

Abstract
Mutations in the catalytic Roc-COR and kinase domains of leucine-rich repeat kinase 2 (LRRK2) are a common cause of familial Parkinson’s disease (PD). LRRK2 mutations cause PD with age-related penetrance and clinical features identical to late-onset sporadic PD. Biochemical studies support an increase in LRRK2 kinase activity and a decrease in GTPase activity for kinase domain and Roc-COR mutations, respectively. Strong evidence exists that LRRK2 toxicity is kinase-dependent leading to extensive efforts to identify selective and brain-permeable LRRK2 kinase inhibitors for clinical development. Cell and animal models of PD indicate that LRRK2 mutations affect vesicular trafficking, autophagy, protein synthesis and cytoskeletal function. Although some of these biological functions are affected consistently by most disease-linked mutations, others are not and it is currently unclear how mutations that produce variable effects on LRRK2 biochemistry and function all commonly result in the degeneration and death of dopamine neurons. LRRK2 is typically present in Lewy bodies and its toxicity in mammalian models appears to be dependent on the presence of α-synuclein, which is elevated in human iPS-derived dopamine neurons from patients harboring LRRK2 mutations. Here, we summarize biochemical and functional studies of LRRK2 and its mutations and focus on aberrant vesicular trafficking and protein synthesis as two leading mechanisms underlying LRRK2-linked disease.
Introduction
LRRK2 (also known as dardarin) is a large protein with functional GTPase and kinase domains flanked by multiple protein interaction domains (Figure 1). LRRK2 missense mutations are the most common known genetic cause of Parkinson’s disease (PD). Many mutations spanning the length of LRRK2 have been associated with PD, although only mutations in the enzymatic GTPase and kinase domains segregate with familial disease suggesting an importance of these enzymatic activities to disease development (Figure 1). The G2019S mutation in LRRK2’s kinase domain is particularly prevalent, constituting 4% of familial cases and 1% of sporadic cases worldwide (Healy et al., 2008). Common genetic variants at the LRRK2 locus have also been identified in GWAS studies, further implicating LRRK2 in sporadic PD (Satake et al., 2009; Simon-Sanchez et al., 2009). The penetrance of LRRK2 mutations in PD is age-related, although incomplete even at advanced age suggesting the importance of gene-environment or genetic background influences on mutant LRRK2 toxicity. Disease symptoms typically begin with late onset and, at least for the G2019S mutation, are usually clinically and pathologically indistinguishable from sporadic disease, including the presence of Lewy bodies in most cases (Giasson et al., 2006; Ross et al., 2006; Taylor et al., 2006). Interestingly, patients with other segregating LRRK2 mutations (R1441C, Y1699C, I2020T) frequently do not exhibit Lewy body pathology and sometimes present other findings such as neurofibrillary tau tangles (Kett and Dauer, 2012) raising the possibility that these mutations cause disease via distinct pathogenic mechanisms. The reported presence of tau-positive neurofibrillary tangles and not Lewy body disease in one G2019S LRRK2 carrier may, however, refute this hypothesis (Rajput et al., 2006). Mutations in LRRK2 cause autosomal dominant PD and homozygous carriers of the G2019S mutation do not experience higher disease risk or progression than heterozygous carriers.
Figure 1. LRRK2 domains and pathogenic mutations.
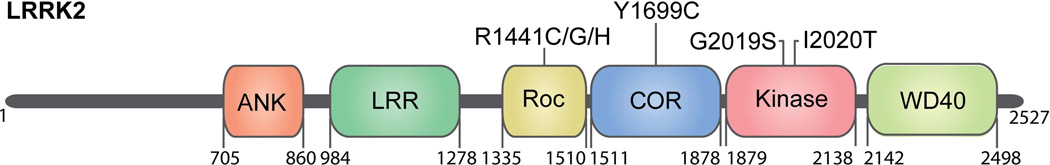
The catalytic core of LRRK2 comprises Ras of complex proteins (Roc), C-terminal of ROC (COR) and kinase domains where all disease-segregating mutations identified are located. R1441 and Y1699 mutations decrease GTPase activity of LRRK2 whereas G2019S and I2020T mutations increase kinase activity. Flanking the catalytic domains are ankyrin-like repeats (ANK), leucine-rich repeats (LRR), and the WD40 domain which are thought to mediate protein-protein interactions, although binding partners for these domains are unknown. These protein interaction domains may additionally influence enzymatic output of the catalytic domains as described in the main text. Numbers indicate amino acid boundaries for the domains and protein length. Mutations that segregate with Parkinson’s disease are annotated at their domain location.
The purpose of this review is to summarize current viewpoints on the role of altered LRRK2 biochemistry and aberrant function in mutant LRRK2 neurotoxicity. Evidence is derived mainly from biochemical studies on LRRK2’s GTPase and kinase activities, and cell or animal disease models utilizing mutant LRRK2 transgenic animals or LRRK2 knockouts. Two leading potential disease mechanisms, alteration in vesicular trafficking and aberrant protein synthesis are explored in detail, and the role of α-synuclein in LRRK2-mediated pathology is also discussed.
LRRK2 mutations implicate kinase and GTPase activities in PD
The prominence of GTPase and kinase domain mutations in PD has driven a large effort to understand how these domains contribute to LRRK2 function and how mutations in LRRK2 affect its biochemical activity in a manner that promotes disease. LRRK2 belongs to the ROCO protein family and contains tandem Ras-of-complex proteins (Roc) GTPase and C-terminal of Roc (COR) domains characteristic of this family (Zimprich et al., 2004) (Figure 1). The GTPase domain of LRRK2 can bind GTP or GDP and purified LRRK2 exhibits low levels of intrinsic GTPase activity. R1441C/G/H mutations may reduce this activity and, at least for R1441H, increase the GTP-binding affinity of the Roc domain (Liao et al., 2014). Crystallographic studies from the Cholorobium tepidum ROCO protein suggest that LRRK2 may homodimerize through its COR domain and function in dimeric form as a GAD (G proteins activated by nucleotide-dependent dimerization) in which both ROC domains are brought close together and participate in the catalytic activity of their counterpart within the dimer (Gotthardt et al., 2008). Consistent with this, the hydrolysis rate of monomeric LRRK2 ROC domain is much lower than that of a full-length dimer (Liao et al., 2014; Liu et al., 2010). However as yet, no crystal structure for the Roc-COR tandem domain has been reported to support this mechanism of GTPase domain regulation. An alternative mechanism of GTPase regulation, through regulatory GTPase-activating proteins (GAP) and guanine nucleotide exchange factors (GEF) similar to other small GTPases has also been postulated. ArfGAP1 is the first LRRK2 GAP identified, and this GAP activity may contribute to LRRK2 toxicity since knock-down of ArfGAP1 or expression of a dominant-negative mutant attenuate neuronal toxicity (Stafa et al., 2012; Xiong et al., 2012). ARHGEF1 is one potential GEF for LRRK2 that interacts with LRRK2 and stimulates its GTPase activity in vitro (Haebig et al., 2010).
LRRK2 serine/threonine kinase activity has been monitored through LRRK2 autophosphorylation as well as phosphorylation of a number of exogenous substrates, some of which are phosphorylated by LRRK2 in vivo. As suggested for GTPase activity, LRRK2 dimerization appears to stimulate kinase activity (Berger et al., 2010; Greggio et al., 2008). It is now well established that the G2019S mutation increases LRRK2 kinase activity (both autophosphorylation and phosphorylation of exogenous kinase substrates). This, considered alongside the effectiveness of specific LRRK2 kinase inhibitors or kinase-dead G2019S/D1994A double mutants to reduce G2019S LRRK2-mediated toxicity provides compelling evidence that LRRK2 toxicity is kinase-dependent, although alternative hypotheses have been suggested (Skibinski et al., 2014). It is less clear, however, if other disease-segregating mutations affect LRRK2 kinase activity. R1441C/G/H, Y1699C and the I2020T kinase domain mutation have been reported to increase kinase activity in some but not all studies, perhaps due to differences in assay methodology and an over reliance on in vitro kinase assays. A recent detailed biochemical characterization of I2020T LRRK2 supports an increase in kinase activity caused by this mutation, through stabilization of the active-state conformation, and increased rate of phosphoryl transfer (Ray et al., 2014). Increased phosphorylation of the LRRK2 substrate Rps15 was observed in transfected cells via kinase domain variants G2019S and I2020T but not via the Roc domain mutants R1441C/G (Martin et al., 2014). This, together with other data from in vitro kinase assays on Roc domain mutants supports the existence of kinase-independent mechanisms of LRRK2 toxicity.
