

Abstract
The mechanisms by which macrophages mediate the enhanced inflammation associated with diabetes complications are not completely understood. We used RNA sequencing to profile the transcriptome of bone marrow macrophages isolated from diabetic db/db mice and identified 1,648 differentially expressed genes compared with control db/+ mice. Data analyses revealed that diabetes promoted a proinflammatory, profibrotic, and dysfunctional alternatively activated macrophage phenotype possibly via transcription factors involved in macrophage function. Notably, diabetes altered levels of several long noncoding RNAs (lncRNAs). Because the role of lncRNAs in diabetes complications is unknown, we further characterized the function of lncRNA E330013P06, which was upregulated in macrophages from db/db and diet-induced insulin-resistant type 2 diabetic (T2D) mice, but not from type 1 diabetic mice. It was also upregulated in monocytes from T2D patients. E330013P06 was also increased along with inflammatory genes in mouse macrophages treated with high glucose and palmitic acid. E330013P06 overexpression in macrophages induced inflammatory genes, enhanced responses to inflammatory signals, and increased foam cell formation. In contrast, small interfering RNA–mediated E330013P06 gene silencing inhibited inflammatory genes induced by the diabetic stimuli. These results define the diabetic macrophage transcriptome and novel functional roles for lncRNAs in macrophages that could lead to lncRNA-based therapies for inflammatory diabetes complications.
Introduction
Macrophages play important roles in the regulation of physiological and pathological processes, including metabolism, inflammation, pathogen response, and tissue damage and repair. Differentiation and polarization of macrophages into classically activated M1 phenotype or alternatively activated M2 phenotype are essential for the plasticity needed to adapt cellular functions in dynamic microenvironments of target tissues and disease states (1,2). In general, M1 macrophages are involved in host defense and inflammation, and M2 macrophages are involved in tissue repair. They exhibit distinct gene profiles that are regulated by specific signaling cascades, transcription factors (TFs), and epigenetic factors (3,4). Recent studies have also demonstrated key roles for microRNAs (miRNAs), such as miR-155 and miR-146a, in regulating macrophage phenotype (5). Dysregulation of macrophage polarization and function plays a central role in the development of several diseases, including diabetes and atherosclerosis (6,7). Increased infiltration of macrophages into various tissues, including adipose, kidney, heart, and vascular tissues, is observed in these disorders (8–11). Diabetes and metabolic disorders can regulate macrophage polarization and alter the expression of genes involved in macrophage functions such as inflammation, phagocytosis, cholesterol transport, and wound healing (2,11–17). Production of proinflammatory mediators by macrophage infiltration into target tissues activates other cell types and further enhances inflammation (6,9,18,19), and this is associated with majority of the complications of diabetes. However, a systematic analysis of how diabetes regulates global coding and noncoding gene expression programs that modulate macrophage phenotype linked to diabetes complications is not available.
Recent studies using high-throughput RNA sequencing (RNA-seq) and profiling of key histone modifications have identified thousands of long noncoding RNAs (lncRNAs), once considered to be transcriptional noise, and also demonstrated that they play key roles in epigenetic mechanisms and gene regulation (20,21). lncRNAs, typically noncoding RNAs (ncRNAs) >200 nucleotides long, can be expressed from intergenic regions, antisense strands, or introns of protein coding genes, or they can be derived by alternate splicing. Major features of lncRNAs include their low expression and ability to form secondary structures that can act as DNA, RNA, and protein binding domains. Several different mechanisms are used by lncRNAs to regulate gene expression at both transcriptional and posttranscriptional levels. Thus they can act as signals to recruit or as decoys to titrate out TFs, as enhancers to guide chromatin modifying enzymes to their genomic target sites, and as competitors that inhibit miRNA binding at the 3′-untranslated regions of target genes (20). Furthermore, some lncRNAs can serve as host genes for miRNAs (short ncRNAs) (22) and thereby downregulate multiple targets of these miRNAs involved in diverse biological processes. Current estimates predict there are thousands of lncRNAs in the human genome, but to date, only a few have been well characterized or their functions identified. Some lncRNAs have recently been linked to various diseases, suggesting their potential to be novel therapeutic targets (23). One report identified lncRNAs in human pancreatic islets, including some affected by type 2 diabetes (T2D) (24). However, their role in macrophages related to diabetic inflammatory complications is not yet known.
We wished to explore the roles of differentially expressed coding transcripts as well as lncRNAs in the molecular mechanisms involved in impaired macrophage function in diabetes. Transcriptome profiling of bone marrow–derived macrophages (BMM) from diabetic db/db mice using RNA-seq followed by bioinformatics analyses revealed that diabetes induced a proinflammatory, dysfunctional polarization, and profibrotic phenotype, and also altered expression of several lncRNAs compared with nondiabetic genetic control db/+. Further examination of one of the differentially expressed lncRNAs, E330013P06 (hereafter referred to as E33) showed that it is significantly upregulated in vivo and in vitro in macrophages under T2D conditions. Furthermore, we found that E33 regulates proinflammatory gene expression and foam cell formation in macrophages, establishing a novel new functional role for lncRNA in processes linked to diabetic inflammatory vascular complications.
Research Design and Methods
Animal Experiments
Experiments were performed with male mice (The Jackson Laboratory, Bar Harbor, ME) using protocols approved by our Institutional Animal Care and Use Committee in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Macrophages were prepared from 10–12-week-old T2D db/db mice (BKS.Cg-m+/+leprdb/J) and age-matched control littermate db/+ mice (16), as well as from type 1 diabetic (T1D) and T2D C57BL/6J mouse models and respective controls. T1D was induced by injecting C57BL/6J mice with streptozotocin (SZ) (50 mg/kg) for 5 consecutive days (25). Macrophages were isolated 4 weeks postdiabetes. In diet-induced T2D models, C57BL/6J mice (8 weeks old) were fed with high-fat diet (60% kcal, Research Diets Inc., D12492i) for 4 weeks and then injected with a single dose of SZ (100 mg/kg) to make them T2D (HFSZ) or with vehicle citrate buffer (HF). Mice maintained on chow diet were also injected with vehicle (SF) as a control group. Macrophages were prepared 4 or more weeks after SZ injections when HFSZ mice exhibited significantly increased blood glucose and insulin resistance. Insulin resistance was determined by glucose tolerance tests in mice fasted for ∼16 h. Mice were weighed, their baseline blood glucose was determined, and they were injected intraperitoneally with glucose solution (2 g/kg). Blood glucose levels were determined at different time intervals up to 2 h using AlphaTRAK glucometer (Abbott Laboratories, Abbott Park, IL).
