

Abstract
Single nucleotide polymorphisms (SNPs) located in the chromosomal region 16p13.13 have been previously associated with risk for several autoimmune diseases, including type 1 diabetes. To identify and localize specific risk variants for type 1 diabetes in this region and understand the mechanism of their action, we resequenced a 455-kb region in type 1 diabetic patients and unaffected control subjects, identifying 93 novel variants. A panel of 939 SNPs that included 46 of these novel variants was genotyped in 3,070 multiplex families with type 1 diabetes. Forty-eight SNPs, all located in CLEC16A, provided a statistically significant association (P < 5.32 × 10−5) with disease, with rs34306440 being most significantly associated (P = 5.74 × 10−6). The panel of SNPs used for fine mapping was also tested for association with transcript levels for each of the four genes in the region in B lymphoblastoid cell lines. Significant associations were observed only for transcript levels of DEXI, a gene with unknown function. We examined the relationship between the odds ratio for type 1 diabetes and the magnitude of the effect of DEXI transcript levels for each SNP in the region. Among SNPs significantly associated with type 1 diabetes, the common allele conferred an increased risk for disease and corresponded to lower DEXI expression. Our results suggest that the primary mechanism by which genetic variation at CLEC16A contributes to the risk for type 1 diabetes is through reduced expression of DEXI.
Introduction
Type 1 diabetes most frequently results from the autoimmune destruction of insulin-producing pancreatic β-cells, resulting in a dependence on exogenous insulin. The disease is complex and arises due to the effects of genetic and environmental factors, as evidenced by incomplete concordance between monozygotic twins (1,2). Before 2007, a small number of genetic loci that contribute to type 1 diabetes risk, including HLA, INS, IL2RA, CTLA4, and PTPN22, were identified through linkage and candidate gene studies (1). With the advent of genome-wide association studies (GWAS), more than 40 additional chromosomal regions have been identified that harbor loci that confer low to moderate risk of developing type 1 diabetes, with odds ratios (OR) of less than 1.3 (3–6). One of these regions is located at chromosome 16p13.13 and is associated with type 1 diabetes and several other autoimmune disorders, including multiple sclerosis (MS), celiac disease, primary biliary cirrhosis, systemic lupus erythematosus, Crohn disease, ulcerative colitis, and primary adrenal insufficiency (4–16).
The region on chromosome 16p13.13 implicated in autoimmunity spans ∼530 kb and contains four known genes. The most significant evidence of an association with type 1 diabetes in this region is observed for two single nucleotide polymorphisms (SNPs), rs12708716 and rs2903692, located in different introns of the gene, CLEC16A, previously known as KIAA0350 (3,5). CLEC16A has no confirmed function but shares primary sequence similarity with C-type lectins and with a Drosophila gene whose product is reported to be involved in endosomal maturation (17,18). In contrast, neighboring genes appear to be stronger candidates for type 1 diabetic pathogenesis based on their known functions. These include CIITA, a transcription factor that is essential for the expression of HLA class II genes (19), and SOCS1, a negative feedback regulator of cytokine signaling (20).The remaining gene in the region is DEXI, a gene of unknown function whose transcript levels are reported to be upregulated in response to dexamethasone, a glucocorticoid drug commonly used as an anti-inflammatory and immunosuppressant medication (21).
Several GWAS (3–6) identified the 16p13.13 region as being associated with type 1 diabetes, and these reported associations have been independently replicated (22–26). Although the association between the 16p13.13 region and type 1 diabetes has been well established, the specific genes or SNPs responsible for the diseased risk in the region have yet to be elucidated. Most of the variants on 16p13.13 associated with type 1 diabetes occur within introns of the CLEC16A gene. It has recently been reported that an intronic region of CLEC16A can physically interact with the putative promoter region of DEXI and thereby modulate DEXI expression in monocytes and in Epstein Barr virus–transformed lymphoblastoid cell lines (27–29). It has also been separately reported that an intronic region of DEXI functions as a distal enhancer for CIITA (30,31). These functional findings suggest a potential model in which genetic variation at intronic sites in CLEC16A could modulate the expression of neighboring genes, which might in turn affect the risk of type 1 diabetes.
In the current study, we resequenced the 16p13.13 region in a subset of type 1 diabetic individuals to identify novel variants. These variants were included in a dense genotyping panel of 939 SNPs to fine map the region associated with type 1 diabetes. We then assessed the relationship of the genotyped SNPs and the expression of the four genes in the region to discern a pattern of regulation associated with diabetes risk.
Research Design and Methods
Subjects
This study was reviewed and approved by the University of Virginia Institutional Review Board. DNA from anonymous type 1 diabetic case subjects and control subjects was obtained from the Virginia Mason Medical Center and Puget Sound Blood Center (Seattle, WA), respectively. DNA from affected sibling-pair families was obtained from the Type 1 Diabetes Consortium (T1DGC) (32) and the Human Biological Data Interchange Repository (HBDI) (33).
