

Abstract
Many protein-misfolding disorders can be modeled in the budding yeast Saccharomyces cerevisiae. Proteins such as TDP-43 and FUS, implicated in amyotrophic lateral sclerosis, and α-synuclein, implicated in Parkinson’s disease, are toxic and form cytoplasmic aggregates in yeast. These features recapitulate protein pathologies observed in patients with these disorders. Thus, yeast are an ideal platform for isolating toxicity suppressors from libraries of protein variants. We are interested in applying protein disaggregases to eliminate misfolded toxic protein conformers. Specifically, we are engineering Hsp104, a hexameric AAA+ protein from yeast that is uniquely capable of solubilizing both disordered aggregates and amyloid and returning the proteins to their native conformations. While Hsp104 is highly conserved in eukaryotes and eubacteria, it has no known metazoan homologue. Hsp104 has only limited ability to eliminate disordered aggregates and amyloid fibers implicated in human disease. Thus, we aim to engineer Hsp104 variants to reverse the protein misfolding implicated in neurodegenerative disorders. We have developed methods to screen large libraries of Hsp104 variants for suppression of proteotoxicity in yeast. As yeast are prone to spontaneous nonspecific suppression of toxicity, a two-step screening process has been developed to eliminate false positives. Using these methods, we have identified a series of potentiated Hsp104 variants that potently suppress the toxicity and aggregation of TDP-43, FUS, and α-synuclein. Here, we describe this optimized protocol, which could be adapted to screen libraries constructed using any protein backbone for suppression of toxicity of any protein that is toxic in yeast.
Keywords: Microbiology, Issue 93, Protein-misfolding disorders, yeast proteinopathy models, Hsp104, proteotoxicity, amyloid, disaggregation
Introduction
Yeast proteinopathy models have been developed for protein-misfolding disorders including amyotrophic lateral sclerosis (ALS) and Parkinson’s disease (PD)1-3. Expression of the proteins TDP-43 and FUS, which misfold in ALS patients, are toxic and mislocalize to form cytoplasmic aggregates in yeast1,2. Similarly, expression of α-synuclein (α-syn), which is implicated in PD, is toxic and mislocalizes to form cytoplasmic aggregates in yeast3. These features recapitulate phenotypes in patients with these disorders4,5. Thus, yeast models provide a useful platform for screening for proteins or small molecules that prevent or reverse these phenotypes2,6-13. We are interested in the development of proteins that are capable of reversing aggregation and toxicity due to TDP-43, FUS, and α-syn. We focus on Hsp104, an AAA+ protein from yeast that is uniquely capable of disaggregating proteins both from amorphous aggregates and amyloid in yeast, yet it has no human homologue14,15. Hsp104 is finely tuned to disaggregate endogenous yeast prions and has only limited ability to disaggregate substrates implicated in human neurodegenerative diseases, which it never ordinarily encounters16,17. Thus, we aim to engineer enhanced versions of Hsp104 that are able to efficaciously disaggregate these human substrates. To do so, we construct large libraries of Hsp104 variants using error-prone PCR; these libraries can be screened using the yeast proteinopathy models17. We have adopted a domain-targeted approach to constructing and screening libraries, as Hsp104 is very large17. We initially focused on the middle domain (MD) of Hsp10417, though similar approaches can be employed to screen other domains. These models enable screening for disaggregase activity directly, as opposed to alternative techniques such as surface display, which is restricted to use for monitoring binding18.
