

Abstract
Background
Sarcomere protein mutations in hypertrophic cardiomyopathy (HCM) induce subtle cardiac structural changes prior to the development of left ventricular hypertrophy (LVH). We have proposed that myocardial crypts are part of this phenotype and independently associated with the presence of sarcomere gene mutations. We tested this hypothesis in genetic HCM pre-LVH (G+LVH−).
Methods and Results
A multi-centre case-control study investigated crypts and 22 other cardiovascular magnetic resonance (CMR) parameters in subclinical HCM to determine their strength of association with sarcomere gene mutation carriage. The G+LVH− sample (n=73) was 29±13 years old and 51% male. Crypts were related to the presence of sarcomere mutations (for ≥1 crypt, β=2.5, 95% confidence interval [CI] 0.5-4.4, p=0.014; for ≥2 crypts, β=3.0, 95%CI 0.8-7.9, p=0.004). In combination with 3 other parameters: anterior mitral valve leaflet (AMVL) elongation (β=2.1, 95%CI 1.7-3.1, p<0.001), abnormal LV apical trabeculae (β=1.6, 95%CI 0.8-2.5, p<0.001), and smaller LV end-systolic volumes (β=1.4, 95%CI 0.5-2.3, p=0.001), multiple crypts indicated the presence of sarcomere gene mutations with 80% accuracy and an area under the curve of 0.85 (95%CI 0.8-0.9). In this G+LVH− population cardiac myosin-binding protein C mutation carriers had twice the prevalence of crypts when compared to the other combined mutations (47 vs. 23%; odds ratio, 2.9; 95%CI 1.1–7.9; p=0.045).
Conclusions
The subclinical HCM phenotype measured by CMR in a multi-center environment and consisting of crypts (particularly multiple), AMVL elongation, abnormal trabeculae and smaller LV systolic cavity, is indicative of the presence of sarcomere gene mutations and highlights the need for further study.
Keywords: hypertrophic cardiomyopathy, genetics, magnetic resonance imaging
Hypertrophic cardiomyopathy (HCM) is a common hereditable heart disease with a population prevalence of 1 in 500 and affected individuals are at risk of adverse outcomes, including sudden cardiac death1 and progression to heart failure. HCM is frequently caused by dominant mutations in sarcomere protein genes; present in 30-60 % of patients.2 Due to the growing availability of advanced imaging, myocardial architectural abnormalities can be investigated in subclinical HCM sarcomere gene mutation carriers before the development of left ventricular hypertrophy (genotype-positive, LVH-negative: G+LVH−). Initially reported separately and in single-center studies, some of the cardiac abnormalities of subclinical HCM (myocardial crypts,3 anterior mitral valve leaflet [AMVL] elongation,4 abnormal LV apical trabeculae5 and smaller LV systolic cavity6) have recently been shown to cluster into an identifiable phenotype by cardiovascular magnetic resonance (CMR)5 with emerging data suggesting that crypts may be an important morphological feature in genetic HCM.3,7 Here we present a multi-center study exploring crypts and other morphological features in subclinical HCM to identify the parameters that are most strongly indicative of the presence of a sarcomere gene mutation before the development of LVH.
Methods
Investigator consortium
A multi-center collaboration established the largest cohort to date, of genotyped, LVH− HCM sarcomere gene mutation carriers (n = 73) with comprehensive CMR imaging. Using a matched case-control design, G+LVH− and healthy controls underwent CMR and electrocardiographic characterization. Twenty-four CMR parameters (2 of which related to crypts) were explored for potential association with the presence of sarcomere mutations. The 12-centers included University College London (UCL, The Heart Hospital) and a further 11 US centers forming part of the HCMNet Hypertrophic Cardiomyopathy clinical network (Supplemental Note 1). The UK component of the G+LVH− population (n = 39) has been previously described.5 HCMNet is comprised of Boston Children’s Hospital, Brigham and Women’s Hospital, Cincinnati Children’s Hospital Medical Center, Cleveland Clinic Florida, Johns Hopkins Hospital, Stanford University Hospital, St. Luke’s Roosevelt Hospital Center, University of Chicago Medical Center, University of Colorado, University of Michigan Cardiovascular Center, Washington University in St. Louis. Imaging data along with all relevant clinical and demographic variables were collected and anonymized by investigators at the respective US and UK institutions. The study protocol was approved by the local institutional review boards. Written informed consent was obtained from all participants or their parents or legal guardians according to the ethics committee guidelines at the respective academic centers.
Study population
Inclusion and exclusion criteria for patients and healthy controls were defined a priori. Inclusion criteria for G+LVH− were: i) maximal LV wall thickness < 13 mm by CMR in subjects ≥18 years; maximal LV wall thickness < 12 mm or z score < 2.5 by echocardiography in subjects < 18 years;8 ii) LV mass within the normal range relative to body surface area (BSA), age and gender; iii) sinus rhythm, no LVH and no pathological Q waves/T-wave inversion on 12-lead electrocardiogram; iv) no causes of secondary LVH such as valve disease, or systemic hypertension that was defined as systolic blood pressure > 140 mmHg and/or diastolic blood pressure > 90 mmHg, or the use of medical therapy for hypertension. Controls had no other medical conditions and were healthy volunteers or relatives without the sarcomere gene mutation. Controls were recruited from 5 centers and matched to unrelated G+LVH− patients on the basis of age (± 8 years), gender, BSA (± 10 %) and ethnicity in a 1:1 ratio. Controls had no personal history of cardiovascular disease, unexplained syncope or systemic hypertension and a normal physical examination. Exclusion criteria for all participants were the presence of conventional contra-indications for CMR, claustrophobia and a high arrhythmogenic burden (e.g. atrial fibrillation, frequent ectopics) that precluded good cine acquisitions.