Isolation, Culture, and Treatment of Mouse Macrophages
BMM were prepared using published protocols with some modifications (26). Briefly, bone marrow was expelled from femurs and tibia, and after lysis of erythrocytes, they were plated in complete medium (CM) consisting of DMEM containing 5.5 mmol/L glucose, 10% FBS, 2 mmol/L glutamine, penicillin/streptomycin antibiotics, 50 μmol/L β-mercaptoethanol, and 10 mmol/L HEPES, pH 7.4, supplemented with either 10 ng/mL of macrophage colony–stimulating factor-1 (M-CSF; R&D Systems, Minneapolis MN) or 20 ng/mL of granulocyte macrophage-stimulating factor (GM-CSF; PeproTech, Rocky Hill, NJ). Cells were allowed to differentiate for 7–8 days before further use. Thioglycolate-elicited peritoneal macrophages (PMs) were isolated as described (27). The mouse macrophage cell line RAW264.7 (from American Type Culture Collection) was used in some experiments and maintained in CM. Where indicated, macrophages were treated with media containing normal (5.5 mmol/L) glucose (NG), high (25 mmol/L) glucose (HG), 200 μmol/L palmitic acid (PA), HG+PA (HP), TNF-α (10 ng/mL), or lipopolysaccharide (LPS; 100 ng/mL). PA stock solutions (10 mmol/L) were prepared in BSA (28), diluted to the indicated concentration in the medium, incubated for 30 min at 37°C, and filtered with 0.2 μm filter before adding to the cells.
Isolation of Human Monocytes
T2D patients and healthy controls (age and sex matched) were recruited at the Baylor College of Medicine (Table 1). Following informed consent, fasting blood (15 mL) was obtained from these subjects, and monocytes isolated following Ficoll density gradient and negative magnetic separation (Miltenyi Biotec, San Diego, CA) as described (29). More than 88% of cells were CD14 positive, and viability was >92%.
Table 1.
Characteristics of healthy controls and T2D patients
| Age (years) | BMI (kg/m2) | BG (mg/dL) | HbA1c (%) | HbA1c (mmol/mol) | |
|---|---|---|---|---|---|
| Control | 37.5 ± 3.8 | 23.9 ± 0.47 | 89.75 ± 3.3 | 5.2 ± 0.07 | 33.3 ± 0.9 |
| T2D | 43.25 ± 6.3 | 26.28 ± 1.1 | 133.8 ± 12.1* | 8.2 ± 0.4* | 66.5 ± 4.3* |
For the isolation of human monocytes, healthy controls (age and sex matched) and T2D patients were recruited at the Baylor College of Medicine. T2D was diagnosed if fasting blood glucose was >126 mg/dL on two occasions or HbA1c >6.5% (48 mmol/mol), and subjects were recruited if they were newly diagnosed or on metformin therapy. All subjects had normal complete blood count; no other chronic diseases; normal kidney, liver, and thyroid function; and no inflammatory disorders. Data represent mean ± SEM.
*P < 0.05 versus control (n = 4).
RNA Isolation and Quantitative Reverse Transcriptase PCR
RNA was extracted using miRNeasy columns (Qiagen, Valencia, CA) from mouse macrophages and mirVana kit (Ambion, Austin TX) from human monocytes, and cDNA synthesis was performed with 0.5–1.0 μg RNA using QuantiTect RT kit (Qiagen). Expression of genes, including lncRNAs E33 and MIR143HG (human equivalent of E33), was analyzed by quantitative real-time PCR using SYBR green reagents (Life Technologies, Carlsbad, CA) or TaqMan assays (Integrated DNA Technologies, Coralville, IA) on an ABI 7500 instrument using indicated gene primers (Supplementary Table 2). miRNA expression was determined by quantitative reverse transcriptase PCR (RT-QPCR) using miScript (Qiagen) or qScript (Quanta Biosciences, Gaithersburg, MD) kits. RT-QPCR data were analyzed by the 2^-ΔΔCt method, normalized against internal controls (Actb or Ppia for mRNA and lncRNA, and U6 or 18S RNA for miRNA). Results were expressed as fold over control samples.
RNA-seq and Data Analysis
Whole transcriptome analysis of RNA from BMMC and BMGM was performed using total RNA depleted of rRNA (RiboMinus kit, Life Technologies). Paired-end libraries were prepared, size selected, gel purified, and sequenced in our Integrative Genomics Core using an Illumina HiSeq 2000 system following manufacturer’s protocols (Illumina Inc., San Diego, CA). Reads were mapped to the mouse genome assembly mm9 using TopHat software (30). Each RefSeq gene’s expression was summarized and normalized by the trimmed mean of M-value method using edgeR bioconductor R package. The total reads for each gene were then scaled by exon length to obtain average coverage. Differentially expressed genes (DEGs) were identified with coverage (≥1 in at least one sample) and fold change (≥1.5). Novel transcripts were identified using published approaches (21,31–33), including ours (22). Specifically, after de novo assembly of all the transcripts using Cufflinks (34), transcripts that mapped to RefSeq genes of mouse and seven other organisms (human, chimp, rat, rabbit, orangutan, rhesus, marmoset) were first filtered out. Filtered transcripts were further assessed for potential open reading frames by using PhyloCSF and searching PfamA/B databases. Multiexonic transcripts with a score of >100 or that encoded PfamA/B protein domains were classified as novel protein-encoding transcripts. Transcripts of at least 200 bp in length containing at least two exons, having a PhyloCSF score of <100 and lacking PfamA/B domains, were defined as novel lncRNAs. Biological functions and network analysis of DEGs were performed using DAVID gene ontology and Ingenuity Pathway Analysis (IPA). TF motif analysis and enrichment were performed using TF affinity prediction (TRAP) method (35) and gene set enrichment analysis, respectively.
E330013P06 Cloning and Lentiviral Transduction of Macrophages
E330013P06 cDNA was cloned downstream of the CMV promoter in the lentiviral expression vector pEZ-LV105 (GeneCopea Inc., Rockville, MD) and verified by DNA sequencing. Plasmid pEZ-LV105 expressing EGFP (LVGFP) was used as vector control. HEK293T cells were cotransfected with lentiviral vector plasmids and three packaging plasmids using Fugene (Promega, Madison, WI). Cell culture supernatants containing lentiviruses were collected 48–96 h posttransfection and concentrated using Lenti-X Concentrator (Clontech, Mountain View, CA). RAW264.7 macrophages were transduced with the indicated lentiviral vectors in the presence of Polybrene (8 μg/mL), and cells resistant to puromycin (2 μg/mL) were selected for further analysis.