GTPases frequently act as molecular switches in signaling pathways to regulate kinases, generating an early interest in LRRK2 kinase self-regulation through GTPase activity analogous to activation of Raf by Ras. Accumulating evidence supports an influence of the GTPase domain on LRRK2 kinase activity, although the exact nature of the relationship between the two domains remains elusive. Studies with purified LRRK2 containing synthetic mutations in the GTPase domain P-loop reveal that an intact ROC GTPase domain capable of binding GTP is required for LRRK2 kinase activity suggesting that GTP binding might regulate kinase activity (Biosa et al., 2013; Ito et al., 2007; Smith et al., 2006; West et al., 2007). A correlation has been suggested between kinase activity and the extent of GTP binding, and to a lesser extent GTP hydrolysis by experiments in which GTP or non-hydrolyzable GTP analogs were added to cell extracts prior to LRRK2 purification and kinase assessment (Biosa et al., 2013; West et al., 2007). Despite this, addition of GDP, GTP or non-hydrolysable GTP analogs (GTPγS or GMPPCP) to purified LRRK2 has no apparent effect on its kinase activity (Biosa et al., 2013; Taymans et al., 2011). These results may be reconciled if one considers the possibility that an upstream GTP-activated protein promotes LRRK2 kinase activity in cells or is co-purified with LRRK2 and phosphorylates it in a GTP-dependent manner during in vitro kinase assays (Taymans et al., 2011). Hence, GTP binding capacity appears to be necessary for functional kinase activity, although the extent of regulatory influence from GTPase activity remains in question. Recent evidence indicates that R1441 mutations may lead to increased kinase activity through a mechanism involving reduced S1444 phosphorylation by protein kinase A and in turn, reduced docking of 14-3-3 proteins to p-S1444. Conversely, genetic studies with kinase-dead mutants show that kinase activity is not required for GTP binding or hydrolysis, although the kinase domain may exert some influence over GTPase activity as LRRK2 autophosphorylates its Roc domain (Greggio et al., 2009; Greggio et al., 2008). Since several PD-linked mutations are reported to increase LRRK2 autophosphorylation, LRRK2 kinase activity could in principle cause toxicity via altered GTPase function at least in part. The effects of Roc domain autophosphorylation on GTP binding or GTPase activity, if any, are largely unknown. One study reported that mutating an autophosphorylation site (T1503) to alanine (phospho-deficient) or aspartate (phospho-mimetic) resulted in substantially reduced GTP binding activity (Webber et al., 2011). Roc domain autophosphorylation and resultant effects on GTPase activity require more detailed examination since one possible scenario, a decrease in hydrolysis of GTP to GDP promoted by autophosphorylation and therefore increased via kinase-enhancing mutations, could suggest a common biochemical effect of both kinase domain and Roc domain mutations, which would reinforce the potential importance of GTP binding/GTPase activity in disease pathogenesis. Indeed, work from our group indicates a significant decrease in GTP hydrolysis rates for G2019S and non-significant decrease for I2020T LRRK2 relative to WT (Xiong et al., 2010; Xiong et al., 2012), while another group also reported a 20% decrease in GTPase activity for N-terminally truncated G2019S LRRK2 with no significant effect on GTP binding (Liu et al., 2011a). Whether these effects are mediated through LRRK2 self-regulation (i.e. autophosphorylation), via phosphorylation of exogenous substrates that affect GTPase activity such as ArfGAP1 or a combination of both remains to be determined.
While efforts to understand the pathogenic output of LRRK2 mutations have focused on its GTPase and kinase activities, it should also be noted that other LRRK2 domains may be important to LRRK2 toxicity, even if only through their effects on these biochemical activities. For example, N-terminally truncated LRRK2 lacking the ankyrin repeat domain exhibits substantially higher kinase activity during in vitro kinase assays, suggesting that this region may somehow modulate kinase activity, possible involving autophosphorylation of ser 910/ser935 which are absent in the truncated form. In contrast, deletion of the C-terminal WD40 domain blocks LRRK2 dimer formation, kinase activity and neurotoxicity in rodent neurons (Jorgensen et al., 2009; Rudenko et al., 2012). Interestingly, a G2385R variant of LRRK2 reported to partially reduce LRRK2 kinase activity (Jaleel et al., 2007; Rudenko et al., 2012) is associated with PD in Asian populations (Funayama et al., 2007; Fung et al., 2006) suggesting either a complex relationship between LRRK2 kinase activity and toxicity or kinase-independent pathogenesis. Regarding the relationship between the WD40 domain and LRRK2 kinase activity, one plausible hypothesis is that this, and perhaps other protein interaction domains of LRRK2 may define its binding partners that ultimately become targets of LRRK2 enzymatic activities (Mills et al., 2014). Examination of putative LRRK2 substrates reveals no consensus substrate sequence, supporting the hypothesis that substrate specificity may be determined by binding to LRRK2’s interaction domains. Binding to the WD40 domain is an attractive possibility considering the proximity of this to LRRK2’s kinase domain and the finding that deletion of the WD40 domain appears to abrogate LRRK2 kinase activity (Jorgensen et al., 2009). Potentially supporting this theory, LRRK2 interacts with and phosphorylates numerous ribosomal proteins including the pathogenic substrate s15 (Martin et al., 2014) and s15 binds predominantly to the WD40 domain of LRRK2. Informatively, the WD40 domain of LRRK2 is predicted to form a canonical β-propeller configuration (Jorgensen et al., 2009), similar to that found in receptor for activated C-kinase 1 (RACK1) which binds to the 40S ribosomal subunit via interaction with several ribosomal proteins. The role of the WD40 domain in LRRK2’s interaction with ribosomes is under investigation.
Looking for clues from LRRK1
Mutations in LRRK1 do not appear to be associated with PD (Haugarvoll et al., 2007; Taylor et al., 2007) although there may be rare variants associated with PD (Schulte et al., 2014). This has lead to the hypothesis that LRRK1 and LRRK2 are divergent in function or expression. LRRK1 and LRRK2 possess the catalytic Roc-Cor-kinase tri-domain, but are divergent in N-terminal repeats and at the C-terminal WD40 domain, both of which influence kinase activity. Although not well characterized, LRRK1 kinase activity appears to be weaker than that of LRRK2, both when comparing autophosphorylation and phosphorylation of the pseudosubstrates LRRKtide and Nictide (Civiero et al., 2012). A lack of robust kinase activity for LRRK1 could indicate functional divergence and is consistent with a central role for kinase activity in LRRK2 toxicity. Both LRRK1 and LRRK2 mRNA have been reported to be widely expressed in adult human brain, with comparative studies indicating relatively strong expression of both in cortical regions and the hippocampus while at least in one study, the striatum demonstrated relatively high levels of LRRK2 but not LRRK1 (Dachsel et al., 2010; Westerlund et al., 2008). Also, other studies have demonstrated significantly higher levels of LRRK2 in the brain relative to LRRK1 (Biskup et al., 2007). Interestingly, LRRK1 has been proposed to regulate endocytosis of EGFR via formation of a LRRK1-Grb2-EGFR complex (Hanafusa et al., 2011), while LRRK2 did not appear to form such a complex in this study suggesting that the two have functionally diverged. While the study of LRRK1 function has considerably lagged behind that of LRRK2, evidence collected to date does not support both LRRKs sharing similar functions. Heterodimerization between overexpressed LRRK2 and LRRK1 has been suggested by co-immunoprecipitation studies (Dachsel et al., 2010; Klein et al., 2009) and if a direct interaction between endogenous LRRKs does in fact occur, one might speculate that heterodimer formation could affect LRRK2 kinase activity since LRRK2 catalytic activity is predominantly found in LRRK2 dimers and not monomers or oligomers. The relevance of heterodimer formation and significance of this in normal LRRK biology or as a potential disease modifier in the context of PD are currently unknown and warrant further study.
Mechanisms of LRRK2 pathobiology
Numerous transgenic vertebrate and invertebrate animal models as well as recent pluripotent stem cell-derived neuron models demonstrate that expression of pathogenic LRRK2 variants leads to PD-related phenotypes. These primarily include degeneration and loss of dopaminergic neurons or neurons of other affected regions as well as locomotor deficits, which in some cases can be rescued by administration of levodopa (Dusonchet et al., 2011; Li et al., 2009; Liu et al., 2008; Martin et al., 2014; Saha et al., 2009; Tong et al., 2009). One caveat is that most rodent LRRK2 transgenic models published to date do not display actual loss of dopaminergic neurons from the SNpc, although they do exhibit deficits in dopamine transmission and in one case axonal pathology of nigrostriatal dopamine tracts (Li et al., 2010; Li et al., 2009; Maekawa et al., 2012; Melrose et al., 2010; Tong et al., 2009). Furthermore, dopamine neuron loss has been demonstrated in invertebrate organisms and through virally-mediated G2019S LRRK2 expression in rodent brain (Dusonchet et al., 2011; Lee et al., 2010). Numerous studies on primary neuronal cultures and intact rodent brain have demonstrated that LRRK2 is important in maintaining neurite length and branching (MacLeod et al., 2006; Smith et al., 2005; West et al., 2007) and may be important in regulating neurite outgrowth, possibly via phosphorylation of ERM (ezrin/radixin/moesin) proteins and consequent maintenance of F-actin homeostasis in filopodia necessary for proper neurite outgrowth (Jaleel et al., 2007; Parisiadou et al., 2009). Expression of pathogenic LRRK2 variants results in neurite dystrophy or loss and ultimately leads to cell death.
