

Abstract
Background
Current evidence suggests a central role for autophagy in many neurodegenerative diseases including Alzheimer’s disease, Huntington’s disease, Parkinson’s disease and amyotrophic lateral sclerosis. Furthermore, it is well admitted that inflammation contributes to the progression of these diseases. Interestingly, crosstalks between autophagy and inflammation have been reported in vitro and at the peripheral level such as in Crohn’s disease. However, the impact of systemic inflammation on autophagic components in the brain remains to be documented. Therefore, this study monitored autophagy markers after acute and chronic lipopolysaccharide (LPS)-induced inflammatory stress in mice.
Results
We showed that acute inflammation, 24 h post-intraperitoneal 10 mg/kg LPS, substantially increased cytokine production (Interleukin(IL)-1β, Tumor necrosis factor (TNF)-α and IL-6), decreased the levels of autophagy markers (Beclin-1, p62 and LC3 II) and reduced p70S6K activation in cortex and hippocampus. In hippocampus, IL-1β levels and LC3 II expression were positively and highly correlated and a negative correlation was noted between TNF-α levels and p70S6K activation. Chronic inflammation by injection of 0.5 mg/kg LPS every three days during three months led to a moderate IL-1β production and decreased TNF-α levels. Interestingly, Beclin-1 and LC3 II levels decreased while those of p62 increased. Cortical IL-1β levels positively correlated with Beclin-1 and LC3 II and on the contrary inversely correlated with p62.
Conclusion
The present study is the first showing links between IL-1β-mediated inflammation and autophagy in the brain. It could open to new therapeutic strategies in brain diseases where regulation impairment of inflammation and autophagy progress with the severity of diseases.
Keywords: Inflammation, Autophagy, IL-1β, Brain, Mouse, Lipopolysaccharide
Background
Autophagy is a major catabolic pathway in eukaryotic cells and is responsible for the degradation in the lysosome of long-lived proteins and altered or unwanted organelles. Autophagy not only plays a crucial role in the maintaining of cell homeostasis and protein quality control but also constitutes an adaptive response to nutritional stress and protects the cells against microbial and viral pathogens and damaged structures. The molecular mechanisms controlling autophagy processes are complex. The mammalian target of rapamycin (mTOR) is an S/T kinase playing a central role in the control of autophagy [1]–[5]. However autophagy can be induced in mTOR-independent manners, in that cases it involves inositol, calcium, 5’-adenosine monophosphate-activated protein kinase (AMPK), Jun kinase (JNK)-beclin-1 complex and Sirtiun-1 [6]–[11]. Through this regulation, autophagy controls cell metabolism, apoptosis, protein secretion and cell-mediated immune responses [12]–[16]. The role of autophagy in inflammatory diseases was initially established through genome-wide association studies (GWAS) showing that polymorphisms in autophagy-associated genes, such as ATG16L1 and IRGM, are linked to Crohn’s disease, the well-known inflammatory bowel disease [17]–[20]. In addition, polymorphisms in autophagy-associated genes have been associated with other inflammatory diseases such as systemic lupus erythematosus [21], asthma [22] and rheumatoid arthritis [23]. Besides, autophagy has been recognized to have an anti-inflammatory action since the production of interleukine (IL)-1β and IL-18 was increased in the absence of functional ATG16L1 (a key protein of the ubiquitin-like conjugation system Atg5-Atg12 ~ Atg16) in a mouse model of Crohn’s disease [24]. Several convergent reports showed that autophagy interferes with inflammasome (complex involved in IL-1β maturation) activation by targeting ubiquitinated aggregates of inflammasome components for destruction [25]–[28]. Conversely, altered proteostasis has been shown recently to activate inflammasome [29]. In addition, NF-κB signalling pathway has been demonstrated to be involved in the induction of autophagy [30],[31] and the pro-inflammatory cytokines IL-1β, TNF-α and IFN-γ were shown to activate autophagy under infectious stimuli [32],[33] contrary to IL-4 and IL-13 [34].
The interconnections between autophagy and inflammation were mainly described at peripheral level in particular in inflammatory bowel diseases [35], type 2 diabete [36], cardiac disorders [37], cystic fibrosis [38]. However, such interconnections remain to be investigated in the central nervous system (CNS) as autophagy alterations and inflammation are also two components of neurodegenerative disorders such as Parkinson’s disease (PD), Alzheimer’s disease (AD), Huntington’s disease (HD), Amyotrophic Lateral Sclerocis (ALS) [39].
Here, we investigated the impact of an inflammatory reaction on the autophagic process in the CNS in vivo. Neuroinflammation was triggered by intraperitoneal lipopolysaccharide (LPS) injection in mice as previously described in rodents [40]–[44]. We showed that according to the inflammatory stress severity (acute versus chronic), neuroinflammation differently altered autophagy in the brain underlying a potential role of cortical IL-1β in the alterations of autophagy in the CNS. These findings reported for the first time relationships between inflammation and autophagy in CNS and could open to new therapeutic strategies in brain diseases where regulation impairment of inflammation and autophagy progress with the severity of diseases.
Results
As indicated, we first studied the impact of a peripheral acute inflammatory stress induced by LPS on cerebral autophagy in wild-type B6C3F1 mice. Preliminary experiments were performed using 2 doses of LPS: either one i.p. injection at 1 mg/kg, and sacrifice 3 h after or 3 i.p. injections at 3 mg/kg per 24 h before sacrifice. However, no statistically significant production of IL-1β, TNF-α and IL-6 was measured compared to saline group mice (Tables 1 and 2). Consequently, we used a higher LPS dose of 10 mg/kg, 3 i.p injections per 24 h, in order to trigger relevant inflammatory response in our experimental settings. This relatively high dose has been previously reported, by other authors, to be lethal 48 h after LPS injection [45]. In our study, animals received 1, 2 or 3 i.p injections and were sacrificed 24 h after LPS injections. In these conditions, no mortality was observed.
Table 1.
Cortical and hippocampus cytokine levels in mice treated with 1 mg/kg of LPS
| |
Cortex |
Hippocampus |
||
|---|---|---|---|---|
| NaCl | LPS | NaCl | LPS | |
|
IL-1β |
28.07 ± 7.53 |
35.05 ± 15.55 |
62.65 ± 16.35 |
40.4 ± 14.80 |
|
TNF-α |
3.57 ± 1.30 |
2.83 ± 0.80 |
6.53 ± 1.42 |
1.47 ± 0.62 |
| IL-6 | 3.06 ± 1.02 | 6.07 ± 1.16 | 3.23 ± 0.88 | 3.05 ± 0.96 |
Levels of IL-1β, TNF-α and IL-6 measured by ELISA in cortex and hippocampus of 3-months old mice treated with one i.p. injection of LPS at 1 mg/kg or saline (0.9% NaCl) and sacrificed after 3 hours. Cytokine levels are expressed in pg/mg of protein. Results are ± SEM of 3 mice per group. ND: not detectable, under the limit of detection.
Table 2.
Cortical and hippocampus cytokine levels in mice treated with 3 mg/kg of LPS
| |
Cortex |
Hippocampus |
||
|---|---|---|---|---|
| NaCl | LPS | NaCl | LPS | |
|
IL-1β |
23.94 ± 6.71 |
18.05 ± 1.44 |
35.1 ± 1.22 |
49.32 ± 8.25 |
|
TNF-α |
1.24 ± 0.29 |
1.78 ± 0.10 |
1.62 ± 0.20 |
1.67 ± 0.56 |
| IL-6 | 3.83 ± 0.95 | 3.56 ± 0.24 | 5.33 ± 0.26 | 3.87 ± 0.08 |
Levels of IL-1β, TNF-α and IL-6 measured by ELISA in cortex and hippocampus of 3-months old mice treated with 3 i.p. injections of LPS at 3 mg/kg or saline (0.9% NaCl) per 24 h before sacrifice. Cytokine levels are expressed in pg/mg of protein. Results are ± SEM of 3 mice per group.
Pro-inflammatory cytokine levels after acute LPS-induced inflammatory stress
In saline group mice, no statistically significant variations of cytokine levels were observed in cortex and hippocampus whatever the design of treatment (see Additional file 1: Table S1 and S2). Therefore, results for NaCl correspond to the mean in each figure.
After 24 h, two or three injections of LPS at a dose of 10 mg/kg increased IL-1β, TNF-α and IL-6 levels in the cortex and in the hippocampus (Figure 1). Cortical IL-1β levels were 5.6- and 13.5-fold higher compared to the corresponding vehicle-injected mice with two and three injections, respectively (Figure 1A). Hippocampal IL-1β levels were 3 and 5 times higher (Figure 1D). Cortical TNF-α levels increased by 131 and 17.5 times with two or three injections, respectively. Hippocampal TNF-α levels were 30 and 8 times higher (Figure 1B and E). For IL-6, cortical levels were 121 or 254 times higher with two or three injections, respectively and hippocampal levels were 40 or 212 times higher, compared to those measured in vehicle-treated mice (Figure 1C and F). Unlike IL-1β and TNF-α, IL-6 levels were significantly increased in the cortex from 6 h (148-fold) and from 4 h in the hippocampus (50-fold).
Figure 1.
