

Abstract
In the middle cerebral artery occlusion model of ischemic injury, inflammation primarily occurs in the infarct and peripheral zones. In the ischemic zone, neurons undergo necrosis and apoptosis, and a large number of reactive microglia are present. In the present study, we investigated the pathological changes in a rat model of middle cerebral artery occlusion. Neuronal necrosis appeared 12 hours after middle cerebral artery occlusion, and the peak of neuronal apoptosis appeared 4 to 6 days after middle cerebral artery occlusion. Inflammatory cytokines and microglia play a role in damage and repair after middle cerebral artery occlusion. Serum intercellular cell adhesion molecule-1 levels were positively correlated with the permeability of the blood-brain barrier. These findings indicate that intercellular cell adhesion molecule-1 may be involved in blood-brain barrier injury, microglial activation, and neuronal apoptosis. Inhibiting blood-brain barrier leakage may alleviate neuronal injury following ischemia.
Keywords: nerve regeneration, middle cerebral artery occlusion, inflammatory reactions, intercellular cell adhesion molecule-1, neurons, blood-brain barrier, microglia, NSFC grant, neural regeneration
Introduction
Ischemic stroke is a very serious disease and can lead to extensive cerebral damage and serious neurological deficits (Flynn et al., 2008). A model of ischemic stroke can be established by transient middle cerebral artery occlusion (Longa et al., 1989). The pathophysiological changes in middle cerebral artery occlusion damage include alterations in the permeability of the blood-brain barrier (Durukan and Tatlisumak, 2007; Valous et al., 2013), neuronal necrosis and apoptosis (Wang et al., 2002; Lawson and Wolf, 2009), the release of inflammatory mediators and intercellular cell adhesion molecule-1 (Zaremba and Losy, 2002), and microglial changes (Graeber and Streit, 2010). However, the sequence of events leading to ischemic injury remains unclear.
Huang et al. (2012) demonstrated that blood-brain barrier function is impaired after middle cerebral artery occlusion injury. The blood-brain barrier is dynamic and regulates leukocyte infiltration from the blood compartment into the brain. Disruption of the blood-brain barrier allows leukocytes to migrate into the brain, leading to neuronal death and apoptosis. Intercellular cell adhesion molecule-1 mediates the adhesion of leukocytes; however, the function of intercellular cell adhesion molecule-1 in blood-brain barrier dysfunction after middle cerebral artery occlusion is unclear. Pan et al. (2013) and Satoh et al. (2011) showed that microglia are involved in neuronal necrosis and apoptosis after middle cerebral artery occlusion, and microglia could release both inflammatory and anti-inflammatory factors at different time points after middle cerebral artery occlusion; however, the deleterious and/or beneficial effects of microglia in the pathological process after middle cerebral artery occlusion are not fully known. As shown in our previous studies (Cao et al., 2011; Zhang et al., 2011), anti-inflammatory factors play an important protective role in rat middle cerebral artery occlusion injury. Many studies have shown that inflammatory factors have an impact on ischemic tissue injury in the brain (Veldhuis et al., 2003; Muir et al., 2007; Brea et al., 2009; Lakhan et al., 2009; Iadecola and Anrather, 2011), but the role of inflammatory factors after middle cerebral artery occlusion is still not clear. In the present study, to provide insight into the processes underlying middle cerebral artery occlusion injury, we examined the relationship between the inflammatory reaction, neuronal necrosis/apoptosis, intercellular cell adhesion molecule-1 changes, blood-brain barrier permeability and microglial activation.
Materials and Methods
Animals
A total of 224 healthy, clean, male, Sprague-Dawley rats, aged 8–9 weeks and weighing 250–300 g, were provided by SJA Laboratory Animal Co., Ltd., Changsha, Hunan Province, China (license No. SCXK (Xiang) 2009-0004). The rats were housed in cages with a 12/12-hour light/dark cycle, regular ventilation, at 18–26°C, and relative humidity between 40–70%. Rats were given food and water ad libitum. Rats were fasted for 12 hours before operation, but drank tap water freely. The investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication No. 85-23, revised 1996), and the protocol was approved by the Institutional Animal Care Committee of Wuhan University in China. All 224 rats were randomly assigned to control (n = 14), sham surgery (n = 14) and model (n = 196) groups.
Establishment of middle cerebral artery occlusion model
A rat infarction model was induced by middle cerebral artery occlusion as previously described (Longa et al., 1989). Rats were anesthetized with 10% chloral hydrate (0.3 mL/100 g) by intraperitoneal injection and placed supine on a desk. The right common carotid artery was exposed through a median neck incision, and, using surgical forceps, the right internal carotid artery and external carotid artery were carefully isolated. The common carotid artery and internal carotid artery were blocked by two micro-artery clamps. The distal end of the external carotid artery was fastened by a 5-0 surgical suture, and then cut off. A 4-cm length of nylon monofilament (Sunbio Biotechnology Company, Beijing, China) was inserted into the internal carotid artery for middle cerebral artery occlusion through the stump of the external carotid artery. The micro-artery clamps of the internal carotid artery were undone, and then the nylon monofilament was advanced approximately 18 to 20 mm, with distance varying according to the animal's weight. The surgical suture was fastened around the intraluminal nylon monofilament in the right external carotid artery to avoid bleeding. The neck incision was then sutured. Two hours later, rats were once more anesthetized, the original incision was re-exposed, and the nylon monofilament was pulled out to establish reperfusion. The neck incision was again sutured (Peng et al., 2007). Anesthesia and vascular dissection only were performed in the sham surgery group. Rats in the control group were routinely fed.
Evaluation of middle cerebral artery occlusion model
Neurological impairment after cerebral ischemia-reperfusion injury was evaluated with a neurobehavioral test scored on a five-point scale, as described previously (Zhang et al., 2006). Neurological scores were evaluated using a modified neurological severity score (de Vasconcelos dos Santos et al., 2010) that evaluates motion, sensation, reflex and balance beam performance. Scoring was as follows: 1–6: mild damage; 7–12: moderate damage; 13–18: severe damage. Middle cerebral artery occlusion rats with scores of 7–12 were used as an ischemic injury model for further experiments.
Weighing and neurological scoring
We evaluated the neurological impairment of rats using the modified neurological severity score and weighed the rats before and 0, 0.5, 1, 2, 4, 6, 12, 24 hours and 2, 4, 6, 10, 14 and 18 days after middle cerebral artery occlusion operation.