Importantly, most of these studies report a strong age-dependency for onset and progression of hallmark PD phenotypes, further supporting the relevance of these cell and animal models to the human disease. Many studies to date have focused predominantly on the common G2019S mutation in LRRK2, which has been shown repeatedly to exert neuronal toxicity through kinase-dependent mechanisms. As might be expected from the central dependency of neurotoxicity on LRRK2 kinase activity, overexpression of wild type LRRK2 also causes toxicity in some models, although to a lesser extent than observed via mutant LRRK2 (Liu et al., 2008; Martin et al., 2014), related to differences in kinase activity and possibly other mechanisms that may differentially regulate kinase activities of wild type and mutant forms of LRRK2 in vivo (Sen et al., 2009; Webber et al., 2011). Abundant insight into normal LRRK2 biology has also been gained from knockout animal models. Taken together, cell and animal models implicate LRRK2 in a diverse array of functions including vesicular trafficking, autophagy, microtubule and cytoskeletal network formation, synaptogenesis and mRNA translation (Figure 2). LRRK2 dimers, which are thought to possess greater kinase activity than monomers (Berger et al., 2010; Sen et al., 2009) are reportedly enriched at membranes, although cell studies with overexpressed LRRK2 suggest no difference between wild type and mutant LRRK2 membrane association (Hatano et al., 2007). The many functions attributed to LRRK2 is not necessarily surprising given the widespread distribution of LRRK2 and its broad interaction with different structures and organelles, including mitochondria and ribosomes. There is strong evidence to support the importance of LRRK2 mutations in altered vesicular trafficking and protein synthesis, which are discussed in more detail below. Genetic models are also beginning to offer some insight on the interplay of LRRK2 with other key PD proteins such as synuclein, which is also discussed further below. Collectively, despite some limitations, these animal and human neuron models will continue to play an essential role in our growing knowledge of LRRK2 biology and LRRK2-mediated toxicity.
Figure 2. LRRK2 functions affected by pathogenic mutations.

Putative functions of LRRK2 that are impacted by one or more pathogenic mutations. LRRK2 has been reported to regulate lysosomal positioning and autophagy (Alegre-Abarrategui et al., 2009; Dodson et al., 2012; Niu et al., 2012; Plowey et al., 2008; Su and Qi, 2013; Wang et al., 2012), synaptic vesicle endocytosis (Matta et al., 2012), synaptogenesis (Parisiadou et al., 2014), cytoskeleton and neurite outgrowth (Jaleel et al., 2007; MacLeod et al., 2006; Parisiadou et al., 2009; Smith et al., 2005; West et al., 2007), protein synthesis (Gehrke et al., 2010; Imai et al., 2008; Martin et al., 2014), golgi sorting and retromer function (Lin et al., 2009; MacLeod et al., 2006; MacLeod et al., 2013; Stafa et al., 2014). Mutations reported to affect each function are indicated in parentheses.
Protein synthesis
Abnormalities in protein synthesis or mRNA metabolism have long been linked to various neurodegenerative diseases, such as FXTAS, ALS or prion-mediated neurodegeneration (Bhakar et al., 2012; Lagier-Tourenne et al., 2012; Polymenidou et al., 2011; Tollervey et al., 2011). Recent studies primarily carried out using Drosophila show that LRRK2 physically and functionally interacts with the protein translation machinery and altered protein synthesis caused by PD-linked LRRK2 mutations is toxic, supporting the hypothesis that dysregulation of mRNA translation has a role in PD pathology (Gehrke et al., 2010; Imai et al., 2008; Martin et al., 2014). The effects of PD-linked LRRK2 mutations on protein synthesis were initially suggested by its genetic interaction with components of the TOR pathway in Drosophila (Imai et al., 2008). Genetic deletion of dLRRK worsened the inhibited cell growth phenotypes caused by the suppression of TOR signaling, through either co-overexpression of TSC1 and TSC2 or constitutively active form of eIF4E-binding protein (4E-BP). Conversely, dLRRK deletion partially suppressed Rheb overexpression-mediated enhanced cell growth (Imai et al., 2008). LRRK2 was proposed to interact with the TOR pathway through phosphorylation of 4E-BP1 in a manner that promotes toxicity, presumably via consequent disruption of 4E-BP1 binding to eIF4E, thus increasing free levels of eIF4E available for translation initiation (Figure 3). However, whether 4E-BP is an in vivo pathogenic target of LRRK2 in mammalian brain is still unclear as neither LRRK2 knockout nor LRRK2 transgenic mice showed alterations of phosphorylated 4E-BP level in brain tissue (Trancikova et al., 2012). A subsequent study reported that pathogenic LRRK2 impacts mRNA translation through negatively regulating miRNA activity (Gehrke et al., 2010). Flies transgenic for mutant human LRRK2 or dLRRK were shown to exhibit increased expression of E2F1 and DP by suppressing the action of let-7 and miR-184, respectively (Gehrke et al., 2010). The mechanism of miRNA disruption is unclear, but thought to involve association of LRRK2 and phospho-4E-BP1 with dAgo1 and reduced dAgo1 stability although further clarification for the role of LRRK2 in miRNA impairment is necessary.
Figure 3. LRRK2 role in mRNA translation.

Schematic illustration of LRRK2 effects on mRNA translation via several proposed mechanisms. LRRK2 may influence translation in an analogous manner to mTOR via phosphorylation of 4E-BP (eIF4E binding protein) which promotes dissociation of 4E-BP from eIF4E thus increasing levels of free eIF4E available for cap-dependent translation initiation. LRRK2 phosphorylates ribosomal protein s15 to stimulate cap-dependent and cap-independent translation. LRRK2 interacts with Eukaryotic translation elongation factor 1A (eEF1A) with unknown consequences and LRRK2 may regulate miRNA function in a miRNA-specific manner involving unclarified mechanisms that may involve binding of LRRK2 and phospho-4EBP to Argonaute (Ago) proteins. The effects of LRRK2 on translation are kinase-dependent and pathogenic mutations that increase kinase activity (e.g. G2019S) cause toxicity at least in part via increased mRNA translation, although the specific mRNA targets affected are yet to be reported.
A role for elevated protein synthesis in PD pathogenesis extends to parkin and Pink1-linked disease. Overexpression of 4E-BP or treatment with the TOR inhibitor rapamycin effectively blocked pathogenic parkin and Pink1 loss-of-function phenotypes in flies, including degeneration of dopamine neurons (Tain et al., 2009). Importantly, while there is good evidence that translational suppression through perturbed TOR signaling is protective against PD-related phenotypes, evidence that LRRK2 or other familial PD gene products act directly on the TOR pathway to affect mRNA translation is still lacking. Indeed, if LRRK2 were to stimulate translation through alternative but parallel pathways, TOR inhibition might still be sufficient to suppress these effects and thereby block toxicity. Recently, alternative hypotheses for the interaction of LRRK2 with other components of the translation machinery have been advanced. LRRK2 has been reported to physically interact with the translation elongation factor eEF1A (Gillardon, 2009). Although its effects in mRNA translation have not been thoroughly addressed, the fact that eEF1A delivers aminoacyl-tRNAs to the ribosomal A-site during protein synthesis suggests a potential role for LRRK2 in translational elongation. In addition, LRRK2 has been reported to affect the expression levels of the proteins centrally involved in protein synthesis. Gene ontology analysis from LRRK2 mouse models revealed that overexpression of LRRK2 G2019S mutant or genetic ablation of LRRK2 can affect expression levels of many genes related to ribosome biosynthesis, mRNA processing or protein synthesis (Nikonova et al., 2012). Most recently, data from our group indicates that LRRK2 affects mRNA translation through direct interaction with the ribosome and phosphorylation of ribosomal protein s15 (Martin et al., 2014). Unbiased proteomic screening and in vitro kinase assays revealed that LRRK2 phosphorylates several ribosomal proteins, several of which show increased phosphorylation in the presence of G2019S and I2020T LRRK2. Blocking the phosphorylation of s15 substantially attenuated pathogenic LRRK2 neurotoxicity in both human neuron and Drosophila models and additionally prevented accelerated age-related locomotor deficits in G2019S LRRK2 transgenic flies. We found that the increased phosphorylation of s15 enhances bulk protein synthesis which is responsible for the neuronal toxicity observed, since treating flies with the protein synthesis inhibitor anisomycin or expressing phospho-deficient T136A s15 blocks the neurotoxicity in LRRK2 transgenic flies (Martin et al., 2014) (Figure 3). This finding provides new insights into the long-suggested relationship between protein synthesis and neurodegeneration, although how s15 phosphorylation can cause increased protein synthesis and why excess protein synthesis kills dopamine neurons is still obscure. Speculatively, increased bulk protein synthesis may deplete neurons of valuable energy stores or incur neuronal stress through increased burden on protein folding/degradation machineries eventually leading to the failure of overall protein quality control. Alternatively, LRRK2 toxicity may be caused by a change in gene-specific translational profiles in which increased bulk protein synthesis impairs the finely-tuned control of translational output under basal conditions but perhaps more importantly also in response to cell stressors or other changes in cell conditions that require strict translation regulation. Why dopaminergic neurons would be selectively susceptible is unclear but could be related to cell type-specific translational profiles that may underlie differential effects of pathogenic LRRK2. Of course, the aggregate pathology of LRRK translation effects might be a consequence of both systematic and gene-level abnormalities. Therefore, comprehensive understanding of the molecular effects of LRRK2 mutations in translational control is required to expand our knowledge on PD pathogenesis.