Sequencing of Region to Identify SNPs
We targeted a 455-kb region (10,943,936–11,399,037 bp [Hg19]) on chromosome 16p13.13 for deep sequencing. This region encompassed the four genes (CIITA, CLEC16A, DEXI,and SOCS1) and all SNPs previously reported to be significantly associated with type 1 diabetes. The region was tiled with ∼10-kb PCR fragments with ∼0.5-kb overlaps. Amplifications were done in 48 pools that each contained DNA from 4 individuals (128 type 1 diabetic patients and 64 control subjects). Amplified PCR fragments were pooled in equimolar amounts to create DNA libraries. DNA libraries were prepared for sequencing using Illumina’s Paired-End Sample Preparation Kit (Illumina, San Diego, CA). Sequencing was performed on an Illumina Genome Analyzer IIx (Illumina) using 63 bp-end reads. Sequencing reads were assembled using the Burrows-Wheeler Aligner tool (34). Sequence Alignment/Map Tools (35) was used for conversion, indexing, and aligning of the data using the reference genome (Hg18; National Center for Biotechnology Information ver. 36) (36,37) as well as for SNP identification.
SNP Filtering and Selection
Putative SNPs identified from sequencing were filtered to limit false positives caused by strand bias by including only minor alleles with a frequency rate ≥0.1%, for which the proportion of forward reads was not statistically different from the proportion of reverse reads. The two proportions were only declared not statistically different if the absolute value of the t statistic for the proportions test was <1. The SNP list was then parsed into two groups: annotated SNPs and novel unreported SNPs, defined as lacking a Single Nucleotide Polymorphism Database (dbSNP) ID (http://www.ncbi.nlm.nih.gov/projects/SNP/).
SNPs annotated in dbSNP were excluded from analysis if they had a minor allele frequency (MAF) <5% (except exonic SNPs) or had <50 mapped reads. The annotated SNP list was further filtered to exclude SNPs that had previously been genotyped by the HapMap project (38), leaving 355 annotated SNPs.
Novel variants were filtered to exclude those with a MAF <5% (exonic SNPs were only excluded if MAF <1%) and those with a combination of <50 aligning reads and >20% nonaligning reads, which left 93 novel SNPs.
Genotyping
We designed two custom GoldenGate genotyping assays (Illumina). The first panel consisted of 123 tagging SNPs that captured information on the 505 reported SNPs in the CEU (Utah residents with ancestry from Northern and Western Europe) HapMap (r2 = 0.9), as determined using Haploview 4.2 (39). The second panel consisted of 290 SNPs identified through our pooled sequencing of type 1 diabetic case subjects and control subjects. There were 16 SNPs duplicated on each panel for quality control. The compiled SNP lists were verified for designability using Illumina's GoldenGate Panel Design software (www.illumina.com). Genotyping was performed for 413 SNPs on 1,850 HBDI and 1,750 TIDGC samples. Data were examined in GenomeStudio (Illumina) and exported in PLINK (40) format.
Quality control measures were performed at the SNP and sample level. Signal cloud graphs were manually reviewed for each of the genotyped SNPs, and those with poor clustering were excluded. SNPs with more than five Mendelian transmission errors were identified and excluded from further analyses. SNPs genotyped in duplicate were examined for concordance using PLINK (40); those with ≥5% discordance were excluded. Also excluded were SNPs with significant deviations from Hardy-Weinberg equilibrium, >5% missingness, or <1% MAF.
For sample quality control in GenomeStudio, DNA samples with a genotype call rate of <90% were removed from further analyses. Subjects with more than five Mendelian transmission errors were identified with PLINK and excluded from the analyses. All remaining Mendelian transmission errors were identified and problematic genotypes removed with PedCheck (41).
ImmunoChip Data Set
After the completion of the above-described sequencing and genotyping, genotype data for 1,734 SNPs in the 16p13.13 region from the ImmunoChip data set became available and were added to the analyses (provided by Dr. Stephen Rich) (42). SNPs with significant deviations from Hardy-Weinberg equilibrium or >5% missingness were excluded from subsequent analyses. PedCheck was used to identify Mendelian errors and remove conflicting genotypes. Finally, all duplicate SNPs and SNPs with <1% MAF were removed.
Imputation
After quality control, the data set that was derived from targeted sequencing and tagging included 319 SNPs with genotypes from 749 families containing 3,370 individuals. The ImmunoChip data set included 1,040 SNPs with genotypes from 2,689 families containing 10,746 individuals. There were 252 SNPs and 1,581 individuals in common between the two data sets.