Harvesting of brain tissue samples
Rats in the model group were anesthetized at 0, 0.5, 1, 2, 4, 6, 12, 24 hours and 2, 4, 6, 10, 14 and 18 days after middle cerebral artery occlusion (n = 14 for each time point). The right atrium and right ventricle were cut with surgical scissors. A needle was inserted into the left ventricle, and 0.9% sodium chloride (37°C) was perfused over approximately 5–10 minutes (200 mL) until the perfusate from the right atrium became colorless. Then, 4% paraformaldehyde solution (pH 7.4) was perfused for 20 minutes (about 200 mL). The brain was then taken out and immersed in 4% paraformaldehyde solution for 24 hours. Brain tissue was removed and sliced into seven continuous pieces along the coronal axis, and then embedded in paraffin.
Hematoxylin-eosin staining
Paraffin-embedded specimens were dewaxed, dehydrated and rinsed with tap water. Specimens were stained with hematoxylin for 5 minutes, rinsed with tap water, immersed in 1% hydrochloric acid/ethanol for 2 seconds, and then rinsed with tap water. Specimens were stained with 0.5% eosin aqueous solution for 3 minutes and rinsed with distilled water. The slides were dehydrated and mounted. The specimens were observed by light microscopy (ECLIPSE E200, Nikon, Tokyo, Japan).
Observation of rat brain ultrastructure by transmission electron microscopy
Sprague-Dawley rats were anesthetized and sacrificed at different time points (n = 2 for each time point). Then, 0.9% sodium chloride, 37°C, was perfused for approximately 5–10 minutes (200 mL) until the perfusate from the right atrium became colorless. The brain was rapidly taken out and the cerebral cortex was dissected into 1 mm × 1 mm × 1 mm pieces and placed in 2.5% glutaraldehyde solution at 4°C for 2 hours. After being placed in 1% osmium tetroxide fixative (pH 7.3–7.4) at 4°C for 2 hours, the tissue was successively dehydrated using increasing concentrations of ethanol and acetone. The specimen was embedded with epoxy resin and cut into ultrathin slices (50-60 nm), and then shifted to 150-mesh copper grids. After double staining with 3% acetic acid uranium and lead citrate tannic acid, the specimens were observed under a transmission electron microscope (Hitachi, Tokyo, Japan).
Cerebral infarction volume evaluated by 2,3,5-triphenyltetrazolium chloride (TTC) staining
Six rats with middle cerebral artery occlusion were perfused as above, and the brain was rapidly taken out and placed in the freezer for 20 minutes. The brain was cut into seven continuous pieces (each piece 2-mm-thick) along the coronal axis. After immersion in 1% TTC solution (Solarbio Technology Company, Beijing, China) in the dark, the slices were placed in a 37°C oven for 15 minutes. Normal brain tissue was stained red, while infarcted tissue was white. Tissues were photographed with a digital camera, and Image Pro plus 5.0 software (Media Cybernetics, Rockville, MD, USA) was used to measure the area of infarction. Infarct volume was calculated as V=∑(S1 + S2)d/2, where S1 and S2 respectively represent slices from the cranial and caudal area, and d represents slice thickness. The volume of cerebral infarction (%) was calculated as infarct volume/non infarct cerebral hemispheric volume × 100% (Bochelen et al., 1999).
ELISA for intercellular cell adhesion molecule-1, tumor necrosis factor-alpha and interleukin-1 beta
Rats in the model group were anesthetized at 0, 0.5, 1, 2, 4, 6, 12, 24 hours and 2, 4, 6, 10, 14, 18 days after middle cerebral artery occlusion (n = 14 for each time point). Cardiac puncture was done for blood collection. The serum concentrations of intercellular cell adhesion molecule-1, tumor necrosis factor-alpha and interleukin-1 beta were measured using double sandwich ELISA kits according to instructions (Zhao et al., 2011; Hoffmann et al., 2013; Tan et al., 2013). Briefly, 100 μL standard or sample were added to each well and incubated with mouse anti-rat intercellular cell adhesion molecule-1 monoclonal antibody (1:100; Uscn Life Science Inc., Wuhan, Hubei Province, China), mouse anti-rat tumor necrosis factor-alpha monoclonal antibody (1:100; Uscn Life Science Inc.) or mouse anti-rat interleukin-1 beta monoclonal antibody (1:100; Uscn Life Science Inc.) for 2 hours at 37°C. 100 μL biotin-conjugated rabbit anti-mouse IgG (1:100; Uscn Life Science Inc.) was incubated for 60 minutes at 37°C. After rinsing in PBS, 100 μL avidin conjugated to horseradish peroxidase (1:100; Uscn Life Science Inc.) was added for 30 minutes at 37°C. After rinsing in PBS, 100 μL tetramethylbenzidine was added for 20 minutes at 37°C. After adding 50 μL sulfuric acid, optical density values were measured at 450 nm with a microplate reader (Thermo Fisher Scientific, Waltham, MA, USA). Using protein samples of known concentration, standard curves were drawn, and intercellular cell adhesion molecule-1, tumor necrosis factor-alpha and interleukin-1 beta concentrations were calculated.
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) for apoptosis
Apoptosis was analyzed using the TUNEL assay (in situ cell death detection kit, Roche Group, Basel, Switzerland) as previously described (Peng et al., 2011). Briefly, paraffin-embedded samples were dewaxed and dehydrated. Tissue sections were incubated for 20 minutes at 25°C with proteinase K working liquid (20 μg/mL in 10 mmol/L Tris/HCl, pH 7.4–8). 50 μL of TUNEL reaction mixture was added and incubated for 60 minutes at 37°C in a humidified atmosphere in the dark with a lid. Sample was treated with 50 μL Converter-POD solution in a humidified chamber for 30 minutes at 37°C, and visualized with 100 μL 3,3′-diaminobenzidine for 10 minutes at 25°C. Sample was counterstained with hematoxylin. The slides were dehydrated and mounted. The slides were observed by light microscopy (ECLIPSE E200, Nikon, Tokyo, Japan), and mean optical density of TUNEL-positive cells was measured and analyzed by an image analysis system (Image Pro Plus 5.0 software, Cybernetics, Bethesda, MD, USA).
Iba-1 expression
Samples were dewaxed and dehydrated. For antigen retrieval, samples were placed in citrate buffer (pH 6.0), and heated in the microwave. All samples were treated with 3% H2O2 for 10 minutes, and then with rabbit anti-Iba-1 polyclonal antibody (1:1,000; Wako, Saitama, Japan) overnight at 4°C, with goat anti-rabbit IgG antibody-HRP polymer (1:200; Maxim, Fuzhou, Fujian Province, China) for 30 minutes at 37°C, and then visualized with 100 μL 3,3′-diaminobenzidine for 2–3 minutes. Samples were counterstained with hematoxylin, dehydrated and mounted, and then observed by light microscopy (ECLIPSE E200, Nikon, Tokyo, Japan). Images were analyzed by an image analysis system (ImagePro Plus 5.0 software, Cybernetics). The image analysis system identified and calculated the mean optical density of positive cells (Sasaki et al., 2001).
