

Abstract
Background:
Some reports revealed that rapamycin could reactivate HBV infection. However, the mechanism has not been clearly explained.
Objectives:
In this report, we studied the mechanism by which rapamycin enhances HBV replication and expression by inducing cellular autophagy.
Materials and Methods:
HepG2.2.15 cells were treated with rapamycin to induce autophagy. Autophagosomes were observed by fluorescence microscopy and transmission electron microscopy. Autophagy marker protein LC3-Ⅱ/LC3-Ⅰwas detected by Western blotting. HBV DNA and mRNA were determined by real time PCR and Southern blotting. HBsAg was evaluated by ELISA.
Results:
In HepG2.2.15 cells, HBV DNA and HBsAg increased when host cells were treated with rapamycin and the effect was reversed by autophagy inhibitor, 3-methyladenine (3-MA).
Conclusions:
These results indicated a potential explanation for reactivation of HBV infection when patients with hepatitis receive rapamycin.
Keywords: HBV, Rapamycin, Autophagy
1. Background
It is increasingly obvious that Mammalian target of rapamycin (mTOR) signaling affects most major cellular functions including autophagy. It plays an important role in regulating basic cell behaviors like growth, proliferation and apoptosis. Interestingly, dysregulation of mTOR signaling pathway occurs in many human diseases, such as cancers, obesity, type II diabetes and neurodegeneration (1). Some mTOR inhibitors have been used for various malignancies (2), because of their immunosuppressive properties. Rapamycin is the canonical inhibitor of the mTOR serine/threonine kinase. It was first considered an antifungal agent, but later developed as an immunosuppressant and anticancer medication. Large of evidence pointed out that rapamycin is able to activate autophagy and therefore used as an inducer in the research of autophagy (3). Autophagy is an evolutionarily conserved catabolic pathway where cells deliver their own cytoplasm materials and/or organelles to lysosomes for degradation. It plays a critical role in the homeostatic process of recycling proteins and organelles, and then linked to developmental and pathological conditions (4). Notably, autophagy was also reported to implicate with survival or clearance of various pathogens in host cells (5). Several previous studies investigated the interaction between Hepatitis B virus (HBV) chronic infection and autophagy (4, 6, 7). HBV was discovered to be able to induce autophagy without nutrient starvation and consequently benefit from autophagy activation for its replication (8, 9).
2. Objectives
In this report, we studied the mechanism by which rapamycin enhances HBV replication and expression by inducing cellular autophagy.
3. Materials and Methods
3.1. Chemicals and Antibodies
Rapamycin and 3-methyladenine (3-MA) were obtained from Sigma-Aldrich (St Louis, USA). Anti-Β-actin antibody (ab3280) was from Abcam (Cambridge, UK). Anti-LC3 antibody was obtained from Cell Signaling Technology (Boston, USA). Anti-p62 was purchased from Santa Cruz Biotechnology (CA, USA); Surface antigen (HBsAg) antibody was obtained from Gene Company Limited (Hong Kong, China).
3.2. Cell Culture and Transfection
The hepatocellular carcinoma cell line which stably expressing HBV, HepG2.2.15 was maintained in minimum essential medium (Gibco, USA) containing 10% fetal bovine serum (Hyclone, USA). A final concentration of 200 mg/mL G418 (Gibco, USA) was added to the cell cultures. Transient transfection was performed with pGFP-LC3 plasmids using lipofectamine 2000 (Invitrogen, USA) according to the manufacturer’s instructions.
3.3. HBsAg Assays
HBsAg secretion in the culture supernatants of HepG2.2.15 was measured by HBsAg ELISA kit and performed according to the manufacturer's instruction (Lizhu, Zhuhai, China). HBsAg levels were shown with the value at the optical density of 450 nm (OD 450) using an automatic microtiter plate reader (Thermo Scientific MultiskanGo, USA). Experiments were performed in triplicate independently from each other.