Finally, another clue to the importance of protein synthesis in PD comes from the recent identification of eIF4G1 missense mutations associated with the disease (Chartier-Harlin et al., 2011; Nuytemans et al., 2013). eIF4G1 is a member of the translation initiation complex eIF4F, and plays a central role in eIF4F complex formation by acting as a scaffold for assembly of other complex components eIF4E and eIF4A (Hershey et al., 2012). The impact of eIF4G1 mutations on cap complex assembly and initiation of protein synthesis are currently unknown. Also unknown is whether LRRK2 and eIF4G1 interact in each other’s pathogenic mechanisms.
Excess protein synthesis resulting from the activity of certain oncogenic pathways frequently occurs during tumor development. The effect of pathogenic LRRK2 mutations on mRNA translation is interesting in the context of recent findings demonstrating a reported increase in cancer risk of patients carrying the G2019S mutation (Saunders-Pullman et al., 2010). While there is currently no direct evidence linking LRRK2 mutations to cancer through altered translation, the 12q12 locus in which LRRK2 resides is frequently amplified in human tumors including type 1 papillary renal cell carcinoma, which is driven by amplification of the proto-oncogene MET (encoding hepatocyte growth factor receptor) (Looyenga et al., 2011). A striking correlation was reported between MET and LRRK2 amplifications/overexpression within the same tumor cells, and downregulation of LRRK2 in cultured tumor cells perturbed MET activation and downstream signaling to STAT3 and mTOR. These effects were apparently mediated by a reduction in direct phosphorylation of MET by LRRK2, and considering the impact of mTOR signaling on translation, raise an additional mechanism by which LRRK2 may impact mRNA translation.
Vesicular trafficking
Several studies point toward a role for LRRK2 in multiple aspects of vesicle trafficking, mainly through interaction with trafficking proteins such as endophilin A, Rab7, Rab7L1 and members of the dynamin GTPase superfamily. Evidence that the LRRK2 paralog LRRK1 mediates endocytosis of EGFR (Hanafusa et al., 2011) supports a broad role for LRRKs in vesicle formation and transport. Studies using Drosophila have highlighted potential roles for LRRK2 in multiple aspects of vesicle trafficking including synaptic vesicle recycling, retromer trafficking and lysosomal positioning. LRRK2 was reported to phosphorylate endophilin A (at S75), a protein whose association with presynaptic membranes is required for endocytosis of synaptic vesicles (Matta et al., 2012). It seems that increasing endo A phosphorylation through expression of G2019S LRRK2 or decreasing it through dLRRK null impedes synaptic vesicle endocytosis (Matta et al., 2012). The data fit with a model in which endo A phosphorylation mediates its release from newly-formed vesicles and then subsequent dephosphorylation permits further binding at the presynaptic membrane and hence any alteration of this tightly coordinated phosphorylation/dephosphorylation cycle impedes vesicle recycling. LRRK2 may be enriched at the Golgi complex (MacLeod et al., 2006; Stafa et al., 2014) and overexpression of WT or G2019S LRRK2 results in golgi complex fragmentation in mice (Lin et al., 2009). Genetic interaction studies suggest involvement of LRRK2 with the retromer complex, which mediates retrograde transport of proteins such as acid hydrolase receptors from endosomes to the trans-golgi network (MacLeod et al., 2013). G2019S LRRK2 expression in rat neuronal cultures resulted in lysosomal enlargement and endolysosomal and golgi sorting deficits that were rescued by overexpression of VPS35, a component of the retromer complex with mutations identified in familial PD (MacLeod et al., 2013). Expression of the PD-linked mutant VPS35 failed to rescue these defects. Also consistent with LRRK2-mediated retromer dysfunction, overexpression of Rab7L1, another retromer component implicated in PD suppressed mutant LRRK2 neurite shortening phenotypes in rat neurons and constitutively active Rab7L1 rescued dopamine neuron loss in G2019S LRRK2 transgenic flies. Rab7L1 also localizes to the trans-Golgi network and a recent study suggests that Rab7L1 is part of a complex with LRRK2 that co-operatively promotes autophagy of the trans-Golgi network (Beilina et al., 2014). Interestingly, the fly LRRK2 homolog dLRRK associates with membranes of late endosomes/lysosomes and physically interacts with Rab7. Rab7 promotes perinuclear clustering of lysosomes under certain conditions such as starvation (Korolchuk et al., 2011) and evidence gathered indicates that dLRRK negatively regulates lysosomal transport towards nuclei whereas the G2019S equivalent mutation in dLRRK promotes it (Dodson et al., 2012). Perinuclear lysosomal localization has been reported to promote autophagy through colocalization with autophagosomes as well as lowering mTOR signaling (Korolchuk and Rubinsztein, 2011) suggesting a possible role for LRRK2 in autophagy. Related to the lysosomal/autophagy pathway, pathogenic LRRK2 variants may impact both macroautophagy (autophagy) and chaperone-mediated although there is a lack of consensus regarding the main effects. LRRK2 associates with autophagic vesicles and multi-vesicular bodies in human brain and cultured cells, and both structures were abnormally present in cells expressing R1441C LRRK2 (Alegre-Abarrategui et al., 2009). Disruption of autophagy in midbrain dopamine neurons through Atg7 conditional knockout leads to eventual neuronal death and locomotor deficits in mice (Ahmed et al., 2012; Friedman et al., 2012) highlighting a possible connection between aberrant autophagy and neurodegeneration. Conversely, G2019S LRRK2 was reported to result in augmented autophagy in various cell culture experiments, possibly through a mechanism involving mitochondrial fragmentation following elevated Drp1 phosphorylation (Niu et al., 2012; Plowey et al., 2008; Su and Qi, 2013; Wang et al., 2012). LRRK2 degradation appears to occur through both the UPS and chaperone-mediated autophagy (CMA) although several pathogenic LRRK2 variants are poorly degraded through the CMA and additionally impede uptake of other CMA substrates including α-synuclein, suggesting a potential mechanism of toxicity (Orenstein et al., 2013).