Transfection of Macrophages
Macrophages were detached by incubating with Accutase (eBioscience, San Diego, CA) for 5–10 min at 37°C, washed with PBS, and resuspended at a concentration of 2.5–3.0 × 107/mL in Nucleofection reagent (Lonza, Walkersville, MD). Then, 100 μL of cell suspension was mixed with plasmid DNA or small interfering RNA (siRNA) oligonucleotides (up to 6 μL) and transfected using Nucleofector program Y-001. Transfected cells were plated in dishes for RNA and foam cell formation assays. Specific siRNAs targeting mouse E33 were designed using GPboost algorithm (36).
Foam Cell Formation Assays
Macrophages were incubated in eight-well chamber slides with acetylated LDL fluorescently labeled with 20 μg/mL 1,1'-dioctadecyl–3,3,3′,3′-tetramethyl-indocarbo-cyanine perchlorate (DiI-Ac-LDL, Biomedical Technologies, Stoughton, MA) for 4 h at 37°C. Cells were washed twice, fixed with 4% paraformaldehyde, washed, stained with Hoechst dye and mounted on slides using VECTASHIELD. Fluorescent images of the cells were collected with Pixera 600 digital camera attached to an Olympus BX51 microscope using InStudio software (Pixera Corporation).
Statistical Analysis
Statistical significance was calculated using unpaired Student t tests to compare two groups and ANOVA to compare multiple groups followed by appropriate multiple comparison tests selected by PRISM 6.0 software (GraphPad, San Diego, CA). P values ≤ 0.05 were considered statistically significant.
Data Deposition
The RNA-seq data have been deposited in the Gene Expression Omnibus (GEO) database under accession code GSE54154.
Results
BMM From Diabetic db/db Mice Have a Dysfunctional Phenotype
To examine the effects of diabetes on macrophage polarization, we differentiated BMM from T2D db/db and nondiabetic control db/+ mice in vitro using GM-CSF (BMGM) or M-CSF (BMMC) to obtain M1 and M2 macrophages, respectively, and analyzed the expression of key macrophage markers by RT-QPCR (Supplementary Fig. 1A and B). We then observed that in BMMC from db/db mice (db/dbBMMC; M2), the expression of Il10, Tgfb1, and Arg1 (M2-associated genes) was inhibited, but expression of proinflammatory prostaglandin-endoperoxide synthase 2 (Ptgs2) gene was increased compared with db/+BMMC, indicating that diabetes induced a dysfunctional M2 phenotype (Fig. 1A). On the other hand, db/dbBMGM (M1) exhibited increased expression of the proinflammatory genes Ccl2, Nos2, Ptgs2, and Tnf compared with db/+BMGM (Fig. 1B), suggesting enhanced proinflammatory M1 phenotype. Furthermore, db/dbBMMC also exhibited enhanced expression of inflammatory genes relative to db/+BMMC in response to TNF-α (10 ng/mL) stimulation (Fig. 1C). We also analyzed proinflammatory genes in thioglycolate-elicited PM derived from these mice. The expression of proinflammatory genes (Tnf, Il6, Nos2, Ccl2, and Ptgs2) was increased, and the anti-inflammatory gene (Il10) was decreased in PM from db/db mice compared with db/+ (Fig. 1D), providing additional in vivo relevance. Overall, our data indicate that diabetes promoted a dysfunctional M2 phenotype and enhanced M1 inflammatory response in macrophages.
Figure 1.

Diabetes induces dysfunctional macrophage polarization. A and B: RT-QPCR analysis of gene expression in BMM derived from diabetic db/db mice and control db/+ mice, which were differentiated in vitro with either M-CSF (BMMC; panel A) or GM-CSF (BMGM; panel B). Bone marrow was isolated from 10–12-week-old db/db and db/+ mice. Blood glucose levels were 479 ± 28 mg/dL in db/db mice versus 162 ± 8 mg/dL in db/+ mice. C: TNF-α–induced gene expression in db/+ and db/dbBMMC. BMMC were serum depleted for 4 h and stimulated with TNF-α (10 ng/mL) for 1 h. D: Gene expression analysis of thioglycolate-elicited PMs from db/+ (PMdb/+) and db/db (PMdb/db) mice. Gene expression was analyzed by RT-QPCR and results expressed as fold over db/+ cells. Data are represented as mean ± SEM. *P < 0.05, n = 3–4.
Diabetes Impacts the Transcriptome of BMM
Prior studies using gene arrays have revealed differences between the transcriptome of BMMC and BMGM of normal mice (37), but the impact of diabetes on the macrophage transcriptome genome-wide and in particular on lncRNAs is unknown. We therefore next used RNA-seq to analyze differences in global gene expression patterns in BMM from db/db and db/+ mice. RNA-seq data (Supplementary Table 1) was analyzed using an in-house data analysis pipeline and publicly available databases (Fig. 2A). Results showed that the BMGM transcriptome in both db/+ and db/db mice was enriched with proinflammatory genes (Supplementary Fig. 2A), whereas genes known to be associated with alternatively activated macrophages were elevated in BMMC (Supplementary Fig. 2B), further confirming the M1 and M2 phenotype of BMGM and BMMC, respectively (37). RNA-seq data also revealed that levels of proinflammatory markers (Itgax and Ly6c) were increased in BMGM (Supplementary Fig. 2A), whereas macrophage-specific markers (Emr1 and Mrc1) were elevated in BMMC relative to BMGM (Supplementary Fig. 2B). Furthermore, Csf1 and Csf1r genes were elevated, and GM-CSF receptors (Csf2ra, Csf2rb, and Csf2rb2) were reduced in BMMC compared with BMGM, indicating inhibition of GM-CSF signaling in BMMC as anticipated (Supplementary Fig. 2C). Flow cytometry showed significant inhibition of F4/80 in BMGM (Supplementary Fig. 3A), confirming RNA-seq results that GM-CSF inhibited macrophage phenotype.
Figure 2.

