

Abstract
Two new 2,2’-bipyridine (bpy) derivatives containing ancillary BODIPY chromophores attached at the 5- and 5’-positions (BB3) or 6- and 6’-positions (BB4) were prepared and characterized. In this work, the basic photophysics, electrochemistry and electrogenerated chemiluminescence (ECL) of BB3 and BB4 are compared with those previously reported for a related bpy-BODIPY derivative (BB2) (J. Phys. Chem. C 2011, 115, 17993–18001). Cyclic voltammetry revealed that BB3 and BB4 display reversible 2e− oxidation and reduction waves, which consist of two closely spaced (50 – 70 mV) 1e− events. This redox behavior is consistent with the frontier molecular orbitals calculated for BB3 and BB4 and indicates that the 2,2’-bipyridine spacer of each bpy- BODIPY homologue does not facilitate efficient electronic communication between the tethered indacene units. In the presence of a coreactant such as tri-n-propylamine (TPA) or benzoyl peroxide (BPO), BB3 and BB4 exhibit strong ECL and produce spectra that are very similar to their corresponding photoluminescence profiles. The ECL signal obtained under annihilation conditions, however, is significantly different and is characterized by two distinct bands. One of these bands is centered at ~570 nm and is attributed to emission via an S- or T-route. The second band, occurs at longer wavelengths and is centered around ~740 nm. The shape and concentration dependence of this long-wavelength ECL signal is not indicative of emission from an excimer or aggregate, but rather is suggests that a new emissive species is formed from the bpy-BODIPY luminophores during the annihilation process.
Keywords: Electrochemistry, Electrogenerated Chemiluminescence, Long-Wavelength Emission, BODIPY, Bipyridine Derivatives
Introduction
Electrogenerated chemiluminescence (ECL) involves the generation of oxidized and reduced species, often radical ions, that undergo a fast electron transfer reaction to produce a species in an excited state.1 The various mechanisms by which ECL processes take place have been extensively discussed.2-5 A prime pathway by which ECL is achieved is radical ion annihilation. An alternative mechanism involves consumption of a coreactant, which generates a reductant upon oxidation (or oxidant upon reduction) that reacts with the emission precursor.6 Coreactants can often be used to help elucidate ECL reaction mechanisms by providing a contrasting pathway to that observed for annihilation ECL.7,8 ECL is often produced by directly generating an excited singlet state (S-route) in which the annihilation energy is sufficient to populate the singlet excited state. Alternatively in cases where this energy is not sufficient to populate the singlet state, ECL can be generated by triplet-triplet annihilation (T-route), which provides an alternate route to the emissive species in the singlet excited state.9 Another ECL mechanism involves emission from an excited state dimer such as an excimer or exciplex (Eroute). 10-13 Exciplex emission is often encountered for aromatic molecules, including derivatives of thianthrene,14 stilbene,15 corannulene,16 perylene,17 anthracene,18,19 and naphthalene20 and is characterized by broad, structureless emission bands at long wavelengths.
4,4-Difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY) dyes were discovered in 1968 by Treibs and Kreuzer.21 BODIPY dyes are characterized by strong UV-vis absorption profiles and fluorescence quantum yields that can approach unity.22-24 Moreover, the photophysical properties of BODIPY fluorophores are relatively insensitive to solvent polarity and pH and they are reasonably stable under physiological conditions.25,26 Based on these attractive properties, BODIPY derivatives have found utility as laser dyes,27 photosensitizers,28-30 fluorescent labels for in-vivo imaging31-33 and in optical devices.25 The electrochemical and ECL properties of BODIPY derivatives are also of note, as these compounds often display reversible redox behavior in aprotic solvents and excellent stability upon oxidation or reduction. These attractive electrochemical properties coupled with robust and tunable photophysics, distinguish BODIPY derivatives as excellent candidates for ECL applications.34-36
A number of electrochemical37-39 and ECL studies40-45 of BODIPY dyes have been reported. In a recent study, we described the electrochemical, photophysical and ECL properties of a 2,2’-bipyridine-BODIPY derivative (BB2), in which the meso-position of two fully substituted BODIPY moieties were linked directly to a 2,2’-bipyridine (bpy) spacer at the 4- and 4’-positions. 46 This BODIPY-appended bipyridine derivative was highly fluorescent and produced strong ECL emission in the presence of a coreactant. Given that the electronic properties of bpy derivatives are sensitive to the substitution pattern about the bpy framework, we were curious as to whether the photophysics and ECL emission might be tuned by altering the position to which the BODIPY moieties were attached. To this end, we have developed two new BODIPYappended bipyridine derivatives, in which the BODIPY units were linked to the bpy spacer at either the 5,5’- (BB3) or 6,6’-positions (BB4). These new compounds, which are juxtaposed with our previously reported bpy-BODIPY derivative (BB2) in Chart 1, were thoroughly characterized by a combination of photophysical, electrochemical and ECL methods. Of note is our finding that BB3 and BB4 exhibit a bifurcated ECL signal which is distinct from that simply observed by photoluminescence experiments.
Chart 1.
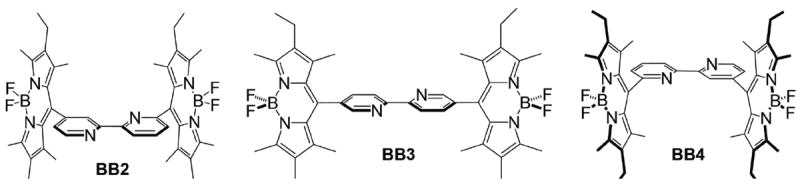
Structures of BODIPY-appended bipyridine derivatives
Experimental
General Materials and Methods
Reactions were performed in oven-dried round-bottomed flasks unless otherwise noted. Reactions that required an inert atmosphere were conducted under a positive pressure of N2 using flasks fitted with Suba-Seal rubber septa or in nitrogen filled glove box. Air and moisture sensitive reagents were transferred using standard syringe or cannula techniques. Reagents and solvents for synthesis were purchased from Sigma Aldrich, Acros, Fisher, Strem, or Cambridge Isotopes Laboratories. Solvents for synthesis were of reagent grade or better and were dried by passage through activated alumina and then stored over 4 Å molecular sieves prior to use.39 Column chromatography was performed with 40-63 μm silica gel with the eluent reported in parentheses. Analytical thin-layer chromatography (TLC) was performed on precoated glass plates and visualized by UV or by staining with KMnO4.
Anhydrous dichloromethane (CH2Cl2, ≥ 99.8%) and tetrahydrofuran (THF, ≥ 99.9%) for electrochemistry, were obtained from Sigma-Aldrich (St. Louis, MO) and transferred directly into an argon atmosphere glovebox (MBraun Inc., Stratham, NH) without further purification. Electrochemical grade tetra-n-butylammonium hexafluorophosphate (TBAPF6) was dried in a vacuum oven at 100 °C prior to transferring directly into an argon atmosphere glove box. Benzoyl peroxide (BPO) and tri-n-propylamine (TPA) were obtained from Sigma-Aldrich and were used as received.