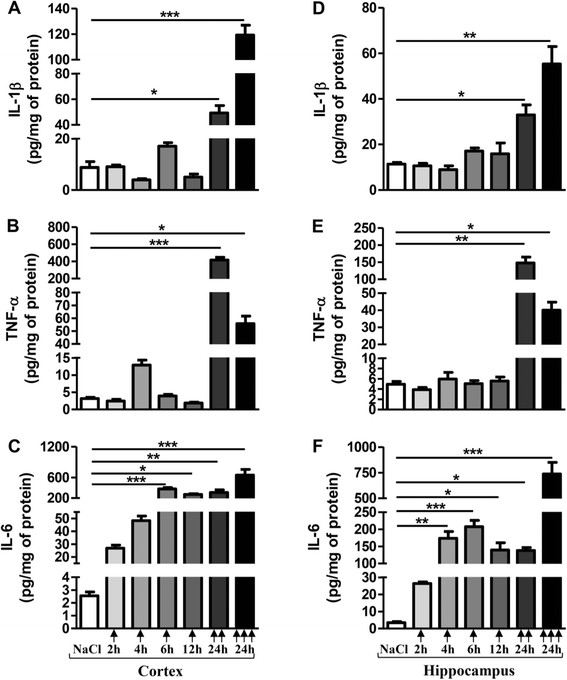
Pro-inflammatory cytokine levels after acute LPS-induced inflammatory stress. IL-1β, TNF-α and IL-6 levels in cortex (A, B, C, respectively) and hippocampus (D, E, F, respectively) of mice treated with a single () or two () or three () i.p. injections of LPS at 10 mg/kg or vehicle (0.9% NaCl) were measured by ELISA assay. Mice were sacrificed 2, 4, 6 or 12 h after a single injection or 24 h after two or three injections. Cytokine levels were expressed in pg/mg protein. Results are mean ± SEM for 6 mice in each group. *p < 0.05, **p < 0.01, ***p < 0.001 compared to 0.9% NaCl-injected mouse group by Kruskal-Wallis test with a Dunns multiple comparison test.
Changes in autophagic factors after acute LPS-induced inflammatory stress
As for inflammatory markers, no statistically significant variations of autophagic markers were observed in cortex and in hippocampus whatever the design of treatment in saline-injected group mice (see Additional file 1: Table S3 and S4). Therefore, results for NaCl correspond to the mean in each panel of Figure 2.
Figure 2.
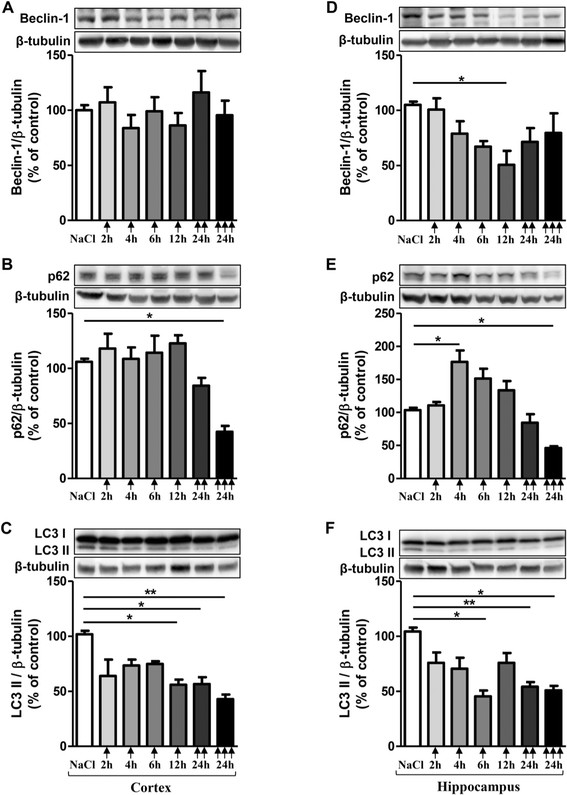
Changes in autophagic factors after acute LPS-induced inflammatory stress. Representative immunoblots showed the immunoreactivity of Beclin-1 (A), p62 (B) LC3 II (C) in cortex and Beclin-1 (D), p62 (E) LC3 II (F) in hippocampus from mice treated with a single () or two () or three () i.p. injections of LPS at 10 mg/kg or vehicle (0.9% NaCl). Mice were sacrificed 2, 4, 6 or 12 h after a single injection and 24 h after two and three injections. Semi-quantitative analysis of immunoblot was performed using Gene Tools software (Syngene, Ozyme France). The immunoreactivity of protein was normalized to β-tubulin immunoreactivity. The results are expressed as arbitrary units (% of 0.9% NaCl-injected mice group set at 100%). Results are mean ± SEM for 6 mice in each group. *p < 0.05, **p < 0.01 compared to 0.9% NaCl-injected mouse group by Kruskal-Wallis test with a Dunns multiple comparison test.
To determine whether autophagy changes occured after a peripheral acute inflammatory stress, Beclin-1, p62, LC3 I and LC3 II were investigated. Beclin-1 is a key component of the class III PI3K (Phosphatidylinositide 3-kinases) complex which is involved in the initiation of autophagosome formation [46]; p62 is an autophagic receptor which recognizes ubiquitinylated proteins and interacts with LC3 II at the forming autophagosome [47]; LC3 is present in free cytoplasmic form as LC3 I which, when is conjugated to phosphatidylethanolamine (through an ubiquitin-like conjugation reaction) of the membrane of autophagosome, produces LC3 II form, a useful marker of autophagic vacuoles [47].
Beclin-1 expression was affected only in hippocampus with a decrease by 52% after 12 h post-injection (Figure 2A and D). A higher inflammatory stress with two or three LPS injections did not significantly change Beclin-1 expression (Figure 2D).
For p62, a single LPS injection induced an increase by 71% after 4 h and return to basal line after 6 and 12 h in the hippocampus (Figure 2E). No changes in p62 levels were observed in the cortex after a single injection (Figure 2B). On the contrary, a decrease in p62 levels of 18-20% with two injections and of 55-60% with three injections in both cortical and hippocampal area were observed (Figure 2B and E).
For LC3 marker, any change was observed for LC3 I both in cortex and hippocampus regardless the time post- LPS injection. However, LC3 II significantly decreased at 12 h in the cortex (45%), at 6 h in the hippocampus (56.5%) and at 24 h after two or three injections (45-58%) in both areas (Figure 2C and F).
Ultrastructure of cells in cortex and hippocampus after systemic LPS administration (two injections per 24 h) was similar to control mice with normal morphology of the mitochondria and the nucleus with evenly distributed chromatin is visible. Cytoplasm is rich in ribosomes and polyribosomes. No accumulation of vesicles was observed in cortex and in hippocampus (Figures 3 and 4).
Figure 3.
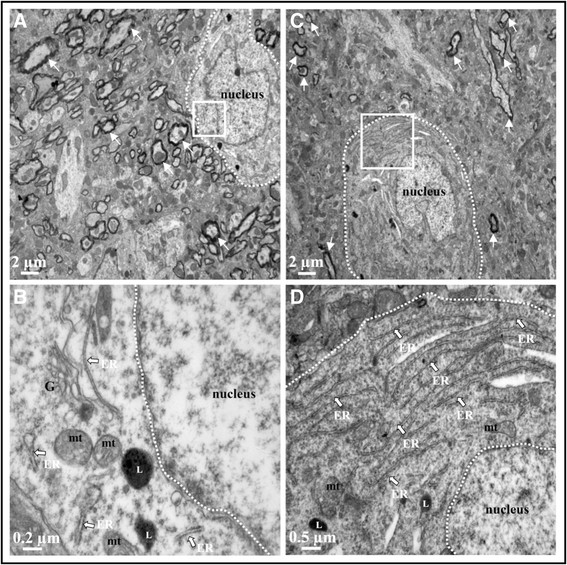
Ultrastructure of cortex after acute LPS-induced inflammation. TEM cortical images of 0.9% NaCl-injected mice (A and B) and 10 mg/kg LPS-treated mice (2 injections per 24 h, images C and D). No alteration of tissue integrity was observed in low magnification images (A and C), myelin rounded axons marked with white arrow. Images B and D represent magnified region of interest of the white square in A and C, respectively. In these magnified images, mitochondria appeared with intact cristae (mt), endoplasmic reticulum (ER) was present with ribosomes and lysosomes (L) were observed. Five sections in each area (cortex and hippocampus) were observed for each mouse brain.
Figure 4.
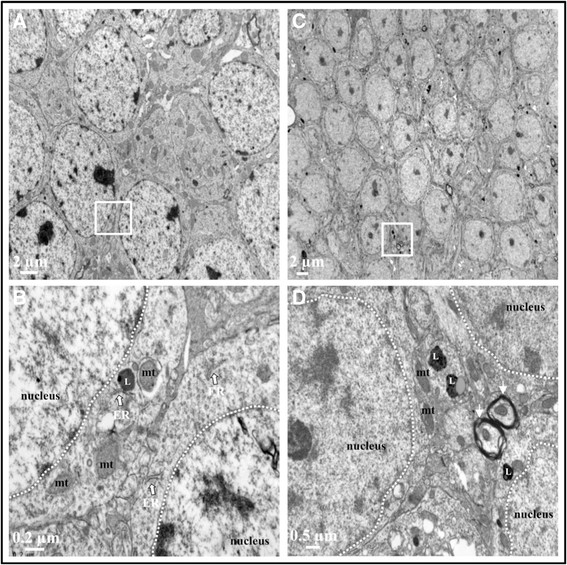
Ultrastructure of hippocampus after acute LPS-induced inflammation. TEM hippocampal images of 0.9% NaCl-injected mice (A andB) and 10 mg/kg LPS-treated mice (2 injections per 24 h, images C and D). No alteration of tissue integrity was observed in low magnification images (A andC). Images b and d represent magnified region of interest of white square in a and c, respectively. In these magnified images, mitochondria appeared with intact cristae (mt), endoplasmic reticulum (ER) was present with ribosomes, myelin rounded axons marked with white arrow and lysosomes (L) were observed. Five sections in each area (cortex and hippocampus) were observed for each mouse brain.
Activation of mTOR signalling pathway after acute LPS-induced inflammatory stress
mTOR activation leads to phosphorylation of various substrates, in particular p70S6K at T389, a ribosomal S6 kinase involved in ribogenesis [48],[49]. Furthermore, mTOR negatively regulates autophagy in several experimental models.