Electrocardiography
Standard 12-lead electrocardiography was performed in the supine position during quiet respiration in all recruited participants. LVH was evaluated with the Romhilt-Estes criteria9,10 (Supplemental Table 1). Within the respective institutions electrocardiograms were analyzed by experienced observers, blinded to clinical information. No G+LVH− had pathological Q waves (defined as duration > 40 ms or depth > ⅓ R wave in ≥ 2 leads) or T wave inversion (defined as ≥ 3 mm in ≥ 2 leads).
Genetic screening
All 73 G+LVH− underwent genetic testing to identify DNA sequence variants in sarcomere genes. Non-synonymous pathogenic and likely pathogenic variants were selected on frequency (< 0.5 % based on the 1000 Genomes Database11), evolutionary conservation, previous reports in the literature, putative functional consequence,12 and cosegregation in the family, when available. G+LVH− were classified as carrying a mutation in one of the following sarcomere genes:13 tropomyosin 1 alpha chain (TPM1); actin, alpha cardiac muscle 1 (ACTC1); myosin-binding protein C, cardiac type (MYBPC3); myosin heavy chain, cardiac muscle beta isoform (MYH7); myosin regulatory light chain 2, ventricular/cardiac muscle isoform (MYL2); myosin light polypeptide 3 (MYL3); troponin T, cardiac muscle (TNNT2); troponin I, cardiac muscle (TNNI3).
Cardiovascular magnetic resonance
CMR consisting of standard clinical scans was performed on all recruited patients and controls using 1.5 and 3-Tesla magnets from different manufacturers (Avanto/Tim Trio, Siemens Medical Solutions, Erlangen, Germany; Signa Excite; General Electric Medical Systems, Waukesha, WI, USA; Achieva, Philips, Amsterdam, The Netherlands). Retrospectively electrocardiographically-gated, breath-held, long- and short-axis cine images were acquired using a steady-state free precession sequence. Scan parameters used for Siemens, General Electric and Philips scanners are provided in Supplemental Table 2. LV volumes, ejection fraction and LV mass were determined according to standardized CMR methods14 at The Heart Hospital and at the National Institutes of Health Clinical Center, with the latter serving as the CMR core lab for HCMNet. Specifically LV end-systolic volume (LVESV; ml) was measured from endocardial tracings applied to the sequential short-axis cines covering the entire ventricle. To adjust for participant gender and age (wide range in this study), measured BSA-indexed LVESVs were converted to normalized ratios (LVESViR; Supplemental Note 2). LV wall thickness was calculated per segment on end-diastolic and end-systolic short-axis cine frames. The structure of the LV was evaluated for crypts by a cardiologist with > 3 years of experience in CMR (G.C.). In the absence of a general consensus on the definition of a crypt, we defined a myocardial crypt as the presence of a focal myocardial defect in diastole, showing at least partial systolic obliteration, and having a depth ≥ 50 % the thickness of the adjacent myocardium. Anything less than this (for example a partial crypt, < 50 % thickness) was not counted. Standard long-axis cines (with cross-validation on the short-axis cines, where appropriate) were used to locate crypts without the modified 2-chamber view. In two equivocal cases the presence or absence of crypts was resolved by consensus. Two readers (G.C. and G.F.) blinded to clinical diagnosis evaluated 50 CMR scans pertaining to a mix of cases and controls for calculation of the intra- and inter-observer variability in crypt rulings (scored as absent, single or multiple crypts). Raw AMVL length was estimated using the method described by Maron et al4 and adjusted for body size by dividing by BSA. Trabeculation was quantified using the technique of fractal analysis. The theory behind the technique is described in Figure 1. Experiments to test dependency of the fractal dimension (FD) on slice-thickness and magnetic field strength are described in Supplemental Note 3. Briefly, FD for an endocardial contour (a 2-dimensional object) is a non-integer value between 1 (least complex) and 2 (most complex) such that previously published5,15 indices for LV maximal apical fractal dimension (FDMaxApical) in healthy controls were 1.199 ± 0.05, and in LV noncompaction, 1.392 ± 0.01. Validation and repeatability experiments for the method have been previously published.5,15
Figure 1.
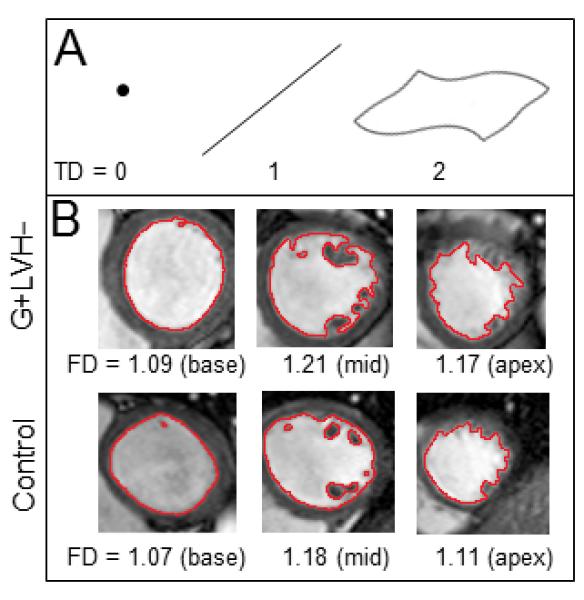
A) Exact mathematical fractals (for example, the popular Mandelbrot set) are complex objects that show scaling self-similarity. Their complexity cannot be efficiently summarized using traditional Euclidean geometry. Fractal geometry is based on a mathematical construct and has the ability to measure complex objects. Real-world biology and some naturally-occurring images may also be complex and exhibit self-similarity within a finite range. As an illustration, perfect points have a TD of 0, straight lines or smooth regular curves have a TD of 1, flat planes have a TD of 2. Biological images tend to have non-linear features of dimension greater than 1: in this case endocardial contours may be regarded as non-linear quasi-fractal forms observed on the 2-dimensional imaging plane. Their FD is therefore any noninteger value between 1 and 2. B) Top row shows 3 exemplar slices from a G+LVH− with corresponding FD. Bottom row shows slices from a control.