Dependence of LRRK2 toxicity on synuclein
PD patients harboring LRRK2 mutations frequently exhibit α-synuclein neuropathology in the form of Lewy bodies, in which LRRK2 is also present. Clues for a role of α-synuclein in LRRK2 toxicity come from studies on human dopamine neuron cultures demonstrating increased α-synuclein levels in G2019S or Y1699C LRRK2 PD patient iPS-derived dopamine neurons (Nguyen et al., 2011; Reinhardt et al., 2013; Skibinski et al., 2014). Informatively, neuronal death resulting from G2019S or Y1699C LRRK2 expression was substantially reduced in primary neurons from α/β/γ-synuclein triple knockout mice (Skibinski et al., 2014). A reciprocal requirement for LRRK2 in α-synuclein-mediated neuropathology is supported by evidence that overexpressed LRRK2 promotes aggregation and toxicity of α-synuclein in A53T transgenic mice while A53T α-synuclein toxicity was blocked in LRRK2 KO mice (Lin et al., 2009). Similarly, injection of human α-synuclein into the substantia nigra of rats induced recruitment of pro-inflammatory myeloid cells and dopamine neuron degeneration in wild type but not LRRK2 knock-out rats (Daher et al., 2014). Despite this, G2019S LRRK2 expression alone did not cause α-synuclein accumulation in the absence of A53T α-synuclein overexpression (Lin et al., 2009) and in a separate study there was a lack of α-synuclein phosphorylation or accumulation in surviving dopamine neurons of SNpc in rat adenoviral vector model of G2019S-LRRK2 toxicity (Dusonchet et al., 2011). Furthermore, A53T α-synuclein-dependent neuropathology in mouse hindbrain was independent of LRRK2 expression suggesting that the genetic interaction may be region-specific (Daher et al., 2012). Based on our current knowledge, two straightforward routes to increased α-synuclein levels and aggregation can be hypothesized. First, the stimulatory action of pathogenic LRRK2 on general mRNA translation could lead to increased translation of α-synuclein mRNA. Given that modulations of the core translation machinery produce variable effects on the translatome at the transcript-specific level (Gkogkas et al., 2013; Thoreen et al., 2012), it is feasible that α-synuclein expression may even be disproportionately affected by pathogenic LRRK2, which would account for α-synuclein pathology in PD patients carrying LRRK2 mutations. Second, pathogenic LRRK2 may promote oligomerization of α-synuclein on the lysosomal surface in a manner that impairs its uptake into the lysosome through chaperone-mediated autophagy and subsequent degradation (Orenstein et al., 2013). Interestingly, loss of LRRK2 may similarly impair autophagic removal of α-synuclein in the kidneys of aged LRRK2 knockout mice as levels of high molecular weight α-synuclein oligomers were observed to fluctuate across age inversely proportional to markers of autophagy function which were substantially reduced by 20 months of age (Tong et al., 2012). While high molecular weight α-synuclein oligomers were increased in 20-month-old mice, they were substantially reduced in 7 month-old mice compared to controls in conjunction with increased LC3I-LC3II conversion and decreased p62. As pronounced morphological abnormalities in the kidneys of LRRK2 knockout mice are already present by 4 months of age, a central involvement for α-synuclein aggregation in LRRK2 knockout mouse kidney pathology is unlikely. While pathogenic LRRK2 toxicity in vertebrates may act through a mechanism involving α-synuclein, this cannot be true for the PD-related phenotypes observed in Drosophila, as invertebrates do not express α-synuclein. This further complicates our interpretation and suggests a potential disconnect in pathogenic mechanisms between invertebrate and vertebrate models of PD. The observation from numerous groups of selective dopamine neuron degeneration and accompanying locomotor dysfunction in Drosophila models of PD would seem to argue against an absolute requirement for α-synuclein in LRRK2 toxicity, and this is potentially further bolstered by the absence of Lewy bodies in some PD patients with mutations in LRRK2 or other familial PD genes.
Conclusions and future directions
A decade of research on LRRK2 has uncovered important clues to PD pathogenesis and potential treatment strategies. LRRK2 possesses both GTPase and kinase activities and work in cell and animal genetic models has revealed a number of diverse biological functions that are impacted by pathogenic mutations. Given the variable effects of mutations in the catalytic core of LRRK2 on GTPase and kinase activities, it is currently difficult to reconcile how all of these mutations impact LRRK2 function to result in a common pathogenic phenotype – one plausible hypothesis is that this may occur via multiple independent physiological consequences that all converge on the death of dopamine-producing neurons. Nevertheless, the established link between LRRK2 kinase activity and toxicity makes the development of LRRK2 kinase inhibitors an attractive prospect and important future goal. Production of potent, specific and brain-permeable LRRK2 kinase inhibitors is already underway. Some of these inhibitors already have demonstrated efficacy in both blocking LRRK2 kinase activity and preventing neurodegenerative phenotypes in various models (Choi et al., 2012; Lee et al., 2010; Liu et al., 2011b) although issues related to kinase specificity are still a concern. While these proof-of-concept studies are encouraging, the development of kinase inhibitors for chronic human use comes with many important safety concerns due to the potential for off-target effects on other kinases and also side-effects of robust LRRK2 kinase inhibition in the CNS or other tissues. An important consideration for the implementation of LRRK2 kinase inhibition is evidence from mouse studies demonstrating the prevalence of pathophysiological changes (darkening, accumulation of lipofuscin granules and age-related atrophy) in kidney of mice expressing kinase-dead LRRK2 or with LRRK2 knockout (Herzig et al., 2011; Tong et al., 2010) causing concern about potential adverse side effects. While not all changes in LRRK2 knockouts may be solely due to loss of kinase activity, the phenotypes observed in mice expressing kinase-dead LRRK2 alone are enough to give pause to the design and implementation of LRRK2 kinase inhibitor therapy in humans. LRRK2 may also have an important immunoregulatory role as a negative regulator of the NFAT family of transcription factors (Liu et al., 2011) Elevated nuclear localization of NFAT and increased occurrence of colitis were observed in LRRK2 knock-out mice (Liu et al., 2011) raising immunological concerns over the use of LRRK2 kinase inhibitors. More information about potential side-effects, the structure of the kinase active site and enzyme kinetics along with a detailed understanding of regulatory mechanisms governing LRRK2 activation is necessary for designing safe and potent kinase inhibitors to combat PD. Another high priority future goal for the LRRK2 field is to gain a better understanding of the role of GTPase activity in physiological processes identified to be impacted by LRRK2 such as protein synthesis and vesicular trafficking. Most studies have focused on the role of elevated kinase activity, while determination of GTP binding/GTPase activity influence in pathogenic phenotypes has largely been overlooked. This may not be justified given the presence of disease-segregating mutations in the Roc domain that reduce GTPase activity and autophosphorylation sites in the same domain that may impact GTP binding or GTPase activity. Also, considering studies using kinase-enhancing mutations published to date, instances where only partial rescue effects are observed through kinase activity inhibition or via expression of kinase-dead mutants are consistent with other activities of pathogenic LRRK2 on phenotypes examined, with GTPase activity being a primary candidate.
Acknowledgements
This work was supported by grants from the NIH/NINDS NS38377 (VLD and TMD), the JPB Foundation (TMD), 2013-MSCRFII-0105-00 (VLD), a New York Stem Cell Foundation-Druckenmiller Fellowship and 2014-MSCRFF-0610 (IM). The authors acknowledge the joint participation by the Adrienne Helis Malvin Medical Research Foundation and the Diana Helis Henry Medical Research Foundation through its direct engagement in the continuous active conduct of medical research in conjunction with The Johns Hopkins Hospital and the Johns Hopkins University School of Medicine and the Foundation’s Parkinson’s Disease Programs.