3.4. Autophagy Assays
For starvation, HepG2.2.15 cell lines were incubated in serum-free Earle’s balanced salt solution (EBSS; starvation medium; Invitrogen, USA) for indicated hours. For rapamycin treatment, HepG2.2.15 cell lines were incubated in rapamycin medium for indicated hours and concentrations. Autophagy was measured as follows; briefly, the cells were transfected with pGFP-LC3 and maintained in normal, starvation or rapamycin medium on coverslips, and then the autophagic fluorescence dots, which represent the autophagosome were observed and counted under fluorescence microscope. Cells containing three or more fluorescence dots were defined as autophagy-positive cells. A minimum of 100 GFP-positive cells per sample was counted and these independent experiments were performed for three times. The cellular localization pattern of GFP-LC3 was photographed using a Leica TCS SP2 confocal microscope (Leica, Wetzlar, Germany).
3.5. Western Blot Analysis
After transfection and treatment, cells were washed with phosphate-buffered saline (PBS) and lysed with 5* SDS protein loading buffer (100 mM Tris-HCl, pH 6.8, 20% glycerol, 10% mercaptoethanol, 4% SDS and 0.2% bromophenol blue). The cell lysates were centrifuged after boiling for 10 minutes. Supernatants of each sample were loaded to and separated by SDS-PAGE and then transferred to PVDF membranes (Bio-Rad, Hemel Hempstead, USA). The membranes were incubated with the indicated primary antibodies overnight at 4°C after being blocked with 5% non-fat milk for one hour. The membranes were washed with 1 × TBS-Tween and incubated with secondary antibodies (Zhongshan, Shanghai, China). Protein signals were visualized by an enhanced chemiluminescence system (ECL, Biyuntian, and Wuhan, China).
3.6. Transmission Electron Microscopy
Briefly, after transfection with pGFP-LC3 and treatments with rapamycin or starvation, cells were centrifuged and the liquid supernatants were removed. Collected cells were fixed in 2% paraformaldehyde and 0.1% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) at room temperature for one hour. The cell pellets were postfixed with 1% Osmium tetroxide for two hours on ice and rinsed three times with distilled water. Next, fixed cell pellets were dehydrated through an ethanol (ETOH) dilution series up to 100% ETOH and immersed in propylene oxide (PO) for two minutes, three times. Pellets were then infiltrated in a 3:1 PO/Eponate resin mixture overnight and then successively embedded in 100% Eponate resin (Ted Pell Inc., Redding, CA, USA) in beam capsules and allowed to harden at 65 ºC in oven overnight, tissue blocks were ultrathin-sectioned at a 70 nm thickness and placed on 300 mesh copper grids after hardening. Grids were counterstained with saturated urinal acetate and lead citrate and then viewed.
3.7. Real Time PCR/RT-PCR Analysis
Total RNA was extracted from the cells using Trizol reagent (Invitrogen, USA) and cDNA was synthesized from 1 µg of total RNA using Superscript II reverse transcriptase (Invitrogen, USA) according to the manufacturers' instructions. HBV DNA in cells was extracted and quantitated with BIO-RAD CFX 96 (BIO-RAD, USA) system using a SyberGreen real time PCR kit (Roche, Switzerland) with the primers 5′-CCTAGTAGTCAGTTATGTCAAC-3′ (forward) and 5′-TCTATAAGCTGGAGGAGTGCGA-3′ (reverse). Concisely, PCR was run with primary denaturing step at 95℃ for 3 minutes, followed by 35 cycles at 94℃ for 15 seconds and annealing at 55ºC for 30 seconds. Data was collected and real-time analysis was performed.
