

Abstract
Background
Germ cell tumors (GCT) are the most common solid tumors in adolescent and young adult males (age 15 and 35 years) and remain one of the most curable of all solid malignancies. However a subset of patients will have tumors that are refractory to standard chemotherapy agents. The management of this refractory population remains challenging and approximately 400 patients continue to die every year of this refractory disease in the United States.
Methods
Given the preclinical evidence implicating vascular endothelial growth factor (VEGF) signaling in the biology of germ cell tumors, we hypothesized that the vascular endothelial growth factor receptor (VEGFR) inhibitor sunitinib (Sutent) may possess important clinical activity in the treatment of this refractory disease. We proposed a Phase II efficacy study of sunitinib in seminomatous and non-seminomatous metastatic GCT’s refractory to first line chemotherapy treatment (ClinicalTrials.gov Identifier: NCT00912912). Next generation targeted exome sequencing using HiSeq 2000 (Illumina Inc., San Diego, CA, USA) was performed on the tumor sample of the unusual responder.
Results
Five patients are enrolled into this Phase II study. Among them we report here the clinical course of a patient (Patient # 5) who had an exceptional response to sunitinib. Next generation sequencing to understand this patient’s response to sunitinib revealed RET amplification, EGFR and KRAS amplification as relevant aberrations. Oncoscan MIP array were employed to validate the copy number analysis that confirmed RET gene amplification.
Conclusion
Sunitinib conferred clinical benefit to this heavily pre-treated patient. Next generation sequencing of this ‘exceptional responder’ identified the first reported case of a RET amplification as a potential basis of sensitivity to sunitinib (VEGFR2/PDGFRβ/c-kit/ FLT3/RET/CSF1R inhibitor) in a patient with refractory germ cell tumor. Further characterization of GCT patients using biomarkers for clinical response and patient selection is warranted.
Trial registration
ClinicalTrials.gov Identifier: NCT00912912
Keywords: Sunitinib, Germ cell tumor, Phase II trials, Targeted therapy, RET, EGFR, Next generation sequencing, Adolescent and young adult, Vascular endothelial growth factor receptor, VEGF, Unusual responder, Exceptional responder, Outlier responder
Introduction
Germ cell tumors (GCT) are the most common solid tumors in adolescent and young adult males (age 15 and 35 years) and remain one of the most curable of all solid malignancies [1]. Development of cisplatin-based chemotherapy revolutionized the treatment of patients with advanced GCT; and with better temporal integration of surgery, dramatic improvements were obtained for patients presenting with GCT’s. However a subset of patients will have tumors that are refractory to standard chemotherapy agents. The management of this refractory population remains challenging and approximately 400 patients continue to die every year of this refractory disease in the United States [1]–[4]. Given the preclinical evidence implicating vascular endothelial growth factor (VEGF) signaling in the biology of germ cell tumors [5]–[7], we hypothesized that the vascular endothelial growth factor receptor (VEGFR) inhibitor sunitinib (Sutent®) [8] may possess important clinical activity in the treatment of this refractory disease. We proposed a Phase II efficacy study of sunitinib in seminomatous and non-seminomatous metastatic GCT’s refractory to first line chemotherapy treatment (ClinicalTrials.gov Identifier NCT00912912). Five patients are enrolled into this a Phase II study. Among them we report here the clinical course of a patient (Patient # 5) who had a dramatic response to sunitinib. Genome sequencing identified the first reported case of a RET amplification as a potential basis of sensitivity to sutent in a germ cell tumor.
Patients and methods
Inclusion/Exclusion criteria
This was a phase II study of sunitinib in refractory male germ cell tumors with the 12 week progression free survival as the primary endpoint. The major inclusion criteria for the phase II study were progressive metastatic GCTs of gonadal or extragonadal origin in males after failure of front-line therapy and at least one salvage regimen. In addition the patients must have evaluable or measurable disease by clinical or radiological studies. Alternatively, in the absence of radiologically evaluable or measurable disease, two sequentially rising marker values each one week apart attributed by treating physician to germ cell tumor was permitted; either beta HCG above 50 mIU/ml and/or AFP above 20 ng/ml qualified as eligible. A major exclusion was prior VEGFR/PDGFR inhibitor therapy.
Treatment plan
Once registered, treatment was started no later than 7 days. Patients were treated with sunitinib malate 50 mg orally daily for 4 weeks every 6 weeks. A cycle was defined as a planned 6-week treatment interval. It was preferred that patients complete at least 2 cycles of therapy unless there is evidence of rapid disease progression. Response evaluations were performed every 6 weeks according to RECIST criteria [9],[10]. The University of Texas MD Anderson cancer IRB approved this clinical trial and all patients were followed according to the protocol. The trial was an investigator initiated single institutional trial supported by Pfizer inc. (New York, NY) which provided Sunitinib (Sutent®) to the patients enrolled on the trial was closed early due to slow accrual.
Next generation sequencing (T200 platform)
Next generation targeted exomic sequencing was performed for genomic profling.200-500ug of genomic DNA from each sample was sheared by sonication using the Covaris E220 instrument (Covaris). Library preparation utilized the KAPA kit following the “with beads” manufacturer protocol using KAPA HiFi polymerase (6 cycles). The capture included all exons from 202 cancer-related genes. Biotin labeled DNA probes were designed using Roche Nimblegen for capturing target regions (over 5,000 exons in 202 genes) and followed manufacture’s protocol for the capture process. Probes (50–105 bp long) were tiled at a minimum coverage of 2× and balanced across the target regions to ensure uniformity. Captured libraries were sequenced on a HiSeq 2000 (Illumina Inc., San Diego, CA, USA) on a version 3 TruSeq paired end flowcell according to manufacturer’s instructions The resulting BCL files containing the sequence data were converted into “.fastq.gz” files and individual libraries within the samples were demultiplexed using CASAVA 1.8.2 with no mismatches.
Deep sequencing data was aligned to to hg19 using BWA [11]. Duplicate reads were removed via Picard [12]. Single nucleotide variant (SNVs), small indels and and copy number alterations were called using an in-house pipeline or a previously published algorithm, respectively [13]. This supported identifying genome aberrations by next-generation sequencing (NGS) based assays to identify genomic alterations in 200 cancer related genes.
