

Abstract
Background
Syndromic forms of osteosarcoma (OS) account for less than 10% of all recorded cases of this malignancy. An individual OS predisposition is also possible by the inheritance of low penetrance alleles of tumor susceptibility genes, usually without evidence of a syndromic condition. Genetic variants involved in such a non-syndromic form of tumor predisposition are difficult to identify, given the low incidence of osteosarcoma cases and the genetic heterogeneity of patients. We recently mapped a major OS susceptibility QTL to mouse chromosome 14 by comparing alpha-radiation induced osteosarcoma in mouse strains which differ in their tumor susceptibility.
Methods
Tumor-specific allelic losses in murine osteosacoma were mapped along chromosome 14 using microsatellite markers and SNP allelotyping. Candidate gene search in the mapped interval was refined using PosMed data mining and mRNA expression analysis in normal osteoblasts. A strain-specific promoter variant in Rb1 was tested for its influence on mRNA expression using reporter assay.
Results
A common Rb1 allele derived from the BALB/cHeNhg strain was identified as the major determinant of radiation-induced OS risk at this locus. Increased OS-risk is linked with a hexanucleotide deletion in the promoter region which is predicted to change WT1 and SP1 transcription factor-binding sites. Both in-vitro reporter and in-vivo expression assays confirmed an approx. 1.5 fold reduced gene expression by this promoter variant. Concordantly, the 50% reduction in Rb1 expression in mice bearing a conditional hemizygous Rb1 deletion causes a significant rise of OS incidence following alpha-irradiation.
Conclusion
This is the first experimental demonstration of a functional and genetic link between reduced Rb1 expression from a common promoter variant and increased tumor risk after radiation exposure. We propose that a reduced Rb1 expression by common variants in regulatory regions can modify the risk for a malignant transformation of bone cells after radiation exposure.
Keywords: Tumor susceptibility, Germline polymorphism, Promoter activity, Radiation-induced, Bone tumor, Mouse model
Background
The relatively low incidence of osteosarcoma (OS) in the general population increases dramatically in cases of high–penetrance cancer syndromes such as Li-Fraumeni, familial retinoblastoma or Werner- and Rothmund-Thompson syndrome [1]. Aggregation of osteosarcoma in families without evidence of an underlying tumor syndrome is rare, and it is possible that this is due to a chance observation or shared environmental risk factor [2-4]. Nevertheless, association between osteosarcoma and other tumor types in relatives does occur [5,6], suggesting that undiscovered germline factors may contribute to osteosarcoma risk. The existence of congenital, low-penetrance tumor predisposition associated with OS in non-syndromic patients was also concluded in a study of 938 Dutch osteosarcoma patients, in which a 2.4 fold increased risk of developing an additional tumor in another organ was found [7]. In the case of Li-Fraumeni and congenital retinoblastoma syndromes, two high-penetrance cancer-prone conditions with a predisposition for OS, a subset of the affected patients had not been found to carry germline mutations in the prototypical TP53 gene [8] or in the RB1 coding sequence [9]. It is therefore likely that phenocopies of these OS linked syndromes are caused by mutations in other genes affecting the same pathways, or that yet undiscovered hypomorphic allelic variants of established susceptibility genes modify tumor risk [9,10]. The presence of such low-penetrance gene variants would imply that a much larger than previously estimated fraction of tumors may be due to a congenital predisposition [11,12]. Since familial clustering would be very unlikely to occur in such conditions [13], population-based genome-wide association studies (GWAS) or whole-genome sequence analysis in patients might help to map the underlying gene variants, but might be hampered by low statistical power. A recent multi-center GWAS study on osteosacoma identified 2 novel candidate loci for OS susceptibility with odds-ratios of 1.57 and 1.39, respectively [14]. The missing association with established OS risk genes such as TP53, RB1, WRN or RECQL4 in this study, however points to the problem of small patient number and genetic and phenotypic heterogeneity for this tumor type.
Strain differences between genetically defined mouse inbred strains have been used successfully to identify genes and allelic variants which influence predisposition for spontaneous or carcinogen-induced malignancies [15-18]. For osteosarcoma, the only established exogenous carcinogen is ionizing radiation, in particular by incorporation of bone-seeking radio-isotopes in the event of medical or occupational exposure. This observation was confirmed in animal models such as dogs and rodents, and variations in the OS susceptibility between different mouse inbred strains were reported [19]. We recently used a mouse model to map several osteosarcoma susceptibility loci in the murine genome using QTL analysis [20,21]. These studies established that the principal susceptibility locus spans an approximately 46 Mbp interval on the distal part of mouse chromosome 14.
The large number of mapped genes and non-coding transcripts in the D14Mit90 to D14Mit225 interval, together with the general problem of deducing a causal relationship from forward genetics, precluded any inference to a single susceptibility gene at this locus. For this purpose, more precise and complementary mapping strategies are necessary to reduce the number of candidate susceptibility genes such that functional studies are feasible. In the present study we combined mapping of tumor-specific allele-losses across the susceptibility locus with positional-candidate mapping and identified a functional gene polymorphism in Rb1.
Results and discussion
Identification of candidate genes
We have previously shown that an interval on murine chromosome Mmu 14 (72.3 – 118 Mbp) contains the principal locus conferring susceptibility to alpha-radiation induced osteosarcoma in four different mouse strains [20,21]. (compound LOD score 4.4). Due to incomplete penetrance of such a susceptibility gene, confounding causes of death after irradiation, and the inherent stochastic nature of radiation tumorigenesis a more precise mapping of this QTL using linkage-analysis was not feasible. We have therefore applied a complementary method to better define the position of the susceptibility gene, using somatic allelic imbalances mapping across this locus in osteosarcoma of (BALB/cHeNhg × CBA/Ca)F1 mice. After 227Thorium incorporation at the age of 100 days, 17 out of 80 female F1 mice developed osteosarcoma that were suitable for molecular analysis (median latency time = 553 days, range from 290 to 766 days). In 13 out of 17 of these tumors we observed allelic loss or imbalance for at least one marker between D14Mit234 and D14Mit97, i.e. within the principle OS susceptibility locus on chromosome 14 [21] (Figure 1). In 10/13 cases the maternal (BALB/c) allele was affected, whereas the paternal CBA allele was lost in the remaining 3 cases. This preferential loss of 10 BALB/c versus 3 CBA alleles (p = 0.014, relative to a total of 34 parental chromosomes) is consistent with the assumption that tumors in heterozygous F1 gain a growth advantage when they somatically lose an allele that confers relative resistance. We observed before that in the progeny of (BALB/cHeNhg × CBA/Ca) F1 mice backcrossed to female BALB/cHeNhg (further referred to as BBC) the BALB/c allele at this locus is associated with relative OS resistance [21]. The smallest common interval of overlapping allelic imbalances spans 2.654 Mbp (between D14Mit90 and D14Mit 225, Figure 1) and encompasses 12 known genes, 1 protein-coding gene with unknown function, 2 non-coding RNA genes and 1 pseudogene (Additional file 1). To nominate candidate genes from this genomic interval, we used PosMed, a search engine based on the entire biomedical literature to find links between a set of genes and a given phenotype [22]. Here we identified 7 genes in the D14Mit90/D14Mit225 interval (Rb1, Esd, Cysltr2, Htr2a, Fndc3a, Ltm2b, Lpar6) with a potential function in osteosarcoma susceptibility (Additional file 2). Of these 7 genes, only Rb1 is directly associated with osteosarcoma as suggested by 12 studies (p = 5 × 10−53) whereas the other 6 candidates are linked to osteosarcoma indirectly, either via additional genes acting in-trans or through comorbidity (next best p-value 1.1 × 10−20 for Esd). It is also worth mentioning that according to the Mouse Genome Informatics database, some modifier loci for other traits have been mapped to this interval (such as Modor3 for ocular retention, Myci2 for myocardial infarction, Hwq1 for heart weight). But since they are not defined as transcribed sequences, we limited our screen to genes and other transcribed elements as validated in the current mouse genome release (NCBI GRCm38).
