

Abstract
Many animal miRNA loci reside in genomic clusters that generate multicistronic primary-miRNA transcripts. While clusters that contain copies of the same miRNA hairpin are clearly products of local duplications, the evolutionary provenance of clusters with disparate members is less clear. Recently, it was proposed that essentially all such clusters in Drosophila derived from de novo formation of miRNA-like hairpins within existing miRNA transcripts, and that the maintenance of multiple miRNAs in such clusters was due to evolutionary hitchhiking on a major cluster member. However, this model seems at odds with the fact that many such miRNA clusters are composed of well-conserved miRNAs. In an effort to trace the birth and expansion of miRNA clusters that are presently well-conserved across Drosophilids, we analyzed a broad swath of metazoan species, with particular emphasis on arthropod evolution. Beyond duplication and de novo birth, we highlight a diversity of modes that contribute to miRNA evolution, including neofunctionalization of miRNA copies, fissioning of locally duplicated miRNA clusters, miRNA deletion, and miRNA cluster expansion via the acquisition and/or neofunctionalization of miRNA copies from elsewhere in the genome. In particular, we suggest that miRNA clustering by acquisition represents an expedient strategy to bring cohorts of target genes under coordinate control by miRNAs that had already been individually selected for regulatory impact on the transcriptome.
Keywords: cluster, evolution, microRNA
INTRODUCTION
microRNAs (miRNAs) are an abundant class of hairpin-containing transcripts that generate ∼22-nt regulatory RNAs. In animals, a variety of miRNA biogenesis strategies have been documented, but the majority of miRNA species derive from a canonical pathway (Yang and Lai 2011). In brief, a primary-miRNA (pri-miRNA) transcript bearing an inverted repeat is cleaved in the nucleus by the Drosha RNase III enzyme to generate a ∼55–70-nt pre-miRNA hairpin, which is then cleaved in the cytoplasm by the Dicer RNase III enzyme to yield a ∼22-nt miRNA/miRNA* duplex. Following loading of the duplex into an Argonaute protein, one strand (the “mature” miRNA) is preferentially retained, and guides the Argonaute complex to target genes (Meister 2013).
Animal miRNAs have propensity to repress transcripts bearing short complementary sites, primarily within 3′ untranslated regions (3′ UTRs). Although several modes of miRNA:target interaction exist (Bartel 2009), functional targets require as little as Watson–Crick pairing to positions ∼2–8 of the miRNA, also known as the miRNA seed (Lai and Posakony 1997; Lai et al. 1998; Lai 2002; Lewis et al. 2003; Doench and Sharp 2004; Brennecke et al. 2005). As a consequence of these minimal pairing requirements, animal miRNAs generally have large target networks. Conserved miRNAs often have hundreds of conserved target sites (Bartel 2009), and a majority of mammalian mRNAs appear to be under selection for direct regulation by one or more miRNAs (Friedman et al. 2009). Additional functional miRNA targets bear poorly conserved and/or noncanonical sites (Giraldez et al. 2006; Baek et al. 2008; Selbach et al. 2008), which may expand the impact of miRNA regulation. Therefore, the developmental and physiological impact of miRNA regulation appears to be broad and profound, a notion supported by the lethal pleiotropy of core miRNA enzyme knockouts (Bernstein et al. 2003; Giraldez et al. 2006; Smibert et al. 2011) and an expansive literature on the functions of various individual miRNAs (Mendell and Olson 2012; Sun and Lai 2013).
The initial cloning studies of miRNAs showed that a substantial fraction of them were genomically clustered (Lagos-Quintana et al. 2001; Lau et al. 2001; Lee and Ambros 2001). Such clusters often exhibited coregulated accumulation of mature small RNAs, suggesting their derivation from multicistronic pri-miRNA transcripts. This notion was reinforced by subsequent analyses of the genomic locations and expression correlations among expanded catalogs of miRNA annotations (Rodriguez et al. 2004; Baskerville and Bartel 2005), and biochemical studies of the stepwise maturation of polycistronic pri-miRNA transcripts (Lee et al. 2002). In our previous computational analysis of Drosophila miRNAs, we were intrigued by the fact that while many genomic clusters were composed of tandemly duplicated loci, many bore miRNAs of clearly distinct sequences (Lai et al. 2003), raising questions on the evolutionary provenance of miRNA clusters. More recently, several possibilities for cluster genesis were hypothesized (Marco et al. 2013). Besides local duplications, these included that miRNAs from distinct neighboring transcription units might come to be fused (“left-together”), that miRNAs from disparate genomic regions might come to be apposed (“put-together”), or that miRNA hairpins might emerge de novo on an existing pri-miRNA transcript and later be stabilized as a functional miRNA (“new hairpin”) (Fig. 1).
FIGURE 1.
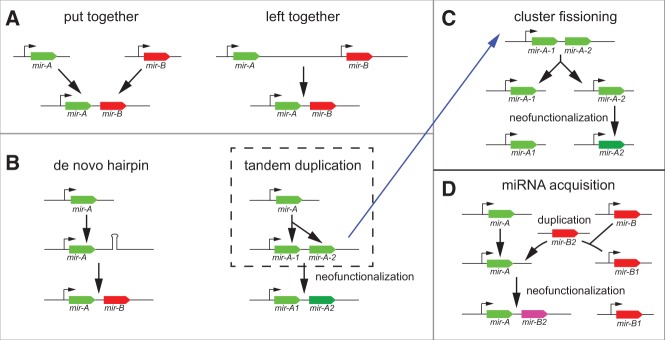
Principles that influence the evolution of miRNA clusters. (A) Two modes were described as hypothetical by Marco et al. (2013), but were concluded not to contribute to the formation of Drosophila miRNA clusters. (B) Two modes that were reported to be the dominant mechanisms that form Drosophila miRNA clusters by Marco et al. (2013). Within the tandem duplication mode, we extend two evolutionary outcomes. First, the copies may be subject to sequence divergence and subsequent neofunctionalization, which can lead to distinct selective pressures on the miRNA copies. (C) An alternate outcome of a local duplication is the fissioning of the cluster and their mobilization into distinct transcription units. This can be accompanied by neofunctionalization of the copies. (D) These evolutionary modes can be combined when a mobilized miRNA copy is transposed into an existing miRNA locus, creating a miRNA cluster. Again, this can be accompanied by neofunctionalization of the copy.
In this study, we re-evaluated how the present-day miRNA clusters that are well-conserved across the Drosophilids (Lai et al. 2003; Ruby et al. 2007) came to adopt their present-day configurations. Thorough evolutionary tracings across the metazoan phylogeny, coupled with reannotation efforts from >100 publicly available small RNA data sets from non-Drosophilid arthropods, provide evidence for several previously underappreciated features of miRNA evolution. In particular, (1) miRNAs can duplicate and subsequently neofunctionalize in sequence, (2) locally duplicated miRNAs can subsequently disperse about the genome, and (3) miRNA clusters can serially acquire members of existing miRNA families. With these concepts in mind, we reassess the emergence, expansion, and modification of the conserved Drosophilid miRNA clusters, and conclude that de novo hairpin birth is a relatively infrequent event among well-conserved miRNA clusters. Instead, the evidence supports that a majority of such miRNA clusters emerged by acquisition and neofunctionalization of extant miRNA copies located elsewhere in the genome. This contrasts with the behavior of testis-restricted Drosophilid miRNA clusters, which exhibit accelerated evolutionary dynamics, including adaptive behavior and a dominant trend for de novo hairpin birth (Mohammed et al. 2014).
The prevailing evolutionary trajectory for conserved miRNA clusters has consequences for interpreting the biological rationale for their genesis and maintenance. Moreover, we draw attention to the analogy between this strategy for miRNA cluster evolution and the acquisition of protein domains via exon shuffling as a means of transforming protein function. We posit that these modes of evolution are predominant because they exploit existing functionalities, at the protein or RNA levels, that have been previously honed and can be recombined in a modular manner.