The two data sets were merged, and missing genotypes were extrapolated by imputation. A reference haplotype population (same individuals from both panels) was generated using the MACH software program (43), and data from 481 parental Caucasian founders whose ethnicity was confirmed using the KING relationship inference software package (44). Imputation was performed using MACH and the reference population. SNPs with r2 ≤ 0.5 (values generated by MACH) were removed. The same quality control steps that were applied to the originally generated genotype data were applied to the imputed data.
Association Testing
The final merged data set was tested for association with type 1 diabetes using the transmission disequilibrium test (TDT) (45) option in PLINK, and the results were visualized using Locus Zoom (46). Tests for residual evidence of disease association were performed using UNPHASED (47) by conditioning on the SNP with the most significant association with type 1 diabetes.
Cell Culturing and RNA Extraction
We procured 33 HapMap Epstein Barr virus–transformed cell lines from the Coriell Institute for Medical Research (Camden, NJ). All individuals were unrelated and healthy. Cells were grown in RPMI media supplemented with 10% FBS, 1% penicillin/streptomycin, and 1% l-glutamine. Cells were split on a regular schedule. Harvesting of cells occurred when cells reached a density of 5 × 10−7 and only if they were 70% viable. Cells were pelleted and stored at −80°C. RNA was isolated using TRIzol (Invitrogen), followed by a DNase treatment (Ambion, Life Technologies).
RT-PCR
RNA was reverse transcribed using random primers (Invitrogen) and Superscript II (Invitrogen), following the manufacturer’s protocol. The cDNA samples, RT control samples, and standards were plated in triplicate using a Biomek FX-Robotic workstation (Beckman Coulter) and air dried. PCR primers were designed based on mRNA transcripts from the National Center for Biotechnology Information database (CIITA III-BQ221200, CIITA IV-BM147267, CLEC16A Isoform I-NM_015226.2, CLEC16A Isoform II-BC112897.1, CLEC16A Isoform III-AK126771.1, DEXI-NM_014015.3, SOCS1-NM_003745.1, GAPDH- NM_002046.3) using Primer3 (48) (Supplementary Table 1). Amplifications were carried out on an ABI 7900 (Life Technologies, Carlsbad, CA) as follows: 95°C for 10 min, 35-40 cycles of 95°C for 15 s, 60° or 61°C for 30 s, 72°C for 30 s, followed by a dissociation curve analysis. The data were examined using ABI’s SDS 2.3 software, and Ct (threshold cycle) and baseline were adjusted for each PCR run.
Expression Quantitative Trait Loci Analysis
Amplification efficiencies were calculated per transcript and compared with that for the control GAPDH gene. Median Ct values were determined from the raw triplicate Ct values. The ΔΔCt values were calculated, correcting for primer efficiencies, and were quantile normalized. The quantile-normalized ΔΔCt along with genotyping data on the 33 HapMap cell lines (ImmunoChip and custom sequencing panel) were used for an expression quantitative trait loci (eQTL) analysis (linear model with quantitative trait) using PLINK. The eQTL results from the custom-sequencing panel and ImmunoChip panels were merged. The data set was filtered to remove all SNPs with a MAF <5% and all duplicate SNPs. The final SNP data set consisted of 915 SNPs and was visualized using LocusZoom. We corrected for multiple hypothesis testing using Bonferroni correction (49). The Bonferroni correction was applied based on the number of independent SNPs where any group of SNPs with pairwise r2 = 1 was considered as independent. A total of 336 independent tests were performed with a corrected P value of 0.05/366 = 1.37 × 10−4.
Results
Resequencing of the 16p13.13 Region Identifies Novel SNPs
A region of 455 kb from 16p13.13 was amplified in 192 individuals (128 type 1 diabetic patients and 64 control individuals) and sequenced to an average depth of coverage of 35×. The region was selected based on capturing the linkage disequilibrium block (D’ ≈ 1) that encompassed the disease-associated SNPs for type 1 diabetes (rs12708716 and rs2903692), which contains the DEXI and CLEC16A gene. We then expanded the region to include the neighboring genes of CIITA and SOCS1 because of their important immunological function. When aligned to the human reference sequence, there were two gaps at coordinates chr16:11,028,710–11,038,336 (Hg19) and chr16:11,340,104–11,350,943 (Hg19), from which no sequence was obtained that passed quality control. When compared with the human reference genome, 962 SNPs were identified, of which 93 had no prior annotation. A genotyping panel spanning the region was designed containing tagging SNPs selected from HapMap and novel SNPs identified from our resequencing (60 of the 93 SNPs were designable and included in the panel).