3.8. Southern Blot Analysis
Intracellular viral nucleocapsid-associated DNA in cells was extracted as described previously (10), resolved by electrophoresis on a 1% agarose gel, and transferred onto a positively charged nylon membrane. All membranes were hybridized with digoxigenin-labeled HBV probes as reported previously (11). Hybridization and detection were performed using the DIG high prime DNA labeling and detection starter kit II (Roche, Switzerland) according to the manufacturer’s instructions and then detected using chemiluminescence (Roche, Switzerland).
3.9. Statistical Analysis
Results were presented as means ± standard deviations. Significance of differences was determined by Student’s T-test and sum rank test. P < 0.05 was considered as statistically significant.
4. Results
4.1. Rapamycin Enhanced HBV Production in HepG2.2.15
HepG2.2.15 cells were used to detect HBV DNA copies and HBsAg release in supernatants (10). As shown in Figure 1 A, HBsAg in supernatants of cells treated with rapamycin increased significantly, with an OD450 value of 6.5; whereas, the level of the control group was only 3.7. To further study the role of rapamycin in HBV replication and expression, intracellular HBV RNA and the HBV DNA copies were detected by real time RT-PCR or PCR. The relative level of HBV mRNA increased to about two fold compared to control group (Figure 1 B). Meanwhile, HBV DNA copies increased significantly from 0.82 × 105 to 2.75 × 106 (Figure 1 C). The results suggested a specific stimulatory effect of rapamycin on HBV production.
Figure 1. Rapamycin Increased the Production of HBV in HepG2.2.15.
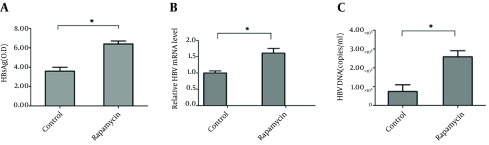
(A) On 48 hours after the treatment, supernatants of cell cultures were collected and HBsAg was detected by ELISA. (B) On 72 hours after the treatment, HBV RNA was measured by Real-time PCR. (C) On 72 hours after the treatment, HBV DNA was determined by Real-time PCR, HepG2.2.15 Cells Were Treated With or Without Rapamycin (50 nM).
4.2. Time and Dose Effect of Rapamycin on HBsAg Release
To observe the time effect of rapamycin on HBV release, HepG2.2.15 cells were treated with rapamycin for various times, from 0 hour to 36 hours, then the supernatants of cell cultures were collected in dated time points and HBsAg levels were determined. HBsAg in cell cultures increased concomitantly with the prolonged time, reaching climax at 36 hours with a value of 6.2 (Figure 2 A). When the treatment time exceeded 36 hours, the concentration of HBsAg began to decrease. Probably because prolonging the treating time more than 36 hours resulted in more cell death, which cancelled the increase of HBsAg secretion. Therefore, we chose 36 hours as the optimal time point and tested various concentrations of rapamycin from 0 nM to 200 nM. The results demonstrated that nearly 60 nM was the optimal concentration (Figure 2 B). A higher concentration than 200 nM resulted in a great hazard on cells and decreased HBsAg level (12).
Figure 2. The Time and Dose Effect of Rapamycin on HBsAg Production.
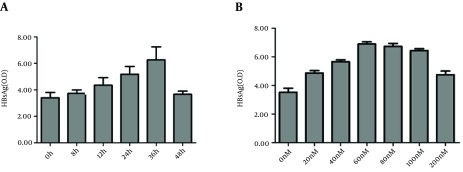
Various concentrations of rapamycin were added into the HepG2.2.15 cells culture and the supernatants of cell cultures were collected at various time points. HBsAg level was assessed by ELISA. (A) Cells were treated with rapamycin (60 nM) and culture mediums were harvested in different time points (0 hour, 8 hours, 12 hours, 24 hours, and 36 hours, 48 hours) to detect HBsAg. (B) Various concentrations of rapamycin were added into the cell cultures and HBsAg levels were detected at 36 hours.
