

Abstract
Structure mapping is a classic experimental approach for determining nucleic acid structure that has gained renewed interest in recent years following advances in chemistry, genomics, and informatics. The approach encompasses numerous techniques that use different means to introduce nucleotide-level modifications in a structure-dependent manner. Modifications are assayed via cDNA fragment analysis, using electrophoresis or next-generation sequencing (NGS). The recent advent of NGS has dramatically increased the throughput, multiplexing capacity, and scope of RNA structure mapping assays, thereby opening new possibilities for genome-scale, de novo, and in vivo studies. From an informatics standpoint, NGS is more informative than prior technologies by virtue of delivering direct molecular measurements in the form of digital sequence counts. Motivated by these new capabilities, we introduce a novel model-based in silico approach for quantitative design of large-scale multiplexed NGS structure mapping assays, which takes advantage of the direct and digital nature of NGS readouts. We use it to characterize the relationship between controllable experimental parameters and the precision of mapping measurements. Our results highlight the complexity of these dependencies and shed light on relevant tradeoffs and pitfalls, which can be difficult to discern by intuition alone. We demonstrate our approach by quantitatively assessing the robustness of SHAPE-Seq measurements, obtained by multiplexing SHAPE (selective 2′-hydroxyl acylation analyzed by primer extension) chemistry in conjunction with NGS. We then utilize it to elucidate design considerations in advanced genome-wide approaches for probing the transcriptome, which recently obtained in vivo information using dimethyl sulfate (DMS) chemistry.
Keywords: next-generation sequencing, RNA structure, structure mapping, genomic big data, high-throughput genomics
INTRODUCTION
RNA is a versatile molecule, capable of performing an array of functions in the context of diverse cellular processes (Sharp 2009). To a large extent, its functionality is dependent on its ability to fold into, and transition between, highly specific complex structures. Structure analysis is thus fundamental to basic RNA research as well as to large-scale engineering efforts to design novel RNAs for a rapidly growing number of biomedical and synthetic biology applications (Chen et al. 2010, 2013; Mali et al. 2013). However, determining structure from sequence remains a challenge. As a result of several recent technological advances, a family of experimental approaches, collectively called structure mapping assays, is emerging as a powerful technique in structural studies that is complementary to other approaches (Weeks 2010).
Structure mapping assays rely on chemicals or enzymes to introduce modifications into an RNA in a structure-dependent fashion (see Fig. 1), so as to glean information about intra- and intermolecular contacts (Weeks 2010). Until recently, sites of modification have been determined by gel or capillary electrophoresis (CE) (Mitra et al. 2008; Karabiber et al. 2013), but these technologies are now being replaced by next-generation sequencing (NGS), thereby allowing probing of a multitude of RNAs in a single experiment (Underwood et al. 2010; Zheng et al. 2010; Mortimer et al. 2012; Silverman et al. 2013; Wan et al. 2013; Ding et al. 2014; Kielpinski and Vinther 2014; Rouskin et al. 2014; Seetin et al. 2014; Siegfried et al. 2014; Talkish et al. 2014). NGS delivers a fundamentally new way of measuring molecular dynamics, namely, via their reduction to the identification and counting of sequences. Once coupled to structural measurements, this “digitalization” has opened up new opportunities for genome-wide structure analysis in vivo (Mortimer et al. 2014) and for massively parallel analysis of RNA libraries in vitro (Qi and Arkin 2014).
FIGURE 1.