RESULTS
Core concepts of miRNA evolution and cluster evolution
Griffiths-Jones and colleagues proposed four models for the formation and evolution of Drosophila miRNA clusters (Fig. 1A,B). They concluded that no evidence could be found for the formation of miRNA clusters by appositions of miRNA genes, either of loci within a local vicinity (“left-together” mode) or of genomically unlinked loci (“put-together” mode). Instead, they reported that the miRNA clusters of Drosophila melanogaster could be accounted for by two evolutionary strategies, namely by tandem duplication or by de novo hairpin birth within existing pri-miRNA transcripts (Marco et al. 2013). They also suggested that once born, miRNA clusters almost never dispersed (e.g., Fig. 1C).
Critical to the interpretation of the phylogenomic history of miRNA clusters is the assignment of ancestral miRNA relationships. The previous study primarily used BLAST criteria to assign homology (Marco et al. 2013). It is clear that mature miRNAs that are identical, or nearly so, share a common ancestry. Among those miRNA clusters whose members are well-conserved across the Drosophilids, several are composed solely of locally duplicated members that are identical or highly related (e.g., the K box miRNA clusters mir-2a-1/2a-2/2b-2 and mir-2c/13a/13b-1, the mir-92a/92b cluster, mir-281-1/2 cluster, and the mir-310/311/312/313 cluster) (Lai et al. 2003; Ruby et al. 2007; Marco et al. 2013). However, many members of seed families fall below the BLAST criteria for inclusion as homologs by this definition. For these miRNAs it may not be easily distinguished whether (1) they were ancestrally related but subsequently diverged, or (2) they evolved from unrelated hairpins but converged upon seed-similar sequences. In the previous study (Marco et al. 2013), all miRNAs below the cutoffs set were inferred to have emerged from independent genomic sequences, which led to the conclusion of frequent de novo hairpin birth.
A concept not previously addressed was the extent to which mobilization of miRNA copies occurs, and whether this might influence miRNA cluster evolution (Fig. 1C). For example, C. elegans retains one ancestral copy of the let-7 miRNA that is nearly identical to canonical fly and vertebrate let-7 (Pasquinelli et al. 2000; Reinhart et al. 2000), but contains additional unlinked paralogs (miR-48/84/241) that are largely unrelated to let-7 outside of their seed regions (Lim et al. 2003). These “let-7 sister” genes function in the same heterochronic pathway as let-7, albeit at an earlier developmental stage (Abbott et al. 2005). This is consistent with the notion of cycles of duplication, dispersal, and neofunctionalization from the ancestral let-7 copy. Similar events may have occurred with the K box family, the largest family of miRNAs among Drosophilid genomes (Lai et al. 1998, 2003; Lagos-Quintana et al. 2001; Lai 2002; Marco et al. 2012). In fruitflies, there are four dispersed genomic loci (harboring 8 K box miRNA copies) of the miR-2/13 subfamily, for which their strongly related sequences make a strong case that they originally derived from a common locus (Supplemental Fig. 1). However, an additional three genomic loci (harboring 5 K box miRNA copies) exist elsewhere, and it seems plausible that these might also have derived from a K box progenitor, but diverged in sequence following their dispersal.
Since the issue of distinguishing the alternate hypotheses of shared ancestry versus convergence of sequence lies at the heart of interpreting miRNA cluster evolution, we attempted to address this question using deep analysis across a large number of metazoan species. Toward this end, it was necessary to analyze a breadth of genomes and bolster their miRNA annotations using several methods. This effort is challenged by the fact that few species have been sampled as deeply with respect to small RNAs as D. melanogaster; thus, some relevant miRNA loci may not yet have been identified in some species clusters. On the other hand, many miRNAs annotated in D. melanogaster were born relatively recently during Drosophilid radiation (Berezikov et al. 2010, 2011; Mohammed et al. 2013). A substantial number of these reside in testis-restricted genomic clusters that evolve according to adaptive dynamics that are not typical of bulk miRNA loci (Lyu et al. 2014; Mohammed et al. 2014). As we recently detailed the evolutionary properties of rapidly evolving testis-restricted miRNA clusters (Mohammed et al. 2014), we focus here on those miRNA clusters whose members are well-conserved among the sequenced fruitflies (i.e., pan-Drosophilid miRNA clusters).
We traced these clusters across 11 arthropod species from varied taxonomic orders, for which small RNA sequencing data sets were available (Supplemental Table 1; Supplemental Fig. 2). We supplemented their miRNA annotations, beyond those available from the miRBase repository, via the following efforts. First, we annotated clear orthologs of known miRNAs not currently deposited in miRBase. Second, in cases of intronic miRNA loci known from Drosophilids, we queried any intronic seed matches in orthologous protein-coding gene hosts in other species for instances that resided on candidate miRNA hairpins. This allowed the possibility of identifying distantly related homologs located within syntenic regions. Third, we performed de novo annotation of miRNAs using available small RNA data, with especial focus on miRNA candidates in the vicinity of known or suspected miRNA clusters. For the latter effort, we downloaded 110 data sets from 11 non-Drosophilid arthropod species and used these for de novo miRNA annotation, focusing on the genomic cluster regions. Altogether, these efforts yielded 105 previously unrecognized (with respect to the current miRBase release 20), high confidence, miRNA loci from homologous, paralogous, and/or progenitor regions of the seven pan-Drosophilid clusters analyzed in this study (Supplemental Table 2).
We used these expanded insect miRNA annotations to trace the emergence and evolution of present-day pan-Drosophilid miRNA clusters. In order to resolve alternative possibilities regarding the histories of several ancient clusters, we also surveyed a selection of outgroup invertebrate and vertebrate species (e.g., nematodes, annelids, molluscs, urchins, vertebrates, and sea anemones, Supplemental Fig. 2). Altogether, these analyses allowed us to reclassify existing modes and to recognize new modes for the prevalent behaviors of miRNA cluster evolution (Supplemental Table 3).
Deep evolutionary analysis of miR-252 family phylogeny provides direct evidence for new modes of miRNA cluster evolution
The evolutionary trajectories that gave rise to the pan-Drosophilid mir-252 and mir-1002/968 loci proved informative, as they illustrate (1) how an ancestral miRNA cluster can be subject to fission and genomic dispersal and (2) how tandemly duplicated miRNAs can diverge their non-seed sequences, demonstrating that overall similarity scores are insufficient to assign miRNA phylogeny accurately. miR-252 is a deeply conserved miRNA with homologs present across both Protostome and Deuterostome species. This miRNA underwent an ancient, local duplication in metazoans, giving rise to the mir-252a/b cluster (Fig. 2A). Even though mir-252a and mir-252b orthologs in several Protostomes and Deuterostomes are adjacent (∼2.9 kb from one another) and share high sequence similarity across most of their mature sequences, their seed regions differ by 1 nt (Fig. 2B). As seed divergence should cause miR-252a/b to recognize largely different target cohorts, we propose that local duplication followed by neofunctionalization facilitated distinct evolutionary pressures that maintained both miRNAs during speciation.
FIGURE 2.

Phylogenetic analysis of the mir-252 superfamily. (A) All Drosophilids contain three members of the mir-252 superfamily, the solo locus mir-252, and a cluster consisting of mir-968 and mir-1002. The mir-252 superfamily is ancient among Protostomes and Deuterostomes, and only recently within Dipteran evolution did all the mir-252 members disperse; in all more basal species, mir-252a/b are located in a cluster. The mir-252b orthologs underwent further duplications within the Lepidopteran/Dipteran lineages, concomitant with the separation of the original mir-252a/b cluster. (B) Sequence alignments that support the orthology of pan-Drosophilid miR-252 to the miR-252a subfamily, and miR-968/miR-1002 to the miR-252b subfamily, based on a distinctive seed neofunctionalization adopted following the original mir-252a/b duplication. Note also that miR-968/miR-1002 are also more divergent than other miR-252b members. (C) Within the sequenced Drosophilids, all three members of the mir-252 superfamily are conserved. However, mir-252 is nearly identical in all species along both mature and star strands, whereas both mir-968 and mir-1002 are still in the process of evolving on both strands. (D) Model for the evolution of mir-252 superfamily leading to the Drosophilid lineage.