Figure 1.
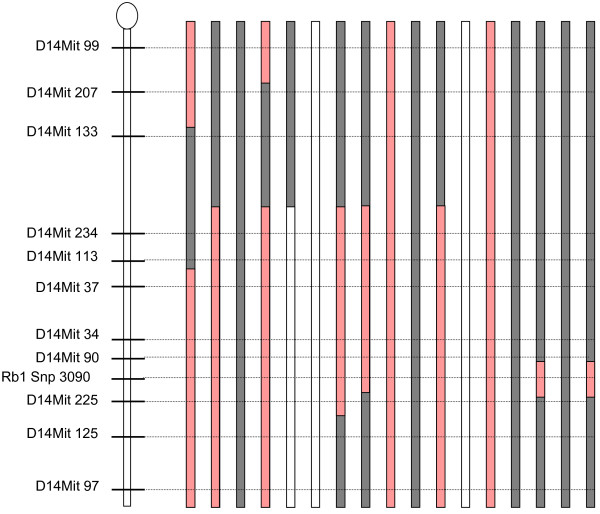
Analysis of allelic imbalance in 17 osteosarcomas induced in (BALB/cHeNhg x CBA/Ca)F1 hybrid mice. Pattern of allelic imbalances at the Rb1 locus and at flanking microsatellite markers are shown as grey bars (retention of heterozygosity), pink bars (reduction or loss of the BALB allele) or white bars (reduction or loss of the CBA allele).
The link between RB1 and osteosarcoma is mainly based on missense and nonsense mutations in the germline which predispose for retinoblastoma in children and OS in adolescents [23]. Following high-dose radiotherapy for primary retinoblastoma in these patients, the risk of a secondary OS is potentiated [24]. We therefore analysed the pattern of allelic imbalances or loss-of-heterozygosity (LOH) across this minimal deleted interval, using polymorphic microsatellite markers and an intragenic SNP at the Rb1 3’UTR (Additional file 3). LOH breakpoints were found between the distal (D14Mit225) markers and Rb1 in three tumors, two of which also had breakpoints in the proximal interval between D14Mit90 and Rb1 (Figure 1).
Expression of candidate genes
We further reasoned that any susceptibility gene for osteosarcoma must be expressed in the relevant target cells in order to influence the tumor risk. We therefore measured the mRNA expression in murine osteoblasts of all genes and none-coding elements as derived from the current release of the mouse genome in this interval (Additional file 1). We found that except for three of them with undetectable expression (Nudt15, Med4, miRNA687hom), the relative expression level in the MC3T3 preosteoblastic cell-line and in-vitro explanted osteoblasts varied between 0.15 fold (Cysltr2) and 3.7 fold (Ltm2b) relative to whole embryo mRNA (Additional file 4).
Rb1 promoter variations
The classical view of genetic predisposition by a germline loss-of-function RB1 allele followed by somatic losses of the wt allele during progression of retinoblastoma is that eventually the tumor expresses the inherited allele which confers high susceptibility in the germline.
We therefore expected to find a gene variation in the CBA allele of Rb1 which can explain the association with the higher OS risk. Sequencing the coding region of Rb1 from the BALB/cHeNhg and CBA/Ca strain, we found only nucleotide variations in the 3’ UTR, but as they did not map to known mRNA stability motifs they were not considered of major functional relevance [25]. Missense and non-sense RB1 mutations, which account for about 80% of heritable retinoblastoma and osteosarcoma cases in patients [26-28], can therefore be ruled out in our case. In addition to coding sequence RB1 mutations associated with highly penetrant phenotypes in humans, a small subset of cases exhibit retinoblastoma predisposition with incomplete penetrance, sometimes caused by mutations that affect RB1 promoter methylation sites [9,29-31]. An association of these low-penetrance gene mutations with OS predisposition, however, was never found, but might have been missed due to the scarcity of such families and the general low OS incidence.
In fact, we found a CBA/Ca specific TCGCCC hexanucleotide deletion in the Rb1 promoter, located 271 nt upstream of the ATG start and 78 nt upstream of the binding sites for Sp1, ATF and E2F in the promoter core (Figure 2a,b) [32]. In-silico analysis using the tool MathInspector [33,34] to analyze changes in TF binding sites predicted that this hexanucleotide InDel could delete a WT1 TF binding site and disrupt a SP1-SP1 module (Additional file 5) [35].
Figure 2.
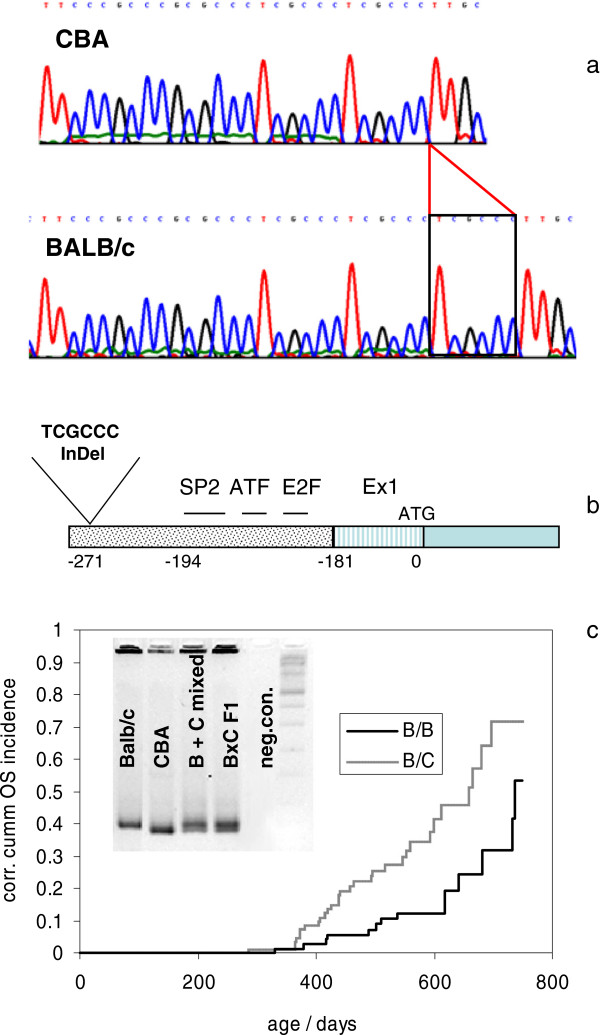
Rb1 Promoter polymorphism between BALB/cHeNhg and CBA/Ca mice. a: Sequence of the Rb1 promoter region between - 267 nt and – 288 nt determined for strains BALB/cHeNhg and CBA/Ca , showing structure of the BALB specific TCGCCC insertion. Base numbering relative to ATG in exon 1. b: Position of the BALB specific hexanucleotide insertion relative to the SP1, ATF and E2F core binding sites according to [32]. c: Association of the Rb1 promoter genotype in BALB/cHeNhg x (BALB/cHeNhg x CBA/Ca) backcrossed mice with osteosarcoma induction. Kaplan-Meier plot of osteosarcoma appearance in 169 BALB/cHeNhg x (BALB/cHeNhg x CBA/Ca) backcrossed mice grouped according to their genotypes at the Rb1 promoter. Mice inheriting the B/C genotype have a significantly shorter latency time and higher overall osteosarcoma incidence than mice with the B/B genotype (p = 0.0007, Log-Rank Test). Of 79 mice inheriting the B/B genotype 14 developed an osteosarcoma, whereas out of 90 mice with the B/C genotype 30 developed an osteosarcoma. Insert: PCR products following amplification of a 122/128 bp fragment (primer 1005f and 1128r) encompassing the site of the BALB/c specific insertion. The insertion specific fragment was used for genotyping all mice of the backcrossed cohort
Impact on Rb1 promoter activity and gene expression