Measurement of blood-brain barrier permeability
Blood-brain barrier permeability was evaluated using Evans blue (ZSGB Biotechnology, Beijing, China) in brain as described by Lenzsér et al. (2007). The rats were anesthetized and injected intravenously with Evans blue (2% solution, 0.4 mL/100 g) for 1 hour. After perfusion with 0.9% sodium chloride (100 mL/100 g), the brain was rapidly taken out, and the cortex was separated on ice and weighed. The specimens were homogenized and immersed in the methanamide solution in a 37°C oven for 48 hours. The samples were centrifuged at 10,000 × g for 20 minutes at 4°C. For the determination of Evans blue, 100 μL supernatant was measured at 620 nm with a microplate reader (Thermo Scientific, Waltham, MA, USA). Based on a standard curve, the dye content of the sample was calculated.
Statistical analysis
Data were analyzed using SPSS 10.0 software (SPSS, Chicago, IL, USA) and statistical data were expressed as the mean ± SD. One-way analysis of variance was used to evaluate differences between groups. Paired t-test was used for intergroup comparisons. In addition, intergroup Pearson correlation analysis was conducted. A value of P < 0.05 was considered statistically significant.
Results
Changes in weight and neurological scores after middle cerebral artery occlusion
To evaluate physical and neurological changes after middle cerebral artery occlusion, weight and modified neurological severity score were monitored. Over time, weight gradually declined until 6 days after middle cerebral artery occlusion compared to the control group and sham surgery group (P < 0.01), and then weight gradually recovered (Figure 1A). The modified neurological severity score was reduced at 10 days compared to the control group and sham surgery group (P < 0.01; Figure 1B). These results suggest that physical health and neurological impairment recovered 10 days after middle cerebral artery occlusion.
Figure 1.
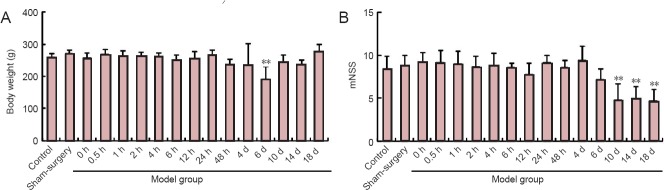
Changes in rat weight (A) and neurological scores (B) after middle cerebral artery occlusion.
Data are expressed as the mean ± SD. One-way analysis of variance was used to evaluate difference between groups. Paired t-test was used for inter-group comparison. **P < 0.01, vs. control and sham surgery groups. h: Hour(s); d: day(s); mNSS: modified neurological severity score.
Changes in infarct volume after middle cerebral artery occlusion
TTC staining showed that the percentage of infarct volume was 31.40 ± 3.36% at 48 hours post operation in the model group (Figure 2).
Figure 2.
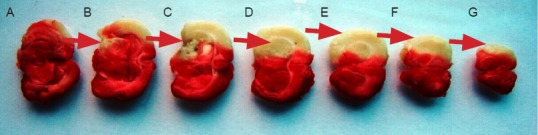
Cerebral infarct area in rats 48 hours after middle cerebral artery occlusion (2,3,5-triphenyltetrazolium chloride staining).
(A–G) The infarcted brain tissue is white. Infarction parts (red arrows) mainly include the midbrain (B), hippocampus (C), basal ganglia (D) and cortex (E–G).
Changes in neural cells in the cerebral infarction area after middle cerebral artery occlusion
Hematoxylin-eosin staining revealed that neural cells were slightly swollen in the middle cerebral artery occlusion group at 6 hours compared with the sham surgery group. Neural cells appeared swollen, with pyknosis at 12 hours after middle cerebral artery occlusion. Neural cells appeared ruptured at 24 hours after middle cerebral artery occlusion (Figure 3).
Figure 3.
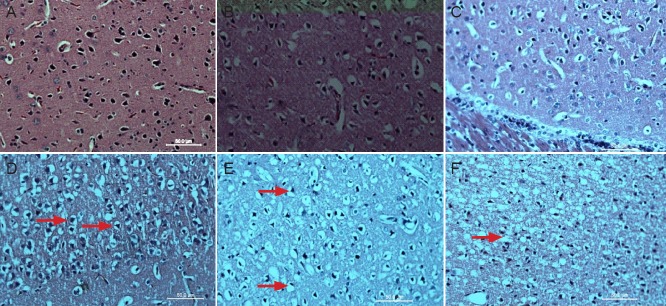
Pathological changes in the cerebral infarction area in rats with cerebral ischemia-reperfusion injury (hematoxylin-eosin staining, light microscopy, × 400).
(A) Sham surgery group. Neural cells are normal looking. (B, C) 0 and 2 hours after middle cerebral artery occlusion, neural cells display no obvi-ous change. (D) 6 hours after middle cerebral artery occlusion, neural cells appeared slightly swollen (arrows). (E) 12 hours after middle cerebral artery occlusion, neural cells appeared swollen, with a bigger volume and nuclear condensation (arrows). (F) 24 hours after middle cerebral artery occlusion, no complete cell membranes are detectable (arrow).
Remarkably, the most characteristic early signs of apoptosis (cell shrinking, chromatin condensation) were observed 4 hours after middle cerebral artery occlusion. Mitochondrial swelling and partial loss of cristae, nuclear membrane retraction, dense cytoplasm pyknosis, which are the hallmarks of the middle stages of apoptosis, were observed. The significant features of the late stages of apoptosis were visible in neural cells in the cerebral ischemic penumbra at 6 days in the middle cerebral artery occlusion group. Separated inner and outer mitochondrial membranes, ruptured cristae, and non-dense matrix spaces were visible. Mitochondrial swelling and loss of membrane cristae gradually recovered with time (Figure 4).
Figure 4.
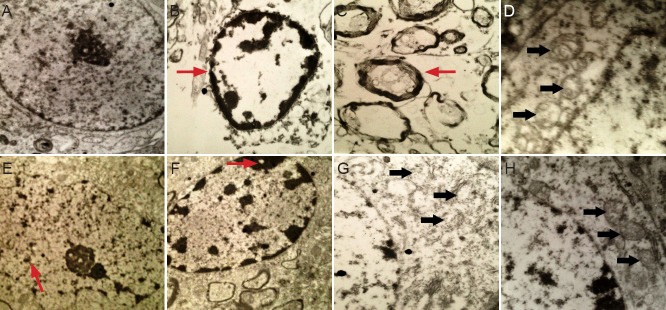
Ultrastructural alterations in the cerebral infarction area in rats with cerebral ischemia-reperfusion injury (electron microscope).