References
- Ahmed I, Liang Y, Schools S, Dawson VL, Dawson TM, Savitt JM. Development and characterization of a new Parkinson's disease model resulting from impaired autophagy. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:16503–16509. doi: 10.1523/JNEUROSCI.0209-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alegre-Abarrategui J, Christian H, Lufino MM, Mutihac R, Venda LL, Ansorge O, Wade-Martins R. LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Human molecular genetics. 2009;18:4022–4034. doi: 10.1093/hmg/ddp346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilina A, Rudenko IN, Kaganovich A, Civiero L, Chau H, Kalia SK, Kalia LV, Lobbestael E, Chia R, Ndukwe K, et al. Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proceedings of the National Academy of Sciences U S A. 2014;111:2626–2631. doi: 10.1073/pnas.1318306111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger Z, Smith KA, Lavoie MJ. Membrane localization of LRRK2 is associated with increased formation of the highly active LRRK2 dimer and changes in its phosphorylation. Biochemistry. 2010;49:5511–5523. doi: 10.1021/bi100157u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakar AL, Dolen G, Bear MF. The pathophysiology of fragile × (and what it teaches us about synapses) Annual review of neuroscience. 2012;35:417–443. doi: 10.1146/annurev-neuro-060909-153138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biosa A, Trancikova A, Civiero L, Glauser L, Bubacco L, Greggio E, Moore DJ. GTPase activity regulates kinase activity and cellular phenotypes of Parkinson's disease-associated LRRK2. Human molecular genetics. 2013;22:1140–1156. doi: 10.1093/hmg/dds522. [DOI] [PubMed] [Google Scholar]
- Biskup S, Moore DJ, Rea A, Lorenz-Deperieux B, Coombes CE, Dawson VL, Dawson TM, West AB. Dynamic and redundant regulation of LRRK2 and LRRK1 expression. BMC neuroscience. 2007;8:102. doi: 10.1186/1471-2202-8-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Dachsel JC, Vilarino-Guell C, Lincoln SJ, Lepretre F, Hulihan MM, Kachergus J, Milnerwood AJ, Tapia L, Song MS, et al. Translation initiator EIF4G1 mutations in familial Parkinson disease. American journal of human genetics. 2011;89:398–406. doi: 10.1016/j.ajhg.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HG, Zhang J, Deng X, Hatcher JM, Patricelli MP, Zhao Z, Alessi DR, Gray NS. Brain Penetrant LRRK2 Inhibitor. ACS medicinal chemistry letters. 2012;3:658–662. doi: 10.1021/ml300123a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civiero L, Vancraenenbroeck R, Belluzzi E, Beilina A, Lobbestael E, Reyniers L, Gao F, Micetic I, De Maeyer M, Bubacco L, et al. Biochemical characterization of highly purified leucine-rich repeat kinases 1 and 2 demonstrates formation of homodimers. PloS one. 2012;7:e43472. doi: 10.1371/journal.pone.0043472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dachsel JC, Nishioka K, Vilarino-Guell C, Lincoln SJ, Soto-Ortolaza AI, Kachergus J, Hinkle KM, Heckman MG, Jasinska-Myga B, Taylor JP, et al. Heterodimerization of Lrrk1-Lrrk2: Implications for LRRK2-associated Parkinson disease. Mechanisms of ageing and development. 2010;131:210–214. doi: 10.1016/j.mad.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daher JP, Pletnikova O, Biskup S, Musso A, Gellhaar S, Galter D, Troncoso JC, Lee MK, Dawson TM, Dawson VL, et al. Neurodegenerative phenotypes in an A53T alpha-synuclein transgenic mouse model are independent of LRRK2. Human molecular genetics. 2012;21:2420–2431. doi: 10.1093/hmg/dds057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daher JP, Volpicelli-Daley LA, Blackburn JP, Moehle MS, West AB. Abrogation of alpha-synuclein-mediated dopaminergic neurodegeneration in LRRK2-deficient rats. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:9289–9294. doi: 10.1073/pnas.1403215111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson MW, Zhang T, Jiang C, Chen S, Guo M. Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Human molecular genetics. 2012;21:1350–1363. doi: 10.1093/hmg/ddr573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusonchet J, Kochubey O, Stafa K, Young SM, Jr, Zufferey R, Moore DJ, Schneider BL, Aebischer P. A rat model of progressive nigral neurodegeneration induced by the Parkinson's disease-associated G2019S mutation in LRRK2. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:907–912. doi: 10.1523/JNEUROSCI.5092-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman LG, Lachenmayer ML, Wang J, He L, Poulose SM, Komatsu M, Holstein GR, Yue Z. Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of alpha-synuclein and LRRK2 in the brain. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:7585–7593. doi: 10.1523/JNEUROSCI.5809-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funayama M, Li Y, Tomiyama H, Yoshino H, Imamichi Y, Yamamoto M, Murata M, Toda T, Mizuno Y, Hattori N. Leucine-rich repeat kinase 2 G2385R variant is a risk factor for Parkinson disease in Asian population. Neuroreport. 2007;18:273–275. doi: 10.1097/WNR.0b013e32801254b6. [DOI] [PubMed] [Google Scholar]
- Fung HC, Chen CM, Hardy J, Singleton AB, Wu YR. A common genetic factor for Parkinson disease in ethnic Chinese population in Taiwan. BMC neurology. 2006;6:47. doi: 10.1186/1471-2377-6-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrke S, Imai Y, Sokol N, Lu B. Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature. 2010;466:637–641. doi: 10.1038/nature09191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Covy JP, Bonini NM, Hurtig HI, Farrer MJ, Trojanowski JQ, Van Deerlin VM. Biochemical and pathological characterization of Lrrk2. Annals of neurology. 2006;59:315–322. doi: 10.1002/ana.20791. [DOI] [PubMed] [Google Scholar]
- Gillardon F. Interaction of elongation factor 1-alpha with leucine-rich repeat kinase 2 impairs kinase activity and microtubule bundling in vitro. Neuroscience. 2009;163:533–539. doi: 10.1016/j.neuroscience.2009.06.051. [DOI] [PubMed] [Google Scholar]
- Gkogkas CG, Khoutorsky A, Ran I, Rampakakis E, Nevarko T, Weatherill DB, Vasuta C, Yee S, Truitt M, Dallaire P, et al. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature. 2013;493:371–377. doi: 10.1038/nature11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotthardt K, Weyand M, Kortholt A, Van Haastert PJ, Wittinghofer A. Structure of the Roc-COR domain tandem of C. tepidum, a prokaryotic homologue of the human LRRK2 Parkinson kinase. The EMBO journal. 2008;27:2239–2249. doi: 10.1038/emboj.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greggio E, Taymans JM, Zhen EY, Ryder J, Vancraenenbroeck R, Beilina A, Sun P, Deng J, Jaffe H, Baekelandt V, et al. The Parkinson's disease kinase LRRK2 autophosphorylates its GTPase domain at multiple sites. Biochemical and biophysical research communications. 2009;389:449–454. doi: 10.1016/j.bbrc.2009.08.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greggio E, Zambrano I, Kaganovich A, Beilina A, Taymans JM, Daniels V, Lewis P, Jain S, Ding J, Syed A, et al. The Parkinson disease-associated leucine-rich repeat kinase 2 (LRRK2) is a dimer that undergoes intramolecular autophosphorylation. The Journal of biological chemistry. 2008;283:16906–16914. doi: 10.1074/jbc.M708718200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haebig K, Gloeckner CJ, Miralles MG, Gillardon F, Schulte C, Riess O, Ueffing M, Biskup S, Bonin M. ARHGEF7 (Beta-PIX) acts as guanine nucleotide exchange factor for leucine-rich repeat kinase 2. PloS one. 2010;5:e13762. doi: 10.1371/journal.pone.0013762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanafusa H, Ishikawa K, Kedashiro S, Saigo T, Iemura S, Natsume T, Komada M, Shibuya H, Nara A, Matsumoto K. Leucine-rich repeat kinase LRRK1 regulates endosomal trafficking of the EGF receptor. Nature communications. 2011;2:158. doi: 10.1038/ncomms1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatano T, Kubo S, Imai S, Maeda M, Ishikawa K, Mizuno Y, Hattori N. Leucine-rich repeat kinase 2 associates with lipid rafts. Human molecular genetics. 2007;16:678–690. doi: 10.1093/hmg/ddm013. [DOI] [PubMed] [Google Scholar]
- Haugarvoll K, Toft M, Ross OA, White LR, Aasly JO, Farrer MJ. Variants in the LRRK1 gene and susceptibility to Parkinson's disease in Norway. Neuroscience letters. 2007;416:299–301. doi: 10.1016/j.neulet.2007.02.020. [DOI] [PubMed] [Google Scholar]
- Healy DG, Falchi M, O'Sullivan SS, Bonifati V, Durr A, Bressman S, Brice A, Aasly J, Zabetian CP, Goldwurm S, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson's disease: a case-control study. Lancet neurology. 2008;7:583–590. doi: 10.1016/S1474-4422(08)70117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershey JW, Sonenberg N, Mathews MB. Principles of translational control: an overview. Cold Spring Harbor perspectives in biology. 2012;4 doi: 10.1101/cshperspect.a011528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig MC, Kolly C, Persohn E, Theil D, Schweizer T, Hafner T, Stemmelen C, Troxler TJ, Schmid P, Danner S, et al. LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Human molecular genetics. 2011;20:4209–4223. doi: 10.1093/hmg/ddr348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Gehrke S, Wang HQ, Takahashi R, Hasegawa K, Oota E, Lu B. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. The EMBO journal. 2008;27:2432–2443. doi: 10.1038/emboj.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito G, Okai T, Fujino G, Takeda K, Ichijo H, Katada T, Iwatsubo T. GTP binding is essential to the protein kinase activity of LRRK2, a causative gene product for familial Parkinson's disease. Biochemistry. 2007;46:1380–1388. doi: 10.1021/bi061960m. [DOI] [PubMed] [Google Scholar]