4.3. Rapamycin Induced Autophagy in HepG2.2.15
Rapamycin was reported to induce autophagy by inhibiting mTOR signal (13-15). To investigate whether autophagy mechanism plays a key role during the process of activation of HBV by rapamycin, autophagy phenomenon was observed by different methods in HepG2.2.15 cells. Since LC3 modification correlated with its relocalization to autophagy vesicles, the relocalization of LC3 from a diffuse cytoplasmic distribution to distinct punctual has been used as indicator of autophagy activation (16). Subcellular localization of GFP-LC3 protein was monitored by confocal microscopy. GFP-LC3 redistribution into discrete dots was dramatically increased by starvation or rapamycin treatment; whereas, autophagic vacuoles were hardly observed in the control group (Figure 3 A). The percentage of GFP-LC3-positive cells containing green punctate dots in the rapamycin-treated cells was increased almost 3.5-fold compared to the control (Figure 3 B). The characteristic ultrastructure of autophagy, double-layer membrane autophagosome, was also observed by transmission electron microscopy in treated HepG2.2.15 (Figure 3 C). Microtubule-associated protein-Light-chain 3 (LC3) changed from a free form (LC3-I) to a phosphatidylethanolamine-conjugated form (LC3-II) when autophagy happened. The accumulation of LC3-II is another commonly used marker of autophagy (17). Rapamycin markedly induced LC3-II expression and reversed the ratios of LC3-I/LC3-II compared to the control by western blot analysis. The level of cytoplastic HBsAg expression was in line with LC3-II expression (Figure 3 D, 3E and 3F). These results showed that rapamycin might increase HBsAg expression by activating autophagy. Detection of P62 expression also showed activation of complete autophagic process induced by rapamycin with a significant decrease of P62, compared to control group, similar to the effect of starvation (Figure 3 G and 3H).
Figure 3. Rapamycin Induced Autophagy Phenomenon in HepG2.2.15.

(A) HepG2.2.15 cells were transfected with pGFP-LC3 and autophagic dots were observed by confocal laser microscopy. (B) Quantitation of the autophagic cells in HepG2.2.15, the cell lines were under treatment of rapamycin of 36 hours, starvation or no treatment (control). (C) Representative electron micrographs of HepG2.2.15 cells under starvation or rapamycin, double membrane organelles and cytoplasmic contain were marked by arrow. (D) HepG2.2.15 cells were treated with rapamycin, starvation or untreated (control) less than 36 hours. The levels of LC3-II and HBsAg protein expression were determined by Western blot analysis. Β-actin expression was examined as a protein loading control. (E) The LC3-II/B-actin ratios were quantified by densitometry analysis using Quantity One software (Bio-Rad). Results represent the mean data from three independent experiments. *, P < 0.05 (F) the HBsAg/B-actin ratios was quantified by densitometry analysis using Quantity One software. Results represent the mean data from three independent experiments.*, P < 0.05. (G) P62 protein expression was determined by Western blot analysis. (H) The P62/B-actin ratio was quantified by densitometry analysis using Quantity One software. Results represent the mean data from three independent experiments. *, P < 0.05.
4.4. Autophagy Inhibitor Abrogated the Effect of Rapamycin on HBV Production
To test the dependence of autophagy induced by rapamycin on HBV production, HBsAg secretion levels in cells supernatants treated with different drugs were detected by ELISA. Compared to control group, HBsAg level increased by 1.2 fold in cells treated with rapamycin. When the autophagy inhibitor 3-MA was added to the culture, HBsAg level decreased significantly compared to rapamycin group, more than a half, even less than the control group (Figure 4 A). Rapamycin treatment resulted in an obvious increase of HBV replication intermediates in HepG2.2.15 cells compared to the control; whereas, 3-MA treatment abrogated the upregulation effect (Figure 4 B). With western blot, LC3-II expression decreased nearly by 40% after the cells were treated with 3-MA, concomitant with a reduction of HBsAg expression (Figure 4 C). Collectively, these results revealed that rapamycin enhanced HBV replication by activating autophagy in HepG2.2.15, and the effect could be blocked by autophagy inhibitor.