(A) Normal-looking neural cells in the sham surgery group (× 8,000). (B, C) Early apoptosis (arrows) 4 hours after middle cerebral artery occlu-sion (× 8,000). (D) Mitochondrial (arrows) changes 24 hours after middle cerebral artery occlusion (× 15,000). (E) Intermediate stages of apopto-sis (arrow) 2 days after middle cerebral artery occlusion (× 8,000). (F) Pyknosis (arrow) 4 days after middle cerebral artery occlusion (× 8,000). (G) Late stage of apoptosis (arrows) 6 days after middle cerebral artery occlusion (× 15,000). (H) Partial mitochondrial (arrows) recovery 18 days after middle cerebral artery occlusion (× 15,000).
Apoptosis in the cerebral infarction area after middle cerebral artery occlusion
No TUNEL-positive cells were visible in the sham surgery or control group. TUNEL-positive cells in the cortical area and infarct zone were observed in the middle cerebral artery occlusion group at 2 hours. The optical density of TUNEL-positive cells was increased compared to the sham surgery and control groups (P < 0.05, P < 0.01). Optical density peaked in the middle cerebral artery occlusion group at 4 days (P < 0.01). The optical density of TUNEL-positive cells was noticeably decreased 6 days after middle cerebral artery occlusion, although there was no statistically significant difference from the sham surgery and control groups (P > 0.05; Figure 5).
Figure 5.

Apoptotic cells in the cerebral infarction area of rats after middle cerebral artery occlusion (TUNEL staining).
(A–F, light microscope, × 400) Cells with yellowish-brown cytoplasm are TUNEL-positive (arrows). (A) Sham surgery group, (B–F) 2, 4 hours, 2, 4, and 6 days after middle cerebral artery occlusion. (G) Mean optical density of TUNEL staining in the infarct zone. Data are expressed as the mean ± SD. One-way analysis of variance was used to evaluate difference between groups. Paired t-test was applied for intergroup comparison. *P < 0.05, **P < 0.01, vs. sham surgery and control groups. TUNEL: Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling; h: hour(s); d: day(s).
Microglial changes in the cerebral penumbra and necrotic area after middle cerebral artery occlusion
To investigate the effect of microglia on neural cells in the cerebral penumbra and necrotic area, changes in microglia were analyzed by immunohistochemical staining for Iba-1. In the sham surgery group, only a few Iba-1-positive microglia were observed in the infarcted region. Over time, the number of Iba-1-positive cells gradually increased. Iba-1-positive cells changed morphologically to acquire a large rounded cell body, characteristic of the amoeboid state and demonstrating activation of the cells. The mean optical density was measured to quantitate the immunohistochemistry data. The optical density of Iba-1-positive cells was significantly higher in the middle cerebral artery occlusion group at 12 hours compared to the sham surgery group (P < 0.05). The peak of the optical density was detected in the middle cerebral artery occlusion group. At 10 days, the optical density of Iba-1-positive cells was significantly higher in the middle cerebral artery occlusion group at 18 days compared to the sham surgery group (P < 0.01; Figure 6).
Figure 6.

Changes in microglia in the cerebral ischemic region of rats after middle cerebral artery occlusion (immunohistochemical staining).
(A–F, light microscope, × 400). Iba-1 is a marker of microglia. Cells with yellowish-brown cytoplasm are positive (arrows). (A) Sham surgery group; (B–F) 6, 12 hours, 4, 10, and 18 days after middle cerebral artery occlusion. (G) Immunohistochemistry for microglia (Iba-1-positive cells) in the corpus callosum and striatum. Data are expressed as the mean ± SD. One-way analysis of variance was used to evaluate differences between groups. Paired t-test was applied for intergroup comparison. *P < 0.05, **P < 0.01, vs. sham surgery and control groups. h: hour(s); d: day(s).
Changes in permeability of the blood-brain barrier following middle cerebral artery occlusion
To examine blood-brain barrier permeability after middle cerebral artery occlusion, we measured the cerebral content of Evans blue by ELISA. Compared to the sham surgery and control groups, the cerebral content of Evans blue was increased in the middle cerebral artery occlusion group at 2 hours (P < 0.05). Evans blue content peaked in the middle cerebral artery occlusion group at 24 hours, and then gradually declined. There were no significant differences in Evans blue content among the middle cerebral artery occlusion, control and sham surgery groups at 10 days (P > 0.05; Figure 7).
Figure 7.
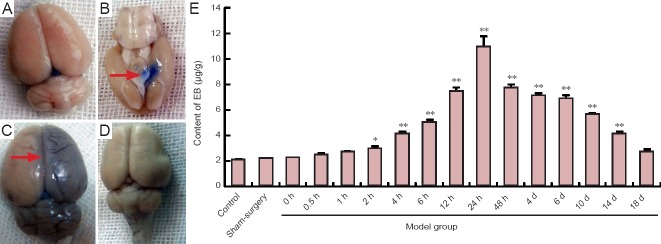
Alterations in the permeability of the blood-brain barrier in rats after middle cerebral artery occlusion.
(A) Sham-surgery group; (B) Evans blue (EB) accumulation 2 hours after middle cerebral artery occlusion (arrow); (C) EB accumulation (arrow) 24 hours after middle cerebral artery occlusion; (D) 10 days after middle cerebral artery occlusion; (E) EB content at different time points. Data are expressed as the mean ± SD. One-way analysis of variance was used to evaluate differences between groups. Paired t-test was applied for intergroup comparisons. *P < 0.05, **P < 0.01, vs. sham surgery and control groups. h: Hour(s); d: day(s).
Changes in intercellular cell adhesion molecule-1, tumor necrosis factor-alpha and interleukin-1 beta expression in rat serum after middle cerebral artery occlusion
Concentrations of intercellular cell adhesion molecule-1, tumor necrosis factor-alpha and interleukin-1 beta in the serum samples were measured by ELISA. Concentration of intercellular cell adhesion molecule-1 was significantly higher in the middle cerebral artery occlusion group at 2 hours compared to the sham surgery and control groups (P < 0.05). The peak concentration was observed at 24 hours. No significant difference in intercellular cell adhesion molecule-1 concentration was detected between the middle cerebral artery occlusion, sham surgery and control groups at 18 days (P > 0.05; Figure 8A). Interestingly, there was a significant positive correlation between the changes in serum intercellular cell adhesion molecule-1 concentration and blood-brain barrier permeability (r = 0.716, P < 0.01; Figure 8B).