We define the miR-252a family to have the UAAGUAG seed, and the mir-252b family (which includes pan-Drosophilid miR-1002 and miR-968, various members of the butterfly miR-2797 family, and the mosquito miR-2943 family) as bearing the UAAGUAC seed (Fig. 2A). Notably, C. elegans encodes two members of the mir-252 superfamily, but both are clearly miR-252b family members (Fig. 2B). Therefore, nematodes appear to have lost miR-252a but gained an extra miR-252b copy. We were not able to date the duplication event that gave rise to the original mir-252a/b cluster, since no outgroup species contains only a single miR-252-superfamily gene. The cluster is notably completely absent from vertebrates, but is preserved in diverse Deuterostome species (Fig. 2A).
Close inspection yielded compelling evidence for a novel trajectory for the evolutionary outcome of local miRNA duplication. In the available Arachnid genomes of ticks (Ixodes scapularis) and mites (Tetranychus urticae), the mir-252a and mir-252b orthologs reside on separate scaffolds. These genomes are highly fragmented and it is thus unclear if they are truly unlinked. However, during arthropod radiation, the mir-252a and mir-252b orthologs clearly became separated by progressively increasing distances (Fig. 2A). The mir-252a/b intercluster distances in basal arthropods are 5–8 kb, while in silkworm Bombyx mori and butterfly Heliconius melpomene, the mir-252 copies reside 12–16 kb apart. These distances are significantly greater than the separation of mir-252 adjacent copies in basal Protostomes and Deuterostomes, which are ∼600 to 2.5 kb apart (Mann–Whitney test P = 0.04, Student's t-test P = 0.07). More strikingly, the mir-252a/b copies dispersed to distant genomic locations in both mosquitoes and fruitflies (Fig. 2A). We conclude that the ancient Bilaterian mir-252 cluster, present already >600 million years ago, started to separate within arthropods, became particularly fragile within Lepidopterans, and dispersed altogether within Dipterans.
Strikingly, we observe that concomitant with their dissociation, mir-252b copies underwent independent rounds of tandem duplication in Bombyx, the mosquito Aedes aegypti, and the Drosophilids (Fig. 2A). We infer this to have happened more than once, since in Bombyx, bmo-mir-2797d is a clear ortholog of mir-252b, whereas three additional copies (bmo-mir-2797a/b/c) differ substantially in their non-seed regions (Fig. 2B). In Aedes, the now genomically independent cluster of mir-252b paralogs, mir-2943-1 and mir-2943-2, underwent tandem duplication and have remained identical. This may have been the progenitor of the present-day pan-Drosophilid mir-252b cluster, bearing the family members mir-1002 and mir-968. Unlike in mosquito, these miR-252b copies have diverged extensively within their non-seed regions, and are substantially different from most other homologs of miR-252a/b sequences (Fig. 2B).
As pan-Drosophilid miR-968 exhibits a 1-nt shift in its dominant 5′ terminus relative to miR-1002 and all other miR-252b homologs, it is likely the neofunctionalized duplicate (Fig. 2B). Curiously, within the Drosophilids, the mature products of both miR-1002 and miR-968 have continued to diverge, indicating that their sequences show reduced conservation unlike the majority of conserved Drosophilid miRNAs (Okamura et al. 2008). In contrast, mir-252 is identical across both mature and star arms in all sequenced Drosophilids (Fig. 2C). It is tempting to speculate that miR-1002 and miR-968 have not fully “settled” into their regulatory networks, even though both are deeply conserved across divergent fruitflies covering ∼60 million years of evolution.
These analyses demonstrate that following local duplications, miRNA copies can neofunctionalize and also disperse about the genome. Moreover, our studies emphasize how critical evaluation of evolutionary phylogeny can reveal ancestral relationships between miRNAs bearing limited sequence similarities. We note that pan-Drosophilid mir-1002/968 were assigned to the “de novo hairpin birth” category by Griffiths-Jones and colleagues (Marco et al. 2013). However, our consideration of their phylogeny supports that not only were mir-1002/968 products of local duplication and divergence from a mir-252b locus, but that this founder gene was in turn genomically displaced following an ancient duplication and divergence event that generated the original mir-252a/b progenitor cluster (Fig. 2D).
Analysis of the miR-279 family generalizes miRNA evolutionary principles
Because these principles of miRNA evolution were germane to our general understanding of miRNA cluster dynamics, we sought to generalize features of the mir-252 family. Relevant to this, the pan-Drosophilid mir-279/996 cluster (Fig. 3A) is comprised of two seed-related loci (GACUAGA), with a third member of this seed family (mir-286) embedded in the mir-309/3/286/4/5/6-1/6-2/6-3 (i.e., mir-306⇉6) cluster. The mature sequences of all three members of the miR-279 family are identical across the sequenced Drosophilids (Okamura et al. 2008), despite substantial variation in their non-seed sequences.
FIGURE 3.

Phylogenetic analysis of the mir-279/996 cluster. (A) These trees highlight the phylogeny of the ancestral, intergenic mir-279 locus present throughout Protostome species, and the derived mir-279/996 cluster that is present in most insects. The ancestral mir-279 locus was duplicated and mobilized into an intron of DNA-polA2 in aphids. Subsequently, the intronic mir-279 was locally duplicated and neofunctionalized in Hymenopterans to generate the mir-279/996 cluster, which was retained within DNA-polA2. The ancestral, intergenic mir-279 copy was subsequently lost in honeybee and in the Dipteran lineage (red Xs), and the mir-279/996 cluster finally mobilized wholesale out of DNA-polA2 intron to an intergenic position away in the pan-Drosophilid ancestor. The numbers of additional clade-specific miR-279-family members are noted on the side, and their evolutionary disposition is documented in Supplemental Figure 3. (B) Alignment of pan-Drosophilid miR-279, miR-996, and miR-286 (sequences identical in all fruitflies) shows that miR-279 and miR-996 share the least primary homology, even though they are clearly the products of a local duplication. (C) Alignments of miR-279 and miR-996 homologs from various species. (D) Model for the evolution of mir-279 copies, including the mir-279/996 cluster.
Simple alignments might suggest that miR-279 and miR-996 are the least related pair in this seed family trio (Fig. 3B). However, the parsimonious explanation for the tandem location of mir-279 and mir-996 is that they formed via local duplication followed by neofunctionalization, akin to mir-1002/968. We sought evidence for this by tracing the ancestry of pan-Drosophilid mir-279 and mir-996. These efforts revealed a complex set of trans- and tandem-duplications of mir-279 family miRNAs throughout invertebrate evolution, including multiple expansion, loss, and mobilization events.
To dissect this scenario, we implemented both “top-down” and “bottom-up” approaches. First, we observed a clearly orthologous pair of mir-279/996 loci in almost all insects, suggesting that we might be able to identify when the cluster was born. Its emergence appears to have occurred within the Endopterygota, since the mir-279/996 cluster is present in all insects from flies to Hymenoptera (bees and ants) but is absent from aphids (Fig. 3A). On the other hand, the miR-279 family is ancient among non-chordate animals, and we identify an orthologous group of deeply conserved, intergenic mir-279 loci present throughout the Lophotrochozoan species, including Capitella teleta and Lottia gigantea (Fig. 3A). This coherent group of miRNAs is clearly identified by neighbor-joining phylogeny reconstruction based on the pairwise alignment of all miR-279 family miRNAs, which we refer to as “ancestral miR-279” (Fig. 3C).