No modification of the mTOR activation was observed after an acute LPS stress (Figure 5A and B). However, the p70S6K activation decreased in time-dependent manner and significantly from 12 h in the cortex (46%) and from 4 h in the hippocampus (46%) after one 10 mg/kg LPS injection (Figure 5C and D). With two and three injections of LPS per 24 h, the p70S6K activation decreased in both brain areas (24-48%). No statistically significant variations of the mTOR and p70S6K activation were observed in cortex and in hippocampus whatever the design of treatment in saline-injected group mice (see Additional file 1: Table S3 and S4). Therefore, results for NaCl correspond to the mean in each panel of Figure 5.
Figure 5.
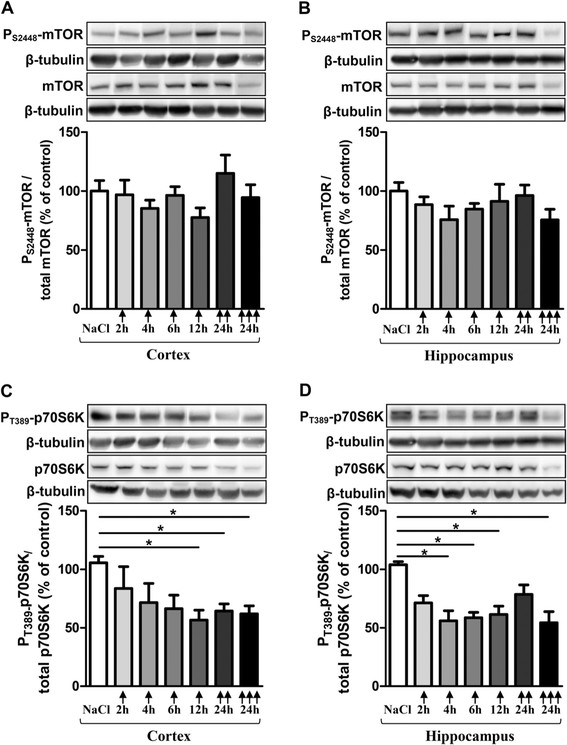
Activation of the mTOR signalling pathway after acute LPS treatment. Representative immunoblots showed the immunoreactivity of mTOR, PS2448-mTOR, p70S6K and PT389-p70S6K in cortex (Aand C) and in hippocampus (Band D) from treated-mice with a single () or two () or three () injections in i.p of LPS at 10 mg/kg or control (0.9% NaCl). Mice were sacrificed 2, 4, 6 or 12 h after a single injection and 24 h after two and three injections. Semi-quantitative analysis of immunoblot was performed using Gene Tools software (Syngene, Ozyme France). The immunoreactivity of protein was normalized to β-tubulin immunoreactivity. The results are expressed as arbitrary units (% of 0.9% NaCl-injected mice group set at 100%). Results are mean ± SEM for 6 mice in each group. *p < 0.01 compared to 0.9% NaCl-injected mouse group by Kruskal-Wallis test with a Dunns multiple comparison test.
Correlations between cytokine levels and expression of autophagic markers after acute LPS-induced inflammatory stress
In our experimental conditions, acute LPS treatment stimulated cytokine production (IL-1β, TNF-α and IL-6), decreased autophagic marker expressions and p70S6K activation without immediate cortical or hippocampal tissue damage as shown by TEM. Spearman correlations were performed between inflammatory and autophagic parameters. In cortex, a positive correlation between IL-6 levels and p70S6K expression was found (rho = 0.88; p = 0.03; n = 6 mice) at 12 h after a single LPS injection. In hippocampus and after two LPS injections per 24 h, two correlations were noted one between IL-1β levels and LC3 II expression (rho = 0.94; p = 0.01; n = 6 mice) and a second between TNF-α levels and p70S6K expression (rho = −0.88; p = 0.03; n = 6 mice).
In this first part, an acute peripheral inflammatory stress affected the cerebral autophagy with a positive correlation between LC3 II and IL-1β levels after two 10 mg/kg LPS injections per 24 h. Moreover, p70S6K expression significantly decreased and the levels of p70S6K were positively correlated to IL-6 levels in the cortex at 12 h and negatively to TNF-α levels in the hippocampus at 24 h with two i.p. LPS injections. In parallel, we also wanted to study the impact of a chronic inflammatory stress in autophagy. Three months old mice received an i.p. dose of 0.5 mg/kg of LPS every 3 days for 3 months. This treatment did not affect the life expectancy of mice compared to control mice (0.9% NaCl as vehicle). The average weight was 31.44 ± 5.63 and 34.53 ± 4.75 mg for control and LPS-treated mice, respectively.
Pro-inflammatory cytokine levels after chronic LPS-induced inflammatory stress
After 3 months of treatment, IL-1β levels significantly increased in cortex (495%) and in hippocampus (367%) compared to control mice (Figure 6A). Surprisingly, TNF-α levels decreased in both brain areas: 79% in cortex and 63% in hippocampus (Figure 6B). For IL-6, no difference was observed in LPS-treated mice versus control mice (Figure 6C).
Figure 6.
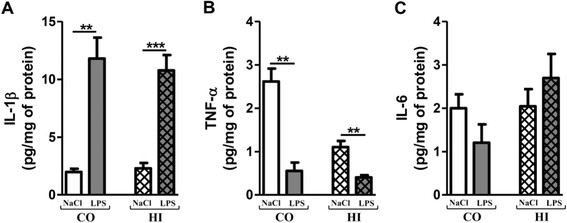
Pro-inflammatory cytokine levels after chronic LPS-induced inflammatory stress. IL1-β (A), TNF-α (B) and IL-6 (C) levels in cortex (CO) and hippocampus (HI) from LPS-treated mice (0.5 mg/kg of LPS every 3 days for 3 months) or control (0.9% NaCl every 3 days for 3 months) by using ELISA assay. Treatment started at 3 months and mice were sacrificed at 6 months of age. Cytokine levels were expressed in pg/mg protein. Results are mean ± SEM for 6 mice in each group. **p < 0.01, ***p < 0.001 compared to control mice by a Mann–Whitney test.
Changes in autophagy markers after chronic LPS-induced inflammatory stress
Chronic LPS-induced inflammatory stress decreased Beclin-1 by 24% and 32% in cortex and hippocampus, respectively (Figure 7A). On the contrary, a robust increase of p62 expression was observed in both areas: 455% in cortex and 208% in hippocampus (Figure 7B). A significant decrease of LC3 II expression was observed in both areas (37.5% in cortex and 45% in hippocampus) without changes in LC3 I levels (Figure 7C).
Figure 7.
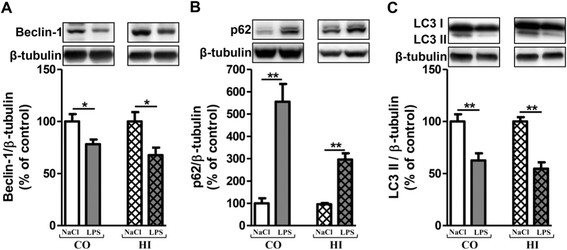
Changes in autophagy markers after chronic LPS-induced inflammatory stress. Representative immunoblots showed the immunoreactivity of Beclin-1 (A), p62 (B) and LC3 II (C) in cortex and hippocampus from LPS-treated mice (0.5 mg/kg of LPS every 3 days for 3 months) or control (0.9% NaCl every 3 days for 3 months). Treatment started at 3 months and mice were sacrificed at 6 months of age. Densities were quantified by using Gene Tools software (Syngene, Ozyme France). Data of each protein were reported to data of the corresponding β-tubulin. The results are expressed as arbitrary units (% expression of 0.9% NaCl-treated mice set at 100%). Results are mean ± SEM for 6 mice in each group. *p < 0.05, **p < 0.01 compared to control mice by Mann–Whitney test.
Similarly to what was observed after acute LPS-induced inflammatory stress, TEM showed that chronic LPS-induced inflammatory stress did not cause major morphological tissue alterations. At the cellular level, mitochondria appeared with a normal shape without alteration and no vacuole accumulations were observed in cortex and in hippocampus (Figure 8).
Figure 8.
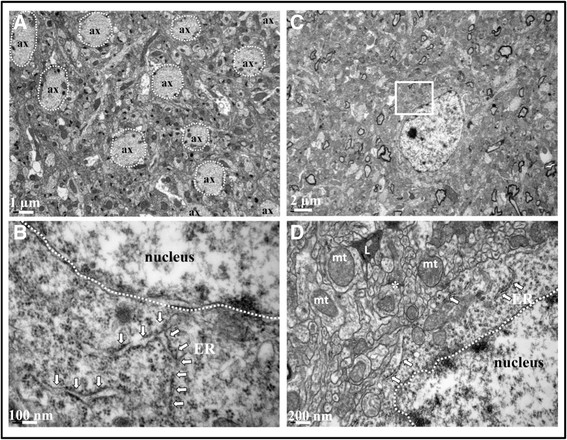
Ultrastructure of cortex and hippocampus after chronic LPS-induced inflammatory stress. TEM was performed in LPS-treated and control mice (n = 3 in each group) in cortex (Aand B) and in hippocampus (Cand D). Low magnification image of cortex (A) showed a regular distribution of axons (ax) and microtubule filament inside. Image d represent magnified region of interest showed in the white square in C. The endoplasmic reticulum (ER) marked with white arrow (B and D) carried ribosomes without abnormalities. Mitochondria (mt) have intact cristae, lysosomes (L) were observed and synapses with pre-synaptic vesicles are marked by (*). Five sections in each area (cortex and hippocampus) were observed for each mouse brain.
Activation of mTOR signaling pathway after chronic LPS-induced inflammatory stress
Similarly to acute LPS-induced inflammatory stress, the mTOR activation was not modified (Figure 9A). However, the p70S6K activation was dramatically reduced in cortex (75%) and hippocampus (74%) (Figure 9B).
Figure 9.
