

Abstract
We report mechanistic aspects of the trapping of thermally (HDDA) generated benzyne derivatives by pendant silyl ether groups, which results in net insertion of the pair of benzyne Csp-hydribized carbon atoms into the silicon–oxygen sigma bond. Cross-over experiments using symmetrical, doubly labeled bis-silyl ether substrates established that the reaction is unimolecular in nature. Competition experiments involving either intramolecular or intermolecular dihydrogen transfer clock reactions (from within a TIPS isopropyl group or cyclooctane, respectively) vs. the silyl ether cyclization were used to gain additional insights. We evaluated effects of the steric bulk of the silyl ether trapping group and of the ring-size of the cyclic ether being formed (furan vs. pyran). These types of competition experiments allow the relative rates of various product-determining steps to be determined. This previously has only rarely been possible because aryne formation is typically rate-limiting, making it challenging to probe the kinetics of subsequent trapping reactions. Solvent effects (polarity of the medium) and computational studies were used to probe the question of stepwise vs. concerted pathways for the Si–O insertion.
Introduction
We recently reported the mild, thermal polycyclization reaction of triynes (cf. 1, Scheme 1) to give complex multi-ring benzenoid products (cf. 2).1 We call this two-stage process an HDDA cascade. The first step involves generation of a reactive benzyne derivative (cf. 3) by a net 4+2 cycloaddition event,2,3 which we have termed a hexadehydro-Diels–Alder (HDDA) reaction.1 This benzyne intermediate is then efficiently (and, in the reaction type that is the subject of this paper, intramolecularly) trapped, resulting in the expeditious construction of highly substituted benzene derivatives (e.g., 2).
Scheme 1.

The hexadehydro-Diels–Alder (HDDA) cascade of triyne 1 gives the highly substituted benzenoid derivative 2 via silyl ether trapping of the HDDA-generated benzyne 3.
Among the hallmarks of this HDDA cascade are: (i) the transformation of triynes like 1 to benzynes like 3 is computed to be exothermic by ca. 50 kcal mol−1 (Δ)1 (cf. ΔErxn = −51.4 kcal mol−1 for the parent ethyne + 1,3-butadiyne to benzyne4); (ii) this thermal mode of benzyne generation is complementary to all previously reported, preparatively practical methods in two important ways—the triyne precursors are not, themselves, benzene derivatives and the aryne is formed in the absence of reagents (e.g., bases or reducing agents) and byproducts (e.g., metal salts, amines, or halide ions) that can affect or, in some instances, interfere with the benzyne trapping reactions; and (iii) a beneficial consequence of point (ii) is that inherent reactivity of benzynes can sometimes be probed more fundamentally and/or that new types of aryne trapping reactions can be uncovered.5,6 An example of this latter feature is the net addition of a silyl ether (like that in 1) across the pair of strained benzyne sp-hybridized carbons to give an o-(trialkylsilyl)aryl ether moiety (like that in 2). Such a process had been previously unknown.1
Results and Discussion
To account for the trapping reaction that produces 2, we suggested1 that the benzyne 3 is intercepted by the silyl ether oxygen atom poised five atoms away and that a transient zwitterionic species like 4 is generated. Migrations of silicon from carbon to oxygen are well known in the form of the Brook rearrangement.7 The opposite process, the reverse (or retro-) Brook rearrangement (i.e., silicon migration from oxygen to carbon), is less common but certainly precedented. 8 These reverse Brook reactions are most often encountered within an anionic manifold of intermediates. In a setting like that of 4 to 2, rearrangement would be expected to be particularly favorable since zwitterionic character is quenched as a result of the silyl group migration. A second possibility is that the reaction mechanism involves zwitterion 4 but is not unimolecular. That is, disproportionation of that intermediate with, e.g., another copy of itself or with the silyl ether moiety in substrate 1 could be responsible for formation of 2. A third possibility is that the oxygen-silicon bond adds in concerted (and intramolecular) fashion to the benzyne π-bond in 3, leading directly to 2. This last possibility is of interest because concerted insertion into a sigma bond with accompanying formation of two new sigma-bonds (e.g., A–B + X=Y → A–X–Y–B in a single step) is a rare type of transformation.