Closer analysis revealed unexpected outcomes regarding the evolutionary fates of the miR-279 ancestral copy, especially with respect to the insect mir-279/996 cluster. For example, C. elegans has four orthologs of miR-279, all of which are slightly derived in sequence relative to those present in the outgroup Lophotrochozoans; two of these (cel-mir-44 and cel-mir-45) retain greater similarity to mir-279/996. Surprisingly, the ancestral miR-279 copy was definitively lost in honeybee, mosquito, and all fruitflies (Fig. 3A). Drosophila miR-279 is indeed very similar in sequence to this ancestral copy, but on the basis of the following observations, we can demonstrate its ancestry as a transduplicated copy that was subject to multiple mobilization events during the course of insect evolution.
The critical informative species is the pea aphid Acyrthosiphon pisum, which bears two genomically separated loci that are highly related to the ancestral miR-279 sequence. One copy is intergenic and the other is located in an intron of the gene coding for DNA polymerase α subunit B enzyme, which is orthologous to human POLA2. Since this essential protein-coding gene is substantially older than the miRNAs, we could use it as a genomic anchor to analyze its relationship to mir-279 members. To test whether miRNA annotation was simply incomplete in Arachnids, we exhaustively searched the introns of Ixodes and Tetranychus POLA2 orthologs for candidate uncloned miR-279 species. To do so, we retrieved all instances of the miR-279 seed and performed secondary structure predictions to identify any associated candidate hairpins, but none were found. Therefore, we conclude that the pan-Drosophilid mir-279/996 cluster first emerged as an aphid mir-279 copy that mobilized into POLA2 (Fig. 3A).
We observe the earliest evidence for duplication and neofunctionalization of mir-279 to yield mir-996 within the Hymenoptera. Thereafter, multiple insects retain the mir-279/996 cluster within POLA2, along with an intergenic copy of the ancestral miR-279 (Fig. 3A). Notably, in these species, the ancestral copy of miR-279 remains more similar in sequence to the copy encoded within POLA2 than to the “miR-996” orthologs (Fig. 3C). We interpret that the selective pressure to maintain miR-996 may be linked to its neofunctionalized sequence, a scenario consistent with the observation that the ancestral, intergenic copy of miR-279 was subsequently independently lost by honeybees as well as by the Dipteran progenitor (Fig. 3A). In contrast, the mir-279/996 cluster is strictly retained in order and sequence across the insects since its birth. Overall, these observations are inconsistent with the previous inference that miRNA clustering is maintained by multiple members that “draft” alongside one critical component that is dominantly under functional selection (Marco et al. 2013). We summarize our interpretation for the general order of events in the evolution of ancestral mir-279 and the Arthropod mir-279/996 cluster (Fig. 3D).
A final surprise is that the mir-279/996 cluster, while maintained in Diptera, has mobilized coordinately to a novel intergenic location. Within the Drosophilids the miRNA cluster resides adjacent to the conserved translation factor EF1gamma, and in D. melanogaster the miRNAs are annotated within the noncoding RNA CR31044, which is a candidate pri-miRNA transcript for these miRNAs. The dipterans encode a clear, single ortholog of POLA2, of which the D. melanogaster copy DNApol-α73 is 2 Mb away from EF1gamma (Fig. 3A). Similar to the rearrangement of the mir-279/996 cluster in Diptera, we identified other clade-specific translocations of mir-279 family copies. For example, we identified a Hymenoptera-specific cluster intronic to the GB18694 protein-coding gene in honeybee Apis mellifera, and a Lepidoptera-specific cluster intronic to the HMEL003294 protein-coding gene in Heliconius (Fig. 3A); these and other clade-specific amplifications are documented in Supplemental Figures 3 and 4. These data indicate a high degree of evolutionary diversification of this family, including multiple loss events as well as multiple duplication and/or mobilization events.
Collectively, these analyses further highlight that miRBase nomenclature may not accurately reflect miRNA orthology or homology. For example, the true phylogeny and evolutionary relationship of miR-279 family members is obscured by the fact that the ancestral, intergenic miR-279 orthologs are frequently named differently from each other, and instead are similar to derived, clustered copies, that some miR-996 orthologs are termed miR-279 genes, and so forth. Therefore, in all subsequent analysis, we assigned evolutionary relationships from first principles, by combining sequence information and/or genomic location, instead of grouping miRNAs by similar miRBase identifiers.
‘miRNA acquisition’ mode for miRNA cluster genesis
The analyses discussed above generalize the points that (1) clearly ancestrally related miRNAs can exhibit substantial divergence, within both seed and non-seed regions (Fig. 1B) and (2) that locally duplicated miRNA clusters can fission and then disperse about the genome (Fig. 1C); indeed, these processes can occur concomitantly. We infer that these processes have contributed to the emergence of miRNA clusters. Beyond the established concept that pri-miRNA transcripts might be birthing grounds for de novo emergence of previously non-hairpin sequence into new miRNAs, we introduce the notion of “miRNA acquisition” by a cluster. In this mode, an existing pri-miRNA transcript expands by the transposition of a fully formed copy of an extant miRNA hairpin (Fig. 1D). Such a copy might transpose directly into another miRNA transcription unit, forming a cluster; or might theoretically proceed through an intermediate genomic location. We describe how this mode has shaped the content of additional pan-Drosophilid miRNA clusters.
Evolution of the mir-285/995/998 families
Cycles of miRNA dispersal and acquisition are well-illustrated by members of the pan-Drosophilid mir-285/995/998 family. These belong to an ancient miRNA family that exists throughout the insect, nematode, and vertebrate clades, including C. elegans mir-49 and mir-83 and multiple copies of human mir-29 (Fig. 4A). The Deuterostome progenitor appears to have undergone a genomic duplication of mir-29 genes that is retained in many present-day descendants, and select protostome lineages also contain a duplication (e.g., Capitella), although this is a rarer situation (Fig. 4A). Most insects contain a specific family member that is most related to vertebrate miR-29 (with miRBase IDs that are sometimes called “miR-29” and other times “miR-285”); we infer these to all be orthologs (Fig. 4B).
FIGURE 4.

Phylogenetic analysis of the mir-285/998/995 family. (A) Drosophilid species contain three members of the seed family miR-285/998/995, of which miR-285 is clearly orthologous to a deeply conserved metazoan family termed miR-29 in vertebrates. mir-285 is intergenic, while mir-998 and mir-995 reside in introns of ancient cell cycle genes, E2f and cdc2c, respectively. mir-998 is also clustered with a member of the ancient K box family, mir-11. Although all Drosophilids retain three members of the miR-29 family, different insects were subject to reciprocal loss of different family members. A parsimonious interpretation of the phylogeny is that a mir-285 copy was acquired by the aphid E2f ortholog, and that another copy was acquired by the Hymenopteran cdc2c ortholog (either from transduplication of mir-285 or from mir-998). Perhaps because of overlapping functions of mir-995 and mir-998, they were not retained in all species; thus we observe loss of Hymenopteran mir-998 and loss of mir-995 from Lepidopterans and some Dipterans (red Xs). We also infer that E2f acquired the K box member mir-11 in the Hymenopteran progenitor. As no equivalent miRNA was acquired by cdc2c, this may explain the greater selection for retention of mir-11 in all insects. (B) Alignments of miR-29 superfamily members. (C) Alignments of miR-11 orthologs. (D) Model for evolution of the mir-285/998/995 family.
Pan-Drosophilid miR-995 and miR-998 emerged more recently during insect evolution, apparently from transpositions of the ancestral mir-29 gene. We can identify an insect-specific origination for these transduplicates because neither of them are present in crustacean (Daphnia pulex) or Arachnid (Ixodes and Tetranychus) outgroups. Conveniently, both mir-995 and mir-998 are located within introns of deeply conserved protein-coding genes, E2f and cdc2c, which allows us to pinpoint when these copies were acquired (Fig. 4A). As well, mir-998 is clustered with the pan-Drosophilid K box family member mir-11 within the E2f intron, which allows us to examine relevance to cluster genesis.