Compound Characterization
1H NMR and 13C NMR spectra were recorded at 25 °C on a Bruker 400 MHz spectrometer. Proton spectra are referenced to the residual proton resonance of the deuterated solvent (CDCl3 = δ 7.26; DMSO-d6 = δ 2.50) and carbon spectra are referenced to the carbon resonances of the solvent (CDCl3 = δ 77.23; DMSO-d6 = δ 40.45). All chemical shifts are reported using the standard δ notation in parts-per-million; positive chemical shifts are to higher frequency from the given reference. LR-GCMS data were obtained using an Agilent gas chromatograph consisting of a 6850 Series GC System equipped with a 5973 Network Mass Selective Detector. Low resolution MS data was obtained using either a LCQ Advantage from Thermofinnigan or a Shimadzu LC/MS-2020 single quadrupole MS coupled with an HPLC system, with dual ESI/APCI source. High-resolution mass spectrometry analyses were either performed by the Mass Spectrometry Laboratory in the Department of Chemistry and Biochemistry at the University of Delaware or at the University of Illinois at Urbana-Champaign.
2,2’-Bipyridine-5,5’-dicarboxylic acid (1)
This compound was prepared using a slightly modified literature method.47 5,5’-dimethyl-2,2’-bipyridine (4.69 g, 0.025 mol) and potassium permanganate (26.2 g, 0.17 mol) were added to 300 mL of water. The resulting mixture was heated at 90 °C with stirring under air for 14 hrs. After cooling the mixture to room temperature, a brown precipitate was removed by filtration and the filtrate was extracted three times with Et2O. The resulting aqueous solution was acidified to a pH of 2 with 1 M HCl, which led to formation of a white precipitate, which was collected by vacuum filtration to deliver the title compound (4.27 g, 70%). 1H NMR (400 MHz, DMSO, 25 °C) δ/ppm: 9.22 (s, 2H), 8.60 (d, J = 8.3 Hz, 2H), 8.49 (s, 2H). HR-ESI-MS [M+H]+ m/z: calc for C12H9N2O4, 245.0562. Found 245.0558.
5,5’-bis(BODIPY)-2,2’-bipyridine (BB3)
This compound was prepared by adapting a literature method.46 2,2’-Bipyridine-4,4’-dicarboxylic acid (1) (0.5 g, 2.23 mmol) was suspended in 20 mL of SOCl2. The resulting mixture was heated to reflux under nitrogen with stirring for 24 hrs, after which time, all solid materials had dissolved to produce a dark yellow solution. The SOCl2 was removed under reduced pressure and the resulting residue was dissolved in 100 mL of CHCl3 and sparged with nitrogen for 45 min. 2,4-Dimethyl-3-ethylpyrrole (2.11 mL, 15.6 mmol) was added to the solution and the reaction was heated at 50 °C under nitrogen for 90 min. After cooling the solution to room temperature, the solvent was removed by rotary evaporation and the resulting residue was dissolved in 200 mL of toluene and CH2Cl2 (95:5) Following the addition of triethylamine (2.48 mL, 17.8 mmol), the resulting dark colored solution was stirred under air for 30 min at room temperature. The solution was then heated to 50 °C and 3.85 mL (31.2 mmol) of BF3•OEt2 was added. The reaction was stirred for an additional 90 min at 50 °C, after which time the solvent was removed under reduced pressure. The resulting residue was purified via flash column chromatography on silica using CH2Cl2 and MeOH (99:1) as the eluent to deliver 746 mg (44%) of the title compound as a brick red powder. 1H NMR (400 MHz, CDCl3, 25 °C), δ/ppm: 8.66 (s, 2H), 8.64 (d, J = 4.0 Hz 2H), 7.83 (d, 2H), 2.56 (s, 12H), 2.32 (q, 8H), 1.40 (s, 12H), 1.00 (t, 12H). 13C NMR HR-ESI-MS [M+H]+ m/z: calc for C44H52B2F4N6, 761.4297. Found 761.4297.
2,2’-Bipyridine-6,6’-dicarboxylic acid (2)
This compound was prepared using the same method used for the preparation of 2,2-bipyridine-5,5’-dicarboxylic acid (1), using 6,6’- dimethyl-2,2’-bipyridine in place of 5,5’-dimethyl-2,2’-bipyridine. The desired product was isolated in 70 % yield. 1H NMR (400 MHz, DMSO, 25 °C), δ/ppm: 8.78 (d, J = 7.7 Hz, 2H), 8.22 (t, J = 8.0 Hz, 2H), 8.17 (d, J = 8.0 Hz, 4H). 13C NMR (101 MHz, DMSO, 25 °C), δ/ppm: 165.92, 154.46, 148.10, 138.94, 125.24, 124.10. HR-ESI-MS [M+H]+ m/z: calc for C12H9N2O4, 245.0562. Found 245.0565.
6,6’-bis(BODIPY)-2,2’-bipyridine (BB4)
This compound was prepared according to the same method used for the preparation of 5,5’-bis(BODIPY)-2,2’-bipyridine (BB3), using 2,2’- Bipyridine-6,6’-dicarboxylic acid (2) in place of 2,2’-bipyridine-5,5’-dicarboxylic acid (1). The desired product was purified via flash column chromatography on silica using CH2Cl2 as the eluent to deliver 610 mg (36%) of the title compound as a brick red powder. 1H NMR (400 MHz, CDCl3 25 °C) δ/ppm: 8.49 (d, J = 8.0 Hz, 2H), 7.92 (t, J = 7.8 Hz, 2H), 7.45 (d, J = 6.8 Hz, 2H), 2.56 (s, 12H), 2.31 (q, J = 7.5 Hz, 8H), 1.27 (s, 13H), 0.99 (t, J = 7.5 Hz, 12H). 13C NMR (101 MHz, CDCl3, 25 °C) δ 156.05, 154.67, 154.16, 138.04, 137.77, 137.26, 132.96, 130.74, 124.82, 121.43, 17.09, 14.68, 12.68, 11.49. HR-ESI-MS [M+H]+ m/z: calc for C44H52B2F4N6, 761.4297. Found 761.4301.
Electrochemical experiments
Electrochemistry experiments were carried out using a three-electrode setup that employed a 0.043 cm2 platinum disk working electrode (WE), a platinum counter electrode (CE), and a silver wire quasi-reference electrode (RE) calibrated using ferrocene as an internal standard (0.342 V vs SCE).48 An L-shaped electrode (disk oriented vertically) was used for the CV measurements and ECL experiments. The working electrode was polished prior to every experiment with 0.3 μm alumina particles dispersed in water, followed by sonication in ethanol and water for several minutes. All glassware was oven-dried for 1 h at 120 °C prior to transferring into an argon atmosphere glove box. All solutions were prepared inside the glove box and sealed in a vacuum-tight electrochemical cell with a Teflon screw cap containing three metal rods for electrode connections. Cyclic voltammetry and chronoamperometry experiments were carried out with a CH Instruments (Austin, TX) model 660 electrochemical workstation. Electrochemistry experiments were carried out in CH2Cl2 containing 0.1 M TBAPF6 as the supporting electrolyte.