To test the impact of these predicted TF binding site alterations on gene expression, we applied both an in-vitro promoter reporter assay as well as direct quantification of Rb1 transcripts in whole embryos of BALB/cHeNhg and CBA/Ca mouse strains. Rb1 promoter fragments encompassing the ubiquitous SP1, ATF and E2F binding sites together with the site of the Δ(TCGCCC) variation from both mouse strains (Additional file 6) were able to activate a CAT reporter gene in an orientation-dependent and reproducible manner (Figure 3a) after transient transfection of ROS17/2.8 rat osteoblasts. Quantification of the reporter activity from the two promoter variants confirmed that the CBA–specific Δ(TCGCCC) deletion produced less CAT-activity (2.42 AU, CI 2.19 – 2.65) when compared to the BALB/c allele (4.38 AU, CI 4.19 – 4.57, Figure 3b). This 1.8 fold difference is highly significant (p = 0.0003, t-test). An inverse orientation of the Rb1 promoter fragment shows only 21% activity and extracts from mock-transfected cells have less than 8.3% basal activity relative to the BALB/c construct. A difference was also found in the average Rb1 gene expression of day 16 whole embryo extracts, being lower in CBA/Ca (183 AU, CI 138 – 228) than in the BALB/cHeNhg strain (232 AU, CI 165 – 299, p = 0.038, t-test, Additional file 7). Whereas the 1.28 fold reduced mRNA expression in whole-embryo might be a more realistic value in view of the natural genomic context of the Rb1-gene, the 1.8 fold lower promoter activity in transfected osteoblasts could reflect some bone-specific regulation of the Rb1 promoter.
Figure 3.
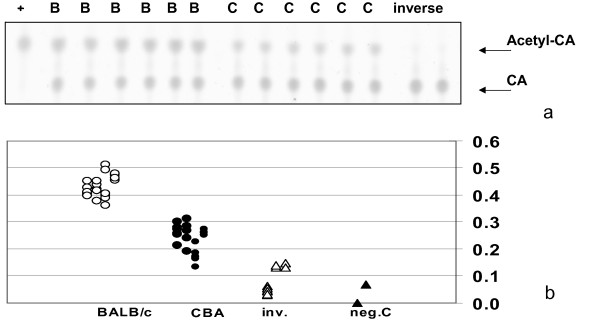
Analysis of transcriptional activity of the variant allele of the Rb1 promoter. a, CAT activity as measured by TLC chromatography of ROS17/2.8 rat osteoblast protein extracts following transient transfection with the BALB/c and the CBA variants of Rb1 promoter fused to CAT reporter constructs. b, Quantification of the CAT reporter signal driven by BALB/c (B) and CBA (C) promoter variants (positive orientation), CBA promoter (inverse orientation) and mock-transfected cells (negC). Transfection efficiency was normalized by simultaneous measurement of luciferase expression from co-transfected RFBpGL3 constructs
It is interesting to note that although the essential TF-binding sites in the core promoter of the Rb1 gene are not affected by this alteration, suggesting that transcription is still possible, two Rb1 control elements map within a 20 bp interval around this hexanucleotide indel [36]. Recently it was shown that these RCEs and other undiscovered sequence elements in-cis of the RB1 core promoter site are crucial for transcriptional fine-tuning in a tissue-specific manner. They are also involved in regulating the RB1 protein homeostasis through feedback loops involving all 3 Rb family proteins members, including a RB1 autosuppression [37].
If for practical purposes we consider a 1.5 fold lower Rb1 expression in homozygous CBA/Ca osteoblasts (i.e. the average between the reporter assay and the in-vivo embryo mRNA level), then the overall Rb1-expression from both alleles in heterozygous BALB/CBA osteoblasts would be reduced to about 83% as compared to 100% in homozygous BALB/BALB osteoblasts (after loss of the BALB/c allele in OS, expression in malignant osteoblasts could further drop to approximately 67%).
Δ(TCGCCC) Rb1 promoter variant and the risk for Th227 induced OS
The impact of this reduced Rb1 expression in normal osteoblast cells for the risk to develop osteosarcoma after Th227-incorporation can be inferred from re-genotyping the BALB/cHeNhg × (BALB/cHeNhg × CBA/Ca) backcross [21] for the Rb1 promoter variant (Figure 2c). Out of 169 backcrossed mice, 93 inherited the BALB/CBA genotype at the Rb1 promoter, of which 23 (or 24.7%) developed an OS. Of the 76 mice inheriting the BALB/BALB genotype, only 10 were diagnosed with a tumor (13.2%, p = 0.016, Fisher’s exact test). In addition to increased tumor incidence, the Δ(TCGCCC) promoter variant in the CBA-allele is also linked to a shorter tumor latency (p = 0.0007, Log-Rank test) (Figure 2c).
The potential impact of reduced RB1 expression for the carcinogenic process
The canonical function of RB1 as a prototype tumor-suppressor gene is based on its ability to regulate the G1/S cell-cycle transition by sequestration of E2F-transcription factors. In bilateral retinoblastoma, this gatekeeper function is completely abolished after loss-of-heterozygosity in carriers of a RB1 germline mutation. In osteosarcoma, although frequently affected by RB1 allelic losses [38], complete absence of gene expression is rarely seen, indicating that a residual function of the wt RB1 protein is compatible with or maybe even accelerates OS growth (Additional file 8). A residual expression was also shown in the majority of other cancer cell lines and tumors except retinoblastoma [26,39], suggesting that reduced expression, but not complete absence of RB1 promotes tumorigenesis
Apart from the canonical RB1 function as a key cell cycle regulator, recent studies also point to its involvement in non-canonical pathways such as regulation of pancentromeric heterochromatin [40], mitotic checkpoint control [41] and telomere maintenance [42,43]. We have recently shown that Rb1 haplo-insufficiency results in a more rapid telomere attrition and a higher number of anaphase-bridges and micro-nuclei after gamma irradiation in murine osteoblasts [43].
One could make the point that these non-canonical functions of RB1 are all involved in the maintenance of genome integrity during cell division. An impaired function of either of these mechanisms could lead to the accumulation of genetic or chromosomal defects, in particular those which are transmitted from parental to daughter cells. A defect in the canonical RB1 cell-cycle regulation, on the other hand might disturb the growth rate of a cell itself, but not necessarily increase genomic instability in the progenitor cells. A transmission of chromosomal instability from parent to daughter cells due to an impaired RB1 function was also suggested by discovering its involvement in the regulation of centromeric heterochromatin, a key factor for a proper chromosome segregation during mitosis [44]. In particular the genotoxic stress by ionizing irradiation would require an efficient cellular surveillance mechanism for genetic and chromosomal stability to minimize the risk of malignant transformation [45].
