemistry. 2001;276:8484–8491. doi: 10.1074/jbc.M009654200. [DOI] [PubMed] [Google Scholar]
- 48.Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. Journal of virology. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shin S, Shea LD. Mol Ther. 2010;18:700–706. doi: 10.1038/mt.2009.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.R. R Foundation for Statistical Computing; Vienna, Austria: 2012. URL http://www.R-project.org/ [Google Scholar]
- 51.Smyth GK. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 52.Brazma A, Parkinson H, Sarkans U, Shojatalab M, Vilo J, Abeygunawardena N, Holloway E, Kapushesky M, Kemmeren P, Lara GG, Oezcimen A, Rocca-Serra P, Sansone SA. Nucleic Acids Res. 2003;31:68–71. doi: 10.1093/nar/gkg091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 54.Bolstad BM, Irizarry RA, Astrand M, Speed TP. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 55.Carvalho BS, Irizarry RA. Bioinformatics. 2010;26:2363–2367. doi: 10.1093/bioinformatics/btq431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mevik BWR, Liland KH. R package version 2.3-0. 2011 http://CRAN.R-project.org/package=pls.
- 57.Faith JJ, Hayete B, Thaden JT, Mogno I, Wierzbowski J, Cottarel G, Kasif S, Collins JJ, Gardner TS. PLoS Biol. 2007;5:e8. doi: 10.1371/journal.pbio.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, Dalla Favera R, Califano A. BMC Bioinformatics. 2006;7(Suppl 1):S7. doi: 10.1186/1471-2105-7-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meyer PE, Kontos K, Lafitte F, Bontempi G. EURASIP J Bioinform Syst Biol. 2007 doi: 10.1155/2007/79879,79879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Meyer PE, Lafitte F, Bontempi G. Bmc Bioinformatics. 2008:9. doi: 10.1186/1471-2105-9-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu J, Smith VA, Wang PP, Hartemink AJ, Jarvis ED. Bioinformatics. 2004;20:3594–3603. doi: 10.1093/bioinformatics/bth448. [DOI] [PubMed] [Google Scholar]
- 62.He F, Balling R, Zeng AP. Journal of Biotechnology. 2009;144:190–203. doi: 10.1016/j.jbiotec.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 63.O’Neill F, Madden SF, Aherne ST, Clynes M, Crown J, Doolan P, O’Connor R. Molecular Cancer. 2012:11. doi: 10.1186/1476-4598-11-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xie XH, Lu J, Kulbokas EJ, Golub TR, Mootha V, Lindblad-Toh K, Lander ES, Kellis M. Nature. 2005;434:338–345. doi: 10.1038/nature03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kel AE, Gossling E, Reuter I, Cheremushkin E, Kel-Margoulis OV, Wingender E. Nucleic Acids Research. 2003;31:3576–3579. doi: 10.1093/nar/gkg585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Grant CE, Bailey TL, Noble WS. Bioinformatics. 2011;27:1017–1018. doi: 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.