RNA-seq analysis of gene expression in macrophages from db/+ and db/db mice. A: Data analysis pipeline. B: Scatter plot of differentially expressed RefSeq genes, including small ncRNAs (<200 bp). C: Pie chart showing differentially expressed genes (DEGs) in db/dbBMMC relative to db/+BMMC (excluding small ncRNAs [<200 bp]). D: Bar graph showing fibrotic genes increased in db/dbBMMC compared with db/+ . Data represents log2 ratio (db/dbBMMC-db/+BMMC). E: Top 10 biological processes enriched among DEGs in db/dbBMMC by DAVID analysis. Enrichment score of an annotation cluster refers to the geometric mean of EASE scores (modified Fisher exact P value in form of –log10) of all the biological processes in the cluster. F: Top 10 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways enriched among DEGs (DAVID analysis). G: Top 10 involved diseases enriched in DEGs of db/dbBMMC by IPA. In panels F and G, y-axis represents P values (–log10) from Fisher exact tests with Benjamini-Hochberg (B-H) adjustments. H: Diagrammatic representation of top five TF motifs enriched in promoters (−250 to +50 bp relative to TSS) of upregulated genes. Transcription factor affinity prediction (TRAP) was used to find high-affinity TF binding sites enriched in the promoters (−250 bp to 50 bp relative to TSS) of upregulated genes. I: IPA showing activation of the indicated TFs in db/dbBMMC. AA, amino acid; cov, coverage; CT, connective tissue; CV, cardiovascular; Dev, developmental; Emb, embryonic; GI, gastrointestinal; OIA, organismal injury and abnormalities; SKM, skeletal and muscular; SKS, skeletal system; Vasc, vascular.
Interestingly, when we next compared db/+ and db/db macrophages, we observed that the levels of 1,648 transcripts, including 1,556 RefSeq genes (Fig. 2B), were altered in db/dbBMMC compared with db/+BMMC, indicating diabetes profoundly affected the BMMC transcriptome. In contrast, relatively few genes were altered in db/dbBMGM relative to db/+BMGM (Supplementary Fig. 4). Further analysis of all the DEGs in db/dbBMMC included both known and novel coding as well as long noncoding transcripts (Fig. 2C). Differentially expressed RefSeq genes included growth factors, fibrotic genes, proteases, and adhesion molecules involved in extracellular matrix (ECM) remodeling, chemokines, cytokines, neuroimmune signaling, and lipid metabolism (GEO accession code GSE54154). Most notably, db/dbBMMC exhibited a strong profibrotic phenotype relative to db/+BMMC, with marked upregulation of several profibrotic growth factors, including Ctgf, Tgfb, and fibrotic/ECM genes, including numerous collagens (Fig. 2D). DAVID gene ontology analysis (Fig. 2E and F) and IPA (Fig. 2G and Supplementary Fig. 5) of DEGs also confirmed marked enrichment of signaling pathways relevant to fibrosis (including transforming growth factor-β signaling), inflammation, ECM remodeling, adhesion and growth factor/cytokine signaling, and cardiovascular diseases among the top 10 categories. Motif analysis of upregulated genes by TRAP showed that binding sites for SP1, Klf4, Egr1, MZF1_5–13, and RREB1 TFs were enriched in proximal promoters of DEGs (−250 to +50 bp relative to transcription start site [TSS]) (Fig. 2H). In addition, gene set enrichment analysis also showed significant enrichment of serum response factor (SRF) motif in upregulated gene promoters (±2 kb flanking TSS) (Supplementary Fig. 6). Furthermore, IPA indicated that activities of several TFs including TP53, Smad3, and SRF were increased, whereas others such as Hoxa10 were inhibited (Fig. 2I). These results suggest that diabetes alters multiple transcription programs and skews macrophages toward a proinflammatory and profibrotic phenotype that can contribute to vascular disorders. They also reveal a novel profibrotic and dysfunctional M2 phenotype in diabetic macrophages.
Diabetes Induces Differential Expression of lncRNAs in Macrophages
Bioinformatics analyses of our RNA-seq data (Fig. 3A) identified several known and novel lncRNAs expressed in BMMC. Of these, only 156 known (RefSeq) ncRNAs including 119 lncRNAs (≥200 bp in length) and 52 novel lncRNAs were differentially expressed in db/db relative to db/+BMMC (Fig. 2C). The fold changes of some of these lncRNAs are depicted in Fig. 3B and D (complete list shown in Supplementary Tables 3 and 4). Some of the differentially expressed lncRNAs such as E330013P06 (E33) are host genes for miRNAs (Fig. 3F). Because lncRNAs can regulate nearby genes, we also analyzed genes located within ∼500 kb from the lncRNAs differentially expressed in db/dbBMMC. This revealed that expression of some nearby genes was also affected, suggesting their coregulation with lncRNAs in db/dbBMMC (Fig. 3C and E, and Supplementary Tables 2 and 3). Interestingly, IPA indicated that these nearby genes have functions related to inflammation and endocrine disorders (Fig. 3G). These results demonstrate for the first time that diabetes can alter the expression of lncRNAs in macrophages, which, in turn, might affect the expression of genes involved in macrophage function.
Figure 3.
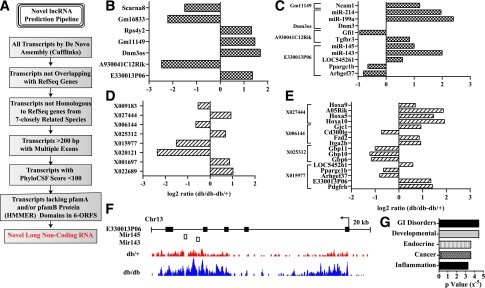
Identification of differentially expressed lncRNAs in macrophages from db/db mice. A: Pipeline used to identify novel lncRNAs in BMMC. B and C: Representative known (RefSeq) lncRNAs differentially expressed (B) and nearby (±500 kb flanking regions) DEGs (C) in db/dbBMMC. D and E: Novel differentially expressed lncRNAs (>200 bp with at least two exons; D) and nearby genes with altered expression in db/dbBMMC (E). F: Schematic showing the genomic structure of lncRNA E330013P06 and the alignment of RNA-seq reads in db/+BMMC (db/+) and db/dbBMMC (db/db). Arrow indicates the direction of transcription. Closed rectangles are exons, and open rectangles are miRNAs expressed from these genomic locations. G: IPA of differentially expressed nearby genes. GI, gastrointestinal.
Upregulation of lncRNA E330013P06 in Macrophages From Diabetic Mice
Next, we further characterized lncRNA E33, which was significantly upregulated (>twofold) in db/dbBMMC. Most of the other lncRNAs were either expressed in lower amounts and/or difficult to clone/express because of their large sizes. Furthermore, mouse E33 genomic organization is similar to the human gene (MIR143HG) and is a host gene for the miRNAs miR-143 and miR-145 (Fig. 3F) that are implicated in cancer, vascular disease, and insulin resistance (38–40). Moreover, the functions of E33 lncRNA have not been examined in any cell or disease type, including macrophages under diabetic conditions.