Most epidemiological studies indicate that ionizing irradiation can increase the number of solid tumors and leukaemia, without changing the age distribution of their diagnosis. This suggests that the rate of accumulating additional genetic defects during tumor latency is a cell inherent process and only marginally influenced by radiation. The latency time, however shortens dramatically in the event of a congenital tumor predisposition, therefore causing a rise in tumors in children or young adulthood. Regarding their pathobiology, radiation-induced OS with latency times between 10 and 20 years therefore represent tumors of the older patient group. But in combination with an increased genetic susceptibility caused by an inherited low expressing RB1 allele, these tumors might shift to younger age groups.
We hypothesize that a reduced RB1 expression renders normal cells more vulnerable to secondary genetic changes, and that the resulting loss of genome stability is a driving factor for tumorigenesis in itself. Rb1 haploinsufficiency in murine ES cells was shown to increase the rate of genetic instability [46], demonstrating that a reduced gene expression can easily compromise genome maintenance. Such a reduced capability of cells to adequately respond to chromosomal defects could have severe consequences after radiation exposure, considering that radiation-induced genotoxicity is mainly due to the generation of DNA strand breaks.
Structural variants in the human RB1 promoter
The majority of RB1 germline mutations found in congenital retinoblastoma patients cause partial or complete loss of the gene (41%) or frame shift and nonsense mutations (42%) [26-28]. The remaining cases, some of which show incomplete penetrance of the condition, are assumed to carry germline mutations affecting regulatory elements of the RB1 gene. It would therefore be worth systematically testing non-syndromic OS patients for the presence of low-penetrance RB1 promoter variants. Recent efforts to screen the human genome for common variants have shown that the RB1 promoter not only harbours several SNPs, but at least two InDel variants. A hexanucleotide variation (rs2092879) 1.5 kbp upstream of the human RB1 promoter core was shown to alter TF binding and promoter activity in vitro [47], whereas a 23bp InDel variant (rs36230211) is located at a similar distance to the transcription start (221 bp proximal) as the murine InDel variation described here (Figure 4). Considering that the human 23bp InDel removes part of the SP1 site from the core promoter, and that the structure of transcription factor binding sites in this region is highly conserved from mouse to human [32,36] one can assume that carriers of the human InDel variant are also affected by a reduced RB1 expression. The observations that a sufficient RB1 expression is instrumental for cells to prevent structural chromosomal defects from being transmitted to the next cell generation suggests, that a reduced RB1 level might result in a steady accumulation of mutations and genomic instability in normal cells. This would cause a higher incidence of OS in the group of older patients, but at the same time become critical in the event of a genotoxic stress as caused by radiation. It might therefore be interesting to search for the presence of the above mentioned human RB1 promoter variants in older OS patients and in those presenting with a post-radiation, therapy associated tumor.
Figure 4.
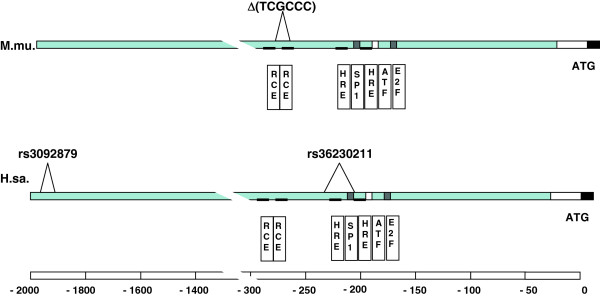
Binding sites for transcription factors and regulatory proteins in the murine and the human Rb1 promoter (below the line) and position of the reported indel polymorphisms (above line). TF binding sites in murine sequence as predicted by MatInspector software [33,34] and as annotated by Zacksenhaus [32] (Genebank Acc.-No. M86180). TF binding sites in human sequence as predicted by [36]. Promoter core consisting of steroid hormone response elements (HRE) and Sp1, ATF and E2F binding sites are highly conserved. Retinoblastoma control elements (RCE) in the human promoter are according to [36] and predicted in the murine promoter using published RCE core sequences [48].
Conclusion
This is the first experimental demonstration of a functional and genetic link between reduced Rb1 expression from a common promoter variant and increased risk for osteosarcoma after radiation exposure in mice. We propose that a reduced Rb1 expression by common variants in regulatory regions can modify the risk for a malignant transformation of bone cells after radiation exposure.
The low phenotypic penetrance of such promoter or regulatory variants spares them from significant selection pressure, and hence increases their chance of becoming fixed and more common in the population. We have shown in this study that in combination with exogenous genomic stress like ionizing radiation these low penetrance variations in RB1 can significantly alter the tumor susceptibility of an organism.
Methods
Mouse breeding and tumor induction
All animal experiments were approved by the state government (license no. RegOB 211-2531-49/00 and 211-2531-56/06) and carried out in accordance with the federal animal welfare guidelines. (BALB/cHeNhg × CBA/Ca)F1 and F1 mice backcrossed to female BALB/cHeNhg (further referred to as BBC) mice were bred from HMGU Neuherberg (formerly GSF) breeding stocks. For tumor induction, 80 (BALB/cHeNhg × CBA/Ca)F1 and 169 BBC mice (all female) were injected i.p. at the age of 100 days with a single dose of 185Bq/g 227Th (as Thorium Citrate) [19]. Mice were housed 5 to a cage and examined 5 days a week for the development of tumors or other life-threatening conditions. Bone tumors were diagnosed radiologically at necropsy followed by histological examination after EDTA decalcification. All malignant diseases were classified by trained clinical and veterinary pathologists, using H&E and van-Gieson staining. Typical histologies and X-ray images of tumors are deposited in the PATHBASE database (http://eulep.pdn.cam.ac.uk, Acc.No. 3410-3414, Acc.No. PB94713 ff).
Statistical evaluation of osteosarcoma induction
Osteosarcoma-free survival curves, corrected for all competing causes of death, were calculated using the Kaplan-Mayer method. Significance of differences were tested with the Statistika 6.0 software (StatSoft, Tulsa OK) using Cox’s F- and LogRank-Test for tumor-free survival, two-tailed Fisher’s exact test for fraction of tumor-bearing mice, one-tailed Chi-square test of a four-field contingency table for the allelic imbalances and t-test for the gene-expression and reporter assays.
DNA extraction, microsatellite genotyping and Rb1 sequencing
DNA for genotyping of mice was extracted from tail-tips and from macro-dissected tumor-sections for measuring allelic imbalances. Genomic PCR was done using primers for polymorphic microsatellite markers and for the Δ(TCGCCC) Rb1 promoter variant (Additional file 9). Reactions were carried out in 96 well plates using a GeneAmp 9700 thermocycler (Applied Biosystems, Foster City, Ca, USA) followed by 90min electrophoresis on 1.5% or 3% agarose/TBE gels.
Analysis of allelic imbalance and LOH in osteosarcoma
Allelotyping of 17 osteosarcomas diagnosed in (BALB/cHeNhg × CBA/Ca)F1 mice and of the paired normal tissue was done for polymorphic microsatellite markers on chromosome 14 (see above) by allel-specific PCR and for a SNP in the Rb1 3’ UTR (3090nt) by genomic sequencing. PCR-amplified microsatellite markers were run for 90min electrophoresis in 3% agarose/TBE/EtBr gels, the PCR products were visualized under UV illumination, captured as 8 bit TIF-files on an electronic DocuGel V system (Mitsubishi, Japan) and quantified using the software package Intelligent Quantifier (BioImage, Jackson MI). To validate linearity and sensitivity of this assay, standard curves of the allelic ratios were generated from BALB/CBA genomic DNA containing different percentages of pure BALB/c or pure CBA/Ca DNA (Additional file 10).