Figure 4. Autophagy Machinery Is Required to Increase HBV Production Stimulated by Rapamycin.
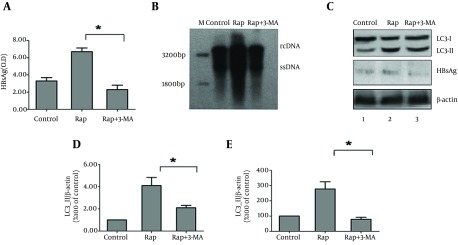
(A) HepG2.2.15 cells were treated with rapamycin for 36 hours and then added with 3-MA (10 mM). After 12 hours, the supernatants of cell cultures were collected and HBsAg was assessed by ELISA. (B) The amounts of HBV replication intermediates in HepG2.2.15 cells were detected by Southern blot. The positions of HBV relaxed circular (RC) and single-stranded (SS) DNA were indicated. (C) The levels of LC3-II, and HBsAg protein expression were determined by Western blot analysis. (D and E) The LC3-II/B-actin ratios and HBsAg/B-actin ratios were quantified by densitometry analysis using Quantity One software. Results represent the mean data from three independent experiments. *, P < 0.05.
5. Discussion
A recent annual nationwide survey confirmed that HBV reactivation related to immunosuppressive therapy is increasing in patients with malignant lymphoma, other hematological malignancies, solid tumors or rheumatologic disease (18). Immunosuppressive drugs such as corticosteroids or azathioprine can induce HBV reactivation in patients carrying HBsAg or anti-HBc, but much less frequently than chemotherapy treatments (19). The mechanism by which chemotherapy drugs reactivate HBV has not yet been clearly elucidated. Rapamycin is one of the newer targeted therapies approved for the management of RCC (20). It is also associated with improved survivals after liver transplantation for HCC. In non-HCC patients, rapamycin lowered their survival rate, showing its beneficial impact for patients with cancer (21). In Europe, product information for rapamycin warns that patients should be evaluated regarding the risk of HBV reactivation, because it occurred in some patients taking rapamycin (22). In this study, our results demonstrated that rapamycin could increase HBV production and HBsAg release by inducing autophagy. Autophagy literally means “to eat oneself”. It plays an important role in lysomal degradation and recycling long-life proteins. Autophagy is reported to be involved in several pathophysiological conditions such as tumor progression, neurodegenerative disorders and myopathies. It is also regarded as a mechanism of innate immunity against invading pathogens. Although the autophagy pathway is emerging as a component of host defense, some viruses have developed strategies to counteract or even use this antiviral mechanism (23). A common pathway was shared by several herpes viruses including HSV-1, KSHV and γHV68 to suppress autophagy (24) and others seem to have coped with the autophagy machinery as proviral host factors to enhance viral replication (23). Regarding HCV, reported data are conflicting. One report suggested that rapamycin may suppress HCV recurrence measured by PCR in HCV-positive liver transplant recipients (25). On the opposite side, some researchers argued autophagy contributed to effective production of HCV particles, although with little effect on intracellular production of HCV mRNA and HCV-related proteins (26). For HBV, recent studies mentioned the importance of autophagy for its production. Two groups reported that autophagy could be induced by HBV and upregulated virus production in certain degree in return (4, 12). One report showed that inhibition of autophagy pathway by 3-MA or siRNA targeting hVps34/ATG7 suppressed the production of HBV DNA (4). Another report showed that inhibition of autophagy by 3-MA or knockdown of Beclin1 or ATG5 decreased HBV DNA replication, but just a slight inhibitory effect on nucleocapsid-associated DNA level, indicating that 3-MA blocks the release of HBsAg (12). This gap might be a consequence of different methods or different cell lines used in the research. In our study, rapamycin