ECL experiments
ECL spectra were generated by annihilation by pulsing the potential with a pulse width of 0.1 s from about 80 mV past the peak potentials, or by stepping from 0 to 80 mV from reduction peak with a step time of 2 s using benzoyl peroxide as a coreactant or by stepping from 0 V to 80 mV from oxidation peak with a step time of 2 s using TPA as a coreactant. ECL spectra were recorded with a Princeton Instruments Spec 10 CCD camera (Trenton, NJ) with an Acton SpectPro-150 monochromator cooled with liquid nitrogen to −100 °C.
ECL-CV simultaneous experiments were carried our prior to spectral measurements to ensure the presence of ECL emission. In this case, a multichannel Eco Chemie Autolab PGSTAT100 (Utrecht, The Netherlands) was used to collect the signal and a photomultiplier tube (Hamamatsu R4220, Tokyo, Japan) was used as a detector. Voltage for the PMT (750 V) was provided by a Kepco power supply (New York, NY), and the signal from the PMT to the potentiostat was transferred using a multimeter (Keithley, Solon, OH). Digital simulations were perfomred using Digisim computer software (Bioanalytical Systems, West Lafayette, IN).
Computations
Geometry optimizations, frequency calculations, and molecular orbital calculations were performed in Gaussian 09 using the B3LYP/6-311G(d) basis set. Only positive frequencies were found for the optimized structures. Molecular orbitals were visualized using Guassview software. All calculations were performed in the gas phase.
Results and discussion
Synthesis
The synthetic protocols used to construct the bpy-BODIPY derivatives 5,5’-bis(BODIPY)-2,2’-bipyridine (BB3) and 6,6’-bis(BODIPY)-2,2’-bipyridine (BB4), are shown in Scheme 1. Preparation of BB3 begins with the conversion of 5,5’-dimethyl-2,2’-bipyridine to the corresponding dicarboxylic acid (1) via oxidation with KMnO4 in water (Scheme 1a). Following purification, the dicarboxylic acid derivative was treated with excess thionyl chloride to generate the corresponding acid chloride derivative. Condensation of this intermediate with 2,4-dimethyl- 3-ethylpyrrole followed by treatment with NEt3 and BF3•OEt2, afforded 5,5’-bis(BODIPY)-2,2’- bipyridine (BB3) in 44% yield. Synthesis of the homologous 6,6’-bis(BODIPY)-2,2’-bipyridine (BB4) derivative was accomplished in 36% yield from dicarboxylic acid 2 using an analogous strategy (Scheme 1b).
Scheme 1.
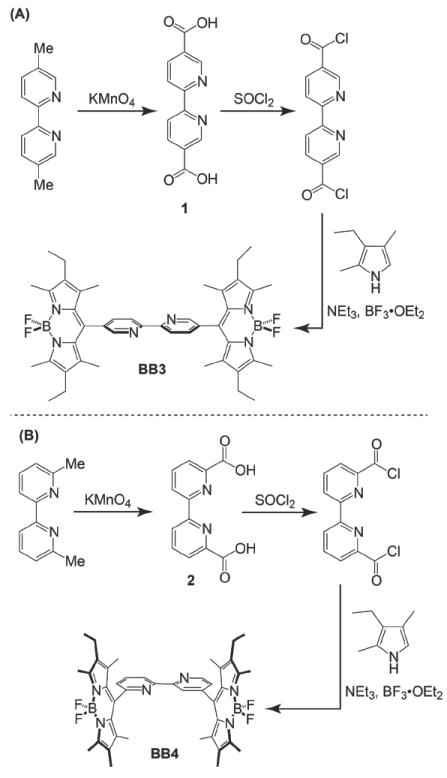
Synthesis of BODIPY-appended bipyridine derivatives BB3 and BB4.
Electrochemistry
The basic redox properties of BB3 and BB4 were probed by cyclic voltammetry (CV). Electrochemical results obtained for both bpy-BODIPY derivatives in CH2Cl2 containing 0.1 M TBAPF6 as the supporting electrolyte are summarized in Table 1 and pertinent CVs are shown in Figure 1. As was previously observed for BB2, both BB3 and BB4 show single redox waves upon oxidation or reduction. Both these waves consist of two closely spaced one-electron (1e−) events, as confirmed by digital simulation. Overall 2e− reduction and oxidation waves were also observed for BB3 and BB4 using a platinum ultramicroelectrode (Figure S1). Fitting of the CV traces was carried out by subtraction of the background current and determining the best fit to the simulation corrected for the measured electrical double layer capacitance and the uncompensated cell resistance (Figures S4–S7).
Table 1.
aRedox potentials recorded for BB2 – BB4 in CH2Cl2.
| E1/2(A/A+) | E1/2(A+/A2+) | E1/2(A/A−) | E1/2(A−/A2−) | D•106 | cΔGann | dΔHann | |
|---|---|---|---|---|---|---|---|
| bBB2 | 1.11 V | 1.15 V | −1.15 V | −1.22 V | 4.0 cm2/s | 2.33 eV | 2.23 eV |
| BB3 | 1.14 V | 1.19 V | −1.17 V | −1.24 V | 4.0 cm2/s | 2.31 eV | 2.21 eV |
| BB4 | 1.12 V | 1.17 V | −1.18 V | −1.24 V | 4.0 cm2/s | 2.30 eV | 2.20 eV |
E1/2 values were obtained by fitting the experimental results to digital simulations and are reported versus SCE.
Data for BB2 is reproduced from reference 46.
ΔGann = E1/2 (A/A+) − E1/2 (A/A−).
ΔHann = ΔGann − 0.1 eV.
Figure 1.
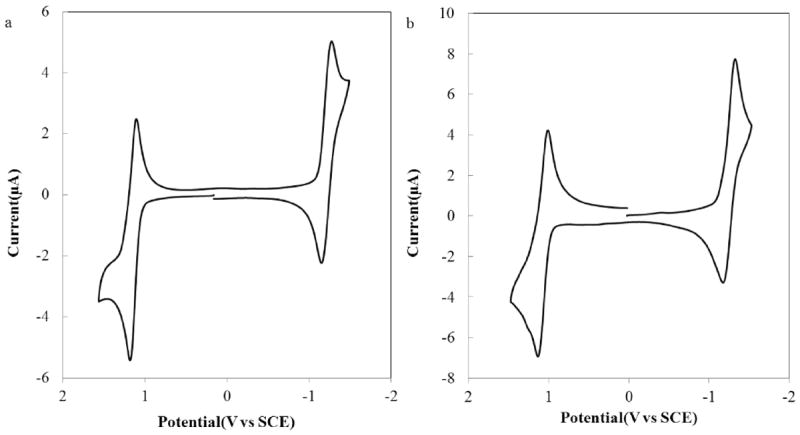
Cyclic voltammograms of CH2Cl2 solutions of (a) 0.59 mM BB3 and (b) 0.68 mM BB4. CVs were recorded using 0.1 M TBAPF6 as the supporting electrolyte using a platinum disk working electrode (A = 0.043 cm2) and a scan rate (ν) of 100 mV/s.