Copy number validation
The validation of the copy number alterations, was done using the OncoScan, a MIParray V3. technology [14].
Results
Five patients are enrolled into this Phase II study; their clinical characteristics are reported in Table 1. Of the 5 patients evaluable for response, one (20%) was free of disease progression for more than 12 weeks. We report the clinical history of the patient that had an unusual response in detail as below.
Table 1.
Characteristics of patients with refractory germ cell tumors enrolled in the sutent phase 2 study and outcomes at presentation
| Pt # | Race | Age ++ | PS | Primary mass | Pathology- Components | Prior Therapies | AFP ng/ml (0–5.0) | B-HCG mIU/ml (0–1) | LDHIU/L (313–618) |
|---|---|---|---|---|---|---|---|---|---|
| 1 |
AA |
17 |
0 |
Testis |
Seminoma;Embryonal, Choriocarcinoma, Immature teratoma, Mature teratoma. |
R orchiectomy, BEP × 4, RPLND,TIP, Gem + Etoposide, Avastin + Gem, Avatin, Taxol, Gem, |
18495.6 |
<1.0 |
2386 |
| 2 |
Asian |
20 |
0 |
Mediastinum |
Yolk sac tumor, Endodermal Sinus Tumor. |
BEP × 3, TIP × 2, Surgery- multiple, ACE × 2 |
2087.9 |
<1.0 |
419 |
| 3 |
White |
36 |
1 |
Testis |
Embryonal Carcinoma (80%) and yolk sac tumor (20%) |
R orchiectomy, BEP × 3, RPLND,TIP × 4, XRT- T10 -L2 , Cisplatin + Epirubicin × 4, XRT to R chest wall, WBRT, Oxaliplatin + Gem, × 4, Oxaliplatin × 8, Gem × 3, etoposide × 3 |
108.9 |
<1.1 |
524 |
| 4 |
White |
19 |
1 |
Retro-peritoneal mass |
Teratoma and Choriocarcinoma |
BEP × 4, TIP × 1, VIP × 5, HD-SCT with carboplatin + etoposide, RPLND, |
1.1 |
227865.8 |
794 |
| 5 | White | 29 and 52* | 1 | Testis | Embryonal Carcinoma (80%) and yolk sac tumor (20%) | R orchiectomy, 20 yrs later BEP × 3, EP × 1, RPLND, TIP × 2, ACE × 1 | 235.5 | 19.8 | 635 |
Abbreviations: ++Age Age at original diagnosis, PS Performance status, *Age at relapse, AA African American, R Right, BEP Bleiomycin, Etoposide, Cisplatin, RPLND Retroperitoneal Lymph node dissection, TIP Taxol, Ifosfamide, Cisplatin, VIP VP-16, Ifosfamide, Cisplatin, ACE Actinomycin, Cyclophosphamide, Etoposide. Gem Gemcitabine.
Patient clinical history
A 52-year-old Caucasian male had been diagnosed at age 29 with a right sided testicular GCT. He underwent a radical, right orchiectomy through an inguinal incision. The primary tumor was classified as a clinical stage I, nonseminomatous GCT. AFP level was above 400 prior to orchiectomy and subsequently normalized appropriately following surgery. He did not receive any adjunctive chemotherapy. For 15 years, serial CT scans of abdomen and pelvis along with serum tumor markers found no evidence of tumor recurrence. However, in February 2009, as a part of work up for abdominal pain, a CT scan of the abdomen and pelvis revealed new and multiple enlarged retroperitoneal lymph nodes (intra-aortocaval and para-aortic). A subsequent CT of the chest showed bilaterally, several small, scattered pulmonary nodules; the largest being a 6 mm × 4 mm, noncalcified nodule in the right, lower lobe. An MRI of the brain found no evidence of intracranial metastases. A scrotal ultrasound revealed a homogenous echo-texture throughout the left testicle without evidence of abnormal masses or scars. Serum alfa-fetoprotein (AFP) and β-hCG levels were elevated at 34.8 ng/ml (normal ranges: 0.0- 5.0) and 22 mIU/ML (0.0- 1.0), respectively.
The patient received standard 3 cycles of bleomycin, etoposide, and cisplatin(BEP) and 1 cycle of etoposide and cisplatin (EP). Serum AFP level a month after his last cycle remained elevated at 56 ng/ml (0.0-5.0). CT scan of the chest, abdomen and pelvis, post-chemotherapy in June 2009, continued to show stable small posterior right lower lobe mass, and a mass in the right retroperitoneum. Because the patient had persistent back pain that did not resolve with the resolution of the retroperitoneal adenopathy, an MRI of the spine was performed in July 2009. This MRI revealed a 2.5 cm enhancing mass in the right retroperitoneum at the level of L2. This mass measured 2.7 × 2.2 cm and comparison to the CT scan done in June 2009 showed an increase in size from 2 × 1.6 cm in just a month. The patient was referred to our institution for further treatment.