G+LVH− = genotype-positive, LVH-negative; FD = fractal dimension; TD = topological dimension.
Statistical analysis
Statistical analysis was performed in R programming language (version 3.0.1, The R Foundation for Statistical Computing). Descriptive data are expressed as mean ± standard deviation except where otherwise stated. Distribution of data was assessed on histograms and using Shapiro-Wilk test. Categorical variables were compared by χ2 or Fisher’s exact tests. Normally distributed continuous variables pertaining to patients and controls were compared using paired t-Test. Univariable conditional logistic regression models were created to estimate the strength of the association between potential predictors and HCM sarcomere gene mutation carriage. These analyses were repeated to account for family relations amongst G+LVH− after randomly retaining only one subject per family (Supplemental Tables 3 and 4). On account of complete separation of the highly significant variable ‘multiple crypts’ conditional logistic regression was not feasible so we performed a Firth-type16 unconditional matched logistic regression as per Heinze et al.17 In this model, significant factors from the univariable analysis were removed one at a time using backwards elimination, starting with the factor that had the largest p value, until all remaining factors had a two-sided p value < 0.05. Additionally a variance inflation factor of < 3 excluded near-multicollinearity. Beta coefficients of model variables were used to compile the prediction rule in this case-control population through an integer score system that assigned a weight based on the magnitude of the coefficient. For the optimal model, we determined the rate of correct classification, i.e. accuracy, using leave one out cross-validation18 and by constructing contingency tables. Optimal threshold values for AMVL length, FDMaxApical, and LVESViR were calculated as the Youden Index derived from the area under the receiver operating characteristics curves. A two-sided p value < 0.05 was considered significant. Cohen’s Kappa was used to compare variability of crypt rulings between readers.
Results
Clinical and demographic information for G+LVH− and controls are provided in Table 1 and provenance information in relation to the individual HCMNet centre is provided in Supplemental Note 1. Of the 73 matched controls, 71 were healthy volunteers and 2 were genotype-negative healthy relatives of HCM probands, the latter with confirmed pathological sarcomere mutations. Children and adolescents (< 18 years) comprised 19 % of the population. The 73 G+LVH− patients were all genotyped and they pertained to 62 unrelated families expressed a total of 44 unique sarcomere gene mutation variants. Prevalence of sarcomere protein gene mutations is shown in Table 1 and a full listing of variants provided in Supplemental Table 1. Of the 24 CMR parameters studied, 11 had significant association with carrier status on univariable logistic regression (Table 2). Differences in AMVL length between G+LVH− and controls persisted for the body size-adjusted variable, and BSA-adjusted AMVL length retained association with genetic status on univariable logistic regression.
Table 1.
Demographic characteristics of G+LVH− and controls and comparison of the 24 CMR functional and morphological parameters studied.
| Variable | G+LVH- (n = 73) |
Controls (n = 73) |
p-Value* |
|---|---|---|---|
| Age (yrs) | 29 (± 13) | 30 (± 11) | — |
| Male/Female | 37/36 | 37/36 | — |
| Ethnicity † | A = 72 | A = 72 | — |
| B = 1 | B = 1 | — | |
| BSA (m2) | 1.79 (± 0.23) | 1.81 (± 0.19) | — |
| Causal gene n (%)‡ | |||
| MYBPC3 | 31 (42) | NA | |
| MYH7 | 23 (31) | NA | |
| TNNT2 | 7 (10) | NA | |
| TNNI3 | 9 (12) | NA | |
| MYL3 | 1 (1) | NA | |
| ACTCl | 3 (4) | NA | |
| LVEDV (ml) | 129 (± 26) | 138 (± 28) | 0.062 |
| LVEDVi (ml/m2) | 72 (± 11) | 76 (± 12) | 0.051 |
| LVESV (ml) | 42 (± 12) | 48 (± 14) | 0.002 |
| LVESVi (ml/m2) | 23 (± 6) | 26 (± 7) | 0.002 |
| LVESViR | 0.81 (± 0.19) | 0.94 (± 0.24) | < 0.001 |
| EF (%) | 68 (± 6) | 66 (± 6) | 0.018 |
| Mass (g) | 110 (± 33) | 117 (± 32) | 0.199 |
| Massi (g/m2) | 61 (± 13) | 64 (± 15) | 0.165 |
| SV (ml) | 87 (± 18) | 90 (± 17) | 0.274 |
| SWTd (mm) | 9.1 (± 1.9) | 8.6 (± 1.4) | 0.061 |
| PWTd (mm) | 7.9 (± 2.3) | 7.2 (± 1.9) | 0.056 |
| SWTs (mm) | 13.1 (± 2.8) | 11.8 (± 1.9) | < 0.001 |
| PWTs (mm) | 13.1 (± 2.8) | 12.1 (± 2.4) | 0.025 |
| SdPdR | 1.21 (± 0.33) | 1.24 (± 0.30) | 0.511 |
| SsSdR | 1.45 (± 0.18) | 1.39 (± 0.21) | 0.105 |
| PsPdR | 1.72 (± 0.40) | 1.74 (± 0.38) | 0.768 |
| LA area (cm2) | 18.4 (± 4.0) | 18.9 (± 2.9) | 0.331 |
| LA areai (cm2/m2) | 10.3 (± 1.7) | 10.6 (± 1.7) | 0.291 |
| AMVL (mm) | 23.1 (± 2.9) | 20.6 (± 3.0) | < 0.0001 |
| AMVL/BSA (mm/m2) | 13.0 (± 2.2) | 11.7 (± 2.3) | 0.0008 |
| FDMaxApical | 1.251 (± 0.073) | 1.195 (± 0.052) | < 0.0001 |
| ≥ 1 Crypt Present (%) | 24 (33) | 2 (3) | < 0.0001 |
| ≥ 2 Crypts Present (%) | 14 (19) | 0 (0) | < 0.0001 |
Characteristics that differed significantly (p < 0.05) between G+LVH− and controls are highlighted in bold. Where matching took place (age, gender, BSA and ethnicity) p values are not significant and represented as —.