Fine Mapping Identifies Statistically Significant Associations With Type 1 Diabetes
Genotypes for 319 SNPs (MAF >1%) were generated from 3,370 members of 749 multiplex type 1 diabetic families. These data were merged with an overlapping data set from the same region derived from ImmunoChip, and imputation was used to fill in the missing genotypes for those subjects genotyped on only one platform.
The merged data set, after imputation, contained genotypes for 939 SNPs on 12,485 individuals (3,070 multiplex families), which included 46 of our novel variants. The family-based TDT (45) was used to test for association of alleles at these SNPs with type 1 diabetes (Fig. 1). A total of 48 SNPs were identified as having alleles significantly associated with type 1 diabetes (P < 5.32 × 10−5; Supplementary Table 2). The strongest evidence for association was obtained at rs34306440 located in intron 20 of CLEC16A (OR 0.838 [95% CI 0.776–0.904], P = 5.74 × 10−6). After conditioning on rs34306440, no residual statistically significant associations were observed (most significant residual effect rs11643024, P = 1.79 × 10−4). All the significantly associated SNPs were located in the introns of CLEC16A (Supplementary Table 2).
Figure 1.
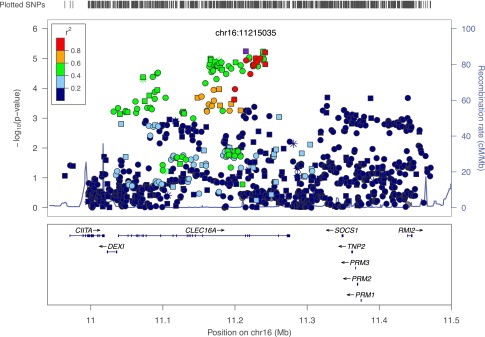
Type 1 diabetes association results on 16p13.13. A total of 939 SNPs (genotyped and imputed) were tested for association with type 1 diabetes in 3,070 multiplex families (12,485 individuals) using the TDT. The log10 of the P value for this test is plotted relative to the physical position on the chromosome for each SNP. The most significant results were obtained for rs34306440 (P = 5.74 × 10−6), marked in purple. The color of all other SNPs indicates pairwise linkage disequilibrium with rs34306440, as measured by the r2 statistic. Symbol shapes indicate those SNPs genotyped only on the custom panels (snowflake), genotyped only on the ImmunoChip (circle), and genotyped on both panels (square). cM/Mb, centimorgan/megabase.
SNPs in CLEC16A Are Associated With Variation in Expression Level of DEXI
An eQTL analysis was performed designating the expression levels of CIITA, DEXI, CLEC16A, and SOCS1 in B-cell lines as phenotypes. Primers that specifically amplified each of the common reported alternative spliced transcripts for the target genes were designed and used to assess transcript levels by quantitative RT-PCR. The full panel of 915 SNPs from the region was then tested for association with quantitative variation in the levels of each of the transcripts. The only significant associations were observed for DEXI transcript and a number of SNPs spanning the CLEC16A gene, many of which had alleles that were modestly correlated (r2 > 0.6). The most statistically significant association was observed at rs7403919 (P = 1.24 × 10−4). A second independent biological replicate focusing on DEXI expression yielded similar results (rs7403919, P = 5.45 × 10−5). A full breakdown of the correlations with rs7403919 and type 1 diabetes associated SNPs based on the HapMap samples can be found in Supplementary Table 3. The rest of the genes tested did not show significant associations. The eQTL plots of all the transcripts tested can be found in Supplementary Fig. 2.
We specifically examined the expression levels of DEXI in relation to three SNPs: rs34306440 (the SNP most strongly associated with type 1 diabetes in the region), rs7403919 (the SNP most strongly associated with variation in DEXI expression in the region), and rs12708716 (most often cited in type 1 diabetes literature; Fig. 2). The risk allele for all three SNPs corresponds to the major allele. Homozygotes for the risk alleles had lower expression of DEXI, whereas the heterozygotes and homozygotes for the alternative allele displayed higher levels of expression. Figure 3 shows the OR for type 1 diabetes versus the linear regression coefficient (β) from the quantile-normalized DEXI expression level for the minor alleles of all 809 SNPs for which these data were available. Disease risk was negatively correlated with DEXI transcript levels (Pearson correlation R = −0.66).
Figure 2.

Expression levels of DEXI grouped by genotypes of SNPs rs7403919 (A), rs12708716 (B), and rs34306440 (C). Alleles associated with type 1 diabetes are in boldface. The centerlines represent the median value, box area represents upper and lower quartiles, the whiskers represent 1 SD from the mean.
Figure 3.

Relationship of risk for type 1 diabetes and DEXI expression. Results are plotted for the minor allele at each of the 809 SNPs in the 16p13.13 region. The ▲ indicates SNPs at which alleles are significantly associated with type 1 diabetes.