- Jaleel M, Nichols RJ, Deak M, Campbell DG, Gillardon F, Knebel A, Alessi DR. LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson's disease mutants affect kinase activity. The Biochemical journal. 2007;405:307–317. doi: 10.1042/BJ20070209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen ND, Peng Y, Ho CC, Rideout HJ, Petrey D, Liu P, Dauer WT. The WD40 domain is required for LRRK2 neurotoxicity. PloS one. 2009;4:e8463. doi: 10.1371/journal.pone.0008463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kett LR, Dauer WT. Leucine-rich repeat kinase 2 for beginners: six key questions. Cold Spring Harbor perspectives in medicine. 2012;2:a009407. doi: 10.1101/cshperspect.a009407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein CL, Rovelli G, Springer W, Schall C, Gasser T, Kahle PJ. Homo- and heterodimerization of ROCO kinases: LRRK2 kinase inhibition by the LRRK2 ROCO fragment. Journal of neurochemistry. 2009;111:703–715. doi: 10.1111/j.1471-4159.2009.06358.x. [DOI] [PubMed] [Google Scholar]
- Korolchuk VI, Rubinsztein DC. Regulation of autophagy by lysosomal positioning. Autophagy. 2011;7:927–928. doi: 10.4161/auto.7.8.15862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korolchuk VI, Saiki S, Lichtenberg M, Siddiqi FH, Roberts EA, Imarisio S, Jahreiss L, Sarkar S, Futter M, Menzies FM, et al. Lysosomal positioning coordinates cellular nutrient responses. Nature cell biology. 2011;13:453–460. doi: 10.1038/ncb2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC, Clutario KM, Ling SC, Liang TY, Mazur C, et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nature neuroscience. 2012;15:1488–1497. doi: 10.1038/nn.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BD, Shin JH, VanKampen J, Petrucelli L, West AB, Ko HS, Lee YI, Maguire-Zeiss KA, Bowers WJ, Federoff HJ, et al. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson's disease. Nature medicine. 2010;16:998–1000. doi: 10.1038/nm.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Patel JC, Wang J, Avshalumov MV, Nicholson C, Buxbaum JD, Elder GA, Rice ME, Yue Z. Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson's disease mutation G2019S. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:1788–1797. doi: 10.1523/JNEUROSCI.5604-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Liu W, Oo TF, Wang L, Tang Y, Jackson-Lewis V, Zhou C, Geghman K, Bogdanov M, Przedborski S, et al. Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson's disease. Nature neuroscience. 2009;12:826–828. doi: 10.1038/nn.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao J, Wu CX, Burlak C, Zhang S, Sahm H, Wang M, Zhang ZY, Vogel KW, Federici M, Riddle SM, et al. Parkinson disease-associated mutation R1441H in LRRK2 prolongs the "active state" of its GTPase domain. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:4055–4060. doi: 10.1073/pnas.1323285111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Parisiadou L, Gu XL, Wang L, Shim H, Sun L, Xie C, Long CX, Yang WJ, Ding J, et al. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson's-disease-related mutant alpha-synuclein. Neuron. 2009;64:807–827. doi: 10.1016/j.neuron.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Dobson B, Glicksman MA, Yue Z, Stein RL. Kinetic mechanistic studies of wild-type leucine-rich repeat kinase 2: characterization of the kinase and GTPase activities. Biochemistry. 2010;49:2008–2017. doi: 10.1021/bi901851y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Kang S, Ray S, Jackson J, Zaitsev AD, Gerber SA, Cuny GD, Glicksman MA. Kinetic, mechanistic, and structural modeling studies of truncated wild-type leucine-rich repeat kinase 2 and the G2019S mutant. Biochemistry. 2011a;50:9399–9408. doi: 10.1021/bi201173d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Hamamichi S, Lee BD, Yang D, Ray A, Caldwell GA, Caldwell KA, Dawson TM, Smith WW, Dawson VL. Inhibitors of LRRK2 kinase attenuate neurodegeneration and Parkinson-like phenotypes in Caenorhabditis elegans and Drosophila Parkinson's disease models. Human molecular genetics. 2011b;20:3933–3942. doi: 10.1093/hmg/ddr312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Lee J, Krummey S, Lu W, Cai H, Lenardo MJ. The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nature immunology. 2011;12:1063–1070. doi: 10.1038/ni.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Wang X, Yu Y, Li X, Wang T, Jiang H, Ren Q, Jiao Y, Sawa A, Moran T, et al. A Drosophila model for LRRK2-linked parkinsonism. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:2693–2698. doi: 10.1073/pnas.0708452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Looyenga BD, Furge KA, Dykema KJ, Koeman J, Swiatek PJ, Giordano TJ, West AB, Resau JH, Teh BT, MacKeigan JP. Chromosomal amplification of leucine-rich repeat kinase-2 (LRRK2) is required for oncogenic MET signaling in papillary renal and thyroid carcinomas. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:1439–1444. doi: 10.1073/pnas.1012500108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52:587–593. doi: 10.1016/j.neuron.2006.10.008. [DOI] [PubMed] [Google Scholar]
- MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, McCabe BD, Marder KS, Honig LS, Clark LN, Small SA, et al. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson's disease risk. Neuron. 2013;77:425–439. doi: 10.1016/j.neuron.2012.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa T, Mori S, Sasaki Y, Miyajima T, Azuma S, Ohta E, Obata F. The I2020T Leucine-rich repeat kinase 2 transgenic mouse exhibits impaired locomotive ability accompanied by dopaminergic neuron abnormalities. Molecular neurodegeneration. 2012;7:15. doi: 10.1186/1750-1326-7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin I, Kim JW, Lee BD, Kang HC, Xu JC, Jia H, Stankowski J, Kim MS, Zhong J, Kumar M, et al. Ribosomal protein s15 phosphorylation mediates LRRK2 neurodegeneration in Parkinson's disease. Cell. 2014;157:472–485. doi: 10.1016/j.cell.2014.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta S, Van Kolen K, da Cunha R, van den Bogaart G, Mandemakers W, Miskiewicz K, De Bock PJ, Morais VA, Vilain S, Haddad D, et al. LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron. 2012;75:1008–1021. doi: 10.1016/j.neuron.2012.08.022. [DOI] [PubMed] [Google Scholar]
- Melrose HL, Dachsel JC, Behrouz B, Lincoln SJ, Yue M, Hinkle KM, Kent CB, Korvatska E, Taylor JP, Witten L, et al. Impaired dopaminergic neurotransmission and microtubule-associated protein tau alterations in human LRRK2 transgenic mice. Neurobiology of disease. 2010;40:503–517. doi: 10.1016/j.nbd.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills RD, Mulhern TD, Liu F, Culvenor JG, Cheng HC. Prediction of the repeat domain structures and impact of parkinsonism-associated variations on structure and function of all functional domains of leucine-rich repeat kinase 2 (LRRK2) Human mutation. 2014;35:395–412. doi: 10.1002/humu.22515. [DOI] [PubMed] [Google Scholar]
- Nguyen HN, Byers B, Cord B, Shcheglovitov A, Byrne J, Gujar P, Kee K, Schule B, Dolmetsch RE, Langston W, et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell stem cell. 2011;8:267–280. doi: 10.1016/j.stem.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikonova EV, Xiong Y, Tanis KQ, Dawson VL, Vogel RL, Finney EM, Stone DJ, Reynolds IJ, Kern JT, Dawson TM. Transcriptional responses to loss or gain of function of the leucine-rich repeat kinase 2 (LRRK2) gene uncover biological processes modulated by LRRK2 activity. Human molecular genetics. 2012;21:163–174. doi: 10.1093/hmg/ddr451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu J, Yu M, Wang C, Xu Z. Leucine-rich repeat kinase 2 disturbs mitochondrial dynamics via Dynamin-like protein. Journal of neurochemistry. 2012;122:650–658. doi: 10.1111/j.1471-4159.2012.07809.x. [DOI] [PubMed] [Google Scholar]