Ethnic groups were self-defined and headings are in accordance with UK Office for National Statistics guidance on national standards: A, White; B, Black (or Black British).
There was one multiple mutation carrier (MYBPC3 + TNNI3).
ACTCl = actin, alpha cardiac muscle 1; AMVL = anterior mitral valve leaflet; BSA = body surface area; EF = ejection fraction; LA areai = left atrial area indexed to BSA; LVEDV = left-ventricular end-diastolic volume; LVESVir = BSA-normalized left ventricular end-systolic volume corrected for age and gender; Massi = LV mass indexed to BSA; FDMaxApicai = maximal apical fractal dimension; MYBPC3 = myosin-binding protein C, cardiac type; MYH7 = myosin heavy chain, cardiac muscle beta isoform; MYL3 = myosin light polypeptide 3; NA = not applicable, PsPdR = systolic posterior to diastolic posterior wall thickness ratio; PWTd/s = maximal posterior wall thickness in diastole/systole; SdPdR = diastolic septal to diastolic posterior wall thickness ratio; SsSdR = systolic septal to diastolic septal wall thickness ratio; SV = stroke volume; SWTd/s = maximal septal wall thickness in diastole/systole; TNNT2 = troponin T, cardiac muscle; TNNI3 = troponin I, cardiac muscle.
Table 2.
Univariable logistic regression showing strength of association between the 11 significant predictors and hypertrophic cardiomyopathy sarcomere gene mutation carriage.
| Variable* | Crude OR (95% CI) | p-Value (Wald χ2) |
|---|---|---|
| ≥ 2 Crypts Present (Y/N)† | 35.82 (4.62, 4612) | < 0.001 |
| ≥ 1 Crypt Present (Y/N) | 12.00 (2.84, 50.77) | < 0.001 |
| FDMaxApical‡ | 5.19 (2.35, 11.43) | < 0.001 |
| AMVL (mm) | 1.31 (1.14, 1.51) | < 0.001 |
| LVESViR | 0.05 (0.01, 0.32) | 0.002 |
| SWTs (mm) | 1.33 (1.11, 1.60) | 0.002 |
| AMVL/BSA (mm/m2) | 0.76 (0.64, 0.91) | 0.003 |
| LVESVi (ml/m2) | 0.91 (0.85, 0.97) | 0.004 |
| LVESV (ml) | 0.95 (0.92, 0.98) | 0.004 |
| EF (%) | 1.08 (1.01, 1.16) | 0.023 |
| PWTs (mm) | 1.17 (1.01, 1.36) | 0.032 |
Variables are sorted in descending order of significance of p value.
Firth’s bias-controlled logistic regression was used for variable ‘≥ 2 Crypts Present’ to account for complete separation. Estimates for the other parameters were derived by fitting a univariable conditional logistic regression.
Coefficients are expressed for each 0.1 unit change in FDMaxApical.
CI = confidence interval; χ2 = Chi-squared; OR = odds ratio; Y/N = yes (present)/no (absent). Other abbreviations as in Table 1.
Twenty-four G+LVH− (33 % of 73; Figure 2) and 2 controls (3 % of 73) had ≥ 1 myocardial crypt. Fourteen G+LVH− (19 %) and no controls (0 %) had ≥2 crypts. For G+LVH− 2 crypts were counted in 5 participants; 3 crypts in 8; and 4 crypts in 1. Compared to the other combined mutations in G+LVH− (Figure 3), MYBPC3 mutation carriers had twice the prevalence of crypts (23 % vs. 47 % respectively; odds ratio [OR], 2.88; 95 % confidence interval [CI], 1.05 – 7.91; p = 0.045) and were less likely to demonstrate LV systolic cavity reduction (LVESViiR < 0.803 in 61 % of non-MYBPC3 vs. 33 % in MYBPC3; OR, 0.33; 95 % CI, 0.12 – 0.87; p = 0.032). The relationship between specific mutation variants and crypt prevalence is illustrated in Figure 4. Reproducibility for crypt rulings was high (intra-observer Kappa, 0.92; inter-observer Kappa, 0.85).
Figure 2.