was found to enhance the production of HBV significantly, including HBV RNA, HBV DNA and HBsAg expression and release. The enhancement was brought out by activation of autophagy, which could be abrogated by autophagy inhibitors such as 3-MA. The autophagic machinery is often used in defense against microbes. It may selectively deliver microorganisms to degradative lysosomes for direct degradation, or deliver microbial nucleic acids and antigens to endo/lysosomal compartments for activation of innate and adaptive immunity, so that clear the invading pathogen by an indirect way. However, some pathogens were reported to have the ability to escape autophagy related degradation; nevertheless, using the mechanism of autophagy to benefit their life cycle. For HBV, autophagy may participate in the envelopment of viral particle, or promote its release from cellular membranes (4, 12). As a result, the life cycle of HBV production is accelerated by autophagy. In summary, our study showed that rapamycin could enhance the release of HBsAg and increase HBV DNA production by activating autophagy. Treatment with 3-MA, HBsAg and HBV DNA levels were decreased markedly. Since patients with chronic HBV complicated with other diseases such as cancer or autoimmune disorders often receive immunosuppressive chemotherapy, clinicians should be aware of HBV reactivation. Surveillance of serum markers of HBV should be considered for those patients in the process of chemotherapy.
Footnotes
Authors’ Contributions:Study concept and design: Weixian Chen. Analysis and interpretation of data: Wenjuan Huang, Fengrong Zhao and Xia Li. Drafting of the manuscript: Wenjuan Huang. Critical revision of the manuscript for important intellectual content: Weixian Chen and Ying Huang. Statistical analysis: Sufei Zhu and Qiao He, Wenjuan Huang and Fengrong Zhao were the joint first author and contributed equally for this work.
Funding/Support:The reported work was supported in part by a research grant number (81171628) from the National Natural Science Foundation of China.
References
- 1.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee JT, Lehmann BD, Terrian DM, Chappell WH, Stivala F, Libra M, et al. Targeting prostate cancer based on signal transduction and cell cycle pathways. Cell Cycle. 2008;7(12):1745–62. doi: 10.4161/cc.7.12.6166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284(12):8023–32. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sir D, Tian Y, Chen WL, Ann DK, Yen TS, Ou JH. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc Natl Acad Sci U S A. 2010;107(9):4383–8. doi: 10.1073/pnas.0911373107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Junkins RD, Shen A, Rosen K, McCormick C, Lin TJ. Autophagy enhances bacterial clearance during P. aeruginosa lung infection. PLoS One. 2013;8(8):e20719. doi: 10.1371/journal.pone.0072263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kotsafti A, Farinati F, Cardin R, Cillo U, Nitti D, Bortolami M. Autophagy and apoptosis-related genes in chronic liver disease and hepatocellular carcinoma. BMC Gastroenterol. 2012;12:118. doi: 10.1186/1471-230X-12-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang H, Da L, Mao Y, Li Y, Li D, Xu Z, et al. Hepatitis B virus X protein sensitizes cells to starvation-induced autophagy via up-regulation of beclin 1 expression. Hepatology. 2009;49(1):60–71. doi: 10.1002/hep.22581. [DOI] [PubMed] [Google Scholar]