Figure 8.

Expression of intercellular cell adhesion molecule-1 (ICAM-1), tumor necrosis factor-alpha (TNF-α) and interleukin-1 beta (IL-1β) in serum of rats after middle cerebral artery occlusion.
(A, C, D) Expression of ICAM-1, TNF-α and IL-1β in serum of rats. Data are expressed as the mean ± SD. One-way analysis of variance was used to evaluate differences between groups. Paired t-test was applied for intergroup comparison. *P < 0.05, **P < 0.01, vs. control and sham surgery groups; #P < 0.05, ##P < 0.01, vs. other time points. (B) Correlation between ICAM-1 concentration and permeability of the blood-brain barrier (r = 0.716, P = 0.002). (E) Correlation between TNF-á and IL-1â expression (r = 0.755, P = 0.001). Intergroup Pearson correlation analysis was conducted. h: Hour(s); d: day(s).
The concentration of tumor necrosis factor-alpha was significantly greater in the middle cerebral artery occlusion group at 0.5 hours compared to the sham surgery and control groups (P < 0.05). Twin concentration peaks were observed at 4 hours and 10 days in the middle cerebral artery occlusion group (Figure 8C). The concentration of interleukin-1 beta was significantly higher in the middle cerebral artery occlusion group at 1 hour compared to the sham surgery and control groups (P < 0.05). Twin concentration peaks were observed at 6 hours and 10 days in the middle cerebral artery occlusion group (Figure 8D). There was a significant positive correlation between the changes in tumor necrosis factor-alpha concentration and interleukin-1 beta concentration (r = 0.755, P < 0.01; Figure 8E).
Discusssion
Pathophysiological findings indicate that death and apoptosis of neurons are the main features of acute cerebral damage (Kao et al., 2006). The peripheral region of the ischemic core is known as the ischemic penumbra, in which neurons have the potential to be saved (Brait et al., 2012). Therefore, distinguishing the penumbra region from the ischemic area is critical for formulating therapeutic strategies. The inflammatory response plays an important role in the pathogenesis of middle cerebral artery occlusion injury (Iadecola and Alexander, 2001; Denes et al., 2010; Smith et al., 2013). In particular, neutrophil granulocytes (Matsuo et al., 1995; Barone et al., 1997; Satoh et al., 1999) and macrophages/microglia are the major players in cerebral ischemic infarction (Campanella et al., 2002; Lakhan et al., 2009; Shichita et al., 2009). However, the mechanisms mediating neuronal cell death, the inflammatory response and microglial changes remained unclear. In this study, we sought to clarify these processes.
In this study, hematoxylin-eosin staining showed that neurons underwent no change until 6 hours after middle cerebral artery occlusion, and then necrotic changes appeared at 12 hours, resulting in bursting 24 hours after middle cerebral artery occlusion. The number of microglia increased at 12 hours after middle cerebral artery occlusion, and they gradually became reactive. The results demonstrate that neuronal necrosis occurs earlier than the changes in microglia. Microglia are resident immune cells in the central nervous system. Under normal conditions, microglia display a quiescent phenotype in the brain. Microglia secrete and release growth factors that play important roles in the support, nutrition, protection and homeostasis of neurons (Nakajima and Kohsaka, 2001, 2004; Yilmaz and Granger, 2010; Kettenmann et al., 2011; Shigemoto-Mogami et al., 2014). Following injury, these M2 phenotype microglia transform into the M1 phenotype and produced pro-inflammatory cytokines, phagocytizing dead cells and other debris (Ekdahl et al., 2003; Graeber and Streit, 2010). Hu et al. (2012) demonstrated that microglia are able to regulate neural networks in various ways after cerebral injury. In the present study, microglia began to increase in number 12 hours after middle cerebral artery occlusion. Over time, neuronal injury also worsened. Microglia peaked 10 days after middle cerebral artery occlusion, and neurological impairment improved. Our data suggest that microglia may play a role in regulating both damage and repair events in cerebral ischemia.
Neuronal apoptosis after cerebral ischemic injury is characterized by a decrease in cell size, pyknosis, fragmentation of the nucleus, and changes in the mitochondrial membrane and cristae (Sahara et al., 1999; Horky et al., 2001; Broughton et al., 2009). The early events of apoptosis were observed 4 hours after middle cerebral artery occlusion. The hallmarks of the intermediate stage of apoptosis were observed 24 hours to 4 days after middle cerebral artery occlusion. The late stages of apoptosis were seen 6 days after middle cerebral artery occlusion. In addition, TUNEL-positive cells were more numerous at 2 hours after middle cerebral artery occlusion compared to the sham surgery and control groups. The peak of TUNEL-positive cells was detected 4 days after middle cerebral artery occlusion. The number of TUNEL-positive cells declined to normal 6 days after middle cerebral artery occlusion. We show that neuronal apoptosis occurs later than the inflammatory response. Over time, the inflammatory response became more intense, and neuronal apoptosis became more evident. Our data show that the inflammatory response promotes neuronal apoptosis.
In this study, tumor necrosis factor-alpha levels quickly rose after middle cerebral artery occlusion, and followed by an increase in interleukin-1 beta and intercellular cell adhesion molecule-1 levels, and their levels continued to increase until 24 hours, 4 and 14 days, respectively, after middle cerebral artery occlusion. In addition, tumor necrosis factor-alpha and interleukin-1 beta levels again rose at 10 days. These double peaks of tumor necrosis factor-alpha and interleukin-1 beta appeared in the model group. Tumor necrosis factor-alpha is a pleiotropic cytokine that is rapidly upregulated in the brain after injury (Feuerstein et al., 1994; Arvin et al., 1996; Barone et al., 1997; Rothwell et al., 1997). Many studies demonstrate that interleukin-1 beta plays a critical role in the brain after damage (Hopkins and Rothwell, 1995; Nicholson and Thornberry, 1997; Rothwell et al., 1997). Intercellular cell adhesion molecule-1 is important in the recruitment of leukocytes during the inflammatory process (Zaremba and Losy, 2002). The inflammatory response and the intercellular cell adhesion molecule-1 response were visible earlier than neuronal necrosis. Over time, the inflammatory response became more intense and neuronal injury more serious.