Using RT-QPCR, we first verified that expression of E33 was significantly increased in db/dbBMMC relative to db/+ (Fig. 4A). Next, we examined its expression in PMs from db/+ and db/db mice, which showed that E33 was significantly increased in PM from db/db mice compared with db/+, further establishing in vivo relevance (Fig. 4B). Because E33 is a host gene for miR-143 and miR-145, we also examined their expression. Levels of miR-143 (Fig. 4C), but not miR-145 (Fig. 4D), were upregulated in db/dbBMMC, indicating these miRNAs may be posttranscriptionally regulated at the level of biogenesis. Recent studies showed that the combination of high-fat (HF) diet and single streptozotocin (SZ) administration can induce insulin resistance and hyperglycemia mimicking T2D (41). We used this mouse model of diet-induced T2D (HFSZ) to verify that observed increases in E33 in db/db versus db/+ mice were not due to genetic differences. HFSZ mice exhibited characteristic features of T2D, including increased body weight (Fig. 4E), blood glucose (Fig. 4F), and insulin resistance (Fig. 4G) compared with standard diet–fed mice (SF). Expression of inflammatory genes Il6, Nos2, and Ptgs2 was significantly increased in BMMC from HF and HFSZ mice (Fig. 4H–J). Furthermore, E33 expression was significantly increased in both BMMC (Fig. 4K) and PM (Fig. 4L) derived from the HFSZ mice compared with SF, as well as in PM from HF mice (Fig. 4L). Increased levels of E33 in the PMs of these T2D mice further supports the in vivo relevance.
Figure 4.

Increased expression of E330013P06 in macrophages from diabetic mice and monocytes from T2D patients. RT-QPCR analysis shows that E33 is significantly increased in (A) BMMC (n = 6) and (B) PM (n = 6) from db/db compared with db/+ mice. RT-QPCR of miR-143 (C) and miR-145 (D) in db/+ and db/dbBMMC (n = 5). Gene expression was analyzed by RT-QPCR and results expressed as fold over macrophages from db/+ mice. Data are represented as mean ± SEM (*P < 0.05). Body weight (E), blood glucose levels (mg/dL) in basal (F), and after intraperitoneal injection of glucose (G) in the control (SF), HF, and diet-induced T2D (HFSZ) mice. C57BL/6 mice were maintained on SF or HF diet for 4 weeks. Then half the HF mice were injected with a single dose of SZ (100 mg/kg) to make them T2D (HFSZ). Glucose tolerance tests were performed 4 weeks later. H–K: Increased expression of inflammatory genes and E33 in BMMC from HF and HFSZ mice relative to SF mice. L: E33 expression in PM from indicated mice. BMMC (n = 5–7) and PM (n = 5–6). Gene expression was analyzed by RT-QPCR and results expressed as fold over macrophages from SF mice. Data are represented as mean ± SEM (*P < 0.05). M: RT-QPCR analysis of PM from SZ-induced T1D mice (SZ) showed no changes in E33 levels relative to controls (NS) mice (mean ± SEM; n = 7–14). Blood glucose levels were 527.1 ± 114 mg/dL in SZ (n = 14) versus 186.4 ± 42.6 mg/dL in control NS mice (n = 7). N: Increased expression of MIR143HG (human equivalent of E33) in monocytes from T2D patients versus healthy controls (control). RT-QPCR results were expressed as fold over control samples. Mean ± SEM (*P < 0.05, n = 4 each). GTT, glucose tolerance test.
On the other hand, examination of E33 levels in PMs derived from SZ-induced T1D mice revealed no significant changes macrophages from T1D mice (SZ) versus controls (NS) (Fig. 4M). These results demonstrate that insulin resistance and T2D can induce E33 expression in macrophages. In order to further determine the relevance to human diabetes, we determined the levels of MIR143HG (RefSeq NR_105060.1), the human equivalent of E33, in monocytes from T2D patients. Results showed significantly increased expression of MIR143HG in monocytes from T2D patients versus healthy controls (Fig. 4N).
Sustained Overexpression of E33 Increases Proinflammatory and Proatherogenic Gene Expression in RAW Macrophages
We next determined the functional relevance of E33 upregulation by using RAW 264.7 (RAW) macrophages stably transduced with lentiviral vectors that express full-length mouse E33 mRNA under the control of a CMV promoter (LVE33). RAW cells transduced with GFP (LVGFP) were used as control. E33 expression was significantly increased in LVE33-transduced RAW cells relative to LVGFP (Fig. 5A). E33 overexpression in RAW cells did not affect cell morphology or growth (data not shown) but clearly altered several inflammatory genes. The anti-inflammatory gene Il10 (Fig. 5B) was downregulated, whereas several proinflammatory genes were upregulated (Fig. 5C–F). The chemokine Ccl2 was inhibited (Fig. 5G), but expression of the proatherogenic scavenger receptor Cd36 (Fig. 5H) was increased by E33 overexpression. We examined if E33 also modulated macrophage responses to proinflammatory signals. LPS (100 ng/mL) dramatically induced the expression of proinflammatory genes in LVGFP as expected, but the expression of Il6, Tnf, Ptgs2, and Ccl2 was further augmented in LVE33 cells treated with LPS (Fig. 5I–L), whereas Il10, Nos2, and Cd36 were not altered (Fig. 5M–O). Furthermore, transient overexpression of E33 in BMMC from normal C57BL/6 mice also increased the expression of inflammatory genes, confirming similar proinflammatory effects in primary mouse macrophages (Supplementary Fig. 7). Overall, the data demonstrate that E33 overexpression can upregulate proinflammatory and proatherogenic genes and also enhance their response to proinflammatory signals in macrophages.
Figure 5.

Regulation of inflammatory genes and foam cell formation in RAW macrophages stably overexpressing E330013P06. A–H: Expression of E33 and the indicated genes in RAW macrophages that either stably overexpressed lncRNA E33 (LVE33) or a control vector that expressed GFP (LVGFP). RAW cells were transduced with LVE33 or LVGFP, and puromycin-resistant cells (2 μg/mL) were used for further analysis by RT-QPCR. Results are expressed as fold increase over LVGFP (GFP). I–O: RAW macrophages stably expressing E33 (LVE33) or GFP (LVGFP) were treated with LPS (100 ng/mL) for 24 h. Expression of indicated genes was analyzed by RT-QPCR. Results are expressed as fold over GFP control. Data in A–O represent mean ± SEM (*P < 0.05, n = 4–9). P: Enhanced foam cell formation in macrophages overexpressing E33. Indicated RAW cells were incubated with Dil-Ac-LDL (panels a and b, red fluorescence) and fluorescent images were collected as described in research design and methods. Blue fluorescence shows nuclear staining with Hoechst dye. Cont, control.