The patient was presented at our multi-disciplinary tumor board. A decision to proceed to retroperitoneal lymph node dissection was jointly made. In August 2009 a complete, non-nerve sparing, bilateral retroperitoneal lymph node dissection extending from the renal vessels superiorly to the crossing of the ureters over the common iliac arteries inferiorly; onto the anterior spinous ligaments posteriorly and laterally to the ureters was completed without complications. Pathologic review of the resected tissue demonstrated residual viable malignant GCT within 1 out 9 precaval lymph nodes and 1 out of 8 retrocaval lymph nodes. The histologic classification of the GCT was 80% embryonal carcinoma and 20% yolk sac tumor. Following recovery from surgery, the patient received 2 cycles of adjuvant chemotherapy consisting of paclitaxel, ifosfamide, and cisplatin (TIP). This treatment was complicated by acute renal failure and ifosfamide induced CNS toxicity. Adjuvant chemotherapy was therefore altered for the third cycle to actinomycin-D, cyclophosphamide, and etoposide (ACE). A CT abdomen and pelvis showed no evidence for recurrent disease. He was followed closely with serial CT scans along with serum tumor markers. In 2010 a new pulmonary nodule (Figure 1A) and rising serum tumor markers were detected. He was then enrolled on the clinical study with sunitinib for chemotherapy-refractory germ cell tumors in June 2010. He received sunitinib 50 mg orally for 4 weeks on and 2 weeks off-cycle. By 09/20010 a decline in serum tumor markers was observed (Figures 2 and 3; The arrow points to date of sunitinib initiation). His most significant response was in serum beta-hCG with a more mild and fluctuating AFP response. He developed hypothyroidism while on treatment with sunitinib, a documented side effect [15] and began levothyroxine therapy with an appropriate response. He otherwise suffered no other treatment related G3 or 4 toxicity related to the sunitinib. Subsequent CT scans show a continued response to the treatment and has not revealed any new disease sites (Figure 1B). He continued to be followed up with clinical and biochemical response with a very good performance status. Unfortunately, in November 2011, he experienced progressive weakness of his legs accompanied by saddle anesthesia, and urinary incontinence. MRI spines showed progressive disease involving the cauda equina and biopsy was consistent with metastatic GCT - immunostain results were positive for cytokeratin, CD30, SALL4, and focally positive for Oct3/4. He was taken off the study at that time, 17 months after starting sunitinib. Because of worsening performance status he elected to receive supportive care and was referred to hospice care.
Figure 1.
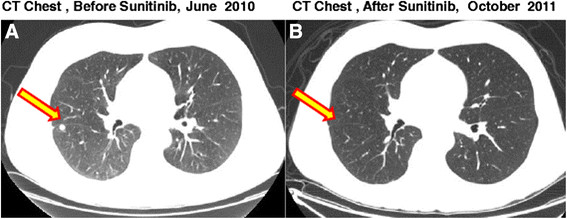
CT scans of the Chest in patient # 5 with metastatic platinum refractory gem cell tumor. A: CT of the Chest showing a metastatic nodule in the right lung before initiation of sutent B. CT of the Chest showing response of the metastatic nodule in the chest.
Figure 2.
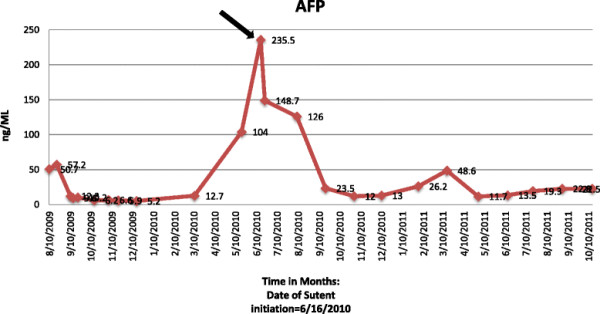
Alfa- feto protein (AFP) tumor marker levels: AFP levels and trends before and after Sutent. Arrow points to initiation of Sutent.
Figure 3.
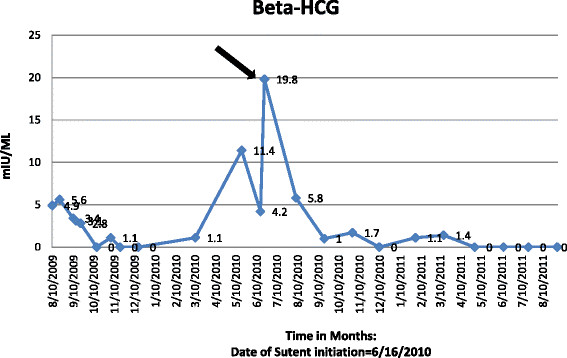
Beta-HCG trends before and after sutent. Arrow points to initiation of sutent.
This patient was enrolled on the unusual responder program at our institution, a program that applies appropriate next generation sequencing and other genomic and proteomic technologies to tumor and germline samples from patients with exceptional responses to chemotherapies and targeted regimens in an attempt to identify potential genetic explanations for the outlier response [16]. Next generation sequencing of unusual responder patient with testicular cancer indicated tumor harboring multiple copy number aberrations. Several genetic alterations were identified in this patient’s tumor. Of these, some of the variants identified represented potential candidates for further exploration to understand this patient’s response to sunitinib. The only alterations were found to have reported relevant information in tumors/cells treated with sunitinib were: RET amplification, PTEN loss, EGFR and KRAS amplification (Table 2). We carried out an extensive literature search in National Library of Medicine’s MEDLINE database on the five alterations and tabulated the levels of evidence/significance of genes relevant to sunitinib based therapy (Table 3). As the next step, to validate these genes an oncoscan MIP array were employed to validate the copy number analysis. The RET amplification, EGFR and KRAS amplification were validated by the second method. The PTEN loss was not validated by PTEN IHC.
Table 2.
Annotation of aberrant genes relevant to sunitinib sensitivity
| CNV | |||
|---|---|---|---|
|
HGNC |
Alteration |
CN |
|
|
RET |
H.AMP |
4.4 |
|
|
KRAS |
H.AMP |
4.2 |
|
|
EGFR |
H.AMP |
4.7 |
|
|
PTEN |
H.DEL |
0.9 |
|
|
SNV | |||
|
HGNC |
Alteration |
Consequence |
Quality |
| AKT1 | c. G529A p. A117T | Likely benign | Low |
Next generation sequencing of unusual responder patient with testicular cancer indicated tumor harboring multiple copy number aberrations. Only 5 alterations were found to have reported relevant information in tumors/cells treated with sunitinib: RET amplification, PTEN loss, EGFR and KRAS amplification.
HGNC – Hugo genome nomenclature committee, CNV, copy number variation, SNV- Single nucleotide variants.
NOTE: Genomic alterations detected may or may not have reported altered protein function.
Table 3.