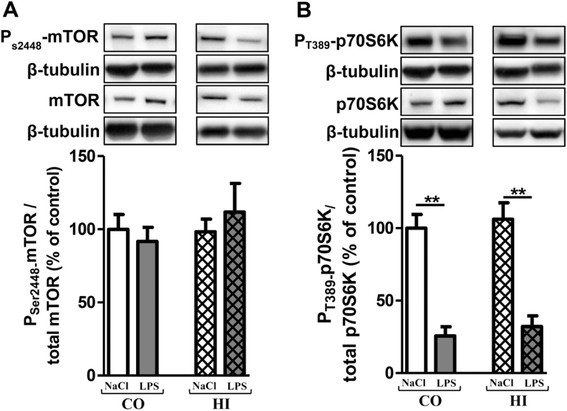
Changes in mTOR and p70S6K activation after chronic LPS-induced inflammatory stress. Representative immunoblots showed the immunoreactivity of mTOR, PS2448-mTOR, p70S6K, PT389-p70S6K in cortex (A) and in hippocampus (B) from LPS-treated mice (0.5 mg/kg of LPS every 3 days for 3 months) or control (0.9% NaCl every 3 days for 3 months). Treatment started at 3 months and mice were sacrificed at 6 months of age. Semi-quantitative analysis of immunoblot was performed using Gene Tools software (Syngene, Ozyme France). The immunoreactivity of protein was normalized to β-tubulin immunoreactivity. The results are expressed as arbitrary units ((% of 0.9% NaCl-injected mice group set at 100%). Results are mean ± SEM for 6 mice in each group. **p < 0.01 compared to control mice by a Mann–Whitney test.
Correlations between levels of cytokine and autophagic markers after chronic LPS treatment
Chronic LPS treatment induced IL-1β production associated with changes of autophagic marker expressions and a great decrease of p70S6K activation without mTOR activation and without tissue morphological alterations. Interestingly, Spearman analysis of data from LPS-treated mice revealed two positive correlations between cortical IL-1β and Beclin-1 and between IL-1β and LC3 II levels. Furthermore, a negative correlation between IL-1β and p62 levels was observed (Table 3). In this LPS mouse group, only IL-1β would control cortical autophagy. Another interesting correlation revealed that levels of beclin-1 expression were negatively correlated with those of p62 (rho = −0.88, p = 0.03). This last correlation would reinforce that the cortical autophagic flux would be induced after a chronic and peripheral LPS treatment. However, any correlation was observed in hippocampus.
Table 3.
Correlation between autophagic factors and IL-1β levels during chronic LPS –induced inflammatory stress
| |
Beclin-1 |
p62 |
LC3 II |
|||
|---|---|---|---|---|---|---|
| rho | p | rho | p | rho | p | |
| IL-1β | 0.88 | 0.03 | - 0.88 | 0.03 | 0.88 | 0.03 |
Spearman correlations were performed between IL-1β and autophagic parameters measured in the cortex of LPS-treated mice (0.5 mg/kg of LPS every 3 days for 3 months). Treatment started at 3 months and mice were sacrificed at 6 months of age (n = 6). In table, rho and p values were indicated. The level of significance was p < 0.05.
Discussion
Alteration of autophagy and excessive inflammatory response are two hallmarks common to various brain diseases such as AD, PD, HD, ALS [13],[39]. However, the relationships between these two defense mechanisms of the body remain unknown in the CNS. Recently, we showed in vitro that IL-1β was involved in the acidic vesicle accumulation in microglia contrary to amyloid peptide [50]. The current study therefore aimed at determining whether an inflammatory reaction could modulate the autophagic process in the CNS.
The i.p. injection of LPS is extensively used to induce brain inflammation [40],[41],[51]. Several studies showed that treatment of mice with LPS induced a central inflammatory response associated with microglial activation, immunomodulatory effects of astrocytes, cyclo-oxygenase-2 and iNOS immunoreactivities and increases in cytokine productions [41],[42],[52]–[55].
However there are various regimens followed in mice. Among the acute treatments, the most frequently encountered are i.p. LPS injection of 0.5, 1 or 5 mg/kg and sacrifice of animals after one hour to ten days. In that issue, several studies only reported the transcriptional analysis of cytokines, in particular IL-1β, IL-6 and TNF-α in brain regions, showing an increase in mRNA expression of these cytokines in hippocampus, choroid plexus [51],[56]–[58]. Here, we used a higher dose (10 mg/kg) because lower LPS doses (i.e. single dose of 1 mg/kg or three doses of 3 mg/kg per 24 h) failed to detect significant increases in cytokine production in the mouse brains by using ELISA as previously reported [59]. In the present study, brain inflammatory response was reflected by a great increase in IL-6 levels started at 4 h in hippocampus and 6 h in cortex after a single injection while levels of IL-1β and TNF-α increased after two or three LPS injections per 24 h.
For the chronic LPS treatment, some authors included in their experimental design a group of mice received a single dose of LPS at 5 mg/kg or 10 mg/kg and sacrificed after 1, 3 or 10 months [41],[54]. Here, 0.5 mg/kg LPS was i.p. injected every three days during three months. Other authors chose twice injections per week for 6 weeks at a dose of 0.5 mg/kg in 4-month-old 3xTg-AD transgenic Alzheimer mouse model [60]. In last study, the monitoring of mRNA expression of IL-1β, IL-6 and TNF-α showed that IL-1β levels were markedly increased in the brains of LPS-treated mice. However, IL-6 and TNF-α expression levels were not significantly altered by LPS treatment. In our experimental conditions, chronically LPS-treated wild-type mice displayed a significant increase in IL-1β whereas TNF-α levels significantly decreased. Therefore, IL-1β could be considered as the critical cytokine in the central inflammatory response induced by peripheral LPS during 3 months. The decrease of TNF-α levels was also observed in brain, liver and serum after a long term time course of single 5 mg/kg LPS injection [54]. For this higher LPS dose, authors demonstrated a role of TNF receptors (TNFR) since in TNFR KO mouse models, LPS-induced TNF-α production was totally inhibited, suggesting a potential downregultion of these receptors in our experimental design. One may also propose that this decrease in TNF-α levels could be due to a modification of TNF-α converting enzyme (TACE) activity, also known as ADAM17 (a disintegrin and metalloprotease-family) and involved in the cleavage of TNF-α precursor to produce mature TNF-α [61]. In accordance with these data, we previously demonstrated that at 18 months of age, APPswePS1dE9 mice displayed a great decrease in TNF-α production [62].
Furthermore, the immuno-tolerance induced by chronic LPS injections could explain the decrease of TNF-α rate in cortex and in hippocampus and maintain a significant higher level of IL-1β. Indeed, it is well established that a first LPS exposure induced an overproduction of cytokines following a modification of gene expression in monocyte/macrophage becoming refractory to secrete some cytokines like TNF-α [48],[63],[64].
Despite this inflammatory response, normal tissue morphology and cell integrity were preserved in both acute and chronic LPS treatment. No signs of cellular damage were visible by TEM after both acute and chronic treatments. Authors showed that acute 1 mg/kg of LPS injection induced no neuronal death (negative Fluorojade B neurons and negative TUNEL neurons) and no rupture of blood brain barrier (BBB) [52],[65]. In addition, maintenance of the mitochondrial architecture critically depends on the induction of autophagy which is essential for regenerating astrocyte mitochondrial networks during inflammation [66]. But ultrastructure of a neuronal cell in the hippocampus 48 h after 1 mg/kg LPS administration showed shrunken and dark cytoplasm, deep invagination of the nuclear envelope into the nucleoplasm and swelling of some mitochondria [51].
The monitoring of autophagy markers, including Beclin-1, p62 and LC3, in mouse brains after systemic LPS-induced inflammatory stress, has never been conducted before. Only two papers described an increased expression of lysosomal cysteine proteases, cathepsins (Cat) C and B [51],[56]. Cat C expression was detected in neurons of cerebral cortex 6 h after 5 mg/kg LPS i.p. injection and 24 h later, Cat C expression was also detected in activated microglial cells throughout the entire brain. The duration of induced Cat C expression in neurons and in microglial cells was ten days and three days, respectively by using in situ hybridation (ISH) and immunohistochemical staining (IHC) [56]. An immunocytochemical analysis of the subcellular localisation of Cat B using post-embedding immunogold methods showed that, 48 h after systemic 1 mg/kg LPS administration, Cat B was translocated from lysosomes to the cytosol and autophagic vacuoles and was also found in the membrane of mitochondria in the hippocampal area.
Our study showed that acute 10 mg/kg LPS treatment induced autophagy early changes in the hippocampus with increased p62 and decreased LC3 II 4 h after injection evolving towards a decrease in the three parameters (Beclin-1, p62 and LC3 II) 12 h after injection. In the cortex, the LC3 II decrease was observed from 12 h. After 24 hr, all markers of autophagy significantly decreased in the cortex and hippocampus. Interestingly, in hippocampus IL-1β levels were positively and strongly (rho = 0.94) correlated to LC3 II expression, indicating a role of this cytokine in LC3 II induction. Several cytokines, including IL-1β are well known as autophagy stimulatory molecules [67]–[69]. Furthermore, the induction of autophagy was associated with a great inactivation of p70S6K (or Ribosomal S6 Kinase 1) without modification of mTOR activation. The T389 site of p70S6K is known to be phosphorylated by the kinase mTOR. However, extensive research on the regulation of the activity of p70S6K studies show direct control by the active Receptor Tyrosine Kinase (RTK)/Phosphoinositide 3-kinase/Phosphoinositide-dependent kinase-1 (PDK1) signaling pathway that the phosphorylation site is not defined yet [48]. In addition, RS6K1 dephosphorylation was an active process of its regulation by protein phosphatase 2 (PP2A) [70],[71]. Here, a negative correlation was observed between p70S6K activation and TNF-α levels in hippocampus, indicating the negative impact of LPS-induced sepsis in this kinase linked to diverse cellular processes, including protein synthesis, mRNA processing, glucose homeostasis, cell growth and survival. Other authors showed that an in vivo sepsis not induced by LPS inhibits mTOR signaling pathways in rat cardiac muscle and that this defect appeared mediated, either directly or indirectly, by the endogenous over production of TNF-α [72].