Our protocol is based on two screening steps (Figure 1). First, Hsp104 variants that suppress the toxicity of the disease substrate in yeast are selected. To do so, the Hsp104 variants and disease-associated substrate are cotransformed into ∆hsp104 yeast. We employ ∆hsp104 yeast to explore Hsp104 sequence space in the absence of wild-type (WT) Hsp10417. Importantly, deletion of Hsp104 does not affect α-syn, FUS, or TDP-43 toxicity in yeast, and expression of Hsp104WT provides minimal rescue1,13,17. The yeast is then plated on inducing media to induce expression of both proteins. Yeast harboring Hsp104 variants that suppress toxicity of the disease-associated substrate confer growth of the colony. These variants are selected for further analysis, while colonies maintaining variants that do not suppress toxicity die. However, false positives are a substantial problem in this screen. Expression of TDP-43, FUS, and α-syn are highly toxic, which creates a strong selective pressure for the appearance of spontaneous genetic suppressors of toxicity unrelated to the Hsp104 variant being expressed. Thus, we have used a secondary screen that is also relatively high throughput to eliminate these nonspecific toxicity suppressors17. In this secondary screen, selected yeast are treated with 5-Fluorootic Acid (5-FOA) to counter select for the Hsp104 plasmid19. The strains are then assessed for substrate (TDP-43, FUS, or α-syn) toxicity via spotting assay to ensure that the toxicity of the substrate is restored after loss of the Hsp104 plasmid. Thus, yeast in which toxicity is restored in this secondary screen presumably originally displayed toxicity suppression due to the presence of the Hsp104 variant. These yeast are designated as ‘hits’ and the Hsp104 plasmid should then be recovered and sequenced to identify the mutations in the Hsp104 gene17 (Figure 1). Any hits should then be reconfirmed by constructing the mutation independently using site-directed mutagenesis and then retesting for toxicity suppression. The potential applications for this protocol are broad. Using these methods, libraries of any type of protein could be screened for variants that suppress toxicity of any substrate protein that is toxic in yeast.
Protocol
1. Library Generation
To construct libraries of Hsp104 using domain-specific error-prone PCR, first amplify the domain of interest with an error prone DNA polymerase20.
Purify the PCR product by gel extraction.
- Perform a megaprimer extension step using a standard site-directed mutagenesis protocol: combine 50 ng template plasmid, 250 ng megaprimer, 200 μM dNTPs, and high-fidelity DNA polymerase in PCR buffer, and dilute to 50 μl total volume with PCR grade water20. Run a standard PCR program. NOTE: Specific primers used will vary based on the particular region of the gene that is to be amplified.
- Following PCR, digest parental template DNA with 1 μl DpnI restriction enzyme for 2 hr at 37 °C.
Transform the library by electroporation or other means in ultracompetent E. coli and purify it by miniprep. NOTE: Library generation varies substantially based upon the aims of a given experiment. Hsp104 is a very large protein, so it is impractical to randomize the entire gene, which is why we utilize domain-specific error prone PCR. Additionally, the structure of Hsp104 remains poorly understood, making the design of directed libraries challenging21. Libraries can be constructed using directed or random approaches to mutagenesis, with the only restriction being that the template plasmid backbone should contain the URA3 gene to allow for 5-FOA counter selection on dextrose media.
2. Transformation of the Hsp104 Library
Integrate the disease-associated substrate into W303aΔhsp104 yeast using a standard lithium acetate/PEG transformation protocol22. Select single colonies and screen them for toxicity to isolate a strain with high toxicity of the disease associated substrate23. NOTE: Use an integrated strain and isolate a single colony to ensure equal expression in all cells. Clone the disease substrate into a plasmid allowing integration of the gene under any marker other than uracil (we use histidine) and the Hsp104 library into the pAG416GAL plasmid (uracil marker)24. To allow for 5-FOA counterselection, ensure that the library is expressed from a plasmid with the uracil marker. Other yeast strains can also be employed. We have noted a similar toxicity suppression phenotype using W303a and BY4741 yeast strains in both WT and Δhsp104 backgrounds (M.E.J. and J.S., unpublished observations).
Transform the Hsp104 library into this strain using the same lithium acetate/PEG transformation protocol22. Scale up the transformation appropriately to maintain the sequence space of the library and to preserve the predicted library size.