Candidate positional mapping using PosMed
The smallest common region affected by allelic imbalances in osteosarcoma from (BALB/cHeNhg × CBA/Ca)F1 mice as defined by flanking microsatellite markers was submitted to a semantic web association study using PosMed [22]. The search parameters were set as: search=“gene”, condition=“genomic interval”, species=“mouse”, keyword=“osteosarcoma susceptibility” genome sequence version=“NCBIm37”. search settings=“all documents”. With a cut-off p-value of 0.01 (with Bonferroni correction), the list of gene hits was ranked in the order of increasing p-value.
qRT-PCR measurement of mRNA expression
mRNA (1μg per sample) was extracted from the MC3T3-E1 preosteoblastic cell line (ATCC code CRL-2593™) and from primary murine osteoblasts grown in-vitro[43] using RNAeasy mini kit (Quiagen GmbH, Hilden, Germany) and reverse transcribed using SuperscriptIII (Life Technologies, Carlsbad, CA). Quantitative RT-PCR was done in 96well plates on a StepOne machine using Fast Sybr Green (both Life Technologies, Carlsbad, CA) and gene specific primers (Additional file 9). Quantification of expression levels was done using the ΔΔCt method with a house keeping gene (TBP) and whole embryo cDNA as a calibrator.
Rb1 mRNA northern blot
Whole embryo RNA was extracted from day 16 p.c. BALB/cHeNhg and CBA/Ca embryos by homogenisation in liquid N2 followed by GTC extraction and phenol/chloroform purification. For each strain, 6 embryos from 3 different pregnant mice were used. Following northern blot of 10μg of each RNA sample, membranes (Roche Diagnostics, Germany) were probed with a 630 bp 32P labelled mouse Rb1 cDNA fragment spanning exon 10 to 13. Signals were quantified using a Storm scanner (Amersham Pharmacia, Uppsala, Sweden) and Image Quant software (Molecular Dynamics, Sunnyvale CA, USA). Relative expression levels (AU) are given as the integrated optical densities of the Rb1 signal normalized to the sum of the 28S and 18S RNA signals.
Prediction of promoter function
A 352bp sequence containing the putative Rb1 promoter from BALB/cHeNhg and CBA/Ca mice, including known upstream regulatory sequences, the 5’-UTR and the initiation codon ATG was analysed for potential TF binding sites using MatInspector software (Genomatix Software GmbH, Munich, Germany) [33,34]. Predefined transcription factor weight matrix descriptions are based on the TRANSFAC-database (all-vertebrates library) with the following default parameter: core similarity = 0.75 and matrix similarity = calculated optimised.
CAT reporter assay
A fragment of the Rb1 promoter was PCR amplified from CBA/Ca (451 bp) and BALB/cHeNhg (457 bp) genomic DNA, using primers 6F and 8.2B, Pwo-polymerase (Peqlab, Erlangen, Germany) and reaction conditions as given in Additional file 9. The fragment was cloned into TOPO TA® vector (Life Technologies, Carlsbad, CA) and directionally sub-cloned into the pCAT3 basic vector (Promega Corp. Madison WI, USA) using SacI and XhoI restriction sites. Plasmids BALB 36, CBA 11 and CBA 14–5 (Additional file 6) were purified using Quiagen plasmid MaxiPrep kit (Quiagen GmbH, Hilden, Germany) yielding a final concentration of 2 μg/μl and OD260/OD280 absorption ratios between 1.68 and 1.73. A luciferase expression vector pGL3® (Promega Corp., Madison WI, USA) under the control of the RFB LTR served as an internal control for transfection efficiency. ROS 17/2.8 osteoblasts at a density of 5*105 cells per 60mm plate were transiently transfected with 2μg Rb1pCAT and 0.2μg pGL3 using FuGene® lipofection (Roche Diagnostics GmbH, Mannheim, Germany). After 48h, cells were harvested using reporter lysis buffer (Promega Corp, Madison WI, USA) and 20μg protein extracts were assayed for CAT activity using the FastCAT yellow® assay ((Life Technologies Molecular Probes, Carlsbad, CA). After thin-layer chromatography, TLC plates (Sigma Aldrich, Germany) were visualized on a Storm laser scanner (Amersham Pharmacia, Uppsala, Sweden) and quantified using Image Quant software (Molecular Dynamics, Sunnyvale CA, USA). Luciferase expression was measured using the luciferase assay system (Promega Corp. Madison WI, USA) and an Auto-Lumat LB953 luminometer (EGG Berthold, Pforzheim, Germany). Promoter activity was expressed in arbitrary units as AU = 1000* ac-CA/((CA + ac-CA)* AUluc), wherein CA and ac-CA are the integral densities of the chloramphenicol and the acetyl-CA signal, respectively, and AUluc is the luciferase signal.
Data access
Histological X-ray images of representational cases of osteosarcoma were deposited to PATHBASE database (http://eulep.anat.cam.ac.uk, acc.No. PB94713 ff). Rb1 promoter sequence variation was deposited to the European Nucleotide Archive (http://www.ebi.ac.uk/ena/) and to the Database of genomic variants archive (http://www.ebi.ac.uk/dgva/, acc.No. pending).
Abbreviations
AU: Arbitrary units; CA: Chloramphenicol; CAT: Chloramphenicol-acetyl-transferase; DMEM: Dulbecco’s modified Eagle medium; FCS: Fetal calf serum; GTC: Guanidiniumthiocyanate; GWAS: genome-wide association study; i.p.: Intraperitoneal; LOD: logarithm of the odds ratio for linkage; LOH: Loss-of-heterozygosity; OD: Optical density; OR: Odds-ratio; OS: Osteosarcoma; QTL: Quantitative trait locus; RCE: Retinoblastoma control element; RFB-LTR: Long terminal repeat of the RFB virus; SNP: Single nucleotide polymorphism; STS: Sequence tagged site; TLC: Thin layer chromatography; TSG: Tumor suppressor gene; TF: Transcription factor; UTR: Untranslated region.
Competing interests
The authors declare that none of the data in this study are affected by competing interests of a third party.
Authors’ contributions
(MR) planned and conducted the animal studies, cloning and reporter assay of the promoter variant, (IGV) measured expression of candidate genes after mapping by PosMed in normal osteoblasts and RB1 in human OS cell lines, (TD) did Rb1 mRNA measurements on microdissectied murine tumors, (VK) performed the LOH/allelic imbalance studies on F1 tumors, (RS) did the in-silico promoter modelling using MathInspector, (MK) performed the tumor histo-pathology, (JF) helped with haplotype analysis and genetic mapping of the murine locus, (MN) provided data on SNP LOH and RB1 mutations of human OS, (MJA) supervised the study and contributed to discussion and conclusion of the paper. All authors read and approved the final manuscript.
Supplementary Material
List of annotated genes on mouse chromosome 14, 72 600 000 Mbp - 75 300 000 Mbp, their function, expression in osteoblasts and evidence for an association with osteosarcoma by PosMed-search.