Based on the results obtained, a differential expression level of autophagy markers between in cortex and in hippocampus was mainly observed after one injection of LPS. Hippocampus responded faster than cortex in particular for Beclin-1, LC3 II and p70S6K. These differences of autophagy levels between these two brain structures were also observed in rats after various acute stresses such as hypoxia-ischemia, oxygen and glucose deprivation or 6-OHDA injection [73]–[75]. Some authors showed a differential brain activity after a peripheral inflammation. It was demonstrated that during sepsis, the BBB lose its structural integrity allowing the cross of peripheral cytokines and macrophages in the brain [76],[77]. The brain can be sense peripheral inflammation through the vagus nerve and the hypothalamic–pituitary–adrenal axis and influence the brain activity, memory, plasticity, neurogenesis [78]. These brain-peripheral immune interactions could also explain the differential expression level of autophagy markers between in cortex and in hippocampus in our study.
Although some authors have examined the level of expression of cathepsins after acute treatment with LPS in mice, no study has yet been published on the autophagic changes in long-term chronic treatment with LPS. This work, however, is very important when taken into consideration the fact that many neurodegenerative diseases are characterized by disturbances both in the regulation of inflammation and autophagy. We observed after 3 months of neuroinflammation induction, an upregulation of p62 coupled to a net decrease in expression of Beclin-1 and LC3 II. Very interestingly, correlation analysis revealed that the IL-1β production after chronic LPS treatment induced autophagic flux. In accordance with our results, it is known that inflammasome, in particular caspase 1 also increases autophagic flux [79]. However, a recent study revealed that inflammatory stimulus in macrophage cell line and in human macrophages activated autophagy and decreased production of IL-1β production [26] due to the degradation of inflammasome complex by autophagy. This feedback would be necessary to counteract and to limit the inflammation reaction due to minor insult. In our in vivo study, this beneficial autophagy feedback could explain that one LPS injection was not sufficient to measured significant IL-1β levels in cortex and hippocampus and required more injections (2 or 3 i.p injections). Interestingly, a chronically administration but not a lower LPS dose could impair the beneficial autophagy feedback against IL-1β production and induce an increase of IL-1β levels in cortex and in hippocampus.
Regarding p62, results showed negative correlation between its levels of expression and those of IL-1β. In other experimental conditions, recent findings showed that the production and secretion of the proinflammatory cytokine IL-1β was significantly enhanced in p62−/− macrophages after infection with Legionella pneumophila. Furthermore, these authors showed that p62 may interact with nucleotide-binding oligomerization domain-like receptor (NLR) family, CARD domain-containing 4 and NLR family, pyrin domain-containing 3 proteins to inhibit their self-dimerization [80]. However, its self-dimerization is a necessary step for its degradation during autophagy [81]. Based on these physical interactions, p62 could accumulate as we observed after a chronic LPS treatment.
Conclusion
An acute and a chronic peripheral inflammatory stress induced by LPS, in particular, with a persistent IL-1β production modified cortical and hippocampus autophagic marker expressions. Chronic inflammatory stress increased p62 and decreased Beclin-1, LC3 II and p70S6K activation without changes of the mTOR activation and any morphological tissue alteration. Moreover, IL-1β levels were positively correlated to Beclin-1 and LC3 II while p62 expression was inversely correlated to IL-1β levels after chronic neuroinflammation. These findings highlighted the induction of central autophagy by IL-1β-mediated inflammation. It is important to note that the rate of this inflammatory factor remains very moderate, less than 15 pg/mg protein in both brain regions studied and therefore this level would be interesting to activate autophagy in neuroinflammatory diseases including neurodegenerative diseases characterized by a great inflammation and accumulation of autophagosomes in advanced stages.
Methods
Chemical products
Sodium fluoride (NaF), phenylmethylsulfonyl fluoride (PMSF), protease and phosphatase inhibitor cocktails, dithiothreitol (DTT), Lipopolysaccharide (LPS), Paraformaldehyde (PFA) and all reagent-grade chemicals for buffers were purchased from Sigma (St Quentin Fallavier, France); Sodium pentobarbital from CEVA, Animal Health (Libourne, France); NuPAGE® LDS 4X LDS Sample Buffer, NuPAGE® Sample Reducing Agent (10X), Novex® 4-20% Tris-glycine Mini gels, NuPAGE® 3-8% Tris-Acetate gels, Novex® Tris-Glycine SDS Running and NuPAGE® Tris-Acetate SDS running buffers, NuPAGE® Antioxidant, Seeblue® Plus2 pre-stained standard, iBlot® Gel Transfer Device (EU), Quant-it® protein assay from Gibco-Invitrogen (Fisher Bioblock Scientific distributor, Illkirch, France); 4X Laemmli sample buffer, 4-15% mini-PROTEAN® TGX™ gels, Tris-glycine running buffer and Trans-Blot® Turbo™ Transfer System from Biorad (Marnes-la-Coquette, France).
For western blot, primary antibodies and secondary anti-rabbit IgG antibody conjugated with Horseradish Peroxydase (HRP) were purchased from Cell Signalling (Ozyme, St Quentin Yvelines, France) excepted p62/SQMT1 from MBL (CliniSciences distributor, Nanterre, France), anti-β tubulin from Sigma (St Quentin Fallavier, France), peroxidase-conjugated anti-mouse IgG from Amersham Biosciences (Orsay, France), IgG- and protease-Free Bovine Serum Albumin (BSA) from Jackson ImmunoResearch Europe Ltd (Interchim distributor, Montluçon, France).
Animals
Adult male and female B6C3F1 mice (3 months, 30.50 ± 0.82 mg in weight) were purchased from Charles River Laboratories (L’Arbresle, France). The use of animals for this study has received the approval of the Ethical and Animal Care Committee at “La direction départementale de la protection de la population (DDPP)” (registration number: 06.12). All animal cares and experimental procedures were conducted in conformity with the French Décret n° 2013–118 1st February 2013 NOR: AGRG1231951D in accordance with European Community guidelines (directive 2010/63/UE for the Care and Use of Laboratory Animals). All efforts were made to minimize animal suffering, as well as, the number of animals used. The animals were housed in a conventional state under adequate temperature (23 ± 3°C) and relative humidity (55 ± 5%) control with a 12/12 h reversed light/dark cycle, and provided with free access to food and water.
Lipopolysaccharide-induced inflammatory stress
LPS (Escherichia coli, serotype 0111:B4) was used to induce an inflammatory response. Two experimental designs were performed. First, an acute treatment with LPS intraperitoneally (i.p.) injected at a dose of 10 mg/kg dissolved in sterile-endotoxin-free 0.9% saline vehicle. Control injections were equivolume vehicle. Mice (6 per group) were sacrificed after 2, 4, 6, 12 h post-injection. Two other groups received either two or three i.p. LPS injections per 24 h before sacrifice (n = 6 in each group). Second a chronic treatment with LPS consisted of an i.p. injection at a dose of 0.5 mg/kg every three days during three months (6 controls versus 6 LPS mice). For this chronic treatment, mice were weighted once a week. The dosage of LPS in both acute and chronic treatments was based on previous studies of LPS-neurotoxicity [52],[54],[82]. For scanning electron microscopy, three mice per group were also included in this study.
Brain tissue preparation
LPS-treated and control (0.9% saline vehicle) mice were transcardially perfused with phosphate buffer saline (154 mM NaCl, 1.54 mM KH2PO4, 2.7 mM Na2HPO4.7H2O, pH 7.3) after deep anesthesia with pentobarbital (80 mg/kg, i.p.). Brains (6 per group for biochemical assays) were rapidly removed and dissected on ice. Cortex and hippocampus were homogenized using 10 up-and-down strokes of a prechilled Teflon-glass homogenizer in 20 volumes of lysis buffer (25 mM Tris–HCl, 150 mM NaCl, 1 mM EDTA, pH 7.4) and supplemented with 50 mM NaF, 1 mM PMSF, protease and phosphatase inhibitor cocktails (50 μL/gr of tissue and 10 μL/mL of lysis buffer, respectively). Lysates were sonicated and centrifuged at 15,000 g for 15 min at 4°C. The resulting supernatants were collected and protein concentrations were determined using Quant-it® protein assay according to the manufacturer’s protocol. Samples were stored at −80°C until ELISA and immunoblotting described below.
Cytokine Enzyme-linked immunosorbent assay (ELISA)
Commercially available ELISA kits were used for measuring mature form of IL-1β (sensitivity: 16 pg/mL), TNF-α (sensitivity: 4 pg/mL) and IL-6 (sensitivity: 2 pg/mL) according to the manufacturers’ instructions (BioLegend, Ozyme distributor, St Quentin Yvelines, France). The range of analysis was between 31.3-2,000 pg/mL for IL-1β and 7.8-500 pg/mL for TNF-α and IL-6. Homogenates from brain tissue (50 mg of tissue/mL) were added in each well of pre-coated plates and all steps were performed at room temperature (RT). The enzymatic reaction was stopped after 15 min incubation with tetramethylbenzidine (TMB) substrate by adding 2 N H2SO4 and the optical density (OD) was read at 450 nm within 30 min, using Multiskan® spectrum spectrophotometer. The cytokine levels were then calculated by plotting the OD of each sample against the standard curve. The intra- and inter-assay reproducibility was >90%. OD values obtained for duplicates that differed from the mean by greater than 10% were not considered for further analysis. For convenience, all results are expressed in pg/mg protein.