Myocardial crypts (black carets) by cardiovascular magnetic resonance in the 24 carriers. Some had ≥ 1 additional crypt visible on a separate cine but single cines per participant are reproduced here.
Views: * 4-chamber; ** left ventricular outflow tract; remainder are 2-chamber.
Figure 3.
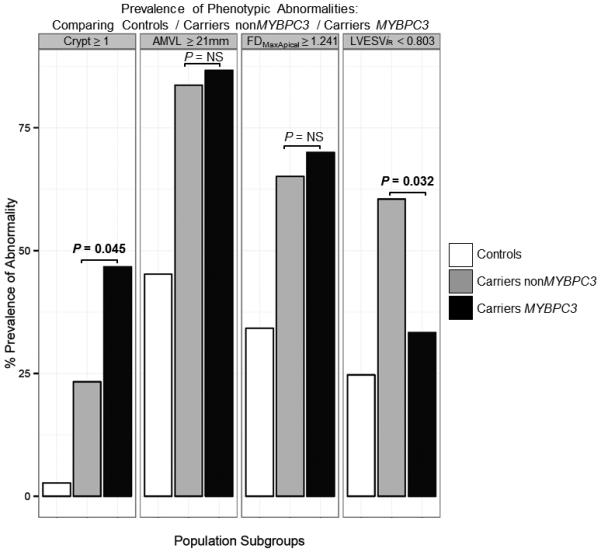
Bar chart comparing the prevalence of the 4 phenotypic abnormalities between controls, carriers bearing non-MYBPC3 mutations and carriers bearing the MYBPC3 mutation.
AMVL = anterior mitral valve leaflet; LVESViR = left ventricular end-systolic volume adjusted for age, body surface area and gender; FDMaxApical = maximal apical fractal dimension; MYBPC3 = myosin-binding protein C, cardiac type; NS = not significant.
Figure 4.

Simplified diagram depicting the major proteins of the thick and thin filaments and the distribution of sarcomere gene mutations expressed in our study population and reported here by DNA change and amino acid change nomenclature. For MYBPC3 only, mutation variants are mapped to individual domains that are displayed in a hypothetical arrangement extending from the thick to the thin filament. This figure is an adaptation of the illustration by Harris et al.13 with the permission of Wolters Kluwer Health).
Key to transcript change per mutation variant: Blue border = missense mutations that cause single amino acid substitutions; Green border = insertions or deletions predicted to cause reading frame shifts (fs); Red border = nonsense mutations predicted to result in premature termination codons (ter); Pink borders = splice site donor/acceptor mutations.
Key to mean crypt prevalence per mutation variant: Mutation fill colors are weighted across a spectrum from white (0 crypts) to deep burgundy (3 crypts).
ACTC1 = actin, alpha cardiac muscle 1; MYH7 = myosin heavy chain, cardiac muscle beta isoform; MYL2 = myosin regulatory light chain 2, ventricular/cardiac muscle isoform; MYL3 = myosin light polypeptide 3; m = position of the MYBPC3 regulatory motif between domains C1 and C2; PA = proline/alanine-rich linker sequence between C0 and C1; TNNT2 = troponin T, cardiac muscle;TNNI3 = troponin I, cardiac muscle. Other abbreviations as in Figure 2.
The final multivariable model consisting of 4 key imaging markers is summarized in Table 3 and Figure 5. The variable ‘≥ 1 crypt’ was also associated with the presence of sarcomere mutations (see Supplemental Table 5) but ‘multiple crypts’ (≥ 2) provided the better model (leave one out cross-validation model accuracy of 79 %). For the prediction rule applied to this case-control population, multiple crypts and elongated AMVL were each assigned a score of ‘2’, while increased LV apical trabeculation and smaller LVESViR were each assigned a score of ‘1’. A combined score of ≥ 3 represented the optimal cut-off for the prediction rule, achieving 79.5 % agreement with genetic diagnosis in this case-control population (positive predictive value: 82.1 %, 95 % CI 70.4 – 90.0; negative predictive value, 77.2 %, 95 % CI 66.1 – 85.6).
Table 3.
Final model by logistic regression using Firth's penalized maximum likelihood method. Derived cut-offs for model variables are also shown. Based on this model a combined prediction score of ≥ 3 denotes G+LVH− status.
| Variable* | β Coefficient | SE of β | Adjusted | χ 2 | p-Value | Prediction | Youden-Derived | % Of Subjects Meeting Cut-off | |
|---|---|---|---|---|---|---|---|---|---|
| (95% CI) | OR | Score | Cut-off | G+LVH− | Controls | ||||
| ≥ 2 Crypts Present | 3.01 | 1.59 (0.79, 7.92) | 20.38 | 8.37 | 0.004 | 2 | ≥ 2 crypts | 19 | 0 |
| AMVL | 2.09 | 0.50 (1.71, 3.13) | 8.08 | 22.08 | < 0.001 | 2 | ≥ 21 mm | 85 | 45 |
| FDMaxApical | 1.61 | 0.45 (0.75, 2.52) | 4.98 | 14.16 | < 0.001 | 1 | ≥ 1.241 | 55 | 16 |
| LVESViR | 1.35 | 0.44 (0.51, 2.26) | 3.87 | 10.25 | 0.001 | 1 | < 0.803 | 55 | 30 |
Figure 5.