In the course of designing primers for RT-PCR analysis of CIITA, several novel alternatively spliced transcripts were observed in publicly available RNA-Seq data (E-MTAB-513). The 3′ untranslated region region of these CIITA transcripts overlapped with the reference transcript from DEXI in the antisense direction. This observation raised the possibility that these genes could exert some regulatory role on each other via antisense inhibition. The transcripts were validated and characterized by amplification and Sanger sequencing and were analyzed by eQTL (Supplementary Fig. 1). There was no significant evidence of association between alleles at any SNP in the 16p13.13 region and variation in the levels of these novel CIITA transcripts in the B-cell lines examined (Supplementary Fig. 2).
Discussion
Chromosomal region 16p13.13 is one of a number of chromosomal regions where there is significant, replicated evidence of allelic association with type 1 diabetes (3–6) as well as with other autoimmune disorders (7–16). Repeated findings of association for multiple autoimmune disorders in a single chromosomal region such as 16p13.13 increases both confidence in the initial findings and the importance of the locus in understanding the underlying mechanism whereby autoimmunity develops. Given the evidence of genetic association at 16p13.13 with multiple diseases that are autoimmune in nature but differ in the target organ, it seems most likely that the effects of this locus would manifest in immune-related cell types, as opposed, for example, to β-cells. However, functional studies that might elucidate the mechanism depend on the ability to specify the relevant gene in the region, if not the specific causative genetic variants. In the current study, we addressed this need for the 16p13.13 region by resequencing the region in a large collection of type 1 diabetic case subjects and unaffected control subjects. The objective was to identify common variants, genotype these variants in a collection of multiplex families with type 1 diabetes, and then separately test for association of these SNPs with disease state and with transcript levels for each of the genes in the region.
There were 46 novel variants from our resequencing that were included among the 939 SNPs that we genotyped in 3,070 multiplex families with type 1 diabetes. Forty-eight of the 939 SNPs displayed a statistically significant association with type 1 diabetes. Alleles at these 48 SNPs were not all highly correlated (pairwise r2 range from 0.4 to 0.9), nor were the SNPs physically clustered. All of these 48 SNPs were located in introns that spanned much of the CLEC16A gene (introns 8–22). Although the most significant disease association result was obtained at a SNP (rs34306440) initially identified from our resequencing effort, the rapid development of independent SNP resources meant that this SNP was also included in the ImmunoChip data set that subsequently became available.
The major allele at rs34306440 was associated with risk for type 1 diabetes. This is not an uncommon finding; the major allele is the risk allele for the most significantly disease-associated SNPs at approximately half of the reported risk loci for type 1 diabetes (3). Given the limited magnitude of the individual effects that each of these loci have on disease risk and the complex genetic nature of most autoimmune diseases, there might not be a significant selection pressure to limit their frequency in the population. There are also precedents, such as the DQ0602 allele at the HLA-DQB1 locus, where the same allele confers risk for one disease (MS) (50) and protection for another (type 1 diabetes) (51).
The broad physical distribution of disease-associated SNPs and their modest correlation raise the possibility that they act through multiple mechanisms or affect multiple genes. Given their intronic locations and prior reports of cis-oriented transcriptional effects in the region, we tested for association between our panel of SNPs and transcripts levels in transformed B cell lines for each of the documented alternatively spliced forms of the four genes, CIITA, DEXI, CLEC16A, and SOCS1. Only DEXI presented statistically significant results. Similar to the disease association results, SNPs associated with DEXI transcript levels appear to be distributed across much of the CLEC16A gene. An examination of the relationship between the OR for the minor allele at each SNP in the region and the corresponding linear regression coefficient for DEXI expression for the same allele indicated that DEXI expression levels were negatively correlated with disease risk (R = −0.66).
Our results suggest that genetic variation over a broad region spanning much of the CLEC16A gene influences the expression of a neighboring gene, DEXI, and that reduced DEXI expression is correlated with an increased risk for type 1 diabetes. This model is consistent with a prior report that sequences in intron 19 of CLEC16A physically interact with the promoter region of DEXI (27), providing a plausible mechanism for how genetic variation in CLEC16A can act at a distance to regulate DEXI expression. Our results suggest that the region of interaction may be large or that distal sequences may act indirectly to affect the stability or structure of the DNA loops formed. Consistent with this, examination of ENCODE data (52) reveals a number of peaks of H3K27 histone acetylation in the same region as the SNPs associated with disease risk and variation in DEXI expression.