Immunoblotting
Samples (40 μg proteins) were prepared for electrophoresis by adding NuPAGE® 4X LDS sample buffer and NuPAGE® Sample Reducing Agent (10X). Samples were then heat-denatured at 100°C for 5 min, loaded into Novex® 4-20% Tris-Glycine mini Gels, run at 150 V for 60 minutes in Novex® Tris-Glycine SDS Running Buffer and in NuPAGE® 3-8% Tris-Acetate Gels, run at 125 V for 120 minutes in NuPAGE® Tris-Acetate SDS running buffer containing NuPAGE antioxidant. Gels were transferred to nitrocellulose membranes using the iBlot® Dry blotting system set at 20 V for 7 min. For LC3 immunoblot, we used Trans-Blot® Turbo™ Transfer System (25 V, 3 min for 0.2 μm nitrocellulose MISI format) after protein gel electrophoresis of samples prepared in 4X Laemmli sample buffer and loaded into 4-15% mini-PROTEAN® TGX™ gels with Tris-glycine SDS running buffer.
Membranes were washed for 10 min in Tris-buffered saline/Tween (TBST: 20 mM Tris–HCl, 150 mM NaCl, pH 7.5, 0.05% Tween 20) and aspecific antigenic sites were blocked by incubating the membranes in TBST containing 5% BSA for 2 h.
Blots were incubated with primary antibody in blocking buffer overnight at 4°C. Antibodies used were rabbit anti-PS2448-mTOR, anti-total mTOR, anti-PT389-p70S6K, anti-total p70S6K, anti-Beclin-1, anti-p62, anti-LC3, all at 1:500 dilution factor. Membranes were washed twice with TBST and then incubated with the HRP-conjugated secondary anti-rabbit IgG antibody (1:1000), during 1 hour at RT. Membranes were washed again and exposed to the chemiluminescence Luminata Forte Western HRP Substrate (Millipore, Saint-Quentin-en-Yvelines, France) followed by signal’s capture with the Gbox system (GeneSnap software, Syngene, Ozyme distributor). After 2 washes in TBST, membranes were probed with mouse antibody against tubulin (1:10000) overnight at 4°C. They were then washed with TBST, incubated with HRP-conjugated secondary antibody anti-mouse (1:1000) for 1 h, exposed to the chemiluminescence Luminata classico substrate (Millipore, Saint-Quentin-en-Yvelines, France) and signals were captured. Automatic image analysis software is supplied with Gene Tools (Syngene, Ozyme distributor). Ratios protein/tubulin were calculated and showed in the corresponding figures. Ratios Phospho-protein/total protein were calculated to evaluate rates of protein activation.
Transmission electron microscopy (TEM)
Three mice in each group were were deeply anesthetized with pentobarbital (80 mg/kg, i.p.) and transcardially perfused with phosphate buffer saline (PBS: 154 mM NaCl, 1.54 mM KH2PO4, 2.7 mM Na2HPO4.7H2O, pH 7.2) and then with paraformaldehyde (PFA 4%). Brains were rapidly removed on ice and thin sagittal sections were isolated and fixed with 3% glutaraldehyde in phosphate buffer saline (0.1 M PBS; pH = 7.4) for 2 h at 4°C. Samples (2 mm3 of tissue in cortex and hippocampus) were then washed three times (3x10 min) in PBS and then post-fixed in 1% osmium tetroxyde in PBS for 1 h at 4°C, processed through a graded acetone series, embedded in Araldite (Fluka, Buchs, Switzerland) and polymerized overnight at 60°C. Thin sections (60 nm) were cut with a diamond knife on Reichert Ultracut S, recovered on Cu grids and contrasted with uranyl acetate (4%) and lead citrate and were observed under a JEOL 1010 transmission electron microscope (Jeol Ltd, Tokyo, Japan). 5 sections in each area (cortex and hippocampus) were observed for each mouse brain.
Statistical analysis
For biochemical analysis, results were expressed as means ± SEM. To compare the two groups of mice in chronic treatment (control versus LPS mice) a mann-Withney’s test was used. Data for multiple variable comparisons were analyzed by a Kruskal-Wallis test with a Dunns multiple comparison test. For correlations between two parameters, we used a Spearman test (GraphPad Instat, GraphPad Software, San Diego, CA, USA). The level of significance was p < 0.05.
Abbreviations
AD: Alzheimer’s disease
ADAM17: A disintegrin and metalloproteinase with a metallopeptidase domain 17
ALS: Amyotrophic lateral sclerosis
AMPK: Adenosine monophosphate kinase
Atg: Autophagy related genes
BBB: Blood brain barrier
BSA: Bovine Serum Albumin
CARD: Caspase recruitment domain
Cat: Cathepsin
CNS: Central nervous system
DTT: Dithiothreitol
ELISA: Enzyme-linked immunosorbent assay
ER: Endoplasmic reticulum
GWAS: Genome-wide association study
HD: Huntington’s disease
HRP: Horseradish peroxidase
i.p.: Intraperitoneal
IFN-γ: Interferon gamma
IHC: Immunohistochemical staining
iNOS: Inducible nitric oxide synthase
IL: Interleukin
ISH: In situ hybridation
JNK: Jun kinase
LC3: Microtubule-associated protein1 light chain 3
LDS: Lithium dodecyl-sulfate
LPS: Lipopolysaccharide
mt: Mitochondria
mTOR: Mammalian target of rapamycin
NaF: Sodium fluoride
NF-κB: Nuclear factor-kappa B
NLR: Nucleotide oligomerization domain receptors
OD: Optical density
PBS: Phosphate buffer saline
p62/SQMT1: Sequestosome 1
PD: Parkinson’s disease
PFA: Paraformaldehyde
PI3K: Phosphatidylinositide 3-kinase
PMSF: Phenylmethylsulfonyl fluoride
PP2A: Protein phosphatase 2A
RT: Room temperature
SDS: Sodium dodecyl-sulfate
TACE: TNFα converting enzyme
TBST: Tris-buffered saline tween
TEM: Transmission electron microscopy
TMB: Tetramethylbenzidine
TNF: Tumor necrosis factor
TNFR: Tumor necrosis factor receptor
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AF performed the research, analyzed the data, their statistical significance and wrote the paper; FT participated in the design of the study and followed the work; NQ and BF carried out TEM; DC participated in brain tissue preparation for biochemical analysis; TJ, ARB and MP followed the research and provided relevant remarks throughout the work; GP conceived of the study, and organized its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Additional file
Supplementary Material
Cortical cytokine levels in saline-treated mice. Table S2. Hippocampal cytokine levels in saline-treated mice. Table S3. Changes in cortical autophagic markers and mTOR signalling pathway in saline-treated mice. Table S4. Changes in hippocampal autophagic markers and mTOR signalling pathway in saline-treated mice. Figure S1. Accumulation of autophagic vesicles in murine primary mixed cell culture.
Contributor Information
Arnaud François, Email: arnaudfrancois85@gmail.com.
Faraj Terro, Email: faraj.terro@unilim.fr.
Nathalie Quellard, Email: microscope.electronique@chu-poitiers.fr.
Béatrice Fernandez, Email: beatrice.fernandez@chu-poitiers.fr.
Damien Chassaing, Email: damien.chassaing@univ-poitiers.fr.
Thierry Janet, Email: thierry.janet@univ-poitiers.fr.
Agnès Rioux Bilan, Email: agnes.rioux-bilan@univ-poitiers.fr.
Marc Paccalin, Email: m.paccalin@chu-poitiers.fr.
Guylène Page, Email: guylene.page@univ-poitiers.fr.
Acknowledgements
This work was supported by a grant from “Ligue Européenne Contre la Maladie d’Alzheimer” (LECMA), by Poitiers University and Poitiers University Hospital. Authors thanked Dr Stéphanie Ragot for her expertise in statistical analysis.