A) The 4 cardiac structural and functional parameters shown to have significant independent association with the presence of sarcomere gene mutations in subclinical hypertrophic cardiomyopathy. B) Receiver operating characteristics (ROC) curve containing the 4 parameters and using as a reference, patient classification according to study criteria for inclusion of carriers and controls, showed an area under the curve (AUC) of 0.85. Diagonal reference line is also provided. C) In this case-control population we used a 2x2 contingency table to calculate the percentage of rulings that agreed with genetic diagnosis and obtained a sensitivity of 75% (95 % confidence intervals [CI] 64 – 84) and specificity, 84%, (95 % CI 73 - 91).
Other abbreviations as in Figure 1.
Discussion
Prior to the development of LVH, HCM sarcomere gene mutation carriers exhibit a subclinical cardiac phenotype that is detectable by CMR in a multi-center environment (3 scanner manufactures, 2 MR field strengths). The combined presence of ≥ 2 myocardial crypts, ≥ 21 mm AMVL length, increased LV apical trabecular complexity with FDMaxApical ≥ 1.241 and smaller LV end-systolic volume with LVESViR < 0.803, is indicative of a subclinical HCM phenotype in sarcomere gene mutation carriers. Of these, myocardial crypts and AMVL elongation emerge as the two parameters that most strongly associate with the presence of sarcomere gene mutations. Our initial observations also suggest that MYBPC3 mutation carriers have a twofold prevalence of crypts and less LV systolic cavity reduction compared to the other combined mutations – intriguing preliminary findings that are hypothesis-generating and merit evaluation in larger future studies.
American and European guidelines on family screening for HCM19,20 recommend genotyping if a pathogenic mutation has been identified in the family. However many probands do not have definitive results with genetic testing21,22 and such data cannot be used to identify at-risk relatives in family screening.23 Further prospective work is needed to explore the added value of CMR imaging rules in these situations.
The definition of myocardial crypts is not yet settled, making comparisons of prevalence between studies difficult. Single myocardial crypts do occur in the non-HCM population, with prevalences of 2.2 % by Srichai et al.,24 6 % by Brouwer et al.,7 7 % by Johansson et al.,25 8.7 % by Petryka et al.,26 and in this study, 2.7%. However, Johansson et al.25 and our study showed how multiple crypts remain an unusual finding in truly healthy hearts (0.8 % and 0% respectively). Erol et al.27 reported a slightly higher prevalence of multiple crypts (49 out of 2093 participants, 2%) but they used contrast computed tomography, acquired a 3-dimensional whole-heart dataset, and studied clinically-referred cardiac patients that included pre-operative evaluation scans and assessment of congenital heart disease. In G+LVH− HCM, multiple myocardial crypts are more prevalent than in the general population: ≥ 2 crypts were identified in 58 % of G+LVH− by Maron et al.28 (11 out of 31), and in 19 % of our G+LVH−. However multiple crypts have also been observed by Brouwer et al.7 in 16 % (9 out of 57) of patients with non-ischaemic (non-HCM) cardiomyopathy, suggesting that crypts in different contexts may have alternative significance.29
Previous studies4,5 have reported on the finding of abnormally elongated AMVL in G+LVH−. Here we show how differences in AMVL length persist even after adjustment for body size. After multiple myocardial crypts, AMVL elongation has the second strongest association with the presence of sarcomere gene mutations. The finding of abnormal trabecular architecture in the apical half of the LV, already described in the UK G+LVH− cohort,5 survived in this multi-center environment where FDMaxApical emerged as the third strongest independent predictor. The mechanism for this remains unclear but our hypothesis is that the hypertrabeculation, like the crypts, represents persistence of the embryological form into adulthood (a neoteny). Other interesting possibilities for the trabecular changes have also been previously discussed.5 The finding of smaller LV systolic cavities in G+LVH− compared to controls similarly accords with published data: compared to controls, G+LVH− had almost 10 ml smaller LVESV30 by CMR and higher ejection fraction31 by echocardiography (71 ± 6 vs. 64 ± 5, p < 0.0001) in the studies by Ho et al. Likewise, Sabe et al.32 showed significantly higher ejection fraction (61 ± 4 vs. 58 ± 4) by echocardiography in G+LVH− compared to controls.
Limitations are that some of the G+LVH− patients were related but we show how findings are independent of the degree of relatedness (see Supplemental Note 4 and Supplemental Tables 4 and 5). Diastolic function and T1 mapping differences30,31 have been observed in subclinical HCM but were not studied here. Future studies should attempt to revisit these parameters systematically and using standardized protocols to build on our present findings. External validation of the CMR imaging rule will help evaluate the fitness of the model and the generalizability of these first multi-center results, independent of any selection bias that may potentially be confounding participant selection in this study. Such validation would also be important to further test the performance of the model and to ensure that the rule is not driven by idiosyncracies in the 73 matched pairs. We used 3 long-axis views, cross-referenced against the short-axis images where appropriate but no modified views for crypt detection. Further work is needed to understand the embryological origins of crypts, the role of both crypts and the other features, in HCM that has not been genetically defined, and the predictive power of the subclinical phenotype regarding both, future LVH development, and clinical outcomes.
In conclusion, the subclinical HCM phenotype measured by CMR in a multi-center environment, and consisting of crypts (particularly multiple), AMVL elongation, abnormal trabeculae and smaller LV systolic cavity, is indicative of the presence of sarcomere gene mutations and highlights the need for further study.
Supplementary Material
Acknowledgements
We thank collaborating US institutions in the HCMNet for Information Management Services and assistance with development and curation of the HCM study and image database.