To probe these mechanistic issues, we prepared the set of three symmetrical tetraynes 6a-c from the diol precursor 5 (Scheme 2). These differ only in the nature of the alkyl groups in the trialkylsilyl moiety of their silyl ethers. Diol 5 was prepared by Cadiot–Chodkiewicz cross-coupling reaction of the known N,N-bispropargyl toluenesulfonamide9 with 4-bromo-3-butyn-1-ol10 in the presence of catalytic CuCl in piperidine.1,11 bis-Silylation of diol 5 gave each of the corresponding bis-TES, -TBS, and -TIPS ethers 6a–c. As we anticipated, each of these substrates smoothly underwent a HDDA cascade to provide the corresponding tricyclic benzofuran derivatives 7a–c when heated alone to the reflux temperature of chloroform. The rate of consumption of each substrate was virtually identical, consistent with the fact that benzyne formation is the rate-limiting step in these HDDA cascade processes. The slightly lower yield of the TIPS-containing product 7c arises from a competing dihydrogen disproportionation reaction that will be discussed later.
Scheme 2.

Preparation of substrates 6a–c and the efficient HDDA cyclization of each to benzofurans 7a–c.
With substrates 6a–c in hand, we designed several experiments to address the question of molecularity of the trapping reaction. First, to distinguish an intra- from an intermolecular process, we carried out a simple crossover/competition study using the pair of substrates 6b and 6c (Scheme 3, panel A). When equimolar amounts of these were heated together in chloroform, the only benzofuran derivatives observed were 7b and 7c. Neither of the analogous products containing one each of the TBS and TIPS groups (i.e., 7d and 7d′) was detected (≤0.5%) by LCMS analysis of the crude product mixture. This result rules out a bimolecular process (i.e., the “second possibility” presented above). A complementary crossover experiment was also performed, this time using the mixed mono-TBS/mono-TIPS silylated substrate 6d,12 which gave 7d and 7d′ (Scheme 3, panel B). Again, there was no evidence of bimolecularity (crossover); that is, none of 7b or 7c was seen by LCMS analysis.
Scheme 3.

Crossover experiments establish unimolecularity.
We then attempted to distinguish between the concerted vs. stepwise mechanisms for silyl ether trapping (Scheme 1 bottom, green vs. orange) by carrying out the reaction in several solvents of varying polarity. In the stepwise mechanism either of the two fundamental events could, in principle, be rate-limiting. The rate constant for the first step in that mechanism, formation of the zwitterion 4 from benzyne 3 (cf. kS1, Scheme 1), would be expected to be significantly increased whereas that of the second (kS2) decreased as the polarity of the medium is increased.
To assess the impact of solvent polarity on the rate, it was desirable to have a competitive trapping reaction that could be used as a benchmark against which to clock the rate of silyl ether trapping. Fortunately, one was at hand. The reaction of bis-TIPS ether 6c gives a byproduct accompanying 7c—namely, the isopropenyl-containing bicyclic arene compound 9 (Scheme 4, panel A). This arises by an intramolecular net redox process in which two hydrogen atoms have been transferred, intramolecularly, from a TIPS alkyl group to the aryne (cf. red arrows in the benzyne intermediate 8). We have recently reported that this type of unprecedented desaturation reaction is facile for bimolecular H2-transfer reactions to benzynes from certain cyclic hydrocarbons and that it proceeds by a concerted pathway in which all six atoms involved in the bond-breaking and -making events are essentially coplanar.6 Although the geometry of the transition structure (TS) 8 might seem unusual at first glance, notice that the hydrogen atoms being transferred occupy positions 9 and 11 of an array that can be mapped onto a bicyclo[5.3.1]undecane skeleton. Indeed, we were able to locate a computed TS geometry [M06-2X/6-31G(d)] for a variant of this transformation in which the benzyne is minimally substituted–namely, 8-TS (Scheme 4, panel B). Two hydrogen atoms are each partially transferred from the isopropyl to the benzyne sp-hybridized carbon atoms. Assuming that this concerted mechanism is operative in 8, one would expect little dependency on solvent polarity, rendering this competitive transformation a good choice as an internal clock reaction.
Scheme 4.

Effect of solvent polarity on the silyl ether trapping using an internal clock reaction. The ratio of intramolecular dihydrogen transfer (6c to 9, panel A) to silyl ether trapping (6c to 7c) processes shows a dependence on solvent polarity (results tabulated in panel C). DFT calculations on a truncated analog led to the identification of the concerted TS 8-TS (panel B, non-participating aliphatic hydrogen atoms removed for clarity). 1H NMR spectrum of the crude product mixture from a typical experiment (panel D), this one for the reaction performed in 1,2-dichloroethane, resulting in a 6:1 ratio of products 7c:9.