Our study only examined expression in a single cell type, B lymphoblastoid cell lines, and only in cell lines derived from unaffected individuals. The possibility cannot be excluded that there may be different, or additional, effects on gene expression associated with the SNP alleles studied here in distinct tissues or specific to affected individuals. However, lower DEXI and/or SOCS1 expression has also been reported in thymic samples for risk alleles associated with MS for SNPs rs12708716, rs7206912, and rs6498169 located in CLEC16A (53). Two of these three SNPs (rs12708716 and rs6498169) were also included in our panel and showed a similar relationship to DEXI expression. Finally, the Genotype-Tissue Expression Portal (GTEx) reports a significant eQTL for rs12708716 in a third tissue type, thyroid; again, with the same allelic association with expression.
Our results and those from prior studies focus attention on DEXI as a candidate gene affecting the risk for type 1 diabetes in the 16p13.13 region. Little is known about the function of DEXI and its predicted 95 amino acid protein product. The protein is well conserved, with orthologous genes readily detectable in multiple species. There is 95% identity between the human and mouse orthologs and 84.5% between human and zebrafish.
There are no identifiable domains in DEXI that might suggest a possible function for the protein. However, Miyaki et al. (54) showed that small interfering RNA–mediated knockdown of DEXI reduced the incidence of apoptosis in cells treated with the chemotherapy agent camptothecin. A role in a basic cellular function, such as apoptosis, would be consistent with the involvement of DEXI in multiple autoimmune diseases with different target tissues.
Supplementary Material
Article Information
Acknowledgments. The authors acknowledge Dr. Stephen Rich from the Center for Public Health Genomics and the Department of Public Health Sciences, University of Virginia, for providing the T1DGC ImmunoChip data. The authors acknowledge the staff of the University of Virginia Center for Public Health Genomics Genome Sciences Laboratory—Emily Farber, Daniel Gallo, Jamie Artale, and Benjamin Artale—for training and maintenance of the genotyping equipment used in the current study and for generation of the ImmunoChip data. The authors thank Dr. Sharon Teraoka, Department of Biochemistry and Molecular Genetics at the University of Virginia, for helpful comments on the manuscript. This study makes use of data generated by the ENCODE Consortium and the ENCODE production laboratories.
Funding. This work was supported by grants from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (R01-DK-46635 and DP3-DK-085678) and the JDRF (33-2008-409) to P.C. This research used resources provided by the Type 1 Diabetes Genetics Consortium, a collaborative clinical study sponsored by the NIDDK, National Institute of Allergy and Infectious Diseases (NIAID), National Human Genome Research Institute (NHGRI), National Institute of Child Health and Human Development (NICHD), and JDRF and supported by U01-DK-062418. This work cites data obtained from a query of the GTEx Portal on 24 March 2014. The GTEx Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health (commonfund.nih.gov/GTEx). Additional funds were provided by the National Cancer Institute (NCI), NHGRI, National Heart, Lung, and Blood Institute, National Institute on Drug Abuse, National Institute of Mental Health, and National Institute of Neurological Disorders and Stroke. Donors were enrolled at Biospecimen Source Sites funded by NCI/Science Applications International Corporation-Frederick (SAIC-F), which subcontracts to the National Disease Research Interchange (10XS170), Roswell Park Cancer Institute (10XS171), and Science Care (X10S172). The Laboratory, Data Analysis, and Coordinating Center (LDACC) was funded through a contract (HHSN268201000029C) to the Broad Institute. Biorepository operations were funded through an SAIC-F subcontract to Van Andel Institute (10ST1035). Additional data repository and project management were provided by SAIC-F (HHSN261200800001E). The Brain Bank was supported by a supplement to University of Miami grant DA006227. Statistical methods development grants were made to the University of Geneva (MH090941), the University of Chicago (MH090951 and MH090937), the University of North Carolina–Chapel Hill (MH090936), and to Harvard University (MH090948). A.P. and J.M. received grant money from the National Institutes of Health, and P.C. and S.O.-G. received grant money from the JDRF and NIDDK.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. M.J.T. designed and performed the experiments, analyzed data, produced Tables and Figures and wrote the manuscript. A.P. analyzed sequencing data, analyzed expression data (eQTL), produced genotype expression plots, and reviewed and edited the manuscript. R.P. assisted genotyping the custom panel based on sequencing data and reviewed and edited the manuscript. M.M. helped design and perform long-range PCR and RT-PCR experiments and reviewed and edited the manuscript. K.L.K. helped design and perform initial genotyping on the 123 custom tagging panel and reviewed and edited the manuscript. X.H. coanalyzed sequencing results and reviewed the manuscript. J.M. coanalyzed sequencing results, coanalyzed the raw ImmunoChip data, and reviewed and critiqued the manuscript. W.-M.C. coanalyzed the raw ImmunoChip data and reviewed and critiqued the manuscript. P.C. helped design and supervise the project and reviewed and edited the manuscript. S.O.-G. helped design and supervise the project, performed experiments, and reviewed and edited the manuscript. M.J.T. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db13-1785/-/DC1.