Tumor necrosis factor-alpha might be a major initiator of inflammation, and interleukin-1 beta might be a critical proinflammatory cytokine that promotes the development of uncontrolled inflammation. Intercellular cell adhesion molecule-1 might promote neuronal necrosis. In addition, we have shown that tumor necrosis factor-alpha and interleukin-1 beta again increase at 10 days after middle cerebral artery occlusion. Concomitantly, neurological impairment alleviated. Our data indicate that tumor necrosis factor-alpha and interleukin-1 beta are possibly protective at 10 days after middle cerebral artery occlusion.
Experimental studies have shown that ischemia and reperfusion of the brain can cause damage to the blood-brain barrier, leading to an increase in permeability (Yang and Rosenberg, 2011; Rosenberg, 2012). Lymphocytes and monocytes/macrophages from the circulation easily traverse the blood-brain barrier and enter the central nervous system, resulting in edema and a neuroinflammatory response, which play critical roles in neurological dysfunction in acute cerebral ischemia (Engelhardt, 2006). We have shown that there is a positive correlation between changes in intercellular cell adhesion molecule-1 concentration and blood-brain barrier permeability. Hao et al. (2000) demonstrated that the expression of intercellular cell adhesion molecule-1 on the endothelial cells played an important role in the rupture of blood-brain barrier. Ishikawa et al. (2003) demonstrated that intercellular cell adhesion molecule-1 mediates the adhesion of leukocytes and endothelial cells after cerebral ischemia-reperfusion. Intercellular cell adhesion molecule-1 participates in the pathology of acute ischemic stroke (Bogoslovsky et al., 2011). Therefore, our data suggest that intercellular cell adhesion molecule-1 might play a critical role in blood-brain barrier disruption.
In summary, inflammatory cytokines and microglia play a role in regulating both damage and repair events in cerebral ischemia after middle cerebral artery occlusion. Intercellular cell adhesion molecule-1 probably plays a role in disrupting the blood-brain barrier. Our results show that changes in tumor necrosis factor-alpha are correlated with changes in interleukin-1 beta, but the changes in intercellular cell adhesion molecule-1 were not correlated with changes in tumor necrosis factor-alpha or interleukin-1 beta. Tumor necrosis factor-alpha may be the first cytokine to induce the downstream expression of interleukin-1 beta. Our data show that microglia play a primarily beneficial role after middle cerebral artery occlusion. Zhang et al. (2012) and Fouda et al. (2013) demonstrated that neuronal injury is lessened by inhibiting expression of inflammatory factors after cerebral ischemia. Thus, suppressing the inflammatory response is a promising therapeutic strategy for the early phase of stroke.
Footnotes
Funding: This project was supported by the National Natural Science Foundation of China, No. 81160148; the Natural Science Foundation of Jiangxi Province, No. 2011033.
Conflicts of interest: None declared.
Copyedited by Patel B, Robens J, Yu J, Qiu Y, Li CH, Song LP, Zhao M
References
- Arvin B, Neville LF, Barone FC, Feuerstein GZ. The role of inflammation and cytokines in brain injury. Neurosci Biobehav Rev. 1996;20:445–452. doi: 10.1016/0149-7634(95)00026-7. [DOI] [PubMed] [Google Scholar]
- Barone FC, Arvin B, White RF, Miller A, Webb CL, Willette RN, Lysko PG, Feuerstein GZ. Tumor necrosis factor-α: a mediator of focal ischemic brain injury. Stroke. 1997;28:1233–1244. doi: 10.1161/01.str.28.6.1233. [DOI] [PubMed] [Google Scholar]
- Bochelen D, Rudin M, Sauter A. Calcineurin inhibitors FK506 and SDZ ASM 981 alleviate the outcome of focal cerebral ischemic/reperfusion injury. J Pharmacol Exp Ther. 1999;288:653–659. [PubMed] [Google Scholar]
- Bogoslovsky T, Spatz M, Chaudhry A, Maric D, Luby M, Frank J, Warach S. Circulating CD133+CD34+ progenitor cells inversely correlate with soluble ICAM-1 in early ischemic stroke patients. J Transl Med. 2011;9:145. doi: 10.1186/1479-5876-9-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brait VH, Arumugam TV, Drummond GR, Sobey CG. Importance of T lymphocytes in brain injury, immunodeficiency, and recovery after cerebral ischemia. J Cereb Blood Flow Metab. 2012;32:598–611. doi: 10.1038/jcbfm.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brea D, Sobrino T, Ramos-Cabrer P, Castillo J. Inflammatory and neuroimmunomodulatory changes in acute cerebral ischemia. Cerebrovasc Dis 27 Suppl. 2009;1:48–64. doi: 10.1159/000200441. [DOI] [PubMed] [Google Scholar]
- Broughton BRS, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke. 2009;40:e331–e339. doi: 10.1161/STROKEAHA.108.531632. [DOI] [PubMed] [Google Scholar]
- Campanella M, Sciorati C, Tarozzo G, Beltramo M. Flow cytometric analysis of inflammatory cells in ischemic rat brain. Stroke. 2002;33:586–592. doi: 10.1161/hs0202.103399. [DOI] [PubMed] [Google Scholar]
- Cao WF, Wang WZ, Gao YQ, Yan Y, Zhang HL, Liu SY, He D, Wu XM. Therapeutic effects of intracerebral transplantation of bone marrow mesenchymal stem cells on cerebral ischemia in rats. Zhongguo Shenjing Jingshen Jibing Zazhi. 2011;37:385–389. [Google Scholar]
- de Vasconcelos dos Santos A, da Costa Reis J, Diaz Paredes B, Moraes L, Jasmin, Giraldi-Guimarães A, Mendez-Otero R. Therapeutic window for treatment of cortical ischemia with bone marrow-derived cells in rats. Brain Res. 2010;1306:149–158. doi: 10.1016/j.brainres.2009.09.094. [DOI] [PubMed] [Google Scholar]
- Denes A, Thornton P, Rothwell NJ, Allan SM. Inflammation and brain injury: acute cerebral ischaemia, peripheral and central inflammation. Brain Behav Immun. 2010;24:708–723. doi: 10.1016/j.bbi.2009.09.010. [DOI] [PubMed] [Google Scholar]