- Nuytemans K, Bademci G, Inchausti V, Dressen A, Kinnamon DD, Mehta A, Wang L, Zuchner S, Beecham GW, Martin ER, et al. Whole exome sequencing of rare variants in EIF4G1 and VPS35 in Parkinson disease. Neurology. 2013;80:982–989. doi: 10.1212/WNL.0b013e31828727d4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, Cortes E, Honig LS, Dauer W, Consiglio A, et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nature neuroscience. 2013;16:394–406. doi: 10.1038/nn.3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisiadou L, Xie C, Cho HJ, Lin X, Gu XL, Long CX, Lobbestael E, Baekelandt V, Taymans JM, Sun L, et al. Phosphorylation of ezrin/radixin/moesin proteins by LRRK2 promotes the rearrangement of actin cytoskeleton in neuronal morphogenesis. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:13971–13980. doi: 10.1523/JNEUROSCI.3799-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisiadou L, Yu J, Sgobio C, Xie C, Liu G, Sun L, Gu XL, Lin X, Crowley NA, Lovinger DM, et al. LRRK2 regulates synaptogenesis and dopamine receptor activation through modulation of PKA activity. Nature neuroscience. 2014;17:367–376. doi: 10.1038/nn.3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plowey ED, Cherra SJ, Liu YJ, 3rd, Chu CT. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. Journal of neurochemistry. 2008;105:1048–1056. doi: 10.1111/j.1471-4159.2008.05217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling SC, Sun E, Wancewicz E, Mazur C, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nature neuroscience. 2011;14:459–468. doi: 10.1038/nn.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajput A, Dickson DW, Robinson CA, Ross OA, Dächsel JC, Lincoln SJ, Cobb SA, Rajput ML, Farrerm MJ. Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology. 2006;67:1506–1518. doi: 10.1212/01.wnl.0000240220.33950.0c. [DOI] [PubMed] [Google Scholar]
- Ray S, Bender S, Kang S, Lin R, Glicksman MA, Liu M. The Parkinson Disease-linked LRRK2 Protein Mutation I2020T Stabilizes an Active State Conformation Leading to Increased Kinase Activity. The Journal of biological chemistry. 2014;289:13042–13053. doi: 10.1074/jbc.M113.537811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt P, Schmid B, Burbulla LF, Schondorf DC, Wagner L, Glatza M, Hoing S, Hargus G, Heck SA, Dhingra A, et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell stem cell. 2013;12:354–367. doi: 10.1016/j.stem.2013.01.008. [DOI] [PubMed] [Google Scholar]
- Ross OA, Toft M, Whittle AJ, Johnson JL, Papapetropoulos S, Mash DC, Litvan I, Gordon MF, Wszolek ZK, Farrer MJ, et al. Lrrk2 and Lewy body disease. Annals of neurology. 2006;59:388–393. doi: 10.1002/ana.20731. [DOI] [PubMed] [Google Scholar]
- Rudenko IN, Kaganovich A, Hauser DN, Beylina A, Chia R, Ding J, Maric D, Jaffe H, Cookson MR. The G2385R variant of leucine-rich repeat kinase 2 associated with Parkinson's disease is a partial loss-of-function mutation. The Biochemical journal. 2012;446:99–111. doi: 10.1042/BJ20120637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S, Guillily MD, Ferree A, Lanceta J, Chan D, Ghosh J, Hsu CH, Segal L, Raghavan K, Matsumoto K, et al. LRRK2 modulates vulnerability to mitochondrial dysfunction in Caenorhabditis elegans. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:9210–9218. doi: 10.1523/JNEUROSCI.2281-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nature genetics. 2009;41:1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- Saunders-Pullman R, Barrett MJ, Stanley KM, Luciano MS, Shanker V, Severt L, Hunt A, Raymond D, Ozelius LJ, Bressman SB. LRRK2 G2019S mutations are associated with an increased cancer risk in Parkinson disease. Movement disorders : official journal of the Movement Disorder Society. 2010;25:2536–2541. doi: 10.1002/mds.23314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte EC, Ellwanger DC, Dihanich S, Manzoni C, Stangl K, Schormair B, Graf E, Eck S, Mollenhauer B, Haubenberger D, et al. Rare variants in LRRK1 and Parkinson's disease. Neurogenetics. 2014;15:49–57. doi: 10.1007/s10048-013-0383-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen S, Webber PJ, West AB. Dependence of leucine-rich repeat kinase 2 (LRRK2) kinase activity on dimerization. The Journal of biological chemistry. 2009;284:36346–36356. doi: 10.1074/jbc.M109.025437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, et al. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nature genetics. 2009;41:1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibinski G, Nakamura K, Cookson MR, Finkbeiner S. Mutant LRRK2 toxicity in neurons depends on LRRK2 levels and synuclein but not kinase activity or inclusion bodies. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34:418–433. doi: 10.1523/JNEUROSCI.2712-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nature neuroscience. 2006;9:1231–1233. doi: 10.1038/nn1776. [DOI] [PubMed] [Google Scholar]
- Smith WW, Pei Z, Jiang H, Moore DJ, Liang Y, West AB, Dawson VL, Dawson TM, Ross CA. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:18676–18681. doi: 10.1073/pnas.0508052102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafa K, Trancikova A, Webber PJ, Glauser L, West AB, Moore DJ. GTPase activity and neuronal toxicity of Parkinson's disease-associated LRRK2 is regulated by ArfGAP1. PLoS genetics. 2012;8:e1002526. doi: 10.1371/journal.pgen.1002526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafa K, Tsika E, Moser R, Musso A, Glauser L, Jones A, Biskup S, Xiong Y, Bandopadhyay R, Dawson VL, et al. Functional interaction of Parkinson's disease-associated LRRK2 with members of the dynamin GTPase superfamily. Human molecular genetics. 2014;23:2055–2077. doi: 10.1093/hmg/ddt600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su YC, Qi X. Inhibition of excessive mitochondrial fission reduced aberrant autophagy and neuronal damage caused by LRRK2 G2019S mutation. Human molecular genetics. 2013;22:4545–4561. doi: 10.1093/hmg/ddt301. [DOI] [PubMed] [Google Scholar]
- Tain LS, Mortiboys H, Tao RN, Ziviani E, Bandmann O, Whitworth AJ. Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nature neuroscience. 2009;12:1129–1135. doi: 10.1038/nn.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JP, Hulihan MM, Kachergus JM, Melrose HL, Lincoln SJ, Hinkle KM, Stone JT, Ross OA, Hauser R, Aasly J, et al. Leucine-rich repeat kinase 1: a paralog of LRRK2 and a candidate gene for Parkinson's disease. Neurogenetics. 2007;8:95–102. doi: 10.1007/s10048-006-0075-8. [DOI] [PubMed] [Google Scholar]
- Taylor JP, Mata IF, Farrer MJ. LRRK2: a common pathway for parkinsonism, pathogenesis and prevention? Trends in molecular medicine. 2006;12:76–82. doi: 10.1016/j.molmed.2005.12.004. [DOI] [PubMed] [Google Scholar]
- Taymans JM, Vancraenenbroeck R, Ollikainen P, Beilina A, Lobbestael E, De Maeyer M, Baekelandt V, Cookson MR. LRRK2 kinase activity is dependent on LRRK2 GTP binding capacity but independent of LRRK2 GTP binding. PloS one. 2011;6:e23207. doi: 10.1371/journal.pone.0023207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature. 2012;485:109–113. doi: 10.1038/nature11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, Konig J, Hortobagyi T, Nishimura AL, Zupunski V, et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nature neuroscience. 2011;14:452–458. doi: 10.1038/nn.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Giaime E, Yamaguchi H, Ichimura T, Liu Y, Si H, Cai H, Bonventre JV, Shen J. Loss of leucine-rich repeat kinase 2 causes age-dependent bi-phasic alterations of the autophagy pathway. Molecular neurodegeneration. 2012;7:2. doi: 10.1186/1750-1326-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Pisani A, Martella G, Karouani M, Yamaguchi H, Pothos EN, Shen J. R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:14622–14627. doi: 10.1073/pnas.0906334106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Yamaguchi H, Giaime E, Boyle S, Kopan R, Kelleher RJ, Shen J., 3rd Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:9879–9884. doi: 10.1073/pnas.1004676107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trancikova A, Mamais A, Webber PJ, Stafa K, Tsika E, Glauser L, West AB, Bandopadhyay R, Moore DJ. Phosphorylation of 4E-BP1 in the mammalian brain is not altered by LRRK2 expression or pathogenic mutations. PloS one. 2012;7:e47784. doi: 10.1371/journal.pone.0047784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Yan MH, Fujioka H, Liu J, Wilson-Delfosse A, Chen SG, Perry G, Casadesus G, Zhu X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Human molecular genetics. 2012;21:1931–1944. doi: 10.1093/hmg/dds003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webber PJ, Smith AD, Sen S, Renfrow MB, Mobley JA, West AB. Autophosphorylation in the leucine-rich repeat kinase 2 (LRRK2) GTPase domain modifies kinase and GTP-binding activities. Journal of molecular biology. 2011;412:94–110. doi: 10.1016/j.jmb.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Choi C, Andrabi SA, Li X, Dikeman D, Biskup S, Zhang Z, Lim KL, Dawson VL, et al. Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Human molecular genetics. 2007;16:223–232. doi: 10.1093/hmg/ddl471. [DOI] [PubMed] [Google Scholar]
- Westerlund M, Belin AC, Anvret A, Bickford P, Olson L, Galter D. Developmental regulation of leucine-rich repeat kinase 1 and 2 expression in the brain and other rodent and human organs: Implications for Parkinson's disease. Neuroscience. 2008;152:429–436. doi: 10.1016/j.neuroscience.2007.10.062. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Coombes CE, Kilaru A, Li X, Gitler AD, Bowers WJ, Dawson VL, Dawson TM, Moore DJ. GTPase activity plays a key role in the pathobiology of LRRK2. PLoS genetics. 2010;6:e1000902. doi: 10.1371/journal.pgen.1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Yuan C, Chen R, Dawson TM, Dawson VL. ArfGAP1 is a GTPase activating protein for LRRK2: reciprocal regulation of ArfGAP1 by LRRK2. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:3877–3886. doi: 10.1523/JNEUROSCI.4566-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]