References
- 1.Concannon P, Rich SS, Nepom GT. Genetics of type 1A diabetes. N Engl J Med 2009;360:1646–1654 [DOI] [PubMed] [Google Scholar]
- 2.Rich SS, Concannon P. Challenges and strategies for investigating the genetic complexity of common human diseases. Diabetes 2002;51(Suppl. 3):S288–S294 [DOI] [PubMed] [Google Scholar]
- 3.Barrett JC, Clayton DG, Concannon P, et al. Type 1 Diabetes Genetics Consortium . Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet 2009;41:703–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wellcome Trust Case Control Consortium . Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 2007;447:661–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hakonarson H, Grant SF, Bradfield JP, et al. A genome-wide association study identifies KIAA0350 as a type 1 diabetes gene. Nature 2007;448:591–594 [DOI] [PubMed] [Google Scholar]
- 6.Todd JA, Walker NM, Cooper JD, et al. Genetics of Type 1 Diabetes in Finland. Wellcome Trust Case Control Consortium . Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet 2007;39:857–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hafler DA, Compston A, Sawcer S, et al. International Multiple Sclerosis Genetics Consortium . Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med 2007;357:851–862 [DOI] [PubMed] [Google Scholar]
- 8.De Jager PL, Jia X, Wang J, et al. International MS Genetics Consortium . Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat Genet 2009;41:776–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dubois PC, Trynka G, Franke L, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet 2010;42:295–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gateva V, Sandling JK, Hom G, et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet 2009;41:1228–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mells GF, Floyd JA, Morley KI, et al. UK PBC Consortium. Wellcome Trust Case Control Consortium 3 . Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat Genet 2011;43:329–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nischwitz S, Cepok S, Kroner A, et al. More CLEC16A gene variants associated with multiple sclerosis. Acta Neurol Scand 2011;123:400–406 [DOI] [PubMed] [Google Scholar]
- 13.Zhang Z, Cheng Y, Zhou X, et al. Polymorphisms at 16p13 are associated with systemic lupus erythematosus in the Chinese population. J Med Genet 2011;48:69–72 [DOI] [PubMed] [Google Scholar]
- 14.Márquez A, Varadé J, Robledo G, et al. Specific association of a CLEC16A/KIAA0350 polymorphism with NOD2/CARD15(-) Crohn’s disease patients. Eur J Hum Genet 2009;17:1304–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McGovern DP, Gardet A, Törkvist L, et al. NIDDK IBD Genetics Consortium . Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat Genet 2010;42:332–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Skinningsrud B, Husebye ES, Pearce SH, et al. Polymorphisms in CLEC16A and CIITA at 16p13 are associated with primary adrenal insufficiency. J Clin Endocrinol Metab 2008;93:3310–3317 [DOI] [PubMed] [Google Scholar]
- 17.Sigrist CJ, Cerutti L, de Castro E, et al. PROSITE, a protein domain database for functional characterization and annotation. Nucleic Acids Res 2010;38:D161–D166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim S, Wairkar YP, Daniels RW, DiAntonio A. The novel endosomal membrane protein Ema interacts with the class C Vps-HOPS complex to promote endosomal maturation. J Cell Biol 2010;188:717–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.LeibundGut-Landmann S, Waldburger JM, Krawczyk M, et al. Mini-review: specificity and expression of CIITA, the master regulator of MHC class II genes. Eur J Immunol 2004;34:1513–1525 [DOI] [PubMed] [Google Scholar]
- 20.Palmer DC, Restifo NP. Suppressors of cytokine signaling (SOCS) in T cell differentiation, maturation, and function. Trends Immunol 2009;30:592–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edgar AJ, Birks EJ, Yacoub MH, Polak JM. Cloning of dexamethasone-induced transcript: a novel glucocorticoid-induced gene that is upregulated in emphysema. Am J Respir Cell Mol Biol 2001;25:119–124 [DOI] [PubMed] [Google Scholar]
- 22.Awata T, Kawasaki E, Tanaka S, et al. Japanese Study Group on Type 1 Diabetes Genetics . Association of type 1 diabetes with two Loci on 12q13 and 16p13 and the influence coexisting thyroid autoimmunity in Japanese. J Clin Endocrinol Metab 2009;94:231–235 [DOI] [PubMed] [Google Scholar]
- 23.Cooper JD, Smyth DJ, Smiles AM, et al. Meta-analysis of genome-wide association study data identifies additional type 1 diabetes risk loci. Nat Genet 2008;40:1399–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cooper JD, Walker NM, Smyth DJ, Downes K, Healy BC, Todd JA, Type I Diabetes Genetics Consortium . Follow-up of 1715 SNPs from the Wellcome Trust Case Control Consortium genome-wide association study in type I diabetes families. Genes Immun 2009;10(Suppl. 1):S85–S94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reddy MV, Wang H, Liu S, et al. Association between type 1 diabetes and GWAS SNPs in the southeast US Caucasian population. Genes Immun 2011;12:208–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu X, Zhu X, Wang X, et al. Intron polymorphism in the KIAA0350 gene is reproducibly associated with susceptibility to type 1 diabetes (T1D) in the Han Chinese population. Clin Endocrinol (Oxf) 2009;71:46–49 [DOI] [PubMed] [Google Scholar]