The first insect for which we could identify a miRNA inserted into either host protein-coding gene was mir-998 into aphid E2f (Fig. 4A). We infer that this event seeded the formation of a miRNA cluster, albeit via a complicated evolutionary route. Going up the phylogenetic tree, we see that all three Hymenopteran species now bear a clear mir-11 ortholog, but lack mir-998 (Fig. 4C). We identified orthologs of mir-11 in the two ant species, and also confirmed the absence of any miR-29-seed hairpin in the introns of Hymenopteran E2f genes. Thereafter, in all Coleopteran, Lepidopteran, and Dipteran species, mir-11 and mir-998 are found clustered within E2f orthologs.
Although we cannot definitively resolve the order of events that formed this cluster, we favor the stepwise scenario in which mir-998 first mobilized into E2f, followed by acquisition of K box miRNA copy within the Endopterygotan progenitor to create the cluster, but that mir-998 was lost along the Hymenopteran branch. This trajectory requires support from the sequencing of additional intermediate genomes, but this interpretation is bolstered by the trajectory of the other mobilized Insect mir-285 copy. We first detect mir-995 within the intron of cdc2c within the Hymenopterans, having annotated orthologs in both ant species de novo (Fig. 4A). However, we could not identify any miR-285-seed-hairpin in aphid cdc2c, thus ruling out that such a miRNA simply has not yet been cloned. Therefore, mir-995 emerged after mir-998 did. Notably, then, the appearance of mir-995 within the Hymenopteran cell cycle gene cdc2c coincides precisely with the disappearance of an ancestral family member from the cell cycle gene E2f (Fig. 4A). These events suggest that the reciprocal loss of mir-998 in Hymenopterans may have been coupled to gain of the similar mir-995 copy.
Interestingly, although mir-995 is conserved among diverse insects, it is also evolutionary volatile, having been definitively lost from the cdc2c intron in silkworm, butterfly, and mosquito, even though it is presently conserved in all 12 sequenced flies. We predict that these miR-285-family miRNAs, acquired by serial rounds of duplication and mobilization into cell cycle genes within insects, may prove to have related biological functions (Fig. 4D). Taken together, these analyses highlight how ancillary evidence such as genomic position guide the interpretation of miRNA cluster evolution, and provide further support to the notion that miRNA content can not only expand but also contract.
Acquisition of multiple seed family classes in the eight member mir-306⇉6 cluster
The evolution of the pan-Drosophilid mir-309/3/286/4/5/6-1,2,3 (i.e., mir-306⇉6) cluster was recently examined by Griffiths-Jones and colleagues, and our analysis agrees with theirs for the species analyzed (Ninova et al. 2014). We extended this analysis by identifying proto-mir-309 clusters in two ant species, for which none of the resident miRNAs had been previously annotated (Fig. 5A). We note that the assignment of cluster relationships across the Arthropods is mostly based on the presence of clustered members of similar seed family members, since the primary miRNA sequences involved usually have diverged extensively (Supplemental Figs. 4–6). However, instead of interpreting this cluster as a series of de novo hairpin births (Marco et al. 2013), based on the breadth of evidence collected in this study, we consider this a prime example of cluster expansion by acquisition of extant miRNA copies.
FIGURE 5.

Phylogenetic analysis of the Drosophilid mir-309/3/286/4/5/6-1,2,3 cluster. (A) The pan-Drosophilid mir-306⇉6 cluster consists of 8 miRNAs in all fruitfly species, comprising members of four different seed families. Although the sequences of the underlying miRNAs is diverging (see Supplemental Figs. 4–6), we assign orthologous clusters in other arthropods based on the presence of multiple members of the cluster. By these criteria, the full cluster existed in the Hymenopteran ancestor, as bees and ants all contain clusters bearing the four seed families. The cluster appears to have rearranged, duplicated, and/or fragmented in other arthropods, such as Tribolium and Aedes. Confident members of the cluster were not identified in Lepidoptera, although many isolated copies of the constituent cluster members exist. The cluster is not clearly identifiable outside of Hymenoptera, although we note instances of a cluster bearing a Brd box and miR-279 family member (along with other unrelated miRNAs) in A. pisum, and a cluster bearing a miR-279 family member and multiple miR-3 family members in T. urticae. (B) Members of the D. melanogaster mir-306⇉6 cluster (red loci) exhibit expression that is restricted to early embryonic stages, whereas other members of the seed families located outside of the mir-306⇉6 cluster are collectively deployed more broadly. Similarly, members of the orthologous H. saltator mir-3791⇉365 cluster are biased for expression in eggs and female bodies, relative to other available tissue type libraries, whereas all other members of these seed families are expressed more broadly.
This cluster is clearly composed of members of four deeply conserved families: (1) mir-286 is a copy of the ancient mir-279 family (Supplemental Fig. 4), (2) mir-4 is a copy of the ancient Brd-box/miR-9 family (Supplemental Fig. 5), (3) mir-5 and the three nearly identical copies of mir-6 are copies of the ancient K-box miRNA family (and likely derived from mir-11, Supplemental Fig. 1), and (4) mir-309 and mir-3 are members of the ancient mir-3 family (and members of the Arthropod mir-3791 and C. elegans mir-36/37/38/39/40/41/42 family, Supplemental Fig. 6). It seems unlikely that the observed degree of homology with four different deeply conserved miRNA families could have occurred by de novo emergence from random sequences. Instead, a parsimonious explanation for the current arrangement at this locus is that it emerged by the assembly of copies of these various ancient miRNA families into a cluster.
The cluster appears to have rearranged during insect evolution (i.e., the two mir-3 family miRNAs are located at opposite ends of progenitor mir-309 cluster in Hymenopterans, but are located adjacent to each other in Drosophilids), was further subject to additional duplications (i.e., the single K box miRNA in the progenitor mir-309 cluster expanded into four in the Drosophilids), and has acquired additional clade-specific miRNAs in certain species (Fig. 5A). Nevertheless, despite changing orders, numbers, and sequences of the miRNAs involved, evidence for the ancestral relationship of these clusters includes the fact that the ant cluster exhibits an egg-biased expression profile analogous to that of the D. melanogaster cluster (Fig. 5B). As Brd box, K box, miR-279 family, and miR-3 family miRNA genes (i.e., the progenitor members of the mir-309 cluster) are collectively expressed much more broadly throughout development (Fig. 5B), this cluster may have been assembled for the purpose of regulating early gene expression.
We were not able to clearly identify early stages in the genesis of the mir-309 cluster, as might be evidenced by a clear stepwise progression in the addition of members to the proto-cluster. There were no more basal Arthropod species bearing a cluster with three-fourth seed members of the present-day mir-309 cluster. We did identify clusters bearing two-fourth seed members in aphids and mites; the former containing a Brd box miRNA clustered with a miR-279 homolog, and the latter bearing a cluster with a miR-279 homolog and multiple miR-3 genes. In both cases, the clusters contained other seed-unrelated miRNAs. The sequencing of additional Arthropods may shed light on the evolutionary origin of this cluster.
De novo emergence of miRNAs in clusters has occurred over widely varying points during evolution
Most of the remaining Drosophila miRNA clusters that bear at least one well-conserved member have plausibly grown via de novo miRNA emergence, although greater certainty regarding some of their evolutionary trajectories may require additional genomes. Clear examples of de novo emergence include the mir-317/277/34 cluster, mir-275/305 cluster, mir-100/let-7/125 cluster, and mir-969/210 cluster (Supplemental Table 3). The provenance of some other cases, including the mir-318/994 cluster, mir-283/304/12, and mir-9c/306/79/9b cluster is potentially ambiguous, and might plausibly involve de novo birth or miRNA acquisition (see also Discussion). The assignment of de novo birth is based on the presence of one or more members of the cluster being selectively present in multiple outgroup species (e.g., mir-34, mir-100, mir-318, mir-210, mir-305, mir-283, and mir-9 are older than other members of these clusters).