Significance of the alterations relevant to sunitinib
| Gene alteration | Interpretation |
|---|---|
|
RET_H.AMP |
RET Amplifications are rare: |
| • Detected in sporadic anaplastic thyroid cancers and radiation-associated thyroid cancers (Nakashima Hum Path 2007) maybe as a result of genomic instability [17]. | |
| • RET amplifications are rare, mainly RET-PTC1 and RET-PTC3 fustions in thyroid cancer (Marina Melillo J Clin Invest 2005) [18]. | |
| Therapy Significance: | |
| • Lung tumors harboring RET amplification and PTEN deletion were sensitive to sunitinib therapy (Jones Genome Biol 2010) [19]. | |
| • Medullary thyroid cancer patient responds to sunitinib without RET alterations (Bugalho Oncologist 2009) [20]. | |
| • Sunitinib inhibits RET/PTC3 fusion phosphorylation causing morphologic reversion of cell transformation (Kim J Clin Endocrinol Metab 2006) [21]. | |
| High level of evidence and Significance |
• Sunitinib selectively inhibits growth of RET/PTC cells (Jeong Cancer Biol Ther 2011) [22]. |
| • Treatment of papillary thyroid cancer patient, but not follicular thyroid carcinoma, with sunitinib resulted in a dramatic reduction in RET phosphorylation and prolong patient survival (Dawson Anticancer Drugs 2008) [23]. | |
|
EGFR_H.AMP |
Therapy Significance: |
| • Sunitinib treatment of glioblastoma multiforme xenograft tumors harboring EGFR amplification and PTEN deletion did not impart any in vivo anti-tumor benefit (Joshi PLoS One 2012) [24]. | |
| Lowest level of significance |
• Sunitinib had minor anti-proliferative effects (IC50 3 μM) in NSCLC cell lines harbor EGFR T790M and KRAS mutations, which are resistant to EGFR inhibitors (Pan J Cancer Res Oncol 2011) [25]. |
|
KRAS_H.AMP |
Amplification of proto-oncogenes (wild-type version of oncogene) does not necessarily suggest the ability of this alteration to transform cell. |
| • However, studies do show that over expression of WT ras proteins leads to morphological transformation of cells (Pulciani Mol Cell Biol 1985) [26]. | |
| • Focal amplification of KRAS is one of the most common amplification events in lung adenocarcinoma (Weir Nature 2007; Shiraishi Cancer Res 1989) [27],[28]. | |
| • Amplification of KRAS is associated with increased protein levels and is quite common in NSCLC (20%; Wagner AJCP 2009) [29]. | |
| Therapy Significance: Mixed data on effecitiveness of Sunitinib in KRAS activated tumors. | |
| • Constitutively active KRAS mutants (G12R) were unresponsive to sunitinib cell growth inhibition in thyroid carcinoma cell lines (Piscazzi J Clin Endocrinol Metab 2012) [30]. | |
| Low level of evidence and significance |
• Sunitinib had minor anti-proliferative effects (IC50 3 μM) in NSCLC cell lines harbor EGFR T790M and KRAS mutations, which are resistant to EGFR inhibitors (Pan J Cancer Res Oncol 2011) [25]. |
| • Sunitinib reduced tumor size and tumor progression, and prolong median survival in KRAS mutant mouse NSCLC model (Gandhi Cancer Prev Res 2009) [31]. | |
|
PTEN_DEL |
Deletion of PTEN in tumors with oncogene amplifications may be a biomarker for increased tumor sensitivity (mixed results below, not conclusive). |
| • Lung tumors harboring RET amplification and PTEN deletion were sensitive to sunitinib therapy (Jones Genome Biol 2010) [19]. | |
| • Sunitinib treatment of glioblastoma multiforme xenograft tumors harboring EGFR amplification and PTEN deletion did not impart any in vivo anti-tumor benefit (Joshi PLoS One 2012) [24]. | |
| Low level of evidence and significance |
• PDGF-driven mouse glioblastoma tumors in PTEN deficient mice had a moderate effect on survival (D’Amico Neurol Res 2012) [32]. |
|
AKT_A117T |
• Mutation has not been previously reported |
| • Sunitinib apoptosis mediated through inhibition of AKT signaling in pediatric medulloblastomas (Yang Mol Cancer Res 2010) [33]. This was likely due to inhibition of upstream RTKs (and no AKT directly). | |
| • Sunitinib decreases phosphorylation of AKT in KIT mutant GIST cell line, as a result of KIT inhibition (Ikezoe Cancer Sci 2006) [34]. | |
| Low level of evidence and significance | • Reduction of Phospo-AKT in AML cancers harboring FLT3 mutations (Fiedler Blood 2005) [35]. |
Sunitinib relevant genes for basis of sensitivity, specifically RET amplification, PTEN loss, EGFR and KRAS amplification and a detailed literature search from National Library of Medicine’s MEDLINE data base.
Discussion
We have reported a patient with platinum refractory GCT who demonstrated clinical and biochemical response to a targeted therapy with sunitinib in a Phase 2 study. Genome sequencing uncovered a RET aberration as a plausible basis for sensitivity of sunitinib. Refractory germ cell tumors that are resistant to cytotoxic chemotherapy are always challenging and outcomes are poor warranting a fresh approach. This phase 2 study was designed based on the rationale that sunitinib was a VEGF inhibitor as there are several strands of evidence that support consideration of a role for VEGFR inhibitors in germ cell tumors. The presence of vascular invasion in Stage I germ cell tumors was associated with a high risk of relapse after orchiectomy [36]. VEGF expression is strongly correlated with microvessel density in primary germ cell tumors of the testis [5],[7] and VEGF receptor mRNA is increased in these tumors [7] suggesting autocrine and/or paracrine signaling loops driven by VEGF may play an important role in the angiogenic progression of these tumors. In a study of patients with seminomatous and non-seminomatous tumors, VEGF expression was increased compared to normal testis in both classes of germ cell tumors [5]. Multivariate analysis in this study indicated microvessel density and VEGF expression alone were predictive of metastases. A pre-clinical study showed a significant inhibition in tumor growth on sunitinib treatment and combination of this with cisplatin enhanced these effects [37]. However, another clinical trial of sunitinib in germ cell tumors was negative [38] and few case reports have reported responses. In the genomic era patient selection assumes significance if genomic aberrations in unusual responders are identified to benefit patient’s with similar molecular profile.