Plate the transformation mixture onto noninducing, selective plates (e.g. SD-His-Ura) using enough plates to ensure growth of a large number of colonies. Use large petri dishes (150 mm) to minimize the number of plates needed. Recovering transformants using plates allows transformation efficiency to be assessed.
Transform Hsp104WT and vector negative controls in parallel.
3. Screening for Suppression of Proteotoxicity
Wash the colonies off the plates using raffinose supplemented dropout media (e.g. SRaff-His-Ura). Use a serological pipette and sterile wooden applicators to loosen the colonies from the plates. Transfer the liquid washes to a 50 ml conical tube and vortex thoroughly to separate any clumps of cells. Dilute to a slightly cloudy culture, OD600 ~0.025. NOTE: Here raffinose serves to relieve the cells of glucose-mediated repression of induction, thus priming the cells for galactose-mediated induction.
Grow the diluted culture overnight in raffinose dropout media with shaking at 30 ºC. Grow the Hsp104WT and vector controls in parallel.
The following morning, plate a range of concentrations onto individual galactose (e.g. SGal-His-Ura) plates (1 μl to 2 ml concentrated culture in 400 μl total volume). Plating a broad range of concentrations will ensure that a plate will have single colonies. The actual concentration required depends upon the toxicity of the disease substrate of interest.
Normalize the vector and Hsp104WT cultures to the OD600 of the library and plate equal volumes to compare growth of the library relative to the controls.
To assess selection stringency, plate the library on glucose (repressing) media. Incubate plates for 2-3 days at 30 ºC, until colonies appear (Figure 2). Assess the resulting colonies and compare to the controls. NOTE: Often both large and small colonies are obtained, but no correlation between colony size and activity is observed. Screen these potential hits using a 5-FOA counter selection technique (Step 4) to minimize false positives carried over to sequencing.
4. 5-FOA Counterselection and Spotting to Eliminate False Positives
Streak out single colonies in duplicate onto double and single dropout media (e.g. SD-His-Ura and SD-His plates) and grow overnight at 30 ºC. Repeat with vector and Hsp104WT controls. Plating on SD-His prior to 5-FOA increases the efficiency of the 5-FOA step by increasing the likelihood that cells will have lost the Hsp104 plasmid when they are plated on 5-FOA.
To prevent plates from drying out, wrap the SD-His-Ura plates in parafilm and store at 4 ºC while performing the 5-FOA screen. These plates will eventually be used for sequencing.
Streak the colonies from the SD-His plates to single colonies on 5-FOA plates (YPD media + 1g/L 5-FOA) and incubate for 1-2 days at 30 ºC until single colonies appear. Here, 5-FOA is converted into a toxic product (5-fluorodeoxyuridine) in cells that contain the URA3 gene. Thus, any cells still harboring the Hsp104 plasmid will die.
Streak the single colonies from the 5-FOA plates in duplicate onto SD-Ura and SD-His plates. Test 3 colonies for each hit to increase the likelihood that at least one colony for each hit will have lost the Hsp104 plasmid. Incubate for 1-2 days at 30 °C. Colonies that have lost the Hsp104 plasmid will grow on SD-His plates but not SD-Ura plates.
Grow the strains that have lost the Hsp104 plasmids for spotting assays. Grow the strains to saturation in raffinose dropout media (e.g. SRaff-His) in 96 DeepWell 2 ml plates overnight at 30 ºC with shaking. Be sure to include the 5-FOA treated control strains.
Prepare plates for spotting assay. Using a multichannel pipette, aliquot 200 µl raffinose dropout media (e.g. SRaff-His) per well of a 96 well plate, reserving columns 1 and 7 for the neat cultures. Aliquot 250 µl of each of the 16 saturated cultures into columns 1 and 7 of the plate.
Serially dilute the cultures 5 fold into each column of the plate, mixing thoroughly with a multichannel pipette. Remove 50 µl of the diluted culture from columns 6 and 12 to ensure the final volume of each well is 200 µl. This is essential to ensure even spotting.