Search for a candidate gene for osteosarcoma susceptibility using the online mapping and annotation tool PosMed. The interval on murine chromosome 14 between D14Mit90 (72.6 Mbp) and D14Mit225 (75.3 Mbp) was analyzed for genes for which literature records have a co-citation with the key-word “Osteosarcoma Susceptibility”). Co-citation of Rb1 with “Osteosarcoma Susceptibility” in 12 publications occurs with highest significance (p = 5 × 10–53), whereas four other genes are only indirectly co-cited with the phenotype and have lower significance (p < 1.2 × 10–20).
Strain polymorphism in the 3’ UTR used for genotyping tumor vs. normal tissue. In the lower panel two representative cases with loss of the BALB- and CBA-alleles are shown, resp.
mRNA expression of mapped candidate genes at the OS susceptibility interval in normal osteoblasts and in a MC3T3 pre-osteoblast cell line. Expression level is relative to TBP housekeeping mRNA. All values are mean +/−2xSE from 3 independent osteoblast cultures.
Position of transcription factor binding sites within the Rb1 promoter sequence as predicted using MatInspector software. The binding sites above the sequence (in black letters) are present in the BALB / CBA consensus sequence, whereas binding sites below the sequence (in red letters) are unique for the BALB allele. Asterisks show TF binding sites as annotated by Zacksenhaus et al. 1993 (Genebank Acc.-No. M86180).
Reporter constructs for in-vitro analysis of Rb1 promoter activity using CAT reporter constructs fused to 452/458 bp long fragments derived from the BALB/c and CBA-variant of the Rb1 promoter. As negative control, an inverse orientated CBA-fragment was used. Base numbering is according to Genebank Acc.-No. M86180.
Rb1 -mRNA expression in BALB/cHeNhg vs. CBA/Ca mouse embryos 16 days p.c. as measured by northern blot.
Expression of RB1-protein in established human OS cell lines (S8a), of Rb1 mRNA in 21 human primary osteosarcoma in comparison with OS cell lines U2OS (Rb1 positive) and SaOS (Rb1 deficient) (S8b) and Rb1 mRNA in murine OS cell lines (S8c). Note that with a few exceptions, all murine and human osteosarcoma maintain a low expression of RB1 mRNA and protein expression.
Primer Sequences and PCR conditions for genomic and qRT-PCR.
Protocol for the quantification of allelic imbalance/loss-of-heterozygosity using genomic PCR of strain - specific microsatellite markers. Quantitative loss or imbalances of BALB- or CBA alleles was calibrated for each of the used markers. For this purpose, pure BALB/c and pure CBA/Ca genomic DNA were mixed in different ratios and used as template for PCR (27, 30, 33 and 35 cycles). After running the PCR products on agarose gel electrophoresis and recording the pattern of EtBr stained BALB- and CBA bands, densitometric measurement of the two signals was done. The ratio of the experimentally determined BALB- and CBA alleles was compared with the relative amount of BALB- and CBA-DNA mixed in the PCR templates and served to draw calibration curves for the later measurement of allelic ratios in the tumor DNA.
Contributor Information
Michael Rosemann, Email: Rosemann@helmholtz-muenchen.de.
Iria Gonzalez-Vasconcellos, Email: Iria.Gonzalez.Vasconcellos@sergas.es.
Tanja Domke, Email: Tanja.Domke@med.uni-muenchen.de.
Virginija Kuosaite, Email: virginija@gmx.net.
Ralf Schneider, Email: ralf.schneider@helmholtz-muench.de.
Markus Kremer, Email: pathologie@khmh.de.
Jack Favor, Email: Favor@helmholtz-muenchen.de.
Michaela Nathrath, Email: michaela.nathrath@lrz.tu-muenchen.de.
Michael J Atkinson, Email: atkinson@helmholtz-muenchen.de.
Acknowledgements
This work was supported by grants from the EU (contract No. 249689, DoReMi), from the German federal government (BMBF 03NUK007) and from the Munich Parents Initiative for Cancer Kids. We are grateful for the skilled technical help from Mrs. Bahar Sanli-Bonazzi, Mrs. Ulrike Reich, Mrs. Eleonore Samson and Mrs. Jacqueline Mueller, and for proof reading and text corrections done by Mrs. Jane Rifkin.
References
- Lindor NM, Greene MH. The Mayo Familial Cancer P. The concise handbook of family cancer syndromes. J Natl Cancer Inst. 1998;90:1039–1071. doi: 10.1093/jnci/90.14.1039. [DOI] [PubMed] [Google Scholar]
- Mulvihill JJ, Gralnick HR, Whang-Peng J, Leventhal BG. Multiple Childhood Osteosarcomas in an American Indian Family with erythroid Macrocytosis and Skeletal Anomalies. Cancer. 1997;40:3115–3122. doi: 10.1002/1097-0142(197712)40:6<3115::aid-cncr2820400655>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Nishida J, Abe M, Shiraishi H, Shimamura T, Tamura G, Satoh T, Ehara S. Familial Occurrence of Telangiectatic Osteosarcoma: Cousin Cases. J Pediatr Orthop. 1994;14:119–122. doi: 10.1097/01241398-199401000-00023. [DOI] [PubMed] [Google Scholar]
- Danckwerth F, Wuisman P, Ritter J, Blasius S, Jurgens H, Ozaki T, Winkelmann W. Osteosarcoma in 2 siblings. A case report. Klin Padiatr. 1995;207:298–301. doi: 10.1055/s-2008-1046555. [DOI] [PubMed] [Google Scholar]
- Neglia JP, Friedman DL, Yasui Y, Mertens AC, Hammond S, Stovall M, Donaldson SS, Meadows AT, Robison LL. Second malignant neoplasms in five-year survivors of childhood cancer: Childhood cancer survivor study. J Natl Cancer Inst. 2001;93:618–629. doi: 10.1093/jnci/93.8.618. [DOI] [PubMed] [Google Scholar]
- Dong C, Hemminki K. Second primary neoplasms in 633,964 cancer patients in Sweden, 1958–1996. Int J Cancer. 2001;93:155–161. doi: 10.1002/ijc.1317. [DOI] [PubMed] [Google Scholar]
- Hauben EI, Arends J, Vandenbroucke JP, van Asperen CJ, Van Marck E, Hogendoorn PC. Multiple primary malignancies in osteosarcoma patients. Incidence and predictive value of osteosarcoma subtype for cancer syndromes related with osteosarcoma. Eur J Hum Genet. 2003;11:611–618. doi: 10.1038/sj.ejhg.5201012. [DOI] [PubMed] [Google Scholar]
- Varley JM, McGown G, Thorncroft M, Santibanez-Koref MF, Kelsey AM, Tricker KJ, Evans DG, Birch JM. Germ-line mutations of TP53 in Li-Fraumeni families: an extended study of 39 families. Cancer Res. 1997;57:3245–3252. [PubMed] [Google Scholar]
- Lohmann DR, Gallie BL. Retinoblastoma: revisiting the model prototype of inherited cancer. Am J Med Genet C: Semin Med Genet. 2004;129c(1):23–28. doi: 10.1002/ajmg.c.30024. [DOI] [PubMed] [Google Scholar]
- Meisner LF, Gilbert E, Ris HW, Haverty G. Genetic mechanisms in cancer predisposition: report of a cancer family. Cancer. 1979;43:679–689. doi: 10.1002/1097-0142(197902)43:2<679::aid-cncr2820430240>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Ponder BA. Inherited predisposition to cancer. Trends Genet. 1990;6:213–228. doi: 10.1016/0168-9525(90)90181-5. [DOI] [PubMed] [Google Scholar]