E33 Overexpression in Macrophages Promotes the Foam Cell Formation
Having shown that E33 upregulates Cd36 expression, we next examined if this is functionally associated with increased uptake of modified LDL. As shown in Fig. 5P, Dil-Ac-LDL uptake was markedly increased in LVE33 (Fig. 5P, panel b) compared with LVGFP cells (Fig. 5P, panel a), suggesting increased foam cell formation. Nuclear staining with Hoechst confirmed similar numbers of cells in both fields (Fig. 5P, panels c and d). Overall, these data reveal a functional role for E33 in enhancing macrophage uptake of modified LDL, a key event in foam cell formation and atherosclerosis.
Regulation of E33 and Inflammatory Genes by HG and PA
Hyperglycemia, insulin resistance, and dyslipidemia, evident in our mouse models in which E33 was upregulated, are key factors involved in diabetes and its complications. Therefore, we examined if HG and free fatty acids such as PA, which is greatly increased in T2D, could regulate E33 expression in macrophages. E33 and key inflammatory genes were examined in db/+BMMC treated with HG (25 mmol/L), PA (200 μmol/L), or HG+PA (HP) for 4 days. Results showed that HP treatment significantly increased E33 expression in db/+BMMC (Fig. 6A). Furthermore, HG and PA also increased the expression of several inflammatory genes (Fig. 6B–F) but showed differences in the patterns of regulation of Cd36 and Nos2. HG inhibited Nos2 expression, whereas PA inhibited Cd36 expression. These results indicate that HG and PA can regulate common genes, including E33, but may impact the expression of others differently. HP also increased E33 expression in BMMC of normal C57BL/6 mice (2.84 ± 0.47 SEM fold vs. NG; n = 2), suggesting it is not specific to only db/+BMMC.
Figure 6.

Regulation of E330013P06 and inflammatory genes in diabetic conditions and its inhibition by E330013P06 gene silencing in macrophages. A–F: BMMC from db/+ mice were treated with NG (5.5 mmol/L), HG (25 mmol/L), 200 μmol/L PA, or HG+PA (HP) for 4 days. Levels of E33 (A) and the indicated inflammatory genes (B–E), as well as Cd36 (F), were determined by RT-QPCR. Results are expressed as fold over gene expression levels in NG. G–L: BMMC from db/db mice were transiently transfected with siRNA targeting E33 (siE33) or a nontargeting control (siNTC). Then, 72 h posttransfection BMMC were stimulated with HG+PA (HP) for another 24 h. The expression of indicated genes was analyzed by RT-QPCR and results expressed as fold over the siNTC transfected cells. Data are represented as mean ± SEM (*P < 0.05; n = 3).
Silencing E33 in db/db Macrophages Inhibits Inflammatory Genes Induced by HG and PA
Next, to test the consequences of silencing E33, we transfected db/dbBMMC with siRNA oligonucleotides containing a nontargeting control (siNTC) or a mixture of three siRNA oligonucleotides targeting E33 (siE33). Three days posttransfection, the cells were treated with HP for 24 h, and gene expression was analyzed. Results showed that siE33 significantly downregulated basal and HP-induced E33 expression (Fig. 6G). E33 gene silencing did not affect basal inflammatory gene expression but significantly inhibited the expression of key inflammatory genes and Cd36 induced by HP (Fig. 6H–L). Together these new results demonstrate that E33 lncRNA is upregulated under diabetic conditions and contributes to the enhanced macrophage inflammatory and atherogenic phenotype and functions. Knockdown of E33 can reverse the diabetic phenotype, at least in part, especially under stimulated conditions.
Discussion
Macrophage differentiation and polarization are associated with the production of proinflammatory and proatherogenic factors. These processes are dysregulated in diabetes and can contribute to enhanced atherosclerosis. In this study, using RNA-seq, we showed that diabetes skewed the macrophage transcriptome toward an increased proinflammatory and profibrotic state, and a dysfunctional, alternatively activated phenotype. These changes were associated with alterations in networks of genes that regulate inflammation and fibrosis. Notably, we observed for the first time that macrophage lncRNA expression levels were also altered. Furthermore, T2D was associated with the increased expression of lncRNA E33, which upregulated the expression of proinflammatory genes, and increased lipid update in macrophages (foam cell formation). Moreover, E33 was upregulated in vitro under metabolically challenged and diabetic conditions (HG and PA), but silencing its expression prevented increased expression of inflammatory genes. These results demonstrate that E33 contributes to a proinflammatory macrophage phenotype, thereby uncovering a lncRNA with a novel functional role in diabetic macrophages.
Imbalances between the M1 and M2 functions of macrophages can result in chronic inflammation and tissue damage (1,2). Our RNA-seq results indicated diabetes had a profound effect on the macrophage transcriptome by enhancing proinflammatory genes and inhibiting genes associated with alternatively activated M2 phenotype. Several profibrotic genes and related TFs were upregulated, which may affect various target tissues related to complications and also lead to adipose tissue remodeling, systemic inflammation, insulin resistance, wound healing defects, and atherosclerotic plaque formation/instability. db/dbBMMC also had lower levels of Nos2, a key enzyme required for phagocytosis (42), suggesting impaired host defense. Furthermore, Csf1 expression was enhanced in db/dbBMMC, which may promote monocyte–vascular smooth muscle cell interactions and foam cell formation (18) to confer atherogenic phenotypes in diabetes. CSF-1 signaling through CSF-1R not only is essential for macrophage survival, but could promote macrophage proliferation in atherosclerotic environments (43). Thus, the diabetes milieu can augment the macrophage inflammatory phenotype and proliferative capacity, which can exacerbate atherosclerosis. In support of this, recent studies demonstrated monocytosis in hyperglycemia (44) and macrophage proliferation in atherosclerosis (45). The key anti-inflammatory and immunomodulatory cytokine Il10 was downregulated in db/dbBMMC, along with interferon γ–induced genes such as guanylate-binding proteins, further suggesting macrophages in diabetes have increased inflammatory activities and defective immune responses. Bioinformatics and in silico modeling revealed that diabetes can affect the function of key TFs involved in cytoskeletal gene programs (SRF) (46) and macrophage differentiation (SP1, Egr1, Klf4, and MZF) (3,47). SRF regulates gene expression by interaction with distal enhancers (46), and our study indicates that this could be affected in diabetes. Overall, the power of RNA-seq reveals enormous changes to the macrophage transcriptome that could lead to multiple pathologies associated with diabetes.