- Durukan A, Tatlisumak T. Acute ischemic stroke: overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol Biochem Behav. 2007;87:179–197. doi: 10.1016/j.pbb.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Ekdahl CT, Claasen J-H, Bonde S, Kokaia Z, Lindvall O. Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci U S A. 2003;100:13632–13637. doi: 10.1073/pnas.2234031100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt B. Molecular mechanisms involved in T cell migration across the blood-brain barrier. J Neural Transm. 2006;113:477–485. doi: 10.1007/s00702-005-0409-y. [DOI] [PubMed] [Google Scholar]
- Feuerstein GZ, Liu T, Barone FC. Cytokines, inflammation, and brain injury: role of tumor necrosis factor-alpha. Cerebrovasc Brain Metab Rev. 1994;6:341–360. [PubMed] [Google Scholar]
- Flynn RW, MacWalter RS, Doney AS. The cost of cerebral ischaemia. Neuropharmacology. 2008;55:250–256. doi: 10.1016/j.neuropharm.2008.05.031. [DOI] [PubMed] [Google Scholar]
- Fouda AY, Kozak A, Alhusban A, Switzer JA, Fagan SC. Anti-inflammatory IL-10 is upregulated in both hemispheres after experimental ischemic stroke: Hypertension blunts the response. Exp Transl Stroke Med. 2013;5:12. doi: 10.1186/2040-7378-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeber MB, Streit WJ. Microglia: biology and pathology. Acta Neuropathol. 2010;119:89–105. doi: 10.1007/s00401-009-0622-0. [DOI] [PubMed] [Google Scholar]
- Hao YL, Pu CQ, Zhu K, Zhang FY, Liu JX, Zhu GM, Zhang SG. The role of endothelium of blood-brain barrier and intercellular adhesion molecule-1 in the course of ischemic cerebral edema in rats. Chin J Neurol. 2000;33:86–89. [Google Scholar]
- Hoffmann MH, Bruns H, Bäckdahl L, Neregård P, Niederreiter B, Herrmann M, Catrina AI, Agerberth B, Holmdahl R. The cathelicidins LL-37 and rCRAMP are associated with pathogenic events of arthritis in humans and rats. Ann Rheum Dis. 2013;72:1239–1248. doi: 10.1136/annrheumdis-2012-202218. [DOI] [PubMed] [Google Scholar]
- Hopkins SJ, Rothwell NJ. Cytokines and the nervous system I: expression and recognition. Trends Neurosci. 1995;18:83–88. [PubMed] [Google Scholar]
- Horky M, Wurzer G, Kotala V, Anton M, Vojtesek B, Vacha J, Wesierska-Gadek J. Segregation of nucleolar components coincides with caspase-3 activation in cisplatin-treated HeLa cells. J Cell Sci. 2001;114:663–670. doi: 10.1242/jcs.114.4.663. [DOI] [PubMed] [Google Scholar]
- Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, Gao Y, Chen J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43:3063–3070. doi: 10.1161/STROKEAHA.112.659656. [DOI] [PubMed] [Google Scholar]
- Huang P, Zhou CM, Qin H, Liu YY, Hu BH, Chang X, Zhao XR, Xu XS, Li Q, Wei XH, Mao XW, Wang CS, Fan JY, Han JY. Cerebralcare Granule® attenuates blood-brain barrier disruption after middle cerebral artery occlusion in rats. Exp Neurol. 2012;237:453–463. doi: 10.1016/j.expneurol.2012.07.017. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Alexander M. Cerebral ischemia and inflammation. Curr Opin Neurol. 2001;14:89–94. doi: 10.1097/00019052-200102000-00014. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa M, Cooper D, Russell J, Salter JW, Zhang JH, Nanda A, Granger DN. Molecular determinants of the prothrombogenic and inflammatory phenotype assumed by the postischemic cerebral microcirculation. Stroke. 2003;34:1777–1782. doi: 10.1161/01.STR.0000074921.17767.F2. [DOI] [PubMed] [Google Scholar]
- Kao TK, Ou YC, Kuo JS, Chen WY, Liao SL, Wu CW, Chen CJ, Ling NN, Zhang YH, Peng WH. Neuroprotection by tetramethylpyrazine against ischemic brain injury in rats. Neurochem Int. 2006;48:166–176. doi: 10.1016/j.neuint.2005.10.008. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- Lakhan SE, Kirchgessner A, Hofer M. Inflammatory mechanisms in ischemic stroke: therapeutic approaches. J Transl Med. 2009;7:97. doi: 10.1186/1479-5876-7-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson C, Wolf S. ICAM-1 signaling in endothelial cells. Pharmacol Rep. 2009;61:22–32. doi: 10.1016/s1734-1140(09)70004-0. [DOI] [PubMed] [Google Scholar]
- Lenzsér G, Kis B, Snipes JA, Gaspar T, Sandor P, Komjati K, Szabo C, Busija DW. Contribution of poly(ADP-ribose) polymerase to postischemic blood-brain barrier damage in rats. J Cereb Blood Flow Metab. 2007;27:1318–1326. doi: 10.1038/sj.jcbfm.9600437. [DOI] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- Matsuo Y, Kihara T, Ikeda M, Ninomiya M, Onodera H, Kogure K. Role of neutrophils in radical production during ischemia and reperfusion of the rat brain: effect of neutrophil depletion on extracellular ascorbyl radical formation. J Cereb Blood Flow Metab. 1995;15:941–947. doi: 10.1038/jcbfm.1995.119. [DOI] [PubMed] [Google Scholar]
- Muir KW, Tyrrell P, Sattar N, Warburton E. Inflammation and ischaemic stroke. Curr Opin Neurol. 2007;20:334–342. doi: 10.1097/WCO.0b013e32813ba151. [DOI] [PubMed] [Google Scholar]
- Nakajima K, Kohsaka S. Microglia: activation and their significance in the central nervous system. J Biochem. 2001;130:169–175. doi: 10.1093/oxfordjournals.jbchem.a002969. [DOI] [PubMed] [Google Scholar]
- Nakajima K, Kohsaka S. Microglia: neuroprotective and neurotrophic cells in the central nervous system. Curr Drug Targets Cardiovasc Haematol Disord. 2004;4:65–84. doi: 10.2174/1568006043481284. [DOI] [PubMed] [Google Scholar]
- Nicholson DW, Thornberry NA. Caspases: killer proteases. Trends Biochem Sci. 1997;22:299–306. doi: 10.1016/s0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- Pan HC, Yang CN, Hung YW, Lee WJ, Tien HR, Shen CC, Sheehan J, Chou CT, Sheu ML. Reciprocal modulation of C/EBP-α and C/EBP-β by IL-13 in activated microglia prevents neuronal death. Eur J Immunol. 2013;43:2854–2865. doi: 10.1002/eji.201343301. [DOI] [PubMed] [Google Scholar]