The small separation observed for the two reductive and oxidative electron transfers events is consistent with there being negligible electronic coupling between the two BODIPY moieties via the bpy spacers of both BB3 and BB4. For two groups that are completely uncoupled, the theoretical difference between the potentials for the first and second electron transfers would be 36 mV due to entropic factors.49 As was the case for BB2, the weak electronic coupling between the two BODIPY groups of BB3 and BB4 is manifest in small separations in potential between the A/A+ and A+/A2+ redox waves of approximately 50 – 70 mV, as judged by the digital simulations described above (Table 1). If the BODIPYs group were more strongly coupled, the two oxidation and reduction waves would be more broadly separated with the A/A2+ and A−/A2− shifted toward more extreme potentials,50 as has been observed for BODIPY derivatives containing directly linked indacene moieties.
DFT calculations carried out for BB3 and BB4 confirm that the BODIPY moieties of both these molecules are effectively insulated from one another, as steric demands force the indacene frameworks to cant significantly with respect to the bipyridine bridges. Orthogonalization of the BODIPY moieties and bpy spacer results in the HOMO and LUMO of both BB3 and BB4 residing on the individual chromophores. These calculations provide no evidence for electronic delocalization onto the bipyridine spacer (Figure 2).
Figure 2.
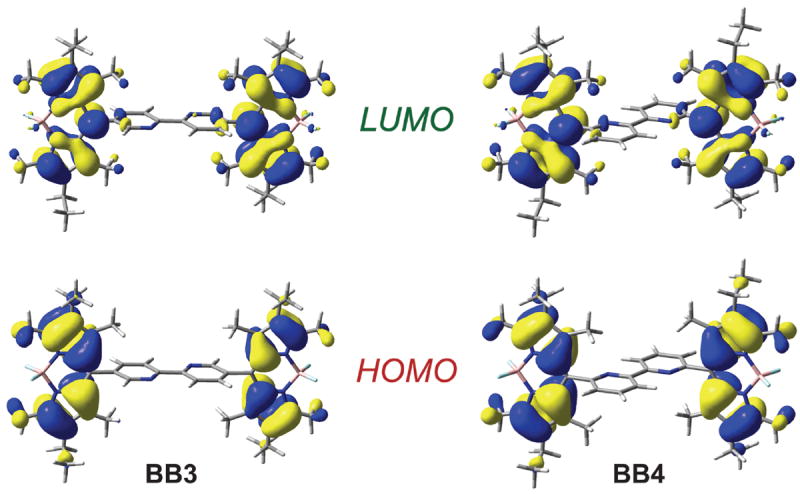
Calculated frontier molecular orbitals for BB3 and BB4 by DFT (B3LYP/6-311G(d)).
Oxidation and reduction of both BB3 and BB4 is reversible, which is consistent with the high stability of the radical cation and anion products formed at the electrode surface. This stability is a result of the BODIPY units of both Bpy-BODIPY homologues being completely substituted along the indacene periphery, which prevents decomposition of the electrochemically generated radical ions. Randles–Sevcik analysis showed that the measured oxidation and reduction peaks vary linearly as a function of the square root of scan rate (ν1/2) (Figures S2 and S3), indicating that these redox processes are diffusion controlled.51 In an effort to obtain better insight into the mechanism and kinetics of both the anodic and cathodic processes, the experimental polarization curves for BB3 and BB4 were digitally simulated (Figure 3 and S4 – S7). Both the cathodic and anodic CVs of BB3 were characterized by two reversible 1e− transfers, with a fast heterogeneous rate constant, (k° > 0.01 cm/s) for both reaction steps (Figures 3a, b; S4 and S5). Virtually identical parameters were also found for BB4 (Figures 3c, d; S6 and S7). The slight differences between simulated and experimental polarization curves may be attributed to weak adsorption of the bpy-BODIPY derivatives to the working electrode. BODIPY redox sites can often be oxidized or reduced by two electron equivalents. As such, the bpy-BODIY constructs considered here, might be expected to be able to donate up to four electrons at sufficiently positive potentials. CV scans recorded for BB3 and BB4 in CH2Cl2 over the potential range of 0.1 – 2.4 V versus SCE, revealed a second irreversible peak at ~2.05 V (Figure 4). This second oxidation wave is more positive than the first 2e− wave by ~0.9 V, which is consistent with that observed for other BODIPY derivatives,28,46 and is presumed to correspond to removal of a third and fourth e− from the bpy-BODIPY derivatives. The reductive window of CH2Cl2 precludes scanning to potentials significantly more negative than the first reduction waves, however, THF, which has a more negative working potential range was used to study the reduction of BB3 and BB4 beyond –1.5 V. An additional reduction wave was observed for BB4 at approximately −2.3 V versus SCE, however, BB3 did not show a new redox wave beyond the first 2e− reduction. This divergent behavior is likely a result of the distinct molecular topologies of the two bpy-BODIPY homologues (Chart 1).
Figure 3.
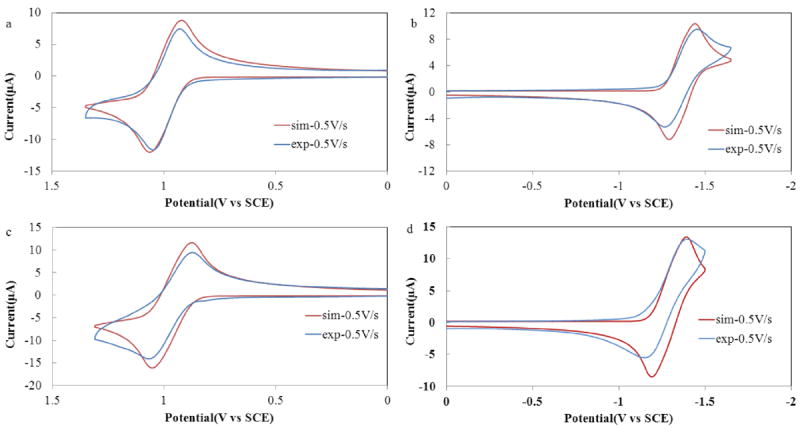
Comparison between simulated (red) and experimental (blue) polarization curves for (a, b) BB3 and (c, d) BB4. Simulations were prepared for an EE mechanism with a heterogeneous ET rate constant of k° > 0.01 cm/s. Experimental polarization curves recorded in CH2Cl2 containing 0.1 M TBAPF6 with a platinum disk working electrode (A = 0.043 cm2) at a scan rate (ν) of 500 mV/s.