- 8.Shlomai A, Paran N, Shaul Y. PGC-1alpha controls hepatitis B virus through nutritional signals. Proc Natl Acad Sci U S A. 2006;103(43):16003–8. doi: 10.1073/pnas.0607837103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290(5497):1717–21. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li J, Lin S, Chen Q, Peng L, Zhai J, Liu Y, et al. Inhibition of hepatitis B virus replication by MyD88 involves accelerated degradation of pregenomic RNA and nuclear retention of pre-S/S RNAs. J Virol. 2010;84(13):6387–99. doi: 10.1128/JVI.00236-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim JW, Lee SH, Park YS, Hwang JH, Jeong SH, Kim N, et al. Replicative activity of hepatitis B virus is negatively associated with methylation of covalently closed circular DNA in advanced hepatitis B virus infection. Intervirology. 2011;54(6):316–25. doi: 10.1159/000321450. [DOI] [PubMed] [Google Scholar]
- 12.Li J, Liu Y, Wang Z, Liu K, Wang Y, Liu J, et al. Subversion of cellular autophagy machinery by hepatitis B virus for viral envelopment. J Virol. 2011;85(13):6319–33. doi: 10.1128/JVI.02627-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cao C, Subhawong T, Albert JM, Kim KW, Geng L, Sekhar KR, et al. Inhibition of mammalian target of rapamycin or apoptotic pathway induces autophagy and radiosensitizes PTEN null prostate cancer cells. Cancer Res. 2006;66(20):10040–7. doi: 10.1158/0008-5472.CAN-06-0802. [DOI] [PubMed] [Google Scholar]
- 14.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22(2):124–31. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carames B, Hasegawa A, Taniguchi N, Miyaki S, Blanco FJ, Lotz M. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann Rheum Dis. 2012;71(4):575–81. doi: 10.1136/annrheumdis-2011-200557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klionsky DJ, Cuervo AM, Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy. 2007;3(3):181–206. doi: 10.4161/auto.3678. [DOI] [PubMed] [Google Scholar]
- 17.Li J, Ni M, Lee B, Barron E, Hinton DR, Lee AS. The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death Differ. 2008;15(9):1460–71. doi: 10.1038/cdd.2008.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oketani M, Ido A, Uto H, Tsubouchi H. Prevention of hepatitis B virus reactivation in patients receiving immunosuppressive therapy or chemotherapy. Hepatol Res. 2012;42(7):627–36. doi: 10.1111/j.1872-034X.2012.00998.x. [DOI] [PubMed] [Google Scholar]
- 19.Manzano-Alonso ML, Castellano-Tortajada G. Reactivation of hepatitis B virus infection after cytotoxic chemotherapy or immunosuppressive therapy. World J Gastroenterol. 2011;17(12):1531–7. doi: 10.3748/wjg.v17.i12.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Costa LJ, Drabkin HA. Renal cell carcinoma: new developments in molecular biology and potential for targeted therapies. Oncologist. 2007;12(12):1404–15. doi: 10.1634/theoncologist.12-12-1404. [DOI] [PubMed] [Google Scholar]
- 21.Rautou PE, Mansouri A, Lebrec D, Durand F, Valla D, Moreau R. Autophagy in liver diseases. J Hepatol. 2010;53(6):1123–34. doi: 10.1016/j.jhep.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 22.Toso C, Merani S, Bigam DL, Shapiro AM, Kneteman NM. Sirolimus-based immunosuppression is associated with increased survival after liver transplantation for hepatocellular carcinoma. Hepatology. 2010;51(4):1237–43. doi: 10.1002/hep.23437. [DOI] [PubMed] [Google Scholar]
- 23.Dreux M, Chisari FV. Viruses and the autophagy machinery. Cell Cycle. 2010;9(7):1295–307. doi: 10.4161/cc.9.7.11109. [DOI] [PubMed] [Google Scholar]
- 24.Sir D, Ou JH. Autophagy in viral replication and pathogenesis. Mol Cells. 2010;29(1):1–7. doi: 10.1007/s10059-010-0014-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brennan DC, Aguado JM, Potena L, Jardine AG, Legendre C, Saemann MD, et al. Effect of maintenance immunosuppressive drugs on virus pathobiology: evidence and potential mechanisms. Rev Med Virol. 2013;23(2):97–125. doi: 10.1002/rmv.1733. [DOI] [PubMed] [Google Scholar]
- 26.Tanida I, Fukasawa M, Ueno T, Kominami E, Wakita T, Hanada K. Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy. 2009;5(7):937–45. doi: 10.4161/auto.5.7.9243. [DOI] [PubMed] [Google Scholar]