Using a 96-bolt replicator tool (frogger), spot the cultures in duplicate onto SD-His and SGal-His plates. Incubate the plates at 30 ºC for 2-3 days.
Select for sequencing strains that exhibit toxicity on the SGal-His plates similar to that of the controls. Discard as false positives strains that are not as toxic as the controls. 5-FOA is also often toxic on its own, so discard strains that exhibit a growth defect on SD-His plates or that exhibit substantially greater toxicity than the disease substrate alone (Figure 3).
5. Sequencing the Hsp104 Variants by Colony PCR
Scrape ~10 μl yeast from the SD-His-Ura plates in 3 μl 20 mM NaOH in PCR strip tubes and lyse the cells by freeze-thaw. Place the strip tubes in a -80 ºC freezer for 10 min or in liquid nitrogen, then incubate at 99 ºC in a thermocycler for 10 min. Dilute the strains to 100 µl with PCR grade water.
Prepare PCR reactions: 5 μl PCR template, 0.5 μl 100 μM each primer, 0.5 μl 10mM dNTPs, 0.25 μl DNA polymerase in PCR buffer, and make up to 25 μl total volume with PCR grade water. Design primers to amplify the randomized region plus approximately 100 bp on either end. Minimize the size of the amplified region to increase the likelihood of successful amplification. Run a standard PCR program.
Analyze samples by agarose gel electrophoresis to confirm amplification of a PCR product of the appropriate size. Repeat PCR if no product is present. NOTE: PCR failure is typically due to too much yeast being used in step 5.1.
Prepare samples for DNA sequencing. Remove PCR primers either by PCR purification or using a PCR product cleanup reagent such as Exonuclease I and Shrimp Alkaline Phosphatase enzyme (ExoSAP-IT) to degrade single stranded DNA. This reagent enzymatically degrades unincorporated primers and dNTPs, allowing for higher throughput.
Sequence the PCR products. Be sure to thoroughly analyze sequencing chromatograms for mutations. Mutations may be observed as a mixture of two nucleotides at a given site (Figure 4), as yeast often harbor multiple plasmids.
Representative Results
We have constructed a library of Hsp104 variants randomized in the middle domain and screened it for suppression of TDP-43 toxicity. The library was transformed and plated onto glucose and galactose plates (Figure 2) to assess the stringency of the screen. Single colonies were selected and the strains were counter selected using 5-FOA to eliminate the Hsp104 plasmid. These strains were then assessed to confirm that toxicity was due to TDP-43 alone, without the Hsp104 variants. Spotting assays of a subset of the variants selected in the initial screen showed that of the 4 colonies selected, 2 displayed Hsp104-mediated TDP-43 toxicity suppression, 1 was a false positive, and 1 displayed enhanced toxicity following 5-FOA treatment (Figure 3). The 2 true hits were then sequenced by colony PCR to identify the middle domain mutations (Figure 4). Once these mutations were identified, the Hsp104 mutation was constructed afresh in the parental Hsp104 plasmid by site-directed mutagenesis to confirm toxicity suppression. Variants selected using these methods were later confirmed to suppress aggregation in yeast, clear preformed aggregates in biochemical assays, and suppress dopaminergic neurodegeneration in a C. elegans model of PD17.
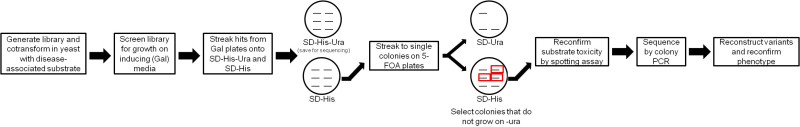 Figure 1: Flow-chart for isolating potentiated Hsp104 variants. Hsp104 libraries (URA3 marker, GAL1 promoter) are transformed in yeast containing the disease substrates (HIS3 marker, GAL1 promoter) and screened for toxicity suppressors by plating on inducing media. Potential hits are then screened again using a 5-FOA counterselection step to eliminate nonspecific toxicity suppressors. Variants selected in this second step are then sequenced by colony PCR. Please click here to view a larger version of this figure.