Figure 4.
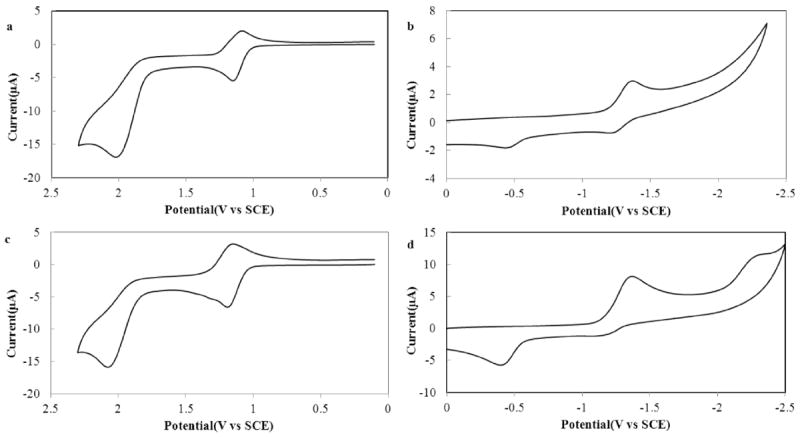
Cyclic voltammograms of (a, b) BB3, and (c, d) of BB4. Anodic scans of (a) 0.59 mM BB3 and (c) 0.68 mM BB4 were recorded in CH2Cl2 containing 0.1 M TBAPF6. Cathodic scans of (b) 0.56 mM BB3 and (d) 0.78 mM BB4 were recorded in THF containing 0.1 M TBAPF6. All polarization curves were recorded using a platinum disk working electrode (A = 0.043 cm2) at a scan rate (ν) of 100 mV/s.
Photophysics
The basic photophysical properties of BB3 and BB4 were assessed in CH2Cl2 (Figure 5) and are summarized in Table 2. Both BB3 and BB4 display optical properties analogous to the previously studied BB2 homologue and other related BODIPY derivatives. UV-vis absorbance and emission spectra for BB3 and BB4 are shown in Figure 5. Both these compounds display strong absorption bands in the visible region centered at approximately 528 nm (S0 → S1 transition) and 371 nm (S0 → S2 transition).25 The molar absorption coefficients of BB3 and BB4 in CH2Cl2 were measured to be 82,000 and 67,000 M−1cm−1, respectively. These relatively high absorptivities are consistent with there being two BODIPY chromophores appended to the bipyridine bridge for both compounds. These molar absorptivities are similar in magnitude to that determined for BB2 (Table 2).46 The bipyridine spacer of BB3 – BB4 causes the major absorbance band observed for each of these derivatives to undergo a hypsochromic shift of approximately 7 nm relative to the analogous BODIPY dye bearing a methyl substituent at the indacene meso-position (PM567).52 These hypsochromic shifts can be attributed to an inductive electron withdrawing effect of the bipyridine spacer.53
Figure 5.
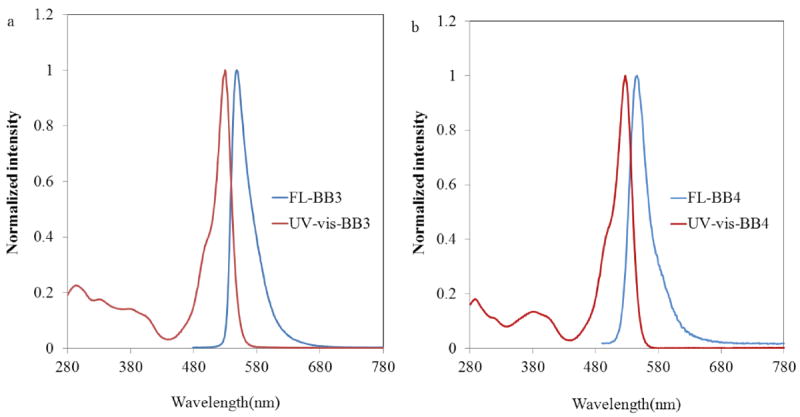
Normalized absorbance (red) and emission (blue) spectra for (a) BB3 and (b) BB4 in CH2Cl2. Emission samples were excited at λex = 480 nm.
Table 2.
Photophysical and ECL properties recorded for BB2 – BB4 in CH2Cl2.
| Absorbance | Emission | Electrogenerated Chemiluminescence | |||||
|---|---|---|---|---|---|---|---|
| λabs | ε (M−1cm−1) | λFI | ΦFI | aE0-0 | λECL | cΦ ECL | |
| bBB2 | 528 nm | 8.4 × 104 | 547 nm | 0.39 | 2.26 eV | 554 nm | —— |
| BB3 | 528 nm | 8.2 × 104 | 545 nm | 0.39 | 2.27 eV | 570 nm, 740 nm | 0.006 |
| BB4 | 528 nm | 6.7 × 104 | 543 nm | 0.47 | 2.28 eV | 571 nm, 741 nm | 0.010 |
E0–0 approximate energy of the first singlet excited state taken as the fluorescence wavelength maximum.
Data for BB2 is reproduced from reference 46.
ECL efficiencies were determined using [Ru(bpy)3]2+ in MeCN as a standard, for which ΦECL = 0.05.
Excitation of BB3 and BB4 leads to a greenish-yellow emission, centered at 545 and 543 nm, respectively (Figure 5, Table 2). The small Stokes shift observed for both bpy-BODIPY derivatives was also observed for BB246 and is typical of many BODIPY dyes.54 Measured quantum yields of fluorescence (ΦFl) for BB3 and BB4 (0.39 and 0.47, respectively) are also similar to that determined for BB2.46 Notably, there was no evidence for dimerization or aggregation of the bpy-BODIPY complexes in solution, as the shape and normalized intensity of the photoluminescence spectra recorded for BB3 and BB4 did not vary as a function of dye concentration.
Electrogenerated Chemiluminescence
Initial ECL studies for BB3 and BB4 were carried out by pulsing between the first reduction and oxidation potentials (80 mV past both the reduction and oxidation waves). ECL spectra obtained via annihilation of the ions and diions generated during these experiments are shown in Figure 6, and associated spectral data is listed in Table 2. Two ECL emission peaks are observed for both bpy-BODIPY homologues. Higher energy ECL bands were observed at ~570 nm, which are similar in energy to those observed in the photoluminescence spectrum. Lower energy ECL signals were also resolved at ~740 nm. By contrast, when the excited states of BB3 or BB4 were generated under oxidizing conditions using tripropylamine (TPA) as a coreactant, or under reducing conditions using benzoyl peroxide (BPO) as a coreactant, only the high-energy ECL peaks (λECL ~ 570 nm) were observed (Figure 7). These ECL emission profiles are very similar to the normal fluorescence spectra observed for BB3 and BB4 when corrected for a small difference inner filter effect (Figure 7).46 Virtually identical results were obtained previously in for BB2, suggesting that all three bpy-BODIPY derivatives produce ECL via the common pathways shown in Scheme 2. 46
Figure 6.