Sunitinib malate is a potent inhibitor of the tyrosine kinase activity of the split kinase domain receptor tyrosine kinases (RTK’s) VEGFR2 and PDGFR, which are involved in angiogenesis, and the RTKs, KIT, the receptor for stem cell factor and FLT3, which are involved in certain solid tumors and hematologic malignancies [8]. NGS sequencing of the unusual responder patient with testicular cancer indicated tumor harboring a RET amplification. RET is reported to be a therapeutic target of Sunitinib. Furthermore, one report indicated lung tumors harboring a RET amplification and PTEN deletion were sensitive to sunitinib therapy [19]. Together these may explain unusual response of testicular cancer patient to sunitinib therapy. Pre-clinical studies have shown that sunitinib inhibits RET/PTC3 fusion phosphorylation causing morphologic reversion of cell transformation and that sunitinib inhibits the RET/PTC3 kinase with an IC50 of 224 nm in vitro[21].
Review of literature revealed a report of a phase 2 study of 10 patients with refractory/metastatic GCT’s using sunitinib [38]. The investigators of that study reported that sunitinib caused a transient decrease in serum tumor markers, but no objective responses were seen in this small study and concluded that it failed to confer any benefit [38]. Four of the five initial patients treated on Feldman et al. trial experienced some tumor marker decline during the four-week “on” period, followed by marker rise during the two-week “off” period. In our study the index patient had a marker drop within the first 4 weeks. There was no considerable time difference in decrease of serum tumor markers between the present study and the referenced study. Sunitinib has been extensively studied in pre-clinical and clinical models of GCT’s [39]. The pre-clinical studies have been translated only with modest benefit in clinical studies. These have been tabulated in Table 4.
Table 4.
Clinical trials and case studies using Sunitinib and Imatinib in relapsed/refractory germ cell tumors
| Drug | Study center/Group | Number | Efficacy | Author | Reference |
|---|---|---|---|---|---|
| Sunitinib |
Canadian Urologic Oncology Group/German |
33 |
3 PR + 1 uPR RR = 13% 6-mo PFS 11% |
K. Oechsle et al. |
[40] |
| Testicular Cancer Study Group cooperative study | |||||
| Sunitinib |
Memorial Sloan Kettering Cancer Center |
10 |
0 RR, 5 SD |
Feldman et al. |
[38] |
| Sunitinib |
Hospital de Dénia, Marina Salud |
1 |
1 SD, B-HCG drop |
Gasent Blesa, J. M.et al. |
[41] |
| Sunitinib |
MD Anderson Cancer Center |
5 |
1 /5PR |
Subbiah et al. |
Journal of Hem/Onc |
| Imatinib |
Indiana University |
6 |
0/6 1 SD 3 mo with > 50% ↓AFP_ |
Einhorn et al. |
[42] |
| Imatinib |
General Hospital Attikon, University of Athens |
1 |
1 CR ~32 mo |
Pectasides, D.et al. |
[43] |
| Imatinib | Central Hospital of Bolzano, Bolzano, Italy | 1 | 1 CR ~ 24 mo | Pedersini R et al. | [44] |
PR- Partial response, PFS – Progression free survival, uPR-unconfirmed partial response, SD- Stable disease, CR – Complete Response.
Identifying the basis of sensitivity can aid in understanding the biology of aggressive GCT’s. Moreover, if validated in other patients with similar biomarker profiles can prevent the patients without the biomarker from futile therapy and help patients with biomarker with the appropriate molecularly matched therapy. A review published about targeted therapies on GCT’s catalogued novel therapies in refractory GCT’s and reports that none of the agents tested including isotretinoin, suramin, arsenic trioxide, thalidomide conferred any benefit in refractory GCT’s [45],[46]. One patient achieved partial response with a bevacizumab based study with high dose ifosfamide, cyclophosphamide and etoposide (HD-ICE) and another patient with c-kit positive seminoma achieved complete response with imatinib [45]. A different study of 6 patients with GCT on imatinib reported no benefit from the drug [45]. Three patients with growing teratoma syndrome in which the tumor expresses high retinoblastoma protein (pRB), who were treated with a cyclin-dependent kinase 4/6 (CDK4/6) inhibitor (PD0332991) reported definite clinical benefit [46],[47].
Novel targeted therapies that include small molecule tyrosine kinase inhibitors and monoclonal antibodies have changed the landscape in the management for several solid tumors. We are learning that only a select population of patients derives benefit from such agents. The challenge will be to identify biomarkers of response and/or resistance and offer a personalized treatment option for such patients [48].
The case presentation is atypical in that the patient had a very late relapse 23 years after his original diagnosis. This brings up a very important aspect in the follow up of GCT that they should have annual evaluations for their whole life even if they reach an asymptomatic stage [49],[50]. The response of GCT to sunitinib is noteworthy for several reasons. This is one of the first molecularly targeted therapy agents to induce a durable response in platinum refractory GCT and the first reported case of a RET aberration in GCT. One of the major limitations of the manuscript is lack of functional validations in cell lines. However, we have validated the exome sequencing findings using a second method OncoScan, a MIParray V3. technology which confirmed the RET amplification in addition to presenting a high level of evidence from a literature search of the National Library of Medicine’s MEDLINE data base (Table 3). Further work is needed to determine the frequency of RET alterations among patients with relapsed GCT, and to functionally validate whether RET amplification confers sunitinib sensitivity. Undoubtedly sunitinib conferred clinical benefit to this heavily pre-treated patient, and the patient tolerated the treatment well without any prohibitive toxicity. Moreover, this novel TKI is oral and conveniently given as a complete outpatient therapy bestowing a good quality of life. It would be interesting to study combination of sunitinib or other novel VEGFR inhibitors with other agents in poor risk patients with GCT with RET amplifications. Further characterization of GCT patients using biomarkers for clinical response and patient selection is warranted.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors contributed to writing the manuscript. VS, FMB, LCP conceived the manuscript. LCP is the primary investigator for the clinical trial. VS, FMB, GBM, KRM, AMB, PR, JFW, and LCP analysed the data. VS, FMB and LCP wrote the paper. All authors read and approved the final manuscript.
Contributor Information
Vivek Subbiah, Email: vsubbiah@mdanderson.org.
Funda Meric-Bernstam, Email: fmeric@mdanderson.org.