Our RNA-seq data also revealed that several lncRNAs are differentially expressed in BMMC from db/db mice compared with db/+. The expression of several genes located within 500 kb from differentially expressed lncRNAs was also altered in db/db macrophages, indicating potential regulation by the lncRNAs. These transcripts included both coding and noncoding genes, including miRNAs. In addition, bioinformatic analysis indicated that the nearby genes were involved in inflammation, highlighting the potential importance of gene regulation by lncRNAs in diabetes. Our hypothesis was that lncRNAs could have a functional role and regulate genes involved in macrophage dysfunction. We therefore further characterized the function of lncRNA E33, which was significantly upregulated in db/dbBMMC and has a genomic organization similar to the human locus. The function and regulation of lncRNA E33 has not been examined in any cell type or disease condition. We found lncRNA E33 was upregulated in macrophages from T2D mice, but not T1D mice. In contrast to T1D, T2D is associated with insulin resistance, hyperglycemia, and dyslipidemia, suggesting E33 expression might be affected by multiple diabetogenic factors, including free fatty acids. This was supported by our data showing that E33 expression was also increased in macrophages from HFSZ and HF mice (Fig. 4K and L) and in macrophages treated with HG+PA (Fig. 6A). Furthermore, MIR143HG expression was higher in monocytes from T2D patients relative to controls (Fig. 4N), demonstrating relevance to human disease. Most notably, we found that E33 overexpression significantly upregulated inflammatory genes and downregulated the anti-inflammatory cytokine Il10. Conversely, gene silencing by siRNAs prevented the increased expression of inflammatory genes induced by HG/PA in db/db macrophages. Interestingly, E33 also upregulated Cd36, a key scavenger receptor involved in oxidized LDL uptake and foam cell formation. Consistently, expressing E33 in macrophages greatly increased their LDL uptake, further supporting a key role for this lncRNA in proatherogenic responses of macrophages.
Because E33 is also a host gene for miR-143/145, effects of increased E33 could also be mediated by these miRNA. However, our E33 expression vector expressed only the exons of E33 lncRNA, and hence it does not express miR-143 or miR-145, which are located in introns. In addition, transfection of macrophages with oligonucleotide mimics of miR-143 (which was increased in db/db mice) did not affect inflammatory genes (data not shown). Therefore, lncRNA E33 regulates macrophage function independently of the miRNAs that are derived from it.
We also showed that E33 gene silencing with siRNAs significantly inhibited HP-induced inflammatory genes, but not basal levels. HP-induced lncRNA E33 might regulate various gene transcription mechanisms that include modulating recruitment/activity of epigenetic enzymes to alter chromatin structure and access to TFs under insulin-resistant conditions. However, it is known that HG alone can regulate inflammatory genes via activation of TFs such as NF-κB through multiple signaling and epigenetic mechanisms (48). Furthermore, PA can also regulate gene expression through activation of nuclear receptors, Toll-like receptors, and NF-κB to promote inflammation in diabetes (8,49,50). Thus novel lncRNA-dependent mechanisms identified in our study could fine-tune the inflammatory phenotype of macrophages in concert with these other pathways activated by the diabetes milieu. Because E33 was not increased in T1D macrophages although proinflammatory genes were elevated relative to nondiabetic mice (data not shown), E33-dependent mechanisms might operate only under T2D conditions. Thus E33, like most ncRNAs, may play a fine-tuning modulatory role, along with the other known factors, to augment macrophage inflammatory states and lipid uptake in T2D.
The dysregulation of lncRNA expression and function is increasingly being implicated in various diseases, and related therapeutic strategies are gradually being explored (23). We have shown for the first time a role for one diabetes-induced lncRNA E33 in macrophage dysfunction, which suggests that, in the future, such lncRNAs can serve as novel therapeutic targets to attenuate inflammation and atherogenic potential of macrophages and concomitant diabetes complications.
Supplementary Material
Article Information
Acknowledgments. The authors are grateful to Dr. H. Gao and J.-H. Wang (Integrative Genomics Core) for assistance with RNA-seq, L. Brown (Analytical Cytometry Core) for flow cytometry, Dr. R. Chandran (Department of Virology) for analysis with FlowJo software, and Dr. M. Morgan (Office of Faculty and Institutional Support) for editing the manuscript (all from the Beckman Research Institute of City of Hope, Duarte, CA).
Funding. These studies were supported by grants from the National Institutes of Health (R01 DK 065073, HL106089, and DK058191 to R.N.).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. M.A.R. conceived the idea, designed the experiments, performed all the experiments, analyzed the data, and wrote and edited the manuscript. Z.C. analyzed RNA-seq data, performed bioinformatics analysis, and wrote and edited the manuscript. J.T.P. and S.P. assisted with macrophage isolation and reviewed the manuscript. M.W., L.L., and Q.Z. performed experiments and analyzed data. K.B. performed experiments, analyzed data, and reviewed the manuscript. A.L. assisted with analysis of RNA-seq data and reviewed the manuscript. X.W. analyzed RNA-seq data. P.S. designed siRNAs and reviewed the manuscript. S.D. provided monocytes from diabetic patients and controls and reviewed the manuscript. R.N. conceived the idea, designed the experiments, analyzed the data, and wrote and edited the manuscript. M.A.R. and R.N. are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Prior Presentation. Parts of this study were presented at Experimental Biology 2014, San Diego, CA, 26–30 April 2014.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db14-0298/-/DC1.
J.T.P. is currently affiliated with the Department of Internal Medicine, College of Medicine, Yonsei University, Seoul, Korea.