Fluorescence (blue) and ECL (red) spectra of (a) BB3 and (b) BB4. ECL emission was generated by oscillating the potential with a pulse width of 0.1 s from ~80 mV past the first oxidation and reduction waves of each Bpy-BODIPY homologue. ECL spectra were recorded in CH2Cl2 containing 0.1 M TBAPF6 with an accumulation time of 2 min.
Figure 7.
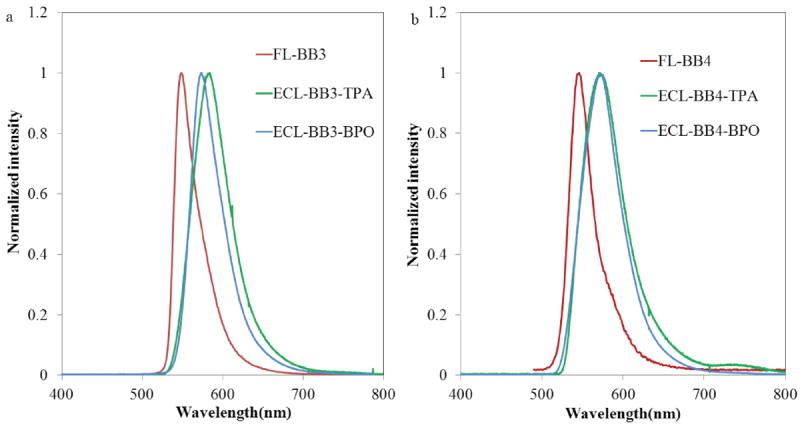
Normalized fluorescence (red) and ECL (blue and green) spectra recorded for 0.5 mM solutions of (a) BB3 and (b) BB4 in CH2Cl2 containing 0.1 M TBAPF6. ECL spectra in blue were generated by pulsing the applied potential from 0 to −1.3 V versus Ag/Ag+ in the presence of 3.5 mM benzoyl peroxide. ECL spectra in green were generated by pulsing the applied potential from 0 to 1.3 V versus Ag/Ag+ in the presence of 25 mM TPA
Scheme 2.
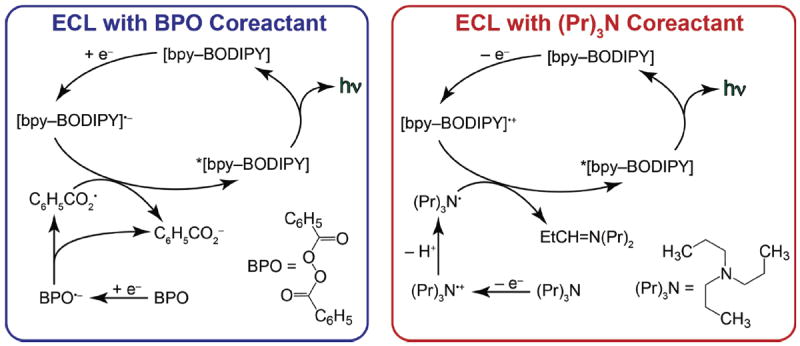
Pathways for ECL generation from bpy-BODIPY derivatives in the presence of either BPO or (Pr)3N coreactants.
In order to clarify the nature of the excited state from which ECL emission is produced for BB3 and BB4, the enthalpy of annihilation (ΔHann) was assessed for both compounds. This parameter was estimated from the reversible standard potentials for reduction and oxidation of each of the Bpy-BODIPY homologues according to the expression ΔHann = ΔGann − T Δ S. The free energy of annihilation (ΔGann) was calculated as the difference between E1/2(A/A+) and E1/2(A/A−) and TΔS was estimated to be 0.1 eV,55 leading to an estimation of ΔHann ~ 2.20 eV. The energies of the first singlet excited state (E0–0) of BB3 and BB4 were also determined based on their maximal fluorescence wavelength (λFl) to be E0–0 = 2.27 eV. The estimated values of ΔHann and E0–0 for BB3 and BB4 differ by less than 0.1 eV, which suggests that the bpy- BODIPY ECL and fluorescence originate from the same excited state. Moreover, these results suggest that ECL signal observed at ~570 nm for BB3 and BB4 is generated through either an Sor T-route (vide supra).
In addition to the ECL signal at ~570 nm, long-wavelength emission at ~740 nm was also observed under annihilation conditions. This long-wavelength signal was not observed in the photoluminescence spectra recorded for BB3 and BB4. As has been observed for previously studied BODIPY derivatives, long-wavelength ECL can be generated via formation of excimers, however, such signals are usually broad and structureless. That the ECL signal observed for BB3 and BB4 at 740 nm is relatively sharp, suggests that this emission is generated via an alternate pathway.
A second pathway by which long-wavelength ECL can be obtained, involves electrochemical production of side-products, which in and of themselves, are efficient ECL luminophores in the red region. Such phenomena have been observed in other ECL studies involving BODIPY derivatives.40,56 In order to determine if a bpy-BODIPY decomposition product was responsible for the long-wavelength ECL in Figure 6, we examined the time and concentration dependence for ECL signal at 740 nm. As shown in Figure 8a, the ratio of ECL signal produced by BB3 at 740 nm versus that at 570 nm (I740/I570) increases with ECL accumulation time. Additionally, when the annihilation ECL experiment was repeated with BB3 solutions of varying concentration (0.1 – 0.8 mM), the signal at 740 nm was maximized at higher concentrations (Figure 8b). When taken together, these experiments suggest that the ECL band at 740 nm is generated by a new species formed from BB3 during the annihilation process. We note that formation of this new species requires the annihilation reaction and is not simply the result of decomposition of one of the radical ions formed during the experiment. Moreover, the rise in ECL intensity at 740 nm is concomitant with a change in the color of the ECL solution from orange to deep purple. This change is irreversible, as the color of the solution does not revert back to orange after completion of the ECL experiment. These observations suggest that the long-wavelength ECL signal is generated from a new emissive product that is irreversibly generated from BB3 during the annihilation experiment. Future investigations will focus on elucidation of these electrochemical products, which may be composed of bpy-BODIPY dimers or oligomers.
Figure 8.