References
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewell JL, Russell RC, Guan KL. Amino acid signalling upstream of mTOR. Nat Rev Mol Cell Biol. 2013;14:133–139. doi: 10.1038/nrm3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Guan KL. Complexity of the TOR signaling network. Trends Cell Biol. 2006;16:206–212. doi: 10.1016/j.tcb.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Korolchuk VI, Renna M, Imarisio S, Fleming A, Williams A, Garcia-Arencibia M, Rose C, Luo S, Underwood BR, Kroemer G, O'Kane CJ, Rubinsztein DC. Complex inhibitory effects of nitric oxide on autophagy. Mol Cell. 2011;43:19–32. doi: 10.1016/j.molcel.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RS, Rosenberg P. Calcium-dependent gene regulation in myocyte hypertrophy and remodeling. Cold Spring Harb Symp Quant Biol. 2002;67:339–344. doi: 10.1101/sqb.2002.67.339. [DOI] [PubMed] [Google Scholar]
- Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, Jahreiss L, Fleming A, Pask D, Goldsmith P, O'Kane CJ, Floto RA, Rubinsztein DC. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat Chem Biol. 2008;4:295–305. doi: 10.1038/nchembio.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30:678–688. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morselli E, Maiuri MC, Markaki M, Megalou E, Pasparaki A, Palikaras K, Criollo A, Galluzzi L, Malik SA, Vitale I, Michaud M, Madeo F, Tavernarakis N, Kroemer G. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 2010;1:e10. doi: 10.1038/cddis.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morselli E, Maiuri MC, Markaki M, Megalou E, Pasparaki A, Palikaras K, Criollo A, Galluzzi L, Malik SA, Vitale I, Michaud M, Madeo F, Tavernarakis N, Kroemer G. The life span-prolonging effect of sirtuin-1 is mediated by autophagy. Autophagy. 2010;6:186–188. doi: 10.4161/auto.6.1.10817. [DOI] [PubMed] [Google Scholar]
- Sarkar S. Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem Soc Trans. 2013;41:1103–1130. doi: 10.1042/BST20130134. [DOI] [PubMed] [Google Scholar]
- Rubinstein AD, Kimchi A. Life in the balance - a mechanistic view of the crosstalk between autophagy and apoptosis. J Cell Sci. 2012;125:5259–5268. doi: 10.1242/jcs.115865. [DOI] [PubMed] [Google Scholar]
- Ghavami S, Shojaei S, Yeganeh B, Ande SR, Jangamreddy JR, Mehrpour M, Christoffersson J, Chaabane W, Moghadam AR, Kashani HH, Hashemi M, Owji AA, Los MJ. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol. 2014;112:24–49. doi: 10.1016/j.pneurobio.2013.10.004. [DOI] [PubMed] [Google Scholar]
- Deretic V. Autophagy as an innate immunity paradigm: expanding the scope and repertoire of pattern recognition receptors. Curr Opin Immunol. 2012;24:21–31. doi: 10.1016/j.coi.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Galluzzi L, Zitvogel L, Kroemer G. Autophagy and cellular immune responses. Immunity. 2013;39:211–227. doi: 10.1016/j.immuni.2013.07.017. [DOI] [PubMed] [Google Scholar]
- Quan W, Lee MS. Role of Autophagy in the Control of Body Metabolism. Endocrinol Metab (Seoul) 2013;28:6–11. doi: 10.3803/EnM.2013.28.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craddock N, Hurles ME, Cardin N, Pearson RD, Plagnol V, Robson S, Vukcevic D, Barnes C, Conrad DF, Giannoulatou E, Holmes C, Marchini JL, Stirrups K, Tobin MD, Wain LV, Yau C, Aerts J, Ahmad T, Andrews TD, Arbury H, Attwood A, Auton A, Ball SG, Balmforth AJ, Barrett JC, Barroso I, Barton A, Bennett AJ, Bhaskar S, Blaszczyk K. et al. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. 2010;464:713–720. doi: 10.1038/nature08979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brest P, Lapaquette P, Souidi M, Lebrigand K, Cesaro A, Vouret-Craviari V, Mari B, Barbry P, Mosnier JF, Hebuterne X, Harel-Bellan A, Mograbi B, Darfeuille-Michaud A, Hofman P. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat Genet. 2011;43:242–245. doi: 10.1038/ng.762. [DOI] [PubMed] [Google Scholar]
- Nguyen HT, Lapaquette P, Bringer MA, Darfeuille-Michaud A. Autophagy and Crohn’s disease. J Innate Immun. 2013;5:434–443. doi: 10.1159/000345129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium WTCC. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos PS, Criswell LA, Moser KL, Comeau ME, Williams AH, Pajewski NM, Chung SA, Graham RR, Zidovetzki R, Kelly JA, Kaufman KM, Jacob CO, Vyse TJ, Tsao BP, Kimberly RP, Gaffney PM, Alarcon-Riquelme ME, Harley JB, Langefeld CD. A comprehensive analysis of shared loci between systemic lupus erythematosus (SLE) and sixteen autoimmune diseases reveals limited genetic overlap. PLoS Genet. 2011;7:e1002406. doi: 10.1371/journal.pgen.1002406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Gupta J, Jyothula SS, Butsch Kovacic M, Biagini Myers JM, Patterson TL, Ericksen MB, He H, Gibson AM, Baye TM, Amirisetty S, Tsoras AM, Sha Y, Eissa NT, Hershey GK. Functional variant in the autophagy-related 5 gene promotor is associated with childhood asthma. PLoS One. 2012;7:e33454. doi: 10.1371/journal.pone.0033454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raychaudhuri S, Thomson BP, Remmers EF, Eyre S, Hinks A, Guiducci C, Catanese JJ, Xie G, Stahl EA, Chen R, Alfredsson L, Amos CI, Ardlie KG, Barton A, Bowes J, Burtt NP, Chang M, Coblyn J, Costenbader KH, Criswell LA, Crusius JB, Cui J, De Jager PL, Ding B, Emery P, Flynn E, Harrison P, Hocking LJ, Huizinga TW, Kastner DL. et al. Genetic variants at CD28, PRDM1 and CD2/CD58 are associated with rheumatoid arthritis risk. Nat Genet. 2009;41:1313–1318. doi: 10.1038/ng.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, Tanaka K, Kawai T, Tsujimura T, Takeuchi O, Yoshimori T, Akira S. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- Harris J, Hartman M, Roche C, Zeng SG, O’Shea A, Sharp FA, Lambe EM, Creagh EM, Golenbock DT, Tschopp J, Kornfeld H, Fitzgerald KA, Lavelle EC. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem. 2011;286:9587–9597. doi: 10.1074/jbc.M110.202911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, Sher A, Kehrl JH. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13:255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Sun B. Negative regulation of NLRP3 inflammasome signaling. Protein Cell. 2013;4:251–258. doi: 10.1007/s13238-013-2128-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuk JM, Jo EK. Crosstalk between autophagy and inflammasomes. Mol Cells. 2013;36:393–399. doi: 10.1007/s10059-013-0298-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JN, Fattah EA, Bhattacharya A, Ko S, Eissa NT. Inflammasome activation by altered proteostasis. J Biol Chem. 2013;288:35886–35895. doi: 10.1074/jbc.M113.514919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criollo A, Niso-Santano M, Malik SA, Michaud M, Morselli E, Marino G, Lachkar S, Arkhipenko AV, Harper F, Pierron G, Rain JC, Ninomiya-Tsuji J, Fuentes JM, Lavandero S, Galluzzi L, Maiuri MC, Kroemer G. Inhibition of autophagy by TAB2 and TAB3. EMBO J. 2011;30:4908–4920. doi: 10.1038/emboj.2011.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng M, Wei X, Wu Z, Li W, Li B, Zhen Y, Chen J, Wang P, Fei Y. NF-kappaB-mediated induction of autophagy in cardiac ischemia/reperfusion injury. Biochem Biophys Res Commun. 2013;436:180–185. doi: 10.1016/j.bbrc.2013.05.070. [DOI] [PubMed] [Google Scholar]
- Pilli M, Arko-Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, Dupont N, Ornatowski W, Jiang S, Bradfute SB, Bruun JA, Hansen TE, Johansen T, Deretic V. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity. 2012;37:223–234. doi: 10.1016/j.immuni.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- Harris J, De Haro SA, Master SS, Keane J, Roberts EA, Delgado M, Deretic V. T helper 2 cytokines inhibit autophagic control of intracellular Mycobacterium tuberculosis. Immunity. 2007;27:505–517. doi: 10.1016/j.immuni.2007.07.022. [DOI] [PubMed] [Google Scholar]
- Randall-Demllo S, Chieppa M, Eri R. Intestinal Epithelium and Autophagy: Partners in Gut Homeostasis. Front Immunol. 2013;4:301. doi: 10.3389/fimmu.2013.00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marselli L, Bugliani M, Suleiman M, Olimpico F, Masini M, Petrini M, Boggi U, Filipponi F, Syed F, Marchetti P. beta-Cell inflammation in human type 2 diabetes and the role of autophagy. Diabetes Obes Metab. 2013;15(Suppl 3):130–136. doi: 10.1111/dom.12152. [DOI] [PubMed] [Google Scholar]
- Pan L, Li Y, Jia L, Qin Y, Qi G, Cheng J, Qi Y, Li H, Du J. Cathepsin S deficiency results in abnormal accumulation of autophagosomes in macrophages and enhances Ang II-induced cardiac inflammation. PLoS One. 2012;7:e35315. doi: 10.1371/journal.pone.0035315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junkins RD, McCormick C, Lin TJ. The emerging potential of autophagy-based therapies in the treatment of cystic fibrosis lung infections. Autophagy. 2014;10:538–547. doi: 10.4161/auto.27750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan CC, Yu JT, Tan MS, Jiang T, Zhu XC, Tan L. Autophagy in aging and neurodegenerative diseases: implications for pathogenesis and therapy. Neurobiol Aging. 2014;35:941–957. doi: 10.1016/j.neurobiolaging.2013.11.019. [DOI] [PubMed] [Google Scholar]
- Nishioku T, Dohgu S, Takata F, Eto T, Ishikawa N, Kodama KB, Nakagawa S, Yamauchi A, Kataoka Y. Detachment of brain pericytes from the basal lamina is involved in disruption of the blood–brain barrier caused by lipopolysaccharide-induced sepsis in mice. Cell Mol Neurobiol. 2009;29:309–316. doi: 10.1007/s10571-008-9322-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian Y, Zhao X, Li M, Zeng S, Zhao B. Various roles of astrocytes during recovery from repeated exposure to different doses of lipopolysaccharide. Behav Brain Res. 2013;253:253–261. doi: 10.1016/j.bbr.2013.07.028. [DOI] [PubMed] [Google Scholar]