Notably, involvement of the de novo birth mechanism does not imply that miRNAs emerged recently. Instead, we observe that fixation of de novo miRNAs can be localized across vastly different spans of evolutionary history. For example, mir-100 orthologs are present in the majority of Bilaterian species and within the basal Cnidarian species Nematostella vectensis (Grimson et al. 2008). This singleton miRNA was likely the source of the three-member cluster, with mir-100 and let-7 emerged later (Fig. 6A), but still quite early during the Bilaterian radiation ∼600 million years ago (MYA) (Erwin et al. 2011; Moran et al. 2014). In contrast, both members of the mir-969/210 cluster are present in all sequenced Drosophilids, but mir-210 alone is found in other arthropods and even throughout vertebrates (Fig. 6B). Therefore, mir-969 was likely born within the Drosophilid ancestor, ∼60 MYA.
FIGURE 6.
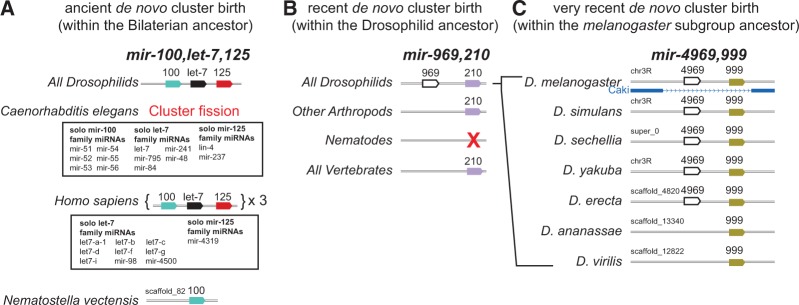
Examples of differing timescales for de novo hairpin birth in miRNA clusters. (A) The present-day mir-100/let-7/mir-125 cluster is found in diverse invertebrate and vertebrate species. The Drosophilid and human clusters are shown as exemplars; note that vertebrates contain multiple “full” cluster copies, as well as additional solo members of this cluster. The cluster appears to have expanded by de novo emergence of let-7 and mir-125 within the Bilaterian ancestor, based on the fact that clear solo mir-100 ortholog and no other cluster members are present in the Nematostella genome. Note that the mir-100/let-7/mir-125 cluster has fragmented in C. elegans, such that its genome contains multiple solo copies of these miRNAs. (B) Example of a more recent de novo miRNA cluster birth event; mir-210 orthologs are found broadly in metazoan genomes (although it was lost from C. elegans). The Drosophilid ancestor gained a clustered mir-969 gene, which is conserved in all sequenced fly genomes. (C) Example of a very recent de novo miRNA cluster birth event. All Drosophilid genomes contain mir-999, but each of the melanogaster-subgroup species (which diverged ∼7–10 MYA) gained mir-4969 adjacent to mir-999.
We can also observe de novo miRNA birth within the Drosophilids (Berezikov et al. 2010). For example, in the mir-999/4969 cluster, mir-999 is conserved in all fruitflies, but mir-4969 emerged within the melanogaster-subgroup, ∼10 MYA (Fig. 6C). Finally, there are numerous instances of clustered miRNAs that were exclusively born recently within individual Drosophila lineages, and these are predominantly restricted to the testis (Mohammed et al. 2014). The accelerated evolutionary dynamics of testis-restricted miRNA clusters appears to represent a special scenario that may be linked to their adaptive evolution (Mohammed et al. 2014). Nevertheless, the ongoing de novo emergence of miRNAs near extant miRNAs throughout the course of metazoan evolution may reflect that existing primary miRNA transcripts are somehow privileged locations for miRNA emergence.
DISCUSSION
Multiple modes of miRNA cluster evolution
Our studies provide evidence that a variety of mechanisms have shaped the content of present-day pan-Drosophilid miRNA clusters, and we discern that multiple types of events have frequently occurred in concert. Beyond established mechanisms of miRNA evolution, which include tandem duplication and de novo miRNA emergence (Ruby et al. 2007; Marco et al. 2013), we provide evidence for novel modes of miRNA cluster evolution. In particular, we witnessed the genomic dispersal of originally locally duplicated miRNAs, and their seeding of novel miRNA clusters, and their acquisition by existing miRNA clusters. Notably, our studies provide evidence that the latter process has been a dominant mechanism that expanded present-day pan-Drosophilid miRNA clusters. We were able to distinguish this from the alternative interpretation of de novo hairpin birth by detailed tracing of evolutionary histories.
We find that miRNA duplicates are often prone to neofunctionalization of sequence, not only outside of seed sequences (K box members, Brd box members, mir-279/996, mir-285 members) but also within seed sequences (mir-252a/b, mir-1002/968, mir-283/304, mir-263a/b, etc, Supplemental Table 3). This may help to explain their preservation within genomes, since they presumably adopt distinct regulatory activities. However, neofunctionalization also obscures the phylogenetic relationships among partially related miRNAs, especially when such altered copies subsequently disperse about the genome (e.g., Figs. 2–4). Reconstructions of miRNA and cluster relationships are further challenged by lineage-specific expansions and/or loss events. We were able to trace many complex histories of current pan-Drosophilid miRNA clusters, but emphasize that the order of some evolutionary events cannot yet be distinguished among the currently available genomes. Nevertheless, we illustrate how collateral information can be gained by leveraging genomic synteny, including with respect to the intronic locations of several miRNA clusters. The latter feature allowed us to pinpoint the birth of several miRNA clusters, and to trace their dynamics within a restricted genomic locale. Moreover, we uncovered an instance where a miRNA cluster that developed within an intronic location (mir-279/996, within DNA-pola73) subsequently migrated away from this position into intergenic space (within Dipterans).
Aspects of some miRNA clusters remain of unresolved heritage. For example, we still lack concrete intermediates that span the putatively stepwise assembly of the complex mir-309⇉6 cluster. As well, both members of the mir-994/mir-318 cluster are present in all Drosophilids, but absent from all other insect species. Thus, more genomes close to fruitflies are needed to clarify if mir-318 transduplicated from the mir-309/mir-3 family and mir-994 emerged subsequently via de novo birth, or if mir-994 is the ancestral cluster member that later acquired mir-318. The evolutionary history of these clusters may be clarified with additional sequenced genomes and especially those of more insects; for example, as proposed for the 5000 arthropod genome initiative (http://www.arthropodgenomes.org/wiki/i5K).
Finally, we comment on two cases of pan-Drosophilid miRNA clusters, which include loci that are seed-identical to other well-conserved miRNAs that derive from opposite hairpin arms. In particular, the mature products of mir-306 (mir-9c/306/79/9b cluster) and mir-275 (mir-275/305 cluster) share their first 8 nt, and both of these miRNAs are present across arthropods (Supplemental Fig. S7). However, mature miR-306 derives from the 5p arm, whereas miR-275 derives from the 3p arm. Similarly, pan-Arthropod miR-12 and Vertebrate miR-496 share their seed regions (Supplemental Fig. S7), but derive from the 5p and 3p hairpin arms, respectively. A reasonable interpretation is that the miR-306/miR-275 and the miR-12/miR-496 “families” actually converged on their seeds. On the other hand, if such highly conserved seed regions imply any evolutionary ancestry, they might support a “miRNA acquisition” mode for the expansion of these clusters. A speculative, but intriguing, notion is that of “hairpin-shifting,” by which miRNA orthologs are proposed to switch from generating a miRNA from one hairpin arm to the other, via alternative folds (de Wit et al. 2009). Again, additional genomes may help to resolve which of these types of events helped form these clusters.