Sources of Funding
This work was funded by: University College London Institute of Life Sciences’ Charlotte and Yule Bogue Research Fellowship; European Union Science and Technology (Research and Innovation) program; National Institutes of Health intramural research program; and Gulbenkian Doctoral Programme for Advanced Medical Education, sponsored by Fundação Calouste Gulbenkian, Fundaçc̃o Champalimaud, Ministério da Saúde and Fundação para a Ciência e Tecnologia, Portugal, and supported by: National Institute for Health Research University College London Hospitals Biomedical Research Center; and National Heart, Lung and Blood Institute at the National Institutes of Health.
Footnotes
Disclosures
None.
References
- 1.Maron BJ, Shirani J, Poliac LC, Mathenge R, Roberts WC, Mueller FO. Sudden death in young competitive athletes. Clinical, demographic, and pathological profiles. JAMA. 1996;276:199–204. [PubMed] [Google Scholar]
- 2.Maron BJ, Seidman JG, Seidman CE. Proposal for contemporary screening strategies in families with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44:2125–32. doi: 10.1016/j.jacc.2004.08.052. [DOI] [PubMed] [Google Scholar]
- 3.Deva DP, Williams LK, Care M, Siminovitch KA, Moshonov H, Wintersperger BJ, Rakowski H, Crean AM. Deep basal inferoseptal crypts occur more commonly in patients with hypertrophic cardiomyopathy due to disease-causing myofilament mutations. Radiology. 2013;269:68–76. doi: 10.1148/radiol.13122344. [DOI] [PubMed] [Google Scholar]
- 4.Maron MS, Olivotto I, Harrigan C, Appelbaum E, Gibson CM, Lesser R, Haas TS, Udelson JE, Manning WJ, Maron BJ. Mitral valve abnormalities identified by cardiovascular magnetic resonance represent a primary phenotypic expression of hypertrophic cardiomyopathy. Circ. 2011;124:40–7. doi: 10.1161/CIRCULATIONAHA.110.985812. [DOI] [PubMed] [Google Scholar]
- 5.Captur G, Lopes L, Patel V, Li C, Bassett P, Syrris P, Sado DM, Maestrini V, Mohun TJ, McKenna WJ, Muthurangu V, Elliott PM, Moon JC. Abnormal cardiac formation in hypertrophic cardiomyopathy - fractal analysis of trabeculae and preclinical gene expression. Circ Cardiovasc Genet. 2014;7:241–8. doi: 10.1161/CIRCGENETICS.113.000362. [DOI] [PubMed] [Google Scholar]
- 6.Kauer F, Van Dalen BM, Michels M, Soliman OII, Vletter WB, Van Slegtenhorst M, Ten Cate FJ, Geleijnse ML. Diastolic abnormalities in normal phenotype hypertrophic cardiomyopathy gene carriers: a study using speckle tracking echocardiography. Echocardiography. 2013;30:558–63. doi: 10.1111/echo.12076. [DOI] [PubMed] [Google Scholar]
- 7.Brouwer WP, Germans T, Head MC, Van der Velden J, Heymans MW, Christiaans I, Houweling AC, Wilde AA, Van Rossum AC. Multiple myocardial crypts on modified long-axis view are a specific finding in pre-hypertrophic HCM mutation carriers. European Heart J Cardiovasc Imaging. 2012;13:292–7. doi: 10.1093/ehjci/jes005. [DOI] [PubMed] [Google Scholar]
- 8.Valente AM, Lakdawala NK, Powell AJ, Evans SP, Cirino AL, Orav EJ, Macrae CA, Colan SD, Ho CY. Comparison of echocardiographic and cardiac magnetic resonance imaging in hypertrophic cardiomyopathy sarcomere mutation carriers without left ventricular hypertrophy. Circ Cardiovasc Genet. 2013;6:230–7. doi: 10.1161/CIRCGENETICS.113.000037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Charron P. Accuracy of European diagnostic criteria for familial hypertrophic cardiomyopathy in a genotyped population. Int J Cardiol. 2003;90:33–8. doi: 10.1016/s0167-5273(02)00534-x. [DOI] [PubMed] [Google Scholar]
- 10.McKenna WJ, Spirito P, Desnos M, Dubourg O, Komajda M. Experience from clinical genetics in hypertrophic cardiomyopathy: proposal for new diagnostic criteria in adult members of affected families. Heart. 1997;77:130–2. doi: 10.1136/hrt.77.2.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lopes LR, Zekavati A, Syrris P, Hubank M, Giambartolomei C, Dalageorgou C, Jenkins S, McKenna W, Plagnol V, Elliott PM. Genetic complexity in hypertrophic cardiomyopathy revealed by high-throughput sequencing. J Med Genet. 2013;50:228–39. doi: 10.1136/jmedgenet-2012-101270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris SP, Lyons RG, Bezold KL. In the thick of it: HCM-causing mutations in myosin binding proteins of the thick filament. Circ Res. 2011;108:751–764. doi: 10.1161/CIRCRESAHA.110.231670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kramer CM, Barkhausen J, Flamm SD, Kim RJ, Nagel E. Standardized cardiovascular magnetic resonance imaging (CMR) protocols, society for cardiovascular magnetic resonance: board of trustees task force on standardized protocols. J Cardiovasc Magn Reson. 2008;10:35. doi: 10.1186/1532-429X-10-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Captur G, Muthurangu V, Cook C, Flett AS, Wilson R, Barison A, Sado DM, Anderson S, McKenna WJ, Mohun TJ, Elliott PM, Moon JC. Quantification of left ventricular trabeculae using fractal analysis. J Cardiovasc Magn Reson. 2013;15:36. doi: 10.1186/1532-429X-15-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen J, Gao S. A solution to separation and multicollinearity in multiple logistic regression. J Data Sci. 2008;6:515–531. [PMC free article] [PubMed] [Google Scholar]