References
- 1.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity 2010;32:593–604 [DOI] [PubMed] [Google Scholar]
- 2.Odegaard JI, Chawla A. Alternative macrophage activation and metabolism. Annu Rev Pathol 2011;6:275–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol 2011;11:750–761 [DOI] [PubMed] [Google Scholar]
- 4.Liao X, Sharma N, Kapadia F, et al. Krüppel-like factor 4 regulates macrophage polarization. J Clin Invest 2011;121:2736–2749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu G, Abraham E. MicroRNAs in immune response and macrophage polarization. Arterioscler Thromb Vasc Biol 2013;33:170–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 2010;72:219–246 [DOI] [PubMed] [Google Scholar]
- 7.Mantovani A, Garlanda C, Locati M. Macrophage diversity and polarization in atherosclerosis: a question of balance. Arterioscler Thromb Vasc Biol 2009;29:1419–1423 [DOI] [PubMed] [Google Scholar]
- 8.Nguyen MT, Favelyukis S, Nguyen AK, et al. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem 2007;282:35279–35292 [DOI] [PubMed] [Google Scholar]
- 9.Wang Y, Harris DC. Macrophages in renal disease. J Am Soc Nephrol 2011;22:21–27 [DOI] [PubMed] [Google Scholar]
- 10.Rocha VZ, Libby P. Obesity, inflammation, and atherosclerosis. Nat Rev Cardiol 2009;6:399–409 [DOI] [PubMed] [Google Scholar]
- 11.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 2007;117:175–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Devaraj S, Jialal I. Low-density lipoprotein postsecretory modification, monocyte function, and circulating adhesion molecules in type 2 diabetic patients with and without macrovascular complications: the effect of alpha-tocopherol supplementation. Circulation 2000;102:191–196 [DOI] [PubMed] [Google Scholar]
- 13.Khan T, Muise ES, Iyengar P, et al. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol 2009;29:1575–1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shanmugam N, Reddy MA, Guha M, Natarajan R. High glucose-induced expression of proinflammatory cytokine and chemokine genes in monocytic cells. Diabetes 2003;52:1256–1264 [DOI] [PubMed] [Google Scholar]
- 15.Li Y, Reddy MA, Miao F, et al. Role of the histone H3 lysine 4 methyltransferase, SET7/9, in the regulation of NF-kappaB-dependent inflammatory genes. Relevance to diabetes and inflammation. J Biol Chem 2008;283:26771–26781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li SL, Reddy MA, Cai Q, et al. Enhanced proatherogenic responses in macrophages and vascular smooth muscle cells derived from diabetic db/db mice. Diabetes 2006;55:2611–2619 [DOI] [PubMed] [Google Scholar]
- 17.Mauldin JP, Srinivasan S, Mulya A, et al. Reduction in ABCG1 in Type 2 diabetic mice increases macrophage foam cell formation. J Biol Chem 2006;281:21216–21224 [DOI] [PubMed] [Google Scholar]
- 18.Meng L, Park J, Cai Q, Lanting L, Reddy MA, Natarajan R. Diabetic conditions promote binding of monocytes to vascular smooth muscle cells and their subsequent differentiation. Am J Physiol Heart Circ Physiol 2010;298:H736–H745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bornfeldt KE, Tabas I. Insulin resistance, hyperglycemia, and atherosclerosis. Cell Metab 2011;14:575–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mercer TR, Mattick JS. Structure and function of long noncoding RNAs in epigenetic regulation. Nat Struct Mol Biol 2013;20:300–307 [DOI] [PubMed] [Google Scholar]
- 21.Guttman M, Amit I, Garber M, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009;458:223–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leung A, Trac C, Jin W, et al. Novel long noncoding RNAs are regulated by angiotensin II in vascular smooth muscle cells. Circ Res 2013;113:266–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wahlestedt C. Targeting long non-coding RNA to therapeutically upregulate gene expression. Nat Rev Drug Discov 2013;12:433–446 [DOI] [PubMed] [Google Scholar]
- 24.Morán I, Akerman I, van de Bunt M, et al. Human β cell transcriptome analysis uncovers lncRNAs that are tissue-specific, dynamically regulated, and abnormally expressed in type 2 diabetes. Cell Metab 2012;16:435–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Putta S, Lanting L, Sun G, Lawson G, Kato M, Natarajan R. Inhibiting microRNA-192 ameliorates renal fibrosis in diabetic nephropathy. J Am Soc Nephrol 2012;23:458–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weischenfeldt J, Porse B. Bone marrow-derived macrophages (BMM): isolation and applications. CSH Protoc 2008;2008:pdb.prot5080PubMed [DOI] [PubMed] [Google Scholar]
- 27.Kanter JE, Kramer F, Barnhart S, et al. Diabetes promotes an inflammatory macrophage phenotype and atherosclerosis through acyl-CoA synthetase 1. Proc Natl Acad Sci U S A 2012;109:E715–E724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wen H, Gris D, Lei Y, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol 2011;12:408–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dasu MR, Devaraj S, Park S, Jialal I. Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care 2010;33:861–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009;25:1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin MF, Jungreis I, Kellis M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics 2011;27:i275–i282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pauli A, Valen E, Lin MF, et al. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res 2012;22:577–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cabili MN, Trapnell C, Goff L, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 2011;25:1915–1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010;28:511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thomas-Chollier M, Hufton A, Heinig M, et al. Transcription factor binding predictions using TRAP for the analysis of ChIP-seq data and regulatory SNPs. Nat Protoc 2011;6:1860–1869 [DOI] [PubMed] [Google Scholar]
- 36.Saetrom P, Snøve O, Jr. A comparison of siRNA efficacy predictors. Biochem Biophys Res Commun 2004;321:247–253 [DOI] [PubMed] [Google Scholar]
- 37.Lacey DC, Achuthan A, Fleetwood AJ, et al. Defining GM-CSF- and macrophage-CSF-dependent macrophage responses by in vitro models. J Immunol 2012;188:5752–5765 [DOI] [PubMed] [Google Scholar]
- 38.Kent OA, Chivukula RR, Mullendore M, et al. Repression of the miR-143/145 cluster by oncogenic Ras initiates a tumor-promoting feed-forward pathway. Genes Dev 2010;24:2754–2759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cordes KR, Sheehy NT, White MP, et al. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 2009;460:705–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jordan SD, Krüger M, Willmes DM, et al. Obesity-induced overexpression of miRNA-143 inhibits insulin-stimulated AKT activation and impairs glucose metabolism. Nat Cell Biol 2011;13:434–446 [DOI] [PubMed] [Google Scholar]
- 41.Chatzigeorgiou A, Halapas A, Kalafatakis K, Kamper E. The use of animal models in the study of diabetes mellitus. In Vivo 2009;23:245–258 [PubMed] [Google Scholar]
- 42.Nathan C, Shiloh MU. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc Natl Acad Sci U S A 2000;97:8841–8848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sieweke MH, Allen JE. Beyond stem cells: self-renewal of differentiated macrophages. Science 2013;342:1242974. [DOI] [PubMed] [Google Scholar]
- 44.Nagareddy PR, Murphy AJ, Stirzaker RA, et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab 2013;17:695–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Randolph GJ. Macrophages in Marseille. Immunity 2013;38:619–621 [DOI] [PubMed] [Google Scholar]
- 46.Sullivan AL, Benner C, Heinz S, et al. Serum response factor utilizes distinct promoter- and enhancer-based mechanisms to regulate cytoskeletal gene expression in macrophages. Mol Cell Biol 2011;31:861–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nagamura-Inoue T, Tamura T, Ozato K. Transcription factors that regulate growth and differentiation of myeloid cells. Int Rev Immunol 2001;20:83–105 [DOI] [PubMed] [Google Scholar]
- 48.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res 2010;107:1058–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jialal I, Kaur H. The Role of Toll-Like Receptors in Diabetes-Induced Inflammation: Implications for Vascular Complications. Curr Diab Rep 2012;12:172–179 [DOI] [PubMed] [Google Scholar]
- 50.Xu HE, Lambert MH, Montana VG, et al. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol Cell 1999;3:397–403 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.