- Hemminki K, Mutanen P. Genetic epidemiology of multistage carcinogenesis. Mutat Res. 2001;473:11–21. doi: 10.1016/s0027-5107(00)00162-7. [DOI] [PubMed] [Google Scholar]
- Ponder BA. Cancer genetics. Nature. 2001;411:336–441. doi: 10.1038/35077207. [DOI] [PubMed] [Google Scholar]
- Savage SA, Mirabello L, Wang Z, Gastier-Foster JM, Gorlick R, Khanna C, Flanagan AM, Tirabosco R, Andrulis IL, Wunder JS, Gokgoz N, Patiño-Garcia A, Sierrasesúmaga L, Lecanda F, Kurucu N, Ilhan IE, Sari N, Serra M, Hattinger C, Picci P, Spector LG, Barkauskas DA, Marina N, de Toledo SR, Petrilli AS, Amary MF, Halai D, Thomas DM, Douglass C, Meltzer PS. Genome-wide association study identifies two susceptibility loci for osteosarcoma. Nat Genet. 2013;45:799–803. doi: 10.1038/ng.2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Qian X, Redman C, Bliskovski V, Ramsay ES, Lowy DR, Mock BA. p16 INK4a gene promoter variation and differential binding of a repressor, the ras-responsive zinc-finger transcription factor, RREB. Oncogene. 2003;22:2285–2295. doi: 10.1038/sj.onc.1206257. [DOI] [PubMed] [Google Scholar]
- Dietrich WF, Lander ES, Smith JS, Moser AR, Gould KA, Luongo C, Borenstein N, Dove W. Genetic identification of Mom-1, a major modifier locus affecting Min-induced intestinal neoplasia in the mouse. Cell. 1993;75:631–639. doi: 10.1016/0092-8674(93)90484-8. [DOI] [PubMed] [Google Scholar]
- Ruivenkamp CA, van Wezel T, Zanon C, Stassen AP, Vlcek C, Csikós T, Klous AM, Tripodis N, Perrakis A, Boerrigter L, Groot PC, Lindeman J, Mooi WJ, Meijjer GA, Scholten G, Dauwerse H, Paces V, van Zandwijk N, van Ommen GJ, Demant P. Ptprj is a candidate for the mouse colon-cancer susceptibility locus Scc1 and is frequently deleted in human cancers. Nat Genet. 2002;31:295–300. doi: 10.1038/ng903. [DOI] [PubMed] [Google Scholar]
- Yu Y, Okayasu R, Weil MM, Silver A, McCarthy M, Zabriskie R, Long S, Cox R, Ullrich RL. Elevated breast cancer risk in irradiated BALB/c mice associates with unique functional polymorphism of the Prkdc (DNA-dependent protein kinase catalytic subunit) gene. Cancer Res. 2001;61:1820–1824. [PubMed] [Google Scholar]
- Luz A, Mueller WA, Linzner U, Strauss PG, Schmidt J, Mueller K, Atkinson MJ, Murray AB, Gossner W, Erfle V, Hofler H. Bone tumor induction after incorporation of short-lived radionuclides. Radiat Environ Biophys. 1991;30:225–227. doi: 10.1007/BF01226625. [DOI] [PubMed] [Google Scholar]
- Rosemann M, Lintrop M, Favor J, Atkinson MJ. Bone tumorigenesis induced by alpha-particle radiation: mapping of genetic Loci influencing predisposition in mice. Radiat Res. 2002;157:426–434. doi: 10.1667/0033-7587(2002)157[0426:btibap]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Rosemann M, Kuosaite V, Kremer M, Favor J, Quintanilla-Martinez L, Atkinson MJ. Multilocus inheritance determines predisposition to alpha-radiation induced bone tumourigenesis in mice. Int J Cancer. 2006;118:2132–2138. doi: 10.1002/ijc.21612. [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Makita Y, Heida N, Asano S, Matsushima A, Ishii M, Mochizuki Y, Masuya H, Wakana S, Kobayashi N, Toyoda T. PosMed (Positional Medline): prioritizing genes with an artificial neural network comprising medical documents to accelerate positional cloning. Nucleic Acids Res. 2009;37:W147–W152. doi: 10.1093/nar/gkp384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friend SH, Bernards R, Rogelj S, Weinberg RA, Rapaport JM, Albert DM, Dryja TP. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323:643–646. doi: 10.1038/323643a0. [DOI] [PubMed] [Google Scholar]
- Wong FL, Boice JD, Abramson DH, Tarone RE, Kleinerman RA, Stovall M, Goldman MB, Seddon JM, Tarbell N, Fraumeni JF, Li FP. Cancer incidence after retinoblastoma: Radiation dose and sarcoma risk. JAMA. 1997;278:1262–1267. doi: 10.1001/jama.278.15.1262. [DOI] [PubMed] [Google Scholar]
- Latchman D. In: Gene regulation. Volume 1. Latchman D, editor. London: Chapmann and Hall; 1994. Regulation of RNA stability; pp. 82–89. [Google Scholar]
- Dunn JM, Phillips RA, Becker AJ, Gallie BL. Identification of germline and somatic mutations affecting the retinoblastoma gene. Science. 1988;241:1797–1800. doi: 10.1126/science.3175621. [DOI] [PubMed] [Google Scholar]
- Blanquet V, Turleau C, Gross-Morand MS, Senamaud-Beaufort C, Doz F, Besmond C. Spectrum of germline mutations in the RB1 gene: a study of 232 patients with hereditary and non hereditary retinoblastoma. Hum Mol Genet. 1995;4:383–388. doi: 10.1093/hmg/4.3.383. [DOI] [PubMed] [Google Scholar]
- Lohmann DR, Brandt B, Hopping W, Passarge E, Horsthemke B. The spectrum of RB1 germ-line mutations in hereditary retinoblastoma. Am J Hum Genet. 1996;58:940–949. [PMC free article] [PubMed] [Google Scholar]
- Sakai T, Ohtani N, McGee TL, Robbins PD, Dryja TP. Oncogenic germ-line mutations in Sp1 and ATF sites in the human retinoblastoma gene. Nature. 1991;353:83–86. doi: 10.1038/353083a0. [DOI] [PubMed] [Google Scholar]
- Cowell JK, Bia B, Akoulitchev A. A novel mutation in the promotor region in a family with a mild form of retinoblastoma indicates the location of a new regulatory domain for the RB1 gene. Oncogene. 1996;12:431–436. [PubMed] [Google Scholar]
- Ohtani-Fujita N, Dryja TP, Rapaport JM, Fujita T, Matsumura S, Ozasa K, Watanabe Y, Hayashi K, Maeda K, Kinoshita S, Matsumura T, Ohnishi Y, Hotta Y, Takahashi R, Kato MV, Ishizaki K, Sasaki MS, Horsthemke B, Minoda K, Sakai T. Hypermethylation in the retinoblastoma gene is associated with unilateral, sporadic retinoblastoma. Cancer Genet Cytogenet. 1997;98:43–49. doi: 10.1016/s0165-4608(96)00395-0. [DOI] [PubMed] [Google Scholar]
- Zacksenhaus E, Gill RM, Phillips RA, Gallie BL. Molecular cloning and characterization of the mouse RB1 promoter. Oncogene. 1993;8:2343–2351. [PubMed] [Google Scholar]
- Quandt K, Frech K, Karas H, Wingender E, Werner T. MatInd and MatInspector: new fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res. 1995;23:4878–4884. doi: 10.1093/nar/23.23.4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner T. Computer-assisted analysis of transcription control regions. Matinspector and other programs. Methods Mol Biol. 2000;132:337–349. doi: 10.1385/1-59259-192-2:337. [DOI] [PubMed] [Google Scholar]
- Hauses M, Tonjes RR, Grez M. The transcription factor Sp1 regulates the myeloid-specific expression of the human hematopoietic cell kinase (HCK) gene through binding to two adjacent GC boxes within the HCK promoter- proximal region. J Biol Chem. 1998;273:31844–31852. doi: 10.1074/jbc.273.48.31844. [DOI] [PubMed] [Google Scholar]