- 27.Davison LJ, Wallace C, Cooper JD, et al. Cardiogenics Consortium . Long-range DNA looping and gene expression analyses identify DEXI as an autoimmune disease candidate gene. Hum Mol Genet 2012;21:322–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nica AC, Montgomery SB, Dimas AS, et al. Candidate causal regulatory effects by integration of expression QTLs with complex trait genetic associations. PLoS Genet 2010;6:e1000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeller T, Wild P, Szymczak S, et al. Genetics and beyond—the transcriptome of human monocytes and disease susceptibility. PLoS ONE 2010;5:e10693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ni Z, Abou El Hassan M, Xu Z, Yu T, Bremner R. The chromatin-remodeling enzyme BRG1 coordinates CIITA induction through many interdependent distal enhancers. Nat Immunol 2008;9:785–793 [DOI] [PubMed] [Google Scholar]
- 31.Reith W, Boss JM. New dimensions of CIITA. Nat Immunol 2008;9:713–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rich SS, Akolkar B, Concannon P, et al. Overview of the Type I Diabetes Genetics Consortium . Genes Immun 2009;(Suppl. 1):S1–S4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lernmark A, Ducat L, Eisenbarth G, et al. Family cell lines available for research. Am J Hum Genet 1990;47:1028–1030 [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, Handsaker B, Wysoker A, et al. 1000 Genome Project Data Processing Subgroup . The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009;25:2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kent WJ, Sugnet CW, Furey TS, et al. The human genome browser at UCSC. Genome Res 2002;12:996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lander ES, Linton LM, Birren B, et al. International Human Genome Sequencing Consortium . Initial sequencing and analysis of the human genome. Nature 2001;409:860–921 [DOI] [PubMed] [Google Scholar]
- 38.International HapMap Consortium . A haplotype map of the human genome. Nature 2005;437:1299–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005;21:263–265 [DOI] [PubMed] [Google Scholar]
- 40.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O’Connell JR, Weeks DE. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet 1998;63:259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cortes A, Brown MA. Promise and pitfalls of the Immunochip. Arthritis Res Ther 2011;13:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol 2010;34:816–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen WM. Robust relationship inference in genome-wide association studies. Bioinformatics 2010;26:2867–2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spielman RS, McGinnis RE, Ewens WJ. Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am J Hum Genet 1993;52:506–516 [PMC free article] [PubMed] [Google Scholar]
- 46.Pruim RJ, Welch RP, Sanna S, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 2010;26:2336–2337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dudbridge F. Likelihood-based association analysis for nuclear families and unrelated subjects with missing genotype data. Hum Hered 2008;66:87–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koressaar T, Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007;23:1289–1291 [DOI] [PubMed] [Google Scholar]
- 49.McIntyre LM, Martin ER, Simonsen KL, Kaplan NL. Circumventing multiple testing: a multilocus Monte Carlo approach to testing for association. Genet Epidemiol 2000;19:18–29 [DOI] [PubMed] [Google Scholar]
- 50.Fernández O, Fernández V, Alonso A, et al. DQB1*0602 allele shows a strong association with multiple sclerosis in patients in Malaga, Spain. J Neurol 2004;251:440–444 [DOI] [PubMed] [Google Scholar]
- 51.Pugliese A, Gianani R, Moromisato R, et al. HLA-DQB1*0602 is associated with dominant protection from diabetes even among islet cell antibody-positive first-degree relatives of patients with IDDM. Diabetes 1995;44:608–613 [DOI] [PubMed] [Google Scholar]
- 52.ENCODE Project Consortium; An integrated encyclopedia of DNA elements in the human genome Nature 2012;489:57–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leikfoss IS, Mero IL, Dahle MK, et al. Multiple sclerosis-associated single-nucleotide polymorphisms in CLEC16A correlate with reduced SOCS1 and DEXI expression in the thymus. Genes Immun 2013;14:62–66 [DOI] [PubMed] [Google Scholar]
- 54.Miyaki Y, Suzuki K, Koizumi K, et al. Identification of a potent epigenetic biomarker for resistance to camptothecin and poor outcome to irinotecan-based chemotherapy in colon cancer. Int J Oncol 2012;40:217–226 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.