Figure 1: Flow-chart for isolating potentiated Hsp104 variants. Hsp104 libraries (URA3 marker, GAL1 promoter) are transformed in yeast containing the disease substrates (HIS3 marker, GAL1 promoter) and screened for toxicity suppressors by plating on inducing media. Potential hits are then screened again using a 5-FOA counterselection step to eliminate nonspecific toxicity suppressors. Variants selected in this second step are then sequenced by colony PCR. Please click here to view a larger version of this figure.
 Figure 2: Screening for toxicity suppressors. Yeast cotransformed with pAG303GAL-TDP-43 and the pAG416GAL-Hsp104 library were plated onto glucose (repressing) or galactose (inducing) media. TDP-43 is highly toxic, so very few Hsp104 variants are capable of suppressing this toxicity, and this screen is very stringent. Single colonies are selected as hits from the galactose plate for the 5-FOA secondary screen. Both large and small colonies are typically obtained, but we have not observed any trends that dictate how colony size correlates with activity.
Figure 2: Screening for toxicity suppressors. Yeast cotransformed with pAG303GAL-TDP-43 and the pAG416GAL-Hsp104 library were plated onto glucose (repressing) or galactose (inducing) media. TDP-43 is highly toxic, so very few Hsp104 variants are capable of suppressing this toxicity, and this screen is very stringent. Single colonies are selected as hits from the galactose plate for the 5-FOA secondary screen. Both large and small colonies are typically obtained, but we have not observed any trends that dictate how colony size correlates with activity.
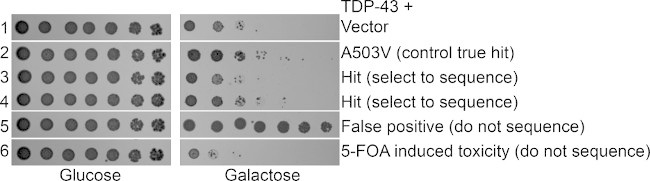 Figure 3: 5-FOA secondary screen. Yeast treated with 5-FOA to counter select for the Hsp104 plasmid were assessed by spotting assay. Strains were grown in SRaff-His media, serially diluted 5 fold, and spotted in duplicate onto SD-His and SGal-His plates. Hsp104A503V is a true hit that we have previously verified and show here as a control along with vector alone17. Rows 3 and 4 are library hits that retain TDP-43 toxicity following loss of the Hsp104 suppressor. Row 5 shows a false positive, where TDP-43 is no longer toxic, so a nonspecific toxicity suppressor is present. Row 6 shows a strain that is more toxic than TDP-43 typically is, possibly due to 5-FOA toxicity. This strain could still be sequenced but may not be a valid hit.
Figure 3: 5-FOA secondary screen. Yeast treated with 5-FOA to counter select for the Hsp104 plasmid were assessed by spotting assay. Strains were grown in SRaff-His media, serially diluted 5 fold, and spotted in duplicate onto SD-His and SGal-His plates. Hsp104A503V is a true hit that we have previously verified and show here as a control along with vector alone17. Rows 3 and 4 are library hits that retain TDP-43 toxicity following loss of the Hsp104 suppressor. Row 5 shows a false positive, where TDP-43 is no longer toxic, so a nonspecific toxicity suppressor is present. Row 6 shows a strain that is more toxic than TDP-43 typically is, possibly due to 5-FOA toxicity. This strain could still be sequenced but may not be a valid hit.