Gordon B Mills, Email: gmills@mdanderson.org.
Kenna R Mills Shaw, Email: KRShaw@mdanderson.org.
Ann Marie Bailey, Email: thomasam98@gmail.com.
Priya Rao, Email: prao@mdanderson.org.
John F Ward, Email: jfward@mdanderson.org.
Lance C Pagliaro, Email: lpagliar@mdanderson.org.
Acknowledgements
The Phase 2 clinical trial was supported by Pfizer and Sutent® was provided by Pfizer.
The next generation sequencing analysis study was supported in part by Sheikh Khalifa Al Nahyan Ben Zayed Institute for Personalized Cancer Therapy, NCATS grant UL1 TR000371(Center for Clinical and Translational Sciences), the Bosarge Foundation, and the MD Anderson Cancer Center Support grant (P30 CA016672).
References
- Kondagunta GV, Motzer RJ. Chemotherapy for advanced germ cell tumors. J Clin Oncol. 2006;24(35):5493–5502. doi: 10.1200/JCO.2006.08.7882. [DOI] [PubMed] [Google Scholar]
- Schmoll HJ, Souchon R, Krege S, Albers P, Beyer J, Kollmannsberger C, Fossa SD, Skakkebaek NE, de Wit R, Fizazi K, Droz JP, Pizzocaro G, Daugaard G, de Mulder PH, Horwich A, Oliver T, Huddart R, Rosti G, Paz Ares L, Pont O, Hartmann JT, Aass N, Algaba F, Bamberg M, Bodrogi I, Bokemeyer C, Classen J, Clemm S, Culine S, de Wit M. et al. European consensus on diagnosis and treatment of germ cell cancer: a report of the European Germ Cell Cancer Consensus Group (EGCCCG) Ann Oncol. 2004;15(9):1377–1399. doi: 10.1093/annonc/mdh301. [DOI] [PubMed] [Google Scholar]
- Sonpavde G, Hutson TE, Roth BJ. Management of recurrent testicular germ cell tumors. Oncologist. 2007;12(1):51–61. doi: 10.1634/theoncologist.12-1-51. [DOI] [PubMed] [Google Scholar]
- Kollmannsberger C, Nichols C, Bokemeyer C. Recent advances in management of patients with platinum-refractory testicular germ cell tumors. Cancer. 2006;106(6):1217–1226. doi: 10.1002/cncr.21742. [DOI] [PubMed] [Google Scholar]
- Fukuda S, Shirahama T, Imazono Y, Tsushima T, Ohmori H, Kayajima T, Take S, Nishiyama K, Yonezawa S, Akiba S, Akiyama S, Ohi Y. Expression of vascular endothelial growth factor in patients with testicular germ cell tumors as an indicator of metastatic disease. Cancer. 1999;85(6):1323–1330. [PubMed] [Google Scholar]
- Arrieta O, Michel Ortega RM, Angeles-Sanchez J, Villarreal-Garza C, Aviles-Salas A, Chanona-Vilchis JG, Arechaga-Ocampo E, Luevano-Gonzalez A, Jimenez MA, Aguilar JL. Serum human chorionic gonadotropin is associated with angiogenesis in germ cell testicular tumors. J Exp Clin Cancer Res. 2009;28:120. doi: 10.1186/1756-9966-28-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viglietto G, Romano A, Maglione D, Rambaldi M, Paoletti I, Lago, Califano D, Monaco C, Mineo A, Santelli G, Manzo G, Botti G, Chiappetta G, Persico MG. Neovascularization in human germ cell tumors correlates with a marked increase in the expression of the vascular endothelial growth factor but not the placenta-derived growth factor. Oncogene. 1996;13(3):577–587. [PubMed] [Google Scholar]
- Chow LQ, Eckhardt SG. Sunitinib: from rational design to clinical efficacy. J Clin Oncol. 2007;25(7):884–896. doi: 10.1200/JCO.2006.06.3602. [DOI] [PubMed] [Google Scholar]
- Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92(3):205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45(2):228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonigro RJ, Grasso CS, Robinson DR, Jing X, Wu YM, Cao X, Quist MJ, Tomlins SA, Pienta KJ, Chinnaiyan AM. Detection of somatic copy number alterations in cancer using targeted exome capture sequencing. Neoplasia. 2011;13(11):1019–1025. doi: 10.1593/neo.111252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cottman M, Schiffman JD. Molecular inversion probes: a novel microarray technology and its application in cancer research. Cancer Genet. 2012;205(7–8):341–355. doi: 10.1016/j.cancergen.2012.06.005. [DOI] [PubMed] [Google Scholar]
- Rini BI, Tamaskar I, Shaheen P, Salas R, Garcia J, Wood L, Reddy S, Dreicer R, Bukowski RM. Hypothyroidism in patients with metastatic renal cell carcinoma treated with sunitinib. J Natl Cancer Inst. 2007;99(1):81–83. doi: 10.1093/jnci/djk008. [DOI] [PubMed] [Google Scholar]
- Meric-Bernstam F, Farhangfar C, Mendelsohn J, Mills GB. Building a personalized medicine infrastructure at a major cancer center. J Clin Oncol. 2013;31(15):1849–1857. doi: 10.1200/JCO.2012.45.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima M, Takamura N, Namba H, Saenko V, Meirmanov S, Matsumoto N, Hayashi T, Maeda S, Sekine I. RET oncogene amplification in thyroid cancer: correlations with radiation-associated and high-grade malignancy. Hum Pathol. 2007;38(4):621–628. doi: 10.1016/j.humpath.2006.10.013. [DOI] [PubMed] [Google Scholar]