- 17.Heinze G, Schemper M. A solution to the problem of separation in logistic regression. Stat Med. 2002;21:2409–19. doi: 10.1002/sim.1047. [DOI] [PubMed] [Google Scholar]
- 18.Torgo L. Predicting algae blooms. Data mining with R. Chapman and Hall/CRC. 2010:39–94. [Google Scholar]
- 19.Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, Seidman CE, Towbin JA, Udelson JE, Yancy CW. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Thorac Cardiovasc Surg. 2011;142:e153–203. doi: 10.1016/j.jtcvs.2011.10.020. [DOI] [PubMed] [Google Scholar]
- 20.Charron P, Arad M, Arbustini E, Basso C, Bilinska Z, Elliott P, Helio T, Keren A, McKenna WJ, Monserrat L, Pankuweit S, Perrot A, Rapezzi C, Ristic A, Seggewiss H, Van Langen I, Tavazzi L. Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2010;31:2715–26. doi: 10.1093/eurheartj/ehq271. [DOI] [PubMed] [Google Scholar]
- 21.Cirino AL, Ho CY. Genetic testing in cardiac disease: from bench to bedside. Nat Clin Pract Cardiovasc Med. 2006;3:462–3. doi: 10.1038/ncpcardio0654. [DOI] [PubMed] [Google Scholar]
- 22.Andersen PS, Havndrup O, Hougs L, Sørensen KM, Jensen M, Larsen LA, Hedley P, Thomsen ARB, Moolman-Smook J, Christiansen M, Bundgaard H. Diagnostic yield, interpretation, and clinical utility of mutation screening of sarcomere encoding genes in Danish hypertrophic cardiomyopathy patients and relatives. Hum Mutat. 2009;30:363–70. doi: 10.1002/humu.20862. [DOI] [PubMed] [Google Scholar]
- 23.Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol. 2012;60:705–15. doi: 10.1016/j.jacc.2012.02.068. [DOI] [PubMed] [Google Scholar]
- 24.Srichai MB, Hecht EM, Kim DC, Jacobs JE. Ventricular diverticula on cardiac CT: more common than previously thought. AJR Am J Roentgenol. 2007;189:204–8. doi: 10.2214/AJR.06.1223. [DOI] [PubMed] [Google Scholar]
- 25.Johansson B, Maceira AM, Babu-Narayan SV, Moon JC, Pennell DJ, Kilner PJ. Clefts can be seen in the basal inferior wall of the left ventricle and the interventricular septum in healthy volunteers as well as patients by cardiovascular magnetic resonance. J Am Coll Cardiol. 2007;50:1294–5. doi: 10.1016/j.jacc.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 26.Petryka J, Baksi J, Prasad SK, Pennell DJ, Kilner PJ. Prevalence of inferobasal myocardial crypts among patients referred for cardiovascular magnetic resonance. Circ Cardiovasc Imaging. 2014;7:259–64. doi: 10.1161/CIRCIMAGING.113.001241. [DOI] [PubMed] [Google Scholar]
- 27.Erol C, Koplay M, Olcay A, Kivrak AS, Ozbek S, Seker M, Paksoy Y. Congenital left ventricular wall abnormalities in adults detected by gated cardiac multidetector computed tomography: clefts, aneurysms, diverticula and terminology problems. Eur J Radiol. 2012;81:3276–81. doi: 10.1016/j.ejrad.2012.03.025. [DOI] [PubMed] [Google Scholar]
- 28.Maron MS, Rowin EJ, Lin D, Appelbaum E, Chan RH, Gibson CM, Lesser JR, Lindberg J, Haas TS, Udelson JE, Manning WJ, Maron BJ. Prevalence and clinical profile of myocardial crypts in hypertrophic cardiomyopathy. Circ Cardiovasc Imaging. 2012;5:441–7. doi: 10.1161/CIRCIMAGING.112.972760. [DOI] [PubMed] [Google Scholar]
- 29.Rowin EJ, Maron MS. Myocardial crypts in hypertrophic cardiomyopathy: the new gang in town. European Heart J Cardiovasc Imaging. 2012;13:281–3. doi: 10.1093/ehjci/jes035. [DOI] [PubMed] [Google Scholar]
- 30.Ho CY, Abbasi SA, Neilan TG, Shah RV, Chen Y, Heydari B, Cirino AL, Lakdawala NK, Orav EJ, González A, López B, Díez J, Jerosch-Herold M, Kwong RY. T1 measurements identify extracellular volume expansion in hypertrophic cardiomyopathy sarcomere mutation carriers with and without left ventricular hypertrophy. Circ Cardiovasc Imaging. 2013;6:415–22. doi: 10.1161/CIRCIMAGING.112.000333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ho C, Sweitzer N, McDonough B, Maron B, Casey S, Seidman J, Seidman C, Solomon S. Assessment of diastolic function with doppler tissue imaging to predict genotype in preclinical hypertrophic cardiomyopathy. Circ. 2002;105:2992–7. doi: 10.1161/01.cir.0000019070.70491.6d. [DOI] [PubMed] [Google Scholar]
- 32.De S, Borowski AG, Wang H, Nye L, Xin B, Thomas JD, Tang WHW. Subclinical echocardiographic abnormalities in phenotype-negative carriers of myosin-binding protein C3 gene mutation for hypertrophic cardiomyopathy. Am Heart J. 2011;162:262–7. doi: 10.1016/j.ahj.2011.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.