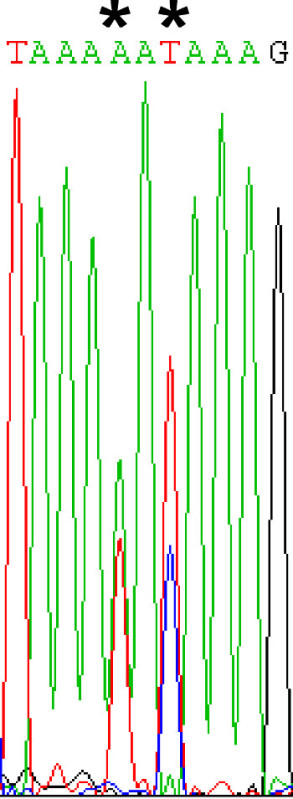 Figure 4. Analyzing sequencing results. Selected yeast often contains multiple plasmids. Therefore, following sequencing of the colony PCR products, care must be taken to ensure any mutations are not overlooked in the chromatograms. In this chromatogram, two potential mutations (denoted by *) could be overlooked. In the first instance, there is a mixture of A and T and in the second, a mixture of T and C.
Figure 4. Analyzing sequencing results. Selected yeast often contains multiple plasmids. Therefore, following sequencing of the colony PCR products, care must be taken to ensure any mutations are not overlooked in the chromatograms. In this chromatogram, two potential mutations (denoted by *) could be overlooked. In the first instance, there is a mixture of A and T and in the second, a mixture of T and C.
Discussion
Here we present our approach to isolating potentiated Hsp104 variants that suppress the toxicity of disease-associated substrates using yeast proteinopathy models. Using this approach, large libraries of variants can be screened in high throughput, with the only limitation being the number of variants that pass the 5-FOA secondary screen. By performing these steps in 96 well format, we routinely screen up to 200 hits at a time in the 5-FOA step over the course of 1-2 weeks. The number of hits obtained during the initial screening step will vary largely depending upon substrate toxicity and the makeup of each particular library. When sequencing hits, it is essential to carefully analyze all sequencing chromatograms since mixtures of plasmids are typically present in each cell.
We have sometimes noted a large percentage of Hsp104WT being isolated as ‘hits’. This is likely due to the presence of spontaneous genetic suppressors or very weak activity. To circumvent this problem, we have found that screening at elevated temperatures (e.g. 34 °C) can decrease the percentage of ‘hits’ that are Hsp104WT. It is also essential to re-clone any sequenced hits to independently assess the activity of any novel variants and to ensure plasmid purity. Once new hits are identified, they can be assessed for suppression of aggregation in yeast and characterized biochemically.
We have used these methods to isolate potentiated Hsp104 variants against TDP-43, FUS, and α-synuclein and verified their activity using pure protein biochemistry assays17. Ultimately, these methods could be used to screen protein libraries against any substrate of interest that is toxic in yeast.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank Sue Lindquist, Aaron Gitler, and Martin Duennwald for kindly sharing reagents. Our studies were supported by: an American Heart Association Post-Doctoral Fellowship (M.E.J); NIH Director's New Innovator Award DP2OD002177, NIH grants R21NS067354, R21HD074510, and R01GM099836, a Muscular Dystrophy Association Research Award (MDA277268), Packard Center for ALS Research at Johns Hopkins University, Target ALS, and an Ellison Medical Foundation New Scholar in Aging Award (J.S.).