- Melillo RM, Castellone MD, Guarino V, De Falco V, Cirafici AM, Salvatore G, Caiazzo F, Basolo F, Giannini R, Kruhoffer M, Orntoft T, Fusco A, Santoro M. The RET/PTC-RAS-BRAF linear signaling cascade mediates the motile and mitogenic phenotype of thyroid cancer cells. J Clin Investig. 2005;115(4):1068–1081. doi: 10.1172/JCI22758. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Jones SJ, Laskin J, Li YY, Griffith OL, An J, Bilenky M, Butterfield YS, Cezard T, Chuah E, Corbett R, Fejes AP, Griffith M, Yee J, Martin M, Mayo M, Melnyk N, Morin RD, Pugh TJ, Severson T, Shah SP, Sutcliffe M, Tam A, Terry J, Thiessen N, Thomson T, Varhol R, Zeng T, Zhao Y, Moore RA, Huntsman DG. et al. Evolution of an adenocarcinoma in response to selection by targeted kinase inhibitors. Genome Biol. 2010;11(8):R82. doi: 10.1186/gb-2010-11-8-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugalho MJ, Domingues R, Borges A. A case of advanced medullary thyroid carcinoma successfully treated with sunitinib. Oncologist. 2009;14(11):1083–1087. doi: 10.1634/theoncologist.2009-0195. [DOI] [PubMed] [Google Scholar]
- Kim DW, Jo YS, Jung HS, Chung HK, Song JH, Park KC, Park SH, Hwang JH, Rha SY, Kweon GR, Lee SJ, Jo KW, Shong M. An orally administered multitarget tyrosine kinase inhibitor, SU11248, is a novel potent inhibitor of thyroid oncogenic RET/papillary thyroid cancer kinases. J Clin Endocrinol Metab. 2006;91(10):4070–4076. doi: 10.1210/jc.2005-2845. [DOI] [PubMed] [Google Scholar]
- Jeong WJ, Mo JH, Park MW, Choi IJ, An SY, Jeon EH, Ahn SH. Sunitinib inhibits papillary thyroid carcinoma with RET/PTC rearrangement but not BRAF mutation. Cancer Biol Ther. 2011;12(5):458–465. doi: 10.4161/cbt.12.5.16303. [DOI] [PubMed] [Google Scholar]
- Dawson SJ, Conus NM, Toner GC, Raleigh JM, Hicks RJ, McArthur G, Rischin D. Sustained clinical responses to tyrosine kinase inhibitor sunitinib in thyroid carcinoma. Anti-Cancer Drugs. 2008;19(5):547–552. doi: 10.1097/CAD.0b013e3282fc6cf7. [DOI] [PubMed] [Google Scholar]
- Joshi AD, Loilome W, Siu IM, Tyler B, Gallia GL, Riggins GJ. Evaluation of tyrosine kinase inhibitor combinations for glioblastoma therapy. PLoS ONE. 2012;7(10):e44372. doi: 10.1371/journal.pone.0044372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan F, Tian J, Zhang X, Zhang Y, Pan Y. Synergistic interaction between sunitinib and docetaxel is sequence dependent in human non-small lung cancer with EGFR TKIs-resistant mutation. J Cancer Res Clin Oncol. 2011;137(9):1397–1408. doi: 10.1007/s00432-011-1009-x. [DOI] [PubMed] [Google Scholar]
- Pulciani S, Santos E, Long LK, Sorrentino V, Barbacid M. Ras gene amplification and malignant transformation. Mol Cell Biol. 1985;5(10):2836–2841. doi: 10.1128/mcb.5.10.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir BA, Woo MS, Getz G, Perner S, Ding L, Beroukhim R, Lin WM, Province MA, Kraja A, Johnson LA, Shah K, Sato M, Thomas RK, Barletta JA, Borecki IB, Broderick S, Chang AC, Chiang DY, Chirieac LR, Cho J, Fujii Y, Gazdar AF, Giordano T, Greulich H, Hanna M, Johnson BE, Kris MG, Lash A, Lin L, Lindeman N. et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450(7171):893–898. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraishi M, Noguchi M, Shimosato Y, Sekiya T. Amplification of protooncogenes in surgical specimens of human lung carcinomas. Cancer Res. 1989;49(23):6474–6479. [PubMed] [Google Scholar]
- Wagner PL, Perner S, Rickman DS, LaFargue CJ, Kitabayashi N, Johnstone SF, Weir BA, Meyerson M, Altorki NK, Rubin MA. In situ evidence of KRAS amplification and association with increased p21 levels in non-small cell lung carcinoma. Am J Clin Pathol. 2009;132(4):500–505. doi: 10.1309/AJCPF10ZUNSOLIFG. [DOI] [PubMed] [Google Scholar]
- Piscazzi A, Costantino E, Maddalena F, Natalicchio MI, Gerardi AM, Antonetti R, Cignarelli M, Landriscina M. Activation of the RAS/RAF/ERK signaling pathway contributes to resistance to sunitinib in thyroid carcinoma cell lines. J Clin Endocrinol Metab. 2012;97(6):E898–E906. doi: 10.1210/jc.2011-3269. [DOI] [PubMed] [Google Scholar]
- Gandhi L, McNamara KL, Li D, Borgman CL, McDermott U, Brandstetter KA, Padera RF, Chirieac LR, Settleman JE, Wong KK. Sunitinib prolongs survival in genetically engineered mouse models of multistep lung carcinogenesis. Cancer Prev Res (Phila) 2009;2(4):330–337. doi: 10.1158/1940-6207.CAPR-08-0213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amico R, Lei L, Kennedy BC, Sisti J, Ebiana V, Crisman C, Christensen JG, Gil O, Rosenfeld SS, Canoll P, Bruce JN. The addition of Sunitinib to radiation delays tumor growth in a murine model of glioblastoma. Neurol Res. 2012;34(3):252–261. doi: 10.1179/1743132812Y.0000000005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Jove V, Xin H, Hedvat M, Van Meter TE, Yu H. Sunitinib induces apoptosis and growth arrest of medulloblastoma tumor cells by inhibiting STAT3 and AKT signaling pathways. Mol Cancer Res. 2010;8(1):35–45. doi: 10.1158/1541-7786.MCR-09-0220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikezoe T, Yang Y, Nishioka C, Bandobashi K, Nakatani H, Taguchi T, Koeffler HP, Taguchi H. Effect of SU11248 on gastrointestinal stromal tumor-T1 cells: enhancement of growth inhibition via inhibition of 3-kinase/Akt/mammalian target of rapamycin signaling. Cancer Sci. 2006;97(9):945–951. doi: 10.1111/j.1349-7006.2006.00263.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedler W, Serve H, Dohner H, Schwittay M, Ottmann OG, O'Farrell AM, Bello CL, Allred R, Manning WC, Cherrington JM, Louie SG, Hong W, Brega NM, Massimini G, Scigalla P, Berdel WE, Hossfeld DK. A phase 1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia (AML) or not amenable to conventional therapy for the disease. Blood. 2005;105(3):986–993. doi: 10.1182/blood-2004-05-1846. [DOI] [PubMed] [Google Scholar]