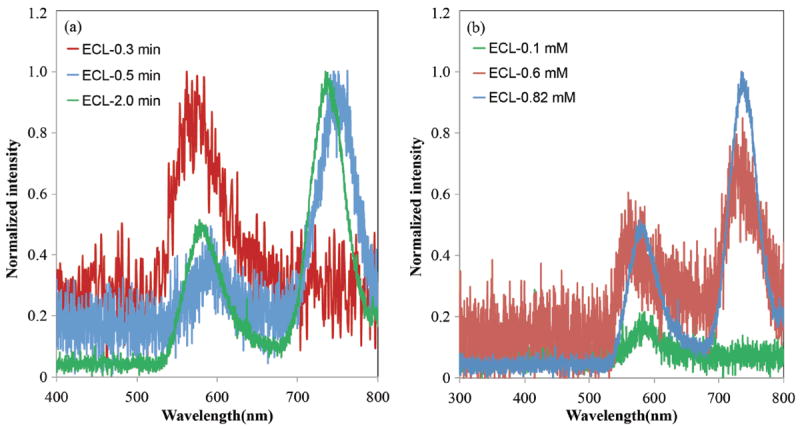
Annihilation ECL spectra recorded (a) as a function of accumulation time for 0.82 mM BB3 and (b) with varying concentrations of BB3.
Summary
Two new 2,2’-bipyridine derivatives containing ancillary BODIPY units at either the 5- and 5’- (BB3) or 6- and 6’-postions (BB4) were prepared and the basic photophysics, electrochemistry and ECL properties of these bpy-BODIPY homologues were investigated. These properties are similar to those observed for a related bpy-BODIPY derivative in which two BODIPY moieties were coupled to a 2,2’-bipyridine spacer at the 4- and 4’-positions (BB2). All three of the bpy-BODIPY constructs are strongly absorbing in the visible region and display high fluorescence quantum yields in CH2Cl2. Cyclic voltammetry showed that the new bpy-BODIPY derivatives (BB3 and BB4) also maintain redox properties that are similar to that observed for BB2. All three systems display reversible 2e− oxidation and reduction waves, which consist of two closely spaced (50 – 70 mV) 1e− events. These observations are consistent with the calculated frontier molecular orbitals for these compounds and indicate that the 2,2’-bipyridine spacer of each bpy-BODIPY homologue does not facilitate efficient electronic communication between the tethered indacene units, regardless of their position on the bpy bridge.
ECL experiments for BB3 and BB4 correlate with the observed electrochemistry. In the presence of a coreactant (TPA or BPO) both of these bpy-BODIPY derivatives exhibit strong ECL and produce spectra that are very similar to their corresponding fluorescence profiles. Similar results have been observed for BB2. Under annihilation conditions, however, the ECL signal obtained from BB3 and BB4 is distinct, and is characterized by two major bands. One of these bands is centered at ~570 nm and is attributed to emission via an S- or T-route. The second band, occurs at longer wavelengths and is centered around ~740 nm. This long-wavelength ECL signal was not observed in the BB3 or BB4 photoluminescence spectra. The shape and concentration dependence of this long-wavelength ECL band was not indicative of luminescence from an excimer or aggregate, but rather is generated by a new species formed from the bpy- BODIPY luminophores during the annihilation process. Future work will be needed to elucidate the structure of these electrochemical products, which may be comprised of bpy-BODIPY dimers, oligomers or more extended assemblies.
Supplementary Material
Acknowledgments
A.J.B acknowledges support from the Robert A. Welch Foundation (F-0021) and the National Science Foundation (CHE-0808927). J.R. was supported through a DuPont Young Professor award. J.R. also thanks the University of Delaware and the Donors of the American Chemical Society’s Petroleum Research Fund for financial support. H.L.Q. thanks The National Science Foundation of China (nos. 20805028, 21027007), the Natural Science Basic Research Plan in Shaanxi Province of China (no. 2013KJXX-73) and the Fundamental Research Funds for the Central Universities (no.GK261001185) for support. NMR and other data were acquired at UD using instrumentation obtained with assistance from the NSF and NIH (NSF-MRI 0421224, NSF-CRIF MU CHE-0840401 and CHE-0541775, NIH P20 RR017716). The authors thank Dr. Fu-Ren F. Fan for valuable discussion.
Footnotes
Supporting Information Available: Electrochemistry and spectroscopic data. This material is available free of charge via the Internet at http://pubs.acs.org.
Contributor Information
Joel Rosenthal, Email: joelr@udel.edu.
Allen J Bard, Email: ajbard@mail.utexas.edu.
References
- 1.Bard AJ, editor. Electrogenerated Chemiluminescence. Dekker; New York: 2004. [Google Scholar]
- 2.Marcus RA. J Chem Phys. 1965;43:2654–2657. [Google Scholar]
- 3.Knight AW, Greenway GM. Analyst. 1994;119:879–890. [Google Scholar]
- 4.Armstrong NR, Wightman RM, Gross EM. Annu Rev Phys Chem. 2001;52:391–422. doi: 10.1146/annurev.physchem.52.1.391. [DOI] [PubMed] [Google Scholar]
- 5.Velasco JG. Electroanalysis. 1991;3:261–271. [Google Scholar]
- 6.Forster RJ, Bertoncello P, Keyes TE. Annu Rev Anal Chem. 2009;2:359–385. doi: 10.1146/annurev-anchem-060908-155305. [DOI] [PubMed] [Google Scholar]
- 7.Richter MM. Chem Rev. 2004;104:3003–3036. doi: 10.1021/cr020373d. [DOI] [PubMed] [Google Scholar]
- 8.Miao WJ. Chem Rev. 2008;108:2506–2553. doi: 10.1021/cr068083a. [DOI] [PubMed] [Google Scholar]
- 9.Omer KM, Ku SY, Wong KT, Bard AJ. Angew Chem Int Ed. 2009;48:9300–9303. doi: 10.1002/anie.200904156. [DOI] [PubMed] [Google Scholar]
- 10.Maloy JT, Bard AJ. J Am Chem Soc. 1971;93:5968–5981. [Google Scholar]
- 11.Keszthelyi CP, Bard AJ. Chem Phys Lett. 1974;24:300–304. [Google Scholar]
- 12.Park SM, Bard AJ. J Am Chem Soc. 1975;97:2978–2985. [Google Scholar]
- 13.Chandros EA, Longwort JW, Visco RE. J Am Chem Soc. 1965;87:3259–3260. [Google Scholar]
- 14.Keszthelyi CP, Tachikawa H, Bard AJ. J Am Chem Soc. 1972;94:1522–1527. [Google Scholar]
- 15.Keszthel CP, Bard AJ. Chem Phys Lett. 1974;24:300–304. [Google Scholar]