References
- Johnson BS, McCaffery JM, Lindquist S, Gitler AD. A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc Natl Acad Sci USA. 2008;105(17):6439–6444. doi: 10.1073/pnas.0802082105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, et al. Molecular Determinants and Genetic Modifiers of Aggregation and Toxicity for the ALS Disease Protein FUS/TLS. PLoS Biol. 2011;9(4) doi: 10.1371/journal.pbio.1000614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outeiro TF, Lindquist S. Yeast Cells Provide Insight into Alpha-Synuclein Biology and Pathobiology. Science. 2003;302(5651):1772–1775. doi: 10.1126/science.1090439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman MS, Trojanowski JQ, Lee VM. Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat Med. 2004;10(10):1055–1063. doi: 10.1038/nm1113. [DOI] [PubMed] [Google Scholar]
- Morimoto RI. Stress, aging, and neurodegenerative disease. N Engl J Med. 2006;355(21):2254–2255. doi: 10.1056/NEJMcibr065573. [DOI] [PubMed] [Google Scholar]
- Elden AC, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466(7310):1069–1075. doi: 10.1038/nature09320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AA, et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science. 2006;313(5785):324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler AD, et al. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nature genetics. 2009;41(3):308–315. doi: 10.1038/ng.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su LJ, et al. Compounds from an unbiased chemical screen reverse both ER-to-Golgi trafficking defects and mitochondrial dysfunction in Parkinson's disease models. Disease models & mechanisms. 2010. pp. 194–208. [DOI] [PMC free article] [PubMed]
- Tardiff DF, et al. Yeast reveal a 'druggable' Rsp5/Nedd4 network that ameliorates alpha-synuclein toxicity in neurons. Science. 2013;342(6161):979–983. doi: 10.1126/science.1245321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardiff DF, Tucci ML, Caldwell KA, Caldwell GA, Lindquist S. Different 8-hydroxyquinolines protect models of TDP-43 protein, alpha-synuclein, and polyglutamine proteotoxicity through distinct mechanisms. The Journal of biological chemistry. 2012;287(6):4107–4120. doi: 10.1074/jbc.M111.308668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, et al. Therapeutic modulation of eIF2alpha phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nature. 2014;46(2):152–160. doi: 10.1038/ng.2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju S, et al. A yeast model of FUS/TLS-dependent cytotoxicity. PLoS Biol. 2011;9(4) doi: 10.1371/journal.pbio.1001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J. Hsp104: a weapon to combat diverse neurodegenerative disorders. Neurosignals. 2008;16(1):63–74. doi: 10.1159/000109760. [DOI] [PubMed] [Google Scholar]
- Vashist S, Cushman M, Shorter J. Applying Hsp104 to protein-misfolding disorders. Biochem Cell Biol. 2010;88(1):1–13. doi: 10.1139/o09-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis ME, et al. Operational Plasticity Enables Hsp104 to Disaggregate Diverse Amyloid and Nonamyloid Clients. Cell. 2012;151(4):778–793. doi: 10.1016/j.cell.2012.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackrel ME, et al. Potentiated Hsp104 Variants Antagonize Diverse Proteotoxic Misfolding Events. Cell. 2014;156(1-2):170–182. doi: 10.1016/j.cell.2013.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittrup KD, Verdine GL. In: Methods in Enzymology. Wittrup KD, Verdine GL, editors. Vol. 503. Academic Press; 2012. pp. 13–14. [Google Scholar]
- Boeke JD, Trueheart J, Natsoulis G, Fink GR. Methods in Enzymology. Vol. 154. Lawrence Grossman Ray Wu: Academic Press; 1987. pp. 164–175. [DOI] [PubMed] [Google Scholar]
- Miyazaki K. eds FrancesH Arnold & George Georgiou. Vol. 231. Humana Press; 2003. Creating Random Mutagenesis Libraries by Megaprimer PCR of Whole Plasmid (MEGAWHOP). Directed Evolution Library Creation: Methods in Molecular Biology; pp. 23–28. [DOI] [PubMed] [Google Scholar]
- Saibil H. Chaperone machines for protein folding, unfolding and disaggregation. Nat Rev Mol Cell Biol. 2013;14(10):630–642. doi: 10.1038/nrm3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protocols. 2007;2(1):31–34. doi: 10.1038/nprot.2007.13. [DOI] [PubMed] [Google Scholar]
- Armakola M, Hart MP, Gitler AD. TDP-43 toxicity in yeast. Methods. 2011;53(3):238–245. doi: 10.1016/j.ymeth.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberti S, Gitler AD, Lindquist S. A suite of Gateway® cloning vectors for high-throughput genetic analysis in Saccharomyces cerevisiae. Yeast. 2007;24(10):913–919. doi: 10.1002/yea.1502. [DOI] [PMC free article] [PubMed] [Google Scholar]