- Albers P, Siener R, Kliesch S, Weissbach L, Krege S, Sparwasser C, Schulze H, Heidenreich A, de Riese W, Loy V, Bierhoff E, Wittekind C, Fimmers R, Hartmann M. Risk factors for relapse in clinical stage I nonseminomatous testicular germ cell tumors: results of the German Testicular Cancer Study Group Trial. J Clin Oncol. 2003;21(8):1505–1512. doi: 10.1200/JCO.2003.07.169. [DOI] [PubMed] [Google Scholar]
- Castillo-Avila W, Piulats JM, Garcia Del Muro X, Vidal A, Condom E, Casanovas O, Mora J, Germa JR, Capella G, Villanueva A, Vinals F. Sunitinib inhibits tumor growth and synergizes with cisplatin in orthotopic models of cisplatin-sensitive and cisplatin-resistant human testicular germ cell tumors. Clin Cancer Res. 2009;15(10):3384–3395. doi: 10.1158/1078-0432.CCR-08-2170. [DOI] [PubMed] [Google Scholar]
- Feldman DR, Turkula S, Ginsberg MS, Ishill N, Patil S, Carousso M, Bosl GJ, Motzer RJ. Phase II trial of sunitinib in patients with relapsed or refractory germ cell tumors. Invest New Drugs. 2010;28(4):523–528. doi: 10.1007/s10637-009-9280-2. [DOI] [PubMed] [Google Scholar]
- Juliachs M, Munoz C, Moutinho CA, Vidal A, Condom E, Esteller M, Graupera M, Casanovas O, Germa JR, Villanueva A, Vinals F. The PDGFRbeta-AKT pathway contributes to CDDP-acquired resistance in testicular germ cell tumors. Clin Cancer Res. 2014;20(3):658–667. doi: 10.1158/1078-0432.CCR-13-1131. [DOI] [PubMed] [Google Scholar]
- Oechsle K, Honecker F, Cheng T, Mayer F, Czaykowski P, Winquist E, Wood L, Fenner M, Glaesener S, Hartmann JT, Chi K, Bokemeyer C, Kollmannsberger C. Preclinical and clinical activity of sunitinib in patients with cisplatin-refractory or multiply relapsed germ cell tumors: a Canadian Urologic Oncology Group/German Testicular Cancer Study Group cooperative study. Ann Oncol. 2011;22(12):2654–2660. doi: 10.1093/annonc/mdr026. [DOI] [PubMed] [Google Scholar]
- Gasent Blesa JM, Grande Pulido E, Laforga Canales J, Alberola Candel V. Final report of the first refractory germ cell tumor treated with Sunitinib Malate. Case Rep Oncol. 2009;2(3):234–241. doi: 10.1159/000260901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einhorn LH, Brames MJ, Heinrich MC, Corless CL, Madani A. Phase II study of imatinib mesylate in chemotherapy refractory germ cell tumors expressing KIT. Am J Clin Oncol. 2006;29(1):12–13. doi: 10.1097/01.coc.0000195086.47548.ef. [DOI] [PubMed] [Google Scholar]
- Pectasides D, Nikolaou M, Pectasides E, Koumarianou A, Valavanis C, Economopoulos T. Complete response after imatinib mesylate administration in a patient with chemoresistant stage IV seminoma. Anticancer Res. 2008;28(4C):2317–2320. [PubMed] [Google Scholar]
- Pedersini R, Vattemi E, Mazzoleni G, Graiff C. Complete response after treatment with imatinib in pretreated disseminated testicular seminoma with overexpression of c-KIT. Lancet Oncol. 2007;8(11):1039–1040. doi: 10.1016/S1470-2045(07)70344-3. [DOI] [PubMed] [Google Scholar]
- Fenner MH, Beutel G, Grunwald V. Targeted therapies for patients with germ cell tumors. Expert Opin Investig Drugs. 2008;17(4):511–522. doi: 10.1517/13543784.17.4.511. [DOI] [PubMed] [Google Scholar]
- Veenstra CM, Vaughn DJ. Third-line chemotherapy and novel agents for metastatic germ cell tumors. Hematol Oncol Clin North Am. 2011;25(3):577–591. doi: 10.1016/j.hoc.2011.03.005. ix. [DOI] [PubMed] [Google Scholar]
- Vaughn DJ, Flaherty K, Lal P, Gallagher M, O’Dwyer P, Wilner K, Chen I, Schwartz G. Treatment of growing teratoma syndrome. N Engl J Med. 2009;360(4):423–424. doi: 10.1056/NEJMc0808558. [DOI] [PubMed] [Google Scholar]
- Kummar S, Chen HX, Wright J, Holbeck S, Millin MD, Tomaszewski J, Zweibel J, Collins J, Doroshow JH. Utilizing targeted cancer therapeutic agents in combination: novel approaches and urgent requirements. Nat Rev Drug Discov. 2011;9(11):843–856. doi: 10.1038/nrd3216. [DOI] [PubMed] [Google Scholar]
- Gerl A, Clemm C, Schmeller N, Hentrich M, Lamerz R, Wilmanns W. Late relapse of germ cell tumors after cisplatin-based chemotherapy. Ann Oncol. 1997;8(1):41–47. doi: 10.1023/a:1008253323854. [DOI] [PubMed] [Google Scholar]
- Baniel J, Foster RS, Gonin R, Messemer JE, Donohue JP, Einhorn LH. Late relapse of testicular cancer. J Clin Oncol. 1995;13(5):1170–1176. doi: 10.1200/JCO.1995.13.5.1170. [DOI] [PubMed] [Google Scholar]