Altogether, miRNA cluster evolution is much more complex, dynamic, and fluid than previously imagined. Moreover, we draw attention to the completely divergent evolutionary behavior of generally somatic miRNA clusters with that of testis-restricted miRNA clusters, which we have shown to be both adaptive as well as prone to exceptionally high rates of de novo hairpin birth (Mohammed et al. 2014). Such findings broadly extend the notion that miRNAs do not evolve at a universal rate, but instead exhibit distinct behavior and flux based on a variety of features. These include clustering, biogenesis mechanism (e.g., Drosha-dependent versus splicing-dependent), genomic positioning, and phylogenetic age (Berezikov et al. 2010; Mohammed et al. 2013).
Analogies of miRNA cluster evolution and exon shuffling during protein evolution
There are many analogies between strategies proposed for the origination and evolution of miRNA genes and protein-coding genes. For individual genes of either type, birth events have been cataloged by genomic duplication or by de novo emergence. Protein-coding genes have further been described to evolve via retrotranspositions from RNA intermediates, gene-fusion, and exon-shuffling. “Gene-fusion” might be considered analogous to the “put-together” miRNA cluster mode, in which genomically disparate miRNA loci become fused into a single operon. This is presumably rare, given that two miRNAs with distinct transcriptional deployment would need to become adapted for precise coexpression.
The “miRNA acquisition” mode that we formulate is analogous to exon-shuffling, also referred to as domain-sharing. This is a dominant mode of protein evolution in which exons encoding specific protein domains are reused and reincorporated in new protein-coding gene contexts (Patthy 1999; Kolkman and Stemmer 2001; Keren et al. 2010). This strategy takes advantage of an evolutionarily honed functionality that can be added “a la carte” to new proteins, without sacrificing the function of the original protein from whence it derived. We suggest that a similar mechanism serves to diversify the functionality of polycistronic miRNA loci.
The miRNA acquisition mode reinterprets the previous view that many such clusters innovated their divergent members by de novo transformation of non-hairpin sequences within existing pri-miRNA transcripts (Marco et al. 2013). In that rubric (the “drift–draft” model), it was proposed that within most miRNA operons there is predominant functional selection of a particular cluster member, which “would dominate the evolutionary fate of the other microRNAs in the cluster,” in which case “the maintenance of the clusters is most likely a by-product of tight genomic linkage” (Marco et al. 2013). We suggest that this scenario does not account for multiple clusters that have not only been preserved across all present-day Drosophilid species, but have also imposed their signatures throughout Drosophilid 3′-UTR evolution (Ruby et al. 2007; Okamura et al. 2008). This comprises powerful evidence that individual members of these clusters have all been individually selected for regulatory functions.
Instead, we infer that these were assembled for the purpose of bringing groups of miRNA seed families under common transcriptional deployment. We envisage this to represent a facile mode of regulatory evolution, since such miRNAs have (1) already achieved satisfactory biogenesis features and (2) have shaped their impact on the transcriptome and thus “come with” networks of regulatory targets. This presents an easier way to rewire regulatory networks of substantial functional impact, compared to truly de novo miRNAs that have emerged from non-hairpin sequence, which would have to fight the gauntlet of achieving reasonable processing, avoiding/purging detrimental targets, and gaining useful/beneficial targets (Chen and Rajewsky 2007; Axtell et al. 2011).
MATERIALS AND METHODS
Small RNA and other genomic data
We first identified publicly accessible, small RNA sequencing data sets for a collection of arthropod species within the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra). This yielded data sets for 11 species within each taxonomic order (Supplemental Table 1). Adapter sequences were removed using the fastx-clipper utility within the FASTX toolkit and the SeqTrimMap program (http://hannonlab.cshl.edu/fastx_toolkit/). Genome assembly sequences and gene annotation for those Arthropod species that sRNAseq data sets were identified for were downloaded from the Ensembl Metazoa genome browser (http://www.ensembl.org) and from other ad hoc locations (Supplemental Table 1). Repeat elements (simple-repeats, transposable elements, etc.) were identified for each genome assembly using the RepeatMasker and Tandem Repeat Finder programs. Repeat elements were soft-masked in the genomes (i.e., converted from an uppercase to lowercase nucleotide) using the twoBitMask program provided by the UCSC Genome Browser. Small RNA sequences were aligned to the reference assemblies using the Bowtie and SAMtools programs (Langmead et al. 2009; Li and Durbin 2009). Known miRNAs’ genomic coordinates, hairpin sequence, and mature and start sequences were downloaded from miRBase (revision 20) (http://www.mirbase.org).
Identification of miRNA orthologs
We used several layers of exhaustive searching to identify orthologs of known D. melanogaster clustered miRNAs. For D. melanogaster miRNAs residing within the sense or antisense strand of protein-coding genes, or for miRNAs adjacent to protein coding genes, we identified confident orthologous protein-coding genes by reciprocal-best TBLASTN search (Altschul et al. 1990). Small RNA reads in the BigWig format were uploaded to the Cornell mirror of the UCSC genome browser (http://genome-mirror.bscb.cornell.edu/) which permitted visualization of read pileups overlapping known miRNAs, putative orthologous protein-coding genes, and 15-kb flanking regions of these loci in order to identify novel miRNAs. Next, we searched all miRBase miRNA sequences to identify seed-matched homologs. For each D. melanogaster mature sequence, we identified its 7-mer seed sequence and searched all miRBase 5p and 3p mature sequences starting at 0, 1, and 2 nt offsets for perfect sequence matches. This offset search approach facilitated the identification of miRNAs with potential seed sequence shifts. Thirdly, we searched all small RNA sequences in a similar manner. The majority of reference genomes examined within this study are still in their first revision and are fragmented into smaller-sized scaffolds. Thus searching the small RNA sequences directly allowed identification of cloned miRNAs and bypassed the limitations of poorer genome assemblies. Finally, we predicted novel miRNAs for all Arthropod species surveyed using miRDeep2 (Friedlander et al. 2012). This exercise allowed us to identify miRNAs adjacent to orthologous clusters.
Phylogeny and alignments
We utilized the NCBI Taxonomy browser to build the phylogeny of species surveyed in this study (http://www.ncbi.nlm.nih.gov/taxonomy). The NCBI taxonomy browser reported polytomy branches with the phylogenies. In order to convert these polytomy nodes into a complete dichotomous tree, we searched the literature for accurate branch assignments. Multiple sequence alignments were performed by the MUSCLE (Edgar 2004) and Fast Statistical Aligner (Bradley et al. 2009) programs and alignments were visualized using the Jalview program (Waterhouse et al. 2009).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Sam Griffiths-Jones for discussion. J.M. was supported in part by the Tri-Institutional Training Program in Computational Biology and Medicine (via NIH training grant 1T32-GM083937). Work in A.S.’s group was supported by a David and Lucile Packard Fellowship for Science and Engineering and NIH grant R01-GM102192. Work in E.C.L.’s group was supported by the Burroughs Wellcome Fund and grants from the Foundation for the National Institutes of Health R01-GM083300 and R01-NS083833.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.046805.114.