Accordingly, we carried out the HDDA cyclization of the bis-TIPS ether 6c in four solvents (Scheme 4, panel C), each of which we have observed to be inert toward HDDA-generated benzynes. These solvents range in relative permittivity (dielectric constant) from 2.2–37.5 Debyes. As the polarity of the medium was increased, we observed a steady increase in the branching ratio (1H NMR analysis of the crude product mixture; cf. Scheme 4, panel D) in favor of formation of the silyl ether trapped product 7c relative to the dihydrogen disproportionation product 9. We interpret the direction of this trend as an argument against the possibility that the rate-limiting event in the silyl ether trapping is the second step in the stepwise mechanism in Scheme 1 (i.e., that associated with the rate constant kS2). That event involves conversion of a zwitterion (4) to a neutral structure (2) and, thus, should be decelerated not accelerated in more polar solvents.
On the other hand, the magnitude of the observed rate enhancement in a more polar solvent medium is small. Under the assumption that the rate of formation of the alkene 9 is the same in each solvent, the fastest to slowest krels for silyl ether cleavage differ only by a factor of ca. 3. The small size of this effect suggests only a minor degree of polarization in the transition structure for the rate-limiting step in the silyl ether trapping process. This is more consistent with an only modestly polarized and early TS, either for an asynchronous concerted process (3 to 2 and kC in Scheme 1) or for zwitterion formation (3 to 4 and kS1 in Scheme 1).
To gain more information relevant to the possibility of single-step insertion into the Si–O sigma bond, we also examined this benzyne trapping process computationally. We studied each of the model benzyne-containing substrates I and VI (Figure 1, panels A and B, respectively). Each comprises a simple trimethylsilyl ether and a mono-substituted benzyne, and the two differ only in the length of the methylene chain (two vs. three CH2s) linking the aryne and silyl ether moieties. Benzyne I leads to the benzofuran product V, the homolog VI to the benzopyran VIII. We used density functional theory (DFT) to map the reaction potential energy surface (PES) for each substrate/product pair. The M06-2X functional was used for all calculations and three different basis sets were used (see Experimental Section). We were able to locate a zwitterionic species as an intermediate only when we applied an implicit (or continuum) solvation model (solvation model density, SMD13) during geometry optimization and, then, only in the five-membered series (cf. the pathway involving I–V). The barrier heights leading away from that zwitterion (III) are quite low, especially so in the forward direction, suggesting that it is a fleeting intermediate. Computational results (energies for I–VIII and geometries for TSs II, IV, and VII) are given in Figure 1. The trends and conclusions are qualitatively the same regardless of the nature of the basis set that was used.
Fig. 1.

Computed energies of minima and transition structures on the potential energy surface for conversion of model benzynes I and VI into silyl ether trapped products V and VIII (panels A and B, respectively). Selected dihedral and internal bond angles are given for TSs II, IV, and VII. [M06-2X/6-31G(d); enthalpic energies (kcal·mol−1) are given in panels A and B. Energies for each species using three basis sets are given in panel C.
The overall transformation for either trapping reaction (I to V or VI to VIII) is highly exothermic (>50 kcal·mol−1). The geometry of each of the initial (and early) TSs II or VII shows only a slight lengthening of the O–Si bond. The major difference in the two (cf. TS geometries in Figure 1) is seen in the dihedral bond angle across the pair of benzyne carbon atoms and the silyl ether oxygen and silicon atoms (∠1234 = 67° in II vs. 30° in VII). TS II was able to settle into the zwitterionic intermediate III, in which this dihedral angle has been significantly reduced to 18°. This then proceeds via the second TS IV (dihedral = 13°) to product V (dihedral = 0°) via a second, low barrier and very exothermic elementary step. The TS VII for the six-membered substrate is very similar to III in both geometry and energy. However, it directly evolves into benzopyran VIII (dihedral = 0°) without indication of encountering an intervening energy minimum species (i.e., an analogous zwitterionic intermediate). It is also of interest that the benzyne moiety in each of TSs II and VII has undergone an appreciably large, induced distortion of geometry in response to the approach of the oxygen nucleophile. Specifically, the two bond angles within the benzyne ring at atoms C1 (∠a12) vs. C2 (∠12b) reveal a large distortion of the benzyne in each (∠12b -∠a12 = 31° for II and 26° for VII). This induced distortion of the transition structure represents an extrapolation of previous explanations of ground-state benzyne distortions that effectively explain the site of greater electrophilic character in unsymmetrical benzynes.14
We have also assessed the dependency of the relative rate of intramolecular silyl ether trapping on the size of the silyl substituent. A different type of dihydrogen transfer competition experiment, this time vis-à-vis the external trapping agent cyclooctane,6 was used. Heating each of 6a–c to 65 °C in cyclooctane led to a product mixture containing both 7a/b/c and the reduced benzenoid product 10a/b/c (Scheme 5). The product ratios reflect the relative ease of trapping by the TES, TBS, and TIPS silyl ethers, respectively, within the benzyne intermediates 11a–c. As expected, the least hindered TES is the fastest. Specifically, the TES ether traps the benzyne ca. 5x faster than the TBS and 20x faster than the bulkiest, TIPS ether (cf. krels in Scheme 5). The magnitude of this effect is consistent with a TS for the product-determining step in which at least some degree of interaction with the silicon center has been established.