- 16.Valenti G, Bruno C, Rapino S, Fiorani A, Jackson EA, Scott LT, Paolucci F, Marcaccio M. J Phys Chem C. 2010;114:19467–19472. [Google Scholar]
- 17.Oyama M, Mitani M, Okazaki S. Electrochem Commun. 2000;2:363–366. [Google Scholar]
- 18.Tachikaw H, Bard AJ. Chem Phys Lett. 1974;26:568–573. [Google Scholar]
- 19.Itaya K, Toshima S. Chem Phys Lett. 1977;51:447–452. [Google Scholar]
- 20.Park SM, Paffett MT, Daub GH. J Am Chem Soc. 1977;99:5393–5399. [Google Scholar]
- 21.Treibs A, Kreuzer FH. Annalen Der Chemie-Justus Liebig. 1968;718:208–223. doi: 10.1002/jlac.19687180118. [DOI] [PubMed] [Google Scholar]
- 22.Qin W, Baruah M, Van der Auweraer M, De Schryver FC, Boens N. J Phys Chem A. 2005;109:7371–7384. doi: 10.1021/jp052626n. [DOI] [PubMed] [Google Scholar]
- 23.Benniston AC, Copley G. Phys Chem Chem Phys. 2009;11:4124–4131. doi: 10.1039/b901383k. [DOI] [PubMed] [Google Scholar]
- 24.Bañuelos J, Arroyo-Córdoba IJ, Valois-Escamilla I, Alvarez-Hernández A, Peña-Cabrera E, Hu R, Zhong, Tang BZ, Esnal I. RSC Adv. 2011;1:677–684. [Google Scholar]
- 25.Loudet A, Burgess K. Chem Rev. 2007;107:4891–4932. doi: 10.1021/cr078381n. [DOI] [PubMed] [Google Scholar]
- 26.Ziessel R, Ulrich G, Harriman A. New J Chem. 2007;31:496–501. [Google Scholar]
- 27.Gomez-Duran CFA, Garcia-Moreno I, Costela A, Martin V, Sastre R, Banuelos J, Arbeloa FL, Arbeloa IL, Pena-Cabrera E. Chem Commun. 2010;46:5103–5105. doi: 10.1039/c0cc00397b. [DOI] [PubMed] [Google Scholar]
- 28.Ulrich G, Ziessel R, Harriman A. Angew Chem Int Ed. 2008;47:1184–1201. doi: 10.1002/anie.200702070. [DOI] [PubMed] [Google Scholar]
- 29.Andrade GA, Pistner AJ, Yap GPA, Lutterman DA, Rosenthal J. ACS Catalysis. 2013;3:1685–1692. doi: 10.1021/cs400332y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leonardi MJ, Topka MR, Dinolfo PH. Inorg Chem. 2012;51:13114–13122. doi: 10.1021/ic301170a. [DOI] [PubMed] [Google Scholar]
- 31.Khatchadourian A, Krumova K, Boridy S, Ngo AT. Biochemistry. 2009;48:5658–5668. doi: 10.1021/bi900402c. [DOI] [PubMed] [Google Scholar]
- 32.Rosenthal J, Lippard SJ. J Am Chem Soc. 2010;132:5536–5537. doi: 10.1021/ja909148v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Royzen M, Wilson JJ, Lippard SJ. J Inorg Biochem. 2013;118:162–170. doi: 10.1016/j.jinorgbio.2012.08.025. [DOI] [PubMed] [Google Scholar]
- 34.Nepomnyashchii AB, Broring M, Ahrens J, Kruger R, Bard AJ. J Phys Chem C. 2010;114:14453–14460. [Google Scholar]
- 35.Nepomnyashchii AB, Bard AJ. Acc Chem Res. 2012;45:1844–1853. doi: 10.1021/ar200278b. [DOI] [PubMed] [Google Scholar]
- 36.Venkatanarayanan A, Martin A, Keyes TE, Forster RJ. Electrochem Commun. 2012;21:46–49. [Google Scholar]
- 37.Krumova K, Cosa G. J Am Chem Soc. 2006;132:17560–17569. doi: 10.1021/ja1075663. [DOI] [PubMed] [Google Scholar]
- 38.Galletta M, Puntoriero F, Campagna S, Chiorboli C, Quesada M, Goeb S, Ziessel R. J Phys Chem A. 2006;110:4348–4358. doi: 10.1021/jp057094p. [DOI] [PubMed] [Google Scholar]
- 39.Lakshmi V, Ravikanth M. Chem Phys Lett. 2013;564:93–97. [Google Scholar]
- 40.Nepomnyashchii AB, Broring M, Ahrens J, Bard AJ. J Am Chem Soc. 2011;133:19498–19504. doi: 10.1021/ja207545t. [DOI] [PubMed] [Google Scholar]
- 41.Nepomnyashchii AB, Broring M, Ahrens J, Bard AJ. J Am Chem Soc. 2011;133:8633–8645. doi: 10.1021/ja2010219. [DOI] [PubMed] [Google Scholar]
- 42.Nepomnyashchii AB, Broring M, Ahrens J, Kruger R, Bard AJ. J Phys Chem C. 2010;114:14453–14460. [Google Scholar]
- 43.Nepomnyashchii AB, Cho S, Rossky PJ, Bard AJ. J Am Chem Soc. 2010;132:17550–17559. doi: 10.1021/ja108108d. [DOI] [PubMed] [Google Scholar]
- 44.Suk J, Omer KM, Bura T, Ziessel R, Bard AJ. J Phys Chem C. 2011;115:15361–15368. [Google Scholar]
- 45.Nepomnyashchii AB, Pistner AJ, Bard AJ, Rosenthal J. J Phys Chem C. 2013;117:5599–5609. doi: 10.1021/jp312166w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rosenthal J, Nepomnyashchii AB, Kozhukh J, Bard AJ, Lippard SJ. J Phys Chem C. 2011;115:17993–18001. doi: 10.1021/jp204487r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Uhlich NA, Sommer P, Bühr C, Schürch S, Reymond J–L, Darbre T. Chem Commun. 2009:6237–6239. doi: 10.1039/b912291e. [DOI] [PubMed] [Google Scholar]
- 48.Sahami S, Weaver MJ. J Electroanal Chem. 1981;122:155–170. [Google Scholar]
- 49.Ammar F, Saveant JM. J Electroanal Chem. 1973;47:215–221. [Google Scholar]
- 50.Itaya K, Bard AJ, Szwarc M. Z Phys Chem Neue Fol. 1978;112:1–9. [Google Scholar]
- 51.Bard AJ, Faulkner LR. Electrochemical Methods: Fundamentals and Applications. John Wiley; New York: 1980. [Google Scholar]
- 52.Lai RY, Bard AJ. J Phys Chem B. 2003;107:5036–5042. [Google Scholar]
- 53.Arbeloa TL, Arbeloa FL, Arbeloa IL, Garcia-Moreno I, Costela A, Sastre R, Amat-Guerri F. Chem Phys Lett. 1999;299:315–321. [Google Scholar]
- 54.Ziessel R, Ulrich G, Harriman A. New J Chem. 2007;31:496–501. [Google Scholar]
- 55.Miao W. In: Handbook of Electrochemistry. Zoski CG, editor. Elsevier; Amsterdam, The Netherlands: 2007. p. 541. [Google Scholar]
- 56.Sartin MM, Camerel F, Ziessel R, Bard AJ. J Phys Chem C. 2008;112:10833–10841. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.