- Gill RM, Hamel PA, Zhe J, Zacksenhaus E, Gallie BL, Phillips RA. Characterization of the human RB1 promoter and of elements involved in transcriptional regulation. Cell Growth Differ. 1994;5:467–474. [PubMed] [Google Scholar]
- Burkhart DL, Ngai LK, Roake CM, Viatour P, Thangavel C, Ho VM, Knudsen ES, Sage J. Regulation of RB transcription in vivo by RB family members. Mol Cell Biol. 2010;30:1729–1745. doi: 10.1128/MCB.00952-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smida J, Baumhoer D, Rosemann M, Walch A, Bielack S, Poremba C, Remberger K, Korsching E, Scheurlen W, Dierkes C, Burdach S, Jundt G, Atkinson MJ, Nathrath M. Genomic alterations and allelic imbalances are strong prognostic predictors in osteosarcoma. Clin Cancer Res. 2010;16:4256–4267. doi: 10.1158/1078-0432.CCR-10-0284. [DOI] [PubMed] [Google Scholar]
- Horowitz JM, Park SH, Bogenmann E, Cheng JC, Yandell DW, Kaye FJ, Minna JD, Dryja TP, Weinberg RA. Frequent inactivation of the retinoblastoma anti-oncogene is restricted to a subset of human tumor cells. Proc Natl Acad Sci U S A. 1990;87:2775–2779. doi: 10.1073/pnas.87.7.2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning AL, Longworth MS, Dyson NJ. Loss of pRB causes centromere dysfunction and chromosomal instability. Genes Dev. 2010;24:1364–1376. doi: 10.1101/gad.1917310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coschi CH, Martens AL, Ritchie K, Francis SM, Chakrabarti S, Berube NG, Dick FA. Mitotic chromosome condensation mediated by the retinoblastoma protein is tumor-suppressive. Genes Dev. 2010;24:1351–1363. doi: 10.1101/gad.1917610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Cao M, Gonzalo S, Dean D, Blasco MA. A role for the Rb family of proteins in controlling telomere length. Nat Genet. 2002;32:415–419. doi: 10.1038/ng1011. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Vasconcellos I, Anastasov N, Sanli-Bonazzi B, Klymenko O, Atkinson MJ, Rosemann M. Rb1 haploinsufficiency promotes telomere attrition and radiation-induced genomic instability. Cancer Res. 2013;73:4247–4255. doi: 10.1158/0008-5472.CAN-12-3117. [DOI] [PubMed] [Google Scholar]
- Sage J, Straight AF. RB's original CIN? Genes Dev. 2010;24:1329–1333. doi: 10.1101/gad.1948010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott KR, Rosemann M. Molecular mechanisms of radiation carcinogenesis and the linear, non-threshold dose response model of radiation risk estimation. Radiat Environ Biophys. 2000;39:79–87. doi: 10.1007/s004110000047. [DOI] [PubMed] [Google Scholar]
- Zheng L, Flesken-Nikitin A, Chen PL, Lee WH. Deficiency of Retinoblastoma gene in mouse embryonic stem cells leads to genetic instability. Cancer Res. 2002;62:2498–2502. [PubMed] [Google Scholar]
- Belanger H, Beaulieu P, Moreau C, Labuda D, Hudson TJ, Sinnett D. Functional promoter SNPs in cell cycle checkpoint genes. Hum Mol Genet. 2005;14:2641–2648. doi: 10.1093/hmg/ddi298. [DOI] [PubMed] [Google Scholar]
- Banchio C, Lingrell S, Vance DE. Sp-1 binds promoter elements that are regulated by retinoblastoma and regulate CTP:phosphocholine cytidylyltransferase-alpha transcription. J Biol Chem. 2007;282:14827–14835. doi: 10.1074/jbc.M700527200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of annotated genes on mouse chromosome 14, 72 600 000 Mbp - 75 300 000 Mbp, their function, expression in osteoblasts and evidence for an association with osteosarcoma by PosMed-search.
Search for a candidate gene for osteosarcoma susceptibility using the online mapping and annotation tool PosMed. The interval on murine chromosome 14 between D14Mit90 (72.6 Mbp) and D14Mit225 (75.3 Mbp) was analyzed for genes for which literature records have a co-citation with the key-word “Osteosarcoma Susceptibility”). Co-citation of Rb1 with “Osteosarcoma Susceptibility” in 12 publications occurs with highest significance (p = 5 × 10–53), whereas four other genes are only indirectly co-cited with the phenotype and have lower significance (p < 1.2 × 10–20).
Strain polymorphism in the 3’ UTR used for genotyping tumor vs. normal tissue. In the lower panel two representative cases with loss of the BALB- and CBA-alleles are shown, resp.
mRNA expression of mapped candidate genes at the OS susceptibility interval in normal osteoblasts and in a MC3T3 pre-osteoblast cell line. Expression level is relative to TBP housekeeping mRNA. All values are mean +/−2xSE from 3 independent osteoblast cultures.
Position of transcription factor binding sites within the Rb1 promoter sequence as predicted using MatInspector software. The binding sites above the sequence (in black letters) are present in the BALB / CBA consensus sequence, whereas binding sites below the sequence (in red letters) are unique for the BALB allele. Asterisks show TF binding sites as annotated by Zacksenhaus et al. 1993 (Genebank Acc.-No. M86180).
Reporter constructs for in-vitro analysis of Rb1 promoter activity using CAT reporter constructs fused to 452/458 bp long fragments derived from the BALB/c and CBA-variant of the Rb1 promoter. As negative control, an inverse orientated CBA-fragment was used. Base numbering is according to Genebank Acc.-No. M86180.
Rb1 -mRNA expression in BALB/cHeNhg vs. CBA/Ca mouse embryos 16 days p.c. as measured by northern blot.
Expression of RB1-protein in established human OS cell lines (S8a), of Rb1 mRNA in 21 human primary osteosarcoma in comparison with OS cell lines U2OS (Rb1 positive) and SaOS (Rb1 deficient) (S8b) and Rb1 mRNA in murine OS cell lines (S8c). Note that with a few exceptions, all murine and human osteosarcoma maintain a low expression of RB1 mRNA and protein expression.
Primer Sequences and PCR conditions for genomic and qRT-PCR.
Protocol for the quantification of allelic imbalance/loss-of-heterozygosity using genomic PCR of strain - specific microsatellite markers. Quantitative loss or imbalances of BALB- or CBA alleles was calibrated for each of the used markers. For this purpose, pure BALB/c and pure CBA/Ca genomic DNA were mixed in different ratios and used as template for PCR (27, 30, 33 and 35 cycles). After running the PCR products on agarose gel electrophoresis and recording the pattern of EtBr stained BALB- and CBA bands, densitometric measurement of the two signals was done. The ratio of the experimentally determined BALB- and CBA alleles was compared with the relative amount of BALB- and CBA-DNA mixed in the PCR templates and served to draw calibration curves for the later measurement of allelic ratios in the tumor DNA.