Scheme 5.

Effect of steric bulk of the silyl group using an external clock reaction. Competition of intramolecular silyl ether trapping vs. bimolecular dihydrogen transfer from cyclooctane.
Finally, we used the cyclooctane external competition experiment to probe one additional feature–namely, the relative ease of furan vs. pyran formation in the silyl ether trapping event. Substrates 6b and 12 differ in the number of methylenes (2 vs. 3, respectively) that tether the TBS ether to the alkyne (Scheme 6).15 Each was heated in cyclooctane, and the ratio of cyclization to reduction products (i.e., 7b to 10b vs. 13 to 14) was measured. The ratios turned out to be identical, suggesting a compensating trade-off between entropic (favoring silyl ether trapping enroute to the furan 7b) and enthalpic effects (lower strain in the TS leading to pyran 13) for these two processes. It is notable that the computed activation enthalpies (ΔH‡) for the reactions passing through TSs II vs. VII (Figure 1) are 3.1 vs. 1.8 kcal·mol−1, respectively. The actual rate for each of these reactions would also be, of course,16 dependent on the mole fraction of each of the reactive conformers I and VI vis-à-vis its associated family of all accessible conformations. The additional rotatable bond in the latter substrate reduces the mole fraction of VI (vs. that of I), thereby slowing its reaction rate to bring it more in line with that of I than is implied by the computed ΔH‡ values alone.
Scheme 6.
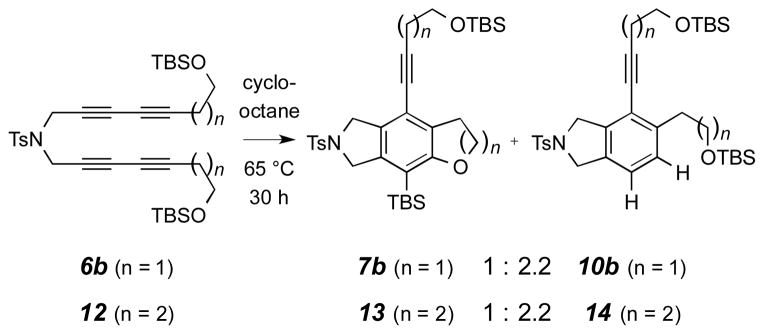
Ring size effects. Competition of intramolecular silyl ether trapping vs. bimolecular dihydrogen transfer from cyclooctane (the external clock reaction).
Conclusions
In summary, we have used various competition and crossover experiments to gain mechanistic insights about the silicon-oxygen bond cleavage event that attends trapping of HDDA-generated benzynes containing a well-disposed, pendant silyl ether. The process is unimolecular. Use of dihydrogen transfer reactions, of both an intra- and intermolecular nature, was instrumental to the design of the competition experiments. These types of competitive reactions allow one to probe the relative rates of various product-determining steps in the reactions of arynes. The rates of trapping reactions are often difficult to unravel because aryne formation is typically rate-limiting, making it challenging to probe the kinetics of the subsequent events. The silyl ether trapping reaction shows a modest rate enhancement in solvents of higher polarity, which we interpret as evidence for only a moderate degree of polarization in the transition structure for the rate-limiting step. Larger alkyl groups on the silyl ether slow the rate of trapping (ca. 20-fold between TES and TIPS). Precursors to benzofuran vs. benzopyran products cyclize at essentially the same rate, suggesting that enthalpic and entropic factors in these two types of tethers are self-compensating. Based on the computational results alone, one would conclude that the silyl ether trapping event in the benzofuran class of precursors occurs in stepwise fashion whereas the benzopyran homologs proceed via a concerted Si–O insertion. However, given the small energetic differences between these two pathways, this conclusion should probably be viewed with a degree of caution. Computed TS geometries suggest that the benzyne distorts in response to the electronic character of the approaching nucleophilic silyl ether oxygen atom. We speculate that this phenomenon may have ramifications extending to many other aryne trapping reactions. Collectively, the studies reported here show how HDDA chemistry can provide access to new fundamental mechanistic insights that are otherwise difficult if not impossible to obtain.