- Datta SC, Opp MR. Lipopolysaccharide-induced increases in cytokines in discrete mouse brain regions are detectable using Luminex xMAP technology. J Neurosci Methods. 2008;175:119–124. doi: 10.1016/j.jneumeth.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semmler A, Okulla T, Sastre M, Dumitrescu-Ozimek L, Heneka MT. Systemic inflammation induces apoptosis with variable vulnerability of different brain regions. J Chem Neuroanat. 2005;30:144–157. doi: 10.1016/j.jchemneu.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Quan N, Sundar SK, Weiss JM. Induction of interleukin-1 in various brain regions after peripheral and central injections of lipopolysaccharide. J Neuroimmunol. 1994;49:125–134. doi: 10.1016/0165-5728(94)90188-0. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I. et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/S0197-4580(00)00124-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–580. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S, Akematsu T, Akira S, Al-Younes HM, Al-Zeer MA, Albert ML, Albin RL, Alegre-Abarrategui J, Aleo MF, Alirezaei M, Almasan A, Almonte-Becerril M, Amano A, Amaravadi R, Amarnath S, Amer AO, Andrieu-Abadie N, Anantharam V. et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton TR, Gout IT. Functions and regulation of the 70 kDa ribosomal S6 kinases. Int J Biochem Cell Biol. 2011;43:47–59. doi: 10.1016/j.biocel.2010.09.018. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Shang YC, Zhang L, Wang S, Maiese K. Mammalian target of rapamycin: hitting the bull’s-eye for neurological disorders. Oxidative Med Cell Longev. 2010;3:374–391. doi: 10.4161/oxim.3.6.14787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francois A, Terro F, Janet T, Bilan AR, Paccalin M, Page G. Involvement of interleukin-1beta in the autophagic process of microglia: relevance to Alzheimer’s disease. J Neuroinflammation. 2013;10:151. doi: 10.1186/1742-2094-10-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czapski GA, Gajkowska B, Strosznajder JB. Systemic administration of lipopolysaccharide induces molecular and morphological alterations in the hippocampus. Brain Res. 2010;1356:85–94. doi: 10.1016/j.brainres.2010.07.096. [DOI] [PubMed] [Google Scholar]
- Chung DW, Yoo KY, Hwang IK, Kim DW, Chung JY, Lee CH, Choi JH, Choi SY, Youn HY, Lee IS, Won MH. Systemic administration of lipopolysaccharide induces cyclooxygenase-2 immunoreactivity in endothelium and increases microglia in the mouse hippocampus. Cell Mol Neurobiol. 2010;30:531–541. doi: 10.1007/s10571-009-9477-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Lee YK, Yuk DY, Choi DY, Ban SB, Oh KW, Hong JT. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflammation. 2008;5:37. doi: 10.1186/1742-2094-5-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begum AN, Jones MR, Lim GP, Morihara T, Kim P, Heath DD, Rock CL, Pruitt MA, Yang F, Hudspeth B, Hu S, Faull KF, Teter B, Cole GM, Frautschy SA. Curcumin structure-function, bioavailability, and efficacy in models of neuroinflammation and Alzheimer’s disease. J Pharmacol Exp Ther. 2008;326:196–208. doi: 10.1124/jpet.108.137455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan K, Wu X, Fan B, Li N, Lin Y, Yao Y, Ma J. Up-regulation of microglial cathepsin C expression and activity in lipopolysaccharide-induced neuroinflammation. J Neuroinflammation. 2012;9:96. doi: 10.1186/1742-2094-9-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray CL, Skelly DT, Cunningham C. Exacerbation of CNS inflammation and neurodegeneration by systemic LPS treatment is independent of circulating IL-1beta and IL-6. J Neuroinflammation. 2011;8:50. doi: 10.1186/1742-2094-8-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsberth C, Hamzic N, Norell M, Blomqvist A. Peripheral lipopolysaccharide administration induces cytokine mRNA expression in the viscera and brain of fever-refractory mice lacking microsomal prostaglandin E synthase-1. J Neuroendocrinol. 2009;21:715–721. doi: 10.1111/j.1365-2826.2009.01888.x. [DOI] [PubMed] [Google Scholar]
- Jaeger LB, Dohgu S, Sultana R, Lynch JL, Owen JB, Erickson MA, Shah GN, Price TO, Fleegal-Demotta MA, Butterfield DA, Banks WA. Lipopolysaccharide alters the blood–brain barrier transport of amyloid beta protein: a mechanism for inflammation in the progression of Alzheimer’s disease. Brain Behav Immun. 2009;23:507–517. doi: 10.1016/j.bbi.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J Neurosci. 2005;25:8843–8853. doi: 10.1523/JNEUROSCI.2868-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- Couturier J, Paccalin M, Lafay-Chebassier C, Chalon S, Ingrand I, Pinguet J, Pontcharraud R, Guillard O, Fauconneau B, Page G. Pharmacological inhibition of PKR in APPswePS1dE9 mice transiently prevents inflammation at 12 months of age but increases Abeta42 levels in the late stages of the Alzheimer’s disease. Curr Alzheimer Res. 2012;9:344–360. doi: 10.2174/156720512800107582. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Arai T, Kondo H, Tanno E, Haga C, Ikeda K. Cell mediators of inflammation in the Alzheimer disease brain. Alzheimer Dis Assoc Disord. 2000;14(Suppl 1):S47–S53. doi: 10.1097/00002093-200000001-00008. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Cataldo AM, Mathews PM. The endosomal-lysosomal system of neurons in Alzheimer’s disease pathogenesis: a review. Neurochem Res. 2000;25:1161–1172. doi: 10.1023/A:1007675508413. [DOI] [PubMed] [Google Scholar]
- Terrazzino S, Bauleo A, Baldan A, Leon A. Peripheral LPS administrations up-regulate Fas and FasL on brain microglial cells: a brain protective or pathogenic event? J Neuroimmunol. 2002;124:45–53. doi: 10.1016/S0165-5728(02)00013-9. [DOI] [PubMed] [Google Scholar]
- Motori E, Puyal J, Toni N, Ghanem A, Angeloni C, Malaguti M, Cantelli-Forti G, Berninger B, Conzelmann KK, Gotz M, Winklhofer KF, Hrelia S, Bergami M. Inflammation-induced alteration of astrocyte mitochondrial dynamics requires autophagy for mitochondrial network maintenance. Cell Metab. 2013;18:844–859. doi: 10.1016/j.cmet.2013.11.005. [DOI] [PubMed] [Google Scholar]
- Deretic V. Autophagy in immunity and cell-autonomous defense against intracellular microbes. Immunol Rev. 2011;240:92–104. doi: 10.1111/j.1600-065X.2010.00995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeisser H, Fey SB, Horowitz J, Fischer ER, Balinsky CA, Miyake K, Bekisz J, Snow AL, Zoon KC. Type I interferons induce autophagy in certain human cancer cell lines. Autophagy. 2013;9:683–696. doi: 10.4161/auto.23921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0 s that spur autophagy and immunity. Immunol Rev. 2012;249:158–175. doi: 10.1111/j.1600-065X.2012.01146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrott LA, Templeton DJ. Osmotic stress inhibits p70/85 S6 kinase through activation of a protein phosphatase. J Biol Chem. 1999;274:24731–24736. doi: 10.1074/jbc.274.35.24731. [DOI] [PubMed] [Google Scholar]
- Petritsch C, Beug H, Balmain A, Oft M. TGF-beta inhibits p70 S6 kinase via protein phosphatase 2A to induce G(1) arrest. Genes Dev. 2000;14:3093–3101. doi: 10.1101/gad.854200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang CH, Pruznak AM, Frost RA. TNFalpha mediates sepsis-induced impairment of basal and leucine-stimulated signaling via S6K1 and eIF4E in cardiac muscle. J Cell Biochem. 2005;94:419–431. doi: 10.1002/jcb.20311. [DOI] [PubMed] [Google Scholar]
- Perez-Rodriguez D, Anuncibay-Soto B, Llorente IL, Perez-Garcia CC, Fernandez-Lopez A. Hippocampus and cerebral cortex present a different autophagic response after oxygen and glucose deprivation in an ex vivo rat brain slice model. Neuropathol Appl Neurobiol. 2014;ᅟ:ᅟ. doi: 10.1111/nan.12152. [DOI] [PubMed] [Google Scholar]
- Weis SN, Toniazzo AP, Ander BP, Zhan X, Careaga M, Ashwood P, Wyse AT, Netto CA, Sharp FR. Autophagy in the brain of neonates following hypoxia-ischemia shows sex- and region-specific effects. Neuroscience. 2014;256:201–209. doi: 10.1016/j.neuroscience.2013.10.046. [DOI] [PubMed] [Google Scholar]
- Zhang S, Xue ZF, Huang LP, Fang RM, He YP, Li L, Fang YQ. Dynamic expressions of Beclin 1 and tyrosine hydroxylase in different areas of 6-hydroxydopamine-induced Parkinsonian rats. Cell Mol Neurobiol. 2013;33:973–981. doi: 10.1007/s10571-013-9964-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks WA. The blood–brain barrier in psychoneuroimmunology. Neurol Clin. 2006;24:413–419. doi: 10.1016/j.ncl.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Roth J, Harre EM, Rummel C, Gerstberger R, Hubschle T. Signaling the brain in systemic inflammation: role of sensory circumventricular organs. Front Biosci. 2004;9:290–300. doi: 10.2741/1241. [DOI] [PubMed] [Google Scholar]
- Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun. 2011;25:181–213. doi: 10.1016/j.bbi.2010.10.015. [DOI] [PubMed] [Google Scholar]
- Sun Q, Gao W, Loughran P, Shapiro R, Fan J, Billiar TR, Scott MJ. Caspase 1 activation is protective against hepatocyte cell death by up-regulating beclin 1 protein and mitochondrial autophagy in the setting of redox stress. J Biol Chem. 2013;288:15947–15958. doi: 10.1074/jbc.M112.426791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuka S, Ishii Y, Matsuyama M, Ano S, Morishima Y, Yanagawa T, Warabi E, Hizawa N. SQSTM1/p62/A170 regulates the severity of Legionella pneumophila pneumonia by modulating inflammasome activity. Eur J Immunol. 2014;44:1084–1092. doi: 10.1002/eji.201344091. [DOI] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- Li G, Liu Y, Tzeng NS, Cui G, Block ML, Wilson B, Qin L, Wang T, Liu B, Liu J, Hong JS. Protective effect of dextromethorphan against endotoxic shock in mice. Biochem Pharmacol. 2005;69:233–240. doi: 10.1016/j.bcp.2004.10.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cortical cytokine levels in saline-treated mice. Table S2. Hippocampal cytokine levels in saline-treated mice. Table S3. Changes in cortical autophagic markers and mTOR signalling pathway in saline-treated mice. Table S4. Changes in hippocampal autophagic markers and mTOR signalling pathway in saline-treated mice. Figure S1. Accumulation of autophagic vesicles in murine primary mixed cell culture.