REFERENCES
- Abbott AL, Alvarez-Saavedra E, Miska EA, Lau NC, Bartel DP, Horvitz HR, Ambros V 2005. The let-7 MicroRNA family members mir-48, mir-84, and mir-241 function together to regulate developmental timing in Caenorhabditis elegans. Dev Cell 9: 403–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ 1990. Basic local alignment search tool. J Mol Biol 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Axtell MJ, Westholm JO, Lai EC 2011. Vive la différence: biogenesis and evolution of microRNAs in plants and animals. Genome Biol 12: 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP 2008. The impact of microRNAs on protein output. Nature 455: 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP 2009. MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskerville S, Bartel DP 2005. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA 11: 241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezikov E, Liu N, Flynt AS, Hodges E, Rooks M, Hannon GJ, Lai EC 2010. Evolutionary flux of canonical microRNAs and mirtrons in Drosophila. Nat Genet 42: 6–9; author reply 9–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezikov E, Robine N, Samsonova A, Westholm JO, Naqvi A, Hung JH, Okamura K, Dai Q, Bortolamiol-Becet D, Martin R, et al. 2011. Deep annotation of Drosophila melanogaster microRNAs yields insights into their processing, modification, and emergence. Genome Res 21: 203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein E, Kim SY, Carmell MA, Murchison EP, Alcorn H, Li MZ, Mills AA, Elledge SJ, Anderson KV, Hannon GJ 2003. Dicer is essential for mouse development. Nat Genet 35: 215–217. [DOI] [PubMed] [Google Scholar]
- Bradley RK, Roberts A, Smoot M, Juvekar S, Do J, Dewey C, Holmes I, Pachter L 2009. Fast statistical alignment. PLoS Comput Biol 5: e1000392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, Stark A, Russell RB, Cohen SM 2005. Principles of microRNA-target recognition. PLoS Biol 3: e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Rajewsky N 2007. The evolution of gene regulation by transcription factors and microRNAs. Nat Rev Genet 8: 93–103. [DOI] [PubMed] [Google Scholar]
- de Wit E, Linsen SE, Cuppen E, Berezikov E 2009. Repertoire and evolution of miRNA genes in four divergent nematode species. Genome Res 19: 2064–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Sharp PA 2004. Specificity of microRNA target selection in translational repression. Genes Dev 18: 504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erwin DH, Laflamme M, Tweedt SM, Sperling EA, Pisani D, Peterson KJ 2011. The Cambrian conundrum: early divergence and later ecological success in the early history of animals. Science 334: 1091–1097. [DOI] [PubMed] [Google Scholar]
- Friedlander MR, Mackowiak SD, Li N, Chen W, Rajewsky N 2012. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res 40: 37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman RC, Farh KK, Burge CB, Bartel DP 2009. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19: 92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraldez AJ, Mishima Y, Rihel J, Grocock RJ, Van Dongen S, Inoue K, Enright AJ, Schier AF 2006. Zebrafish miR-430 promotes deadenylation and clearance of maternal mRNAs. Science 312: 75–79. [DOI] [PubMed] [Google Scholar]
- Grimson A, Srivastava M, Fahey B, Woodcroft BJ, Chiang HR, King N, Degnan BM, Rokhsar DS, Bartel DP 2008. Early origins and evolution of microRNAs and Piwi-interacting RNAs in animals. Nature 455: 1193–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren H, Lev-Maor G, Ast G 2010. Alternative splicing and evolution: diversification, exon definition and function. Nat Rev Genet 11: 345–355. [DOI] [PubMed] [Google Scholar]
- Kolkman JA, Stemmer WP 2001. Directed evolution of proteins by exon shuffling. Nat Biotechnol 19: 423–428. [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T 2001. Identification of novel genes coding for small expressed RNAs. Science 294: 853–858. [DOI] [PubMed] [Google Scholar]
- Lai EC 2002. microRNAs are complementary to 3′ UTR sequence motifs that mediate negative post-transcriptional regulation. Nat Genet 30: 363–364. [DOI] [PubMed] [Google Scholar]
- Lai EC, Posakony JW 1997. The Bearded box, a novel 3′ UTR sequence motif, mediates negative post-transcriptional regulation of Bearded and Enhancer of split Complex gene expression. Development 124: 4847–4856. [DOI] [PubMed] [Google Scholar]
- Lai EC, Burks C, Posakony JW 1998. The K box, a conserved 3′ UTR sequence motif, negatively regulates accumulation of Enhancer of split Complex transcripts. Development 125: 4077–4088. [DOI] [PubMed] [Google Scholar]
- Lai EC, Tomancak P, Williams RW, Rubin GM 2003. Computational identification of Drosophila microRNA genes. Genome Biol 4: R42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau N, Lim L, Weinstein E, Bartel DP 2001. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 294: 858–862. [DOI] [PubMed] [Google Scholar]
- Lee RC, Ambros V 2001. An extensive class of small RNAs in Caenorhabditis elegans. Science 294: 862–864. [DOI] [PubMed] [Google Scholar]
- Lee Y, Jeon K, Lee JT, Kim S, Kim VN 2002. MicroRNA maturation: stepwise processing and subcellular localization. EMBO J 21: 4663–4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB 2003. Prediction of mammalian microRNA targets. Cell 115: 787–798. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim L, Lau N, Weinstein E, Abdelhakim A, Yekta S, Rhoades M, Burge C, Bartel D 2003. The microRNAs of Caenorhabditis elegans. Genes Dev 17: 991–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu Y, Shen Y, Li H, Chen Y, Guo L, Zhao Y, Hungate E, Shi S, Wu CI, Tang T 2014. New microRNAs in Drosophila—birth, death and cycles of adaptive evolution. PLoS Genet 10: e1004096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco A, Hooks K, Griffiths-Jones S 2012. Evolution and function of the extended miR-2 microRNA family. RNA Biol 9: 242–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco A, Ninova M, Ronshaugen M, Griffiths-Jones S 2013. Clusters of microRNAs emerge by new hairpins in existing transcripts. Nucleic Acids Res 41: 7745–7752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister G 2013. Argonaute proteins: functional insights and emerging roles. Nat Rev Genet 14: 447–459. [DOI] [PubMed] [Google Scholar]
- Mendell JT, Olson EN 2012. MicroRNAs in stress signaling and human disease. Cell 148: 1172–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed J, Flynt AS, Siepel A, Lai EC 2013. The impact of age, biogenesis, and genomic clustering on Drosophila microRNA evolution. RNA 19: 1295–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed J, Bortolamiol-Becet D, Flynt AS, Gronau I, Siepel A, Lai EC 2014. Adaptive evolution of testis-specific, recently evolved, clustered miRNAs in Drosophila. RNA 20: 1195–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran Y, Fredman D, Praher D, Li XZ, Wee LM, Rentzsch F, Zamore PD, Technau U, Seitz H 2014. Cnidarian microRNAs frequently regulate targets by cleavage. Genome Res 24: 651–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninova M, Ronshaugen M, Griffiths-Jones S 2014. Fast-evolving microRNAs are highly expressed in the early embryo of Drosophila virilis. RNA 20: 360–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura K, Phillips MD, Tyler DM, Duan H, Chou YT, Lai EC 2008. The regulatory activity of microRNA* species has substantial influence on microRNA and 3′ UTR evolution. Nat Struct Mol Biol 15: 354–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquinelli AE, Reinhart BJ, Slack F, Martindale MQ, Kuroda MI, Maller B, Hayward DC, Ball EE, Degnan B, Müller P, et al. 2000. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature 408: 86–89. [DOI] [PubMed] [Google Scholar]
- Patthy L 1999. Genome evolution and the evolution of exon-shuffling—a review. Gene 238: 103–114. [DOI] [PubMed] [Google Scholar]
- Reinhart BJ, Slack F, Basson M, Pasquinelli A, Bettinger J, Rougvie A, Horvitz HR, Ruvkun G 2000. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403: 901–906. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A 2004. Identification of mammalian microRNA host genes and transcription units. Genome Res 14: 1902–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby JG, Stark A, Johnston WK, Kellis M, Bartel DP, Lai EC 2007. Evolution, biogenesis, expression, and target predictions of a substantially expanded set of Drosophila microRNAs. Genome Res 17: 1850–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N 2008. Widespread changes in protein synthesis induced by microRNAs. Nature 455: 58–63. [DOI] [PubMed] [Google Scholar]
- Smibert P, Bejarano F, Wang D, Garaulet DL, Yang JS, Martin R, Bortolamiol-Becet D, Robine N, Hiesinger PR, Lai EC 2011. A Drosophila genetic screen yields allelic series of core microRNA biogenesis factors and reveals post-developmental roles for microRNAs. RNA 17: 1997–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Lai EC 2013. Adult-specific functions of animal microRNAs. Nat Rev Genet 14: 535–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ 2009. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25: 1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JS, Lai EC 2011. Alternative miRNA biogenesis pathways and the interpretation of core miRNA pathway mutants. Mol Cell 43: 892–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.