Supplementary Material
Acknowledgments
This research was supported by the National Institute of General Medical Sciences (GM65597) and the National Cancer Institute (CA76497) of the U.S. Department of Health and Human Services.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/b000000x/
Notes and references
- 1.Hoye TR, Baire B, Niu D, Willoughby PH, Woods BP. Nature. 2012;490:208–212. doi: 10.1038/nature11518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miyawaki K, Suzuki R, Kawano T, Ueda I. Tetrahedron Lett. 1997;38:3943–3946. [Google Scholar]
- 3.Bradley AZ, Johnson RP. J Am Chem Soc. 1997;119:9917–9918. [Google Scholar]
- 4.Ajaz A, Bradley AZ, Burrell RC, Li WH, Daoust KJ, Bovee LB, DiRico KJ, Johnson RP. J Org Chem. 2011;76:9320–9328. doi: 10.1021/jo201567d. [DOI] [PubMed] [Google Scholar]
- 5.a) Karmakar R, Mamidipalli P, Yun SY, Lee D. Org Lett. 2013;15:1938–1941. doi: 10.1021/ol4005905. [DOI] [PubMed] [Google Scholar]; b) Yun SY, Wang KP, Lee NK, Mamidipalli P, Lee D. J Am Chem Soc. 2013;135:4668–4671. doi: 10.1021/ja400477r. [DOI] [PubMed] [Google Scholar]; c) Wang KP, Yun SY, Mamidipalli P, Lee D. Chem Sci. 2013;4:3205–3211. [Google Scholar]
- 6.Niu D, Willoughby PH, Baire B, Woods BP, Hoye TR. Nature. 2013;501:531–534. doi: 10.1038/nature12492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brook AG. Acc Chem Res. 1974;7:77–84. [Google Scholar]
- 8.Li JJ. Name reactions for homologations. Part II. Wiley; New York: 2009. pp. 406–437. [Google Scholar]
- 9.Oppolzer W, Pimm A, Stammen B, Hume WE. Helv Chim Acta. 1997;80:623–639. [Google Scholar]
- 10.Montierth JM, DeMario DR, Kurth MJ, Schore NE. Tetrahedron. 1998;54:11741–11748. [Google Scholar]
- 11.Baire B, Niu D, Willoughby PH, Woods BP, Hoye TR. Nature Protocols. 2013;8:501–508. doi: 10.1038/nprot.2013.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Substrate 6d was prepared by i) partial silylation of diol 5 with TBS-Cl (1.5 equiv), ii) isolation of the mono-TBS ether, and iii) a second silylation with TIPS-Cl (see Supporting Information).
- 13.Marenich AV, Olson RM, Kelly CP, Cramer CJ, Truhlar DG. J Chem Theory Comput. 2007;3:2011–2033. doi: 10.1021/ct7001418. [DOI] [PubMed] [Google Scholar]
- 14.a) Hamura T, Ibusuki Y, Sato K, Matsumoto T, Osamura Y, Suzuki K. Org Lett. 2003;5:3551–3554. doi: 10.1021/ol034877p. [DOI] [PubMed] [Google Scholar]; b) Garr AN, Luo D, Brown N, Cramer CJ, Buszek KR, VanderVelde D. Org Lett. 2010;12:96–99. doi: 10.1021/ol902415s. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Cheong PHY, Paton RS, Bronner SM, Im GYJ, Garg NK, Houk KN. J Am Chem Soc. 2010;132:1267–1269. doi: 10.1021/ja9098643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heating 12 alone in CHCl3 (EtOH-free) showed that it was a well-behaved substrate; the benzopyran derivative 13 was formed cleanly (see Supporting Information).
- 16.a) “A rate is a rate constant times a concentration.”16bHoye TR, Ryba TD. J Am Chem Soc. 2005;127:8256–8257. doi: 10.1021/ja051604b.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.