- Peng HY, Du JR, Zhang GY, Kuang X, Liu YX, Qian ZM, Wang CY. Neuroprotective effect of Z-ligustilide against permanent focal ischemic damage in rats. Biol Pharm Bull. 2007;30:309–312. doi: 10.1248/bpb.30.309. [DOI] [PubMed] [Google Scholar]
- Peng YR, Ding YF, Wei YJ, Shu B, Li YB, Liu XD. Caudatin-2,6-dideoxy-3-O-methy-β-d-cymaropyranoside 1 induced apoptosis through caspase 3-dependent pathway in human hepatoma cell line SMMC7721. Phytother Res. 2011;25:631–637. doi: 10.1002/ptr.3312. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA. Neurological diseases in relation to the blood-brain barrier. J Cereb Blood Flow Metab. 2012;32:1139–1151. doi: 10.1038/jcbfm.2011.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell N, Allan S, Toulmond S. The role of interleukin 1 in acute neurodegeneration and stroke: pathophysiological and therapeutic implications. J Clin Invest. 1997;100:2648–2652. doi: 10.1172/JCI119808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahara S, Aoto M, Eguchi Y, Imamoto N, Yoneda Y, Tsujimoto Y. Acinus is a caspase-3-activated protein required for apoptotic chromatin condensation. Nature. 1999;401:168–173. doi: 10.1038/43678. [DOI] [PubMed] [Google Scholar]
- Sasaki Y, Ohsawa K, Kanazawa H, Kohsaka S, Imai Y. Iba1 is an actin-cross-linking protein in macrophages/microglia. Biochem Biophys Res Commun. 2001;286:292–297. doi: 10.1006/bbrc.2001.5388. [DOI] [PubMed] [Google Scholar]
- Satoh K, Niwa M, Binh NH, Nakashima M, Kobayashi K, Takamatsu M, Hara A. Increase of galectin-3 expression in microglia by hyperthermia in delayed neuronal death of hippocampal CA1 following transient forebrain ischemia. Neurosci Lett. 2011;504:199–203. doi: 10.1016/j.neulet.2011.09.015. [DOI] [PubMed] [Google Scholar]
- Satoh S, Kobayashi T, Hitomi A, Ikegaki I, Suzuki Y, Shibuya M, Yoshida J, Asano T. Inhibition of neutrophil migration by a protein kinase inhibitor for the treatment of ischemic brain infarction. Jpn J Pharmacol. 1999;80:41–48. doi: 10.1254/jjp.80.41. [DOI] [PubMed] [Google Scholar]
- Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, Takada I, Iwaki T, Okada Y, Iida M, Cua DJ, Iwakura Y, Yoshimura A. Pivotal role of cerebral interleukin-17-producing gammadeltaT cells in the delayed phase of ischemic brain injury. Nat Med. 2009;15:946–950. doi: 10.1038/nm.1999. [DOI] [PubMed] [Google Scholar]
- Shigemoto-Mogami Y, Hoshikawa K, Goldman JE, Sekino Y, Sato K. Microglia enhance neurogenesis and oligodendrogenesis in the early postnatal subventricular zone. J Neurosci. 2014;34:2231–2243. doi: 10.1523/JNEUROSCI.1619-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CJ, Lawrence CB, Rodriguez-Grande B, Kovacs KJ, Pradillo JM, Denes A. The immune system in stroke: clinical challenges and their translation to experimental research. J Neuroimmune Pharmacol. 2013;8:867–887. doi: 10.1007/s11481-013-9469-1. [DOI] [PubMed] [Google Scholar]
- Tan S, Wang G, Guo Y, Gui D, Wang N. Preventive effects of a natural anti-inflammatory agent, astragaloside IV, on ischemic acute kidney injury in rats. Evid Based Complement Alternat Med 2013. 2013:284025. doi: 10.1155/2013/284025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valous NA, Lahrmann B, Zhou W, Veltkamp R, Grabe N. Multistage histopathological image segmentation of Iba1-stained murine microglias in a focal ischemia model: methodological workflow and expert validation. J Neurosci Methods. 2013;213:250–262. doi: 10.1016/j.jneumeth.2012.12.017. [DOI] [PubMed] [Google Scholar]
- Veldhuis WB, Derksen JW, Floris S, van der Meide PH, de Vries HE, Schepers J, Vos IMP, Dijkstra CD, Kappelle LJ, Nicolay K, Bär PR. Interferon-beta blocks infiltration of inflammatory cells and reduces infarct volume after ischemic stroke in the rat. J Cereb Blood Flow Metab. 2003;23:1029–1039. doi: 10.1097/01.WCB.0000080703.47016.B6. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Deng HY, Maier CM, Sun GH, Yenari MA. Mild hypothermia reduces ICAM-1 expression, neutrophil infiltration and microglia/monocyte accumulation following experimental stroke. Neuroscience. 2002;114:1081–1090. doi: 10.1016/s0306-4522(02)00350-0. [DOI] [PubMed] [Google Scholar]
- Yang Y, Rosenberg GA. Blood-brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke. 2011;42:3323–3328. doi: 10.1161/STROKEAHA.110.608257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz G, Granger DN. Leukocyte recruitment and ischemic brain injury. Neuromolecular Med. 2010;12:193–204. doi: 10.1007/s12017-009-8074-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaremba J, Losy J. Adhesion molecules of immunoglobulin gene superfamily in stroke. Folia Morphol (Warsz) 2002;61:1–6. [PubMed] [Google Scholar]
- Zhang C, Li Y, Chen J, Gao Q, Zacharek A, Kapke A, Chopp M. Bone marrow stromal cells upregulate expression of bone morphogenetic proteins 2 and 4, gap junction protein connexin-43 and synaptophysin after stroke in rats. Neuroscience. 2006;141:687–695. doi: 10.1016/j.neuroscience.2006.04.054. [DOI] [PubMed] [Google Scholar]
- Zhang HL, Xie XF, Wu LF, Wang CD, Deng P, Yang HY, Kong WG, Wu XM. Therapeutic effect of intravenous injection of bone marrow mesenchymal stem cells on cerebral ischemia in rats. Zhongfeng yu Shenjing Jibing Zazhi. 2011;28:115–119. [Google Scholar]
- Zhang L, Dong LY, Li YJ, Hong Z, Wei WS. The microRNA miR-181c controls microglia-mediated neuronal apoptosis by suppressing tumor necrosis factor. J Neuroinflammation. 2012;9:211. doi: 10.1186/1742-2094-9-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Wang F, Gui B, Hua F, Qian Y. Prophylactic lithium alleviates postoperative cognition impairment by phosphorylating hippocampal glycogen synthase kinase-3β (Ser9) in aged rats. Exp Gerontol. 2011;46:1031–1036. doi: 10.1016/j.exger.2011.09.002. [DOI] [PubMed] [Google Scholar]