Overview of chemical structure mapping followed by next-generation sequencing. Reagent molecules preferentially react with unconstrained nucleotides to modify them. Reverse transcriptase (RT) traverses the RNA and drops off upon encountering the first modification. RT may occasionally drop off prior to the modification in what is termed natural drop off. Sequencing of the resulting cDNA fragments reveals the sites of modification. When RT starts at a single predetermined primer binding site, one can control two parameters: average degree of modification, which depends on reagent concentration and reaction duration, and number of sequenced fragments, which depends on choice of sequencing coverage. Stochasticity in the composition of sequencing readouts arises from randomness in modification patterns, transcription termination events, and fragment sequencing.
The coupling of structure mapping to sequencing is conceptually simple (see Fig. 1). First, a library of fragments that terminate at the sites of modification is constructed. Their subsequent sequencing reveals their identities, in contrast to estimation of their length by electrophoresis. In practice, however, performing multiplexed mapping requires a careful balance between the extent of modification that is applied to the RNAs in a sample and the depth of sequencing to be performed to detect modifications. Moreover, the degree of multiplexing and the relative abundances of the RNAs affect the nature of this balance, and therefore, experiment design requires making a series of nontrivial decisions that can greatly affect outcomes.
In this study, we perform the first systematic quantitative investigation of the effects of controllable experimental parameters on performance of NGS-based mapping assays via a series of modeling and simulation studies. Our results quantify input–output relationships, elucidate their complexity, and shed light on relevant tradeoffs and pitfalls. Simulations rely on stochastic models of the modification process and fragment generation dynamics. Since NGS readouts are in fact “molecular counters,” we are able to directly link an experiment's molecular dynamics to data variation (or quality)—a link that is missing in electrophoresis-based quantification. Recent advances in genomics thus present new opportunities for informatics-assisted design methodology.
Our analysis leads to a roadmap for rational experiment design, where quantification by simulations guides parameter optimization rather than intuition or heuristics. The roadmap involves the incorporation of prior structure profiling from small-scale studies, and we have developed an in silico framework that exploits this paradigm to allow for experiment design of large-scale multiplexed experiments as well as for evaluation of data analysis schemes. In what follows, we first devise it and demonstrate its utility in the context of SHAPE (selective 2′-hydroxyl acylation analyzed by primer extension) chemistry (Merino et al. 2005) and its recent multiplexing in conjunction with NGS, dubbed SHAPE-Seq (Mortimer et al. 2012). We then broaden its scope to encompass key features of nascent techniques, which further leverage NGS advances to enable probing of entire transcriptomes with multiple pertinent chemical reagents. As these breakthroughs propel the field into an era of ribonomic big data, we discuss data intricacies and subtleties, with a forward-looking perspective on the role that solid informatics infrastructure can play in accelerating progress. We anticipate this work will provide a quantitative basis for intuition that is needed to guide experimental design, and that it will be of particular use to the many experimentalists that will soon adopt current and forthcoming techniques as sequencing becomes cheaper and as the biochemical assays needed for in vivo and in vitro studies become mainstream.
RESULTS
In silico analysis of large-scale chemical mapping
We use a stochastic model of a SHAPE experiment and the sequencing that follows it (see Fig. 1) to generate SHAPE-Seq data in silico for RNA sequences with predetermined SHAPE profiles. The generated data undergo analysis by a method we previously developed (Aviran et al. 2011a), which uses a model and adjoined maximum-likelihood estimation (MLE) algorithm to infer the degrees of chemical modification by the SHAPE reagent at each nucleotide. This corrects for numerous biases, which distort the sought structural information to yield noisy and convoluted measurements of it. See Materials and Methods for experiment, model, and statistical inference details.
The primary outcome of data analysis is a set of point estimates that quantify the intensities of reaction between each nucleotide and the SHAPE reagent (see, for example, Fig. 2A). These are called SHAPE reactivities, and they can be used either independently or in conjunction with algorithms to infer RNA structural dynamics (Low and Weeks 2010). The basis for such structural inference is strong correlation between low SHAPE reactivities and nucleotide participation in base-pairing or other tertiary structure interactions (Vicens et al. 2007; Bindewald et al. 2011; Sükösd et al. 2013). In this paper, however, we limit attention to evaluating statistical uncertainty in reactivity estimates, with no further quantification of its subsequent impact on uncertainty in structure prediction. In doing so, we eliminate additional sources of variation which these computational and/or knowledge-based methods inevitably introduce (Eddy 2014) and can thus focus solely on understanding interexperiment variability.
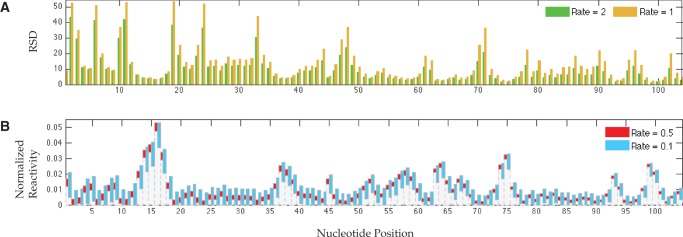
FIGURE 2.

Relative standard deviations (RSD) of ML reactivity estimates computed from 100,000 SHAPE-Seq simulations at a range of hit rates. (A) Target normalized SHAPE reactivity profile of the P546 domain of the bI3 group I intron, following omission of negligible reactivities. RSD per nucleotide values are grouped for hit rates 3 and 2 (B), 2 and 1 (C), and 0.5 and 0.1 (D). Vertical scale for the low-rate plot is >5 times the scale for high-rate plots.
We initialized simulations with two sets of values per RNA, determined by previous SHAPE measurements. One set comprised normalized relative SHAPE reactivities (also called normalized SHAPE profile), and the other comprised the propensities of reverse transcriptase (RT) to drop off at each nucleotide in the absence of chemical modification (see Materials and Methods for data and experiment descriptions). Throughout this study, we considered these to be the true inherent structural properties of the RNA, and we kept them fixed. Nonetheless, while a normalized profile is inherent to an RNA, it is not directly measured by a SHAPE experiment. Rather, reaction intensities are being measured, or more precisely, for each nucleotide, one assesses the fraction of molecules in which it is modified—a measure that depends on a tunable reagent concentration and/or reaction duration, which we lump together into a notion of concentration. It is thus a parameter that modulates the reactivity profile that we estimated. To simulate changes in concentration, we defined a hit rate parameter corresponding to the average number of modifications (see Materials and Methods). It captures the overall degree of modification, or the average number of modifications per molecule, also termed hit kinetics. Notably, for a given RNA sequence, the rate could range from small fractions of 1 to >1, depending primarily on the concentration. Since the relative relations between nucleotide reactivities should remain unchanged, we scaled the normalized profile by the hit rate to obtain a true SHAPE profile per given modification condition (see Materials and Methods). A second controllable feature is the total volume of data collected, which is the number of sequencing reads analyzed. It is a function of a chosen sequencing coverage depth and of the amount of RNA subjected to modification and reverse transcription. In simulations, we modified the total number of reads that we generated for the control and the experiment. Since each read provides evidence on a single molecule's fate, this is the effective number of probed molecules.
Once the hit rate and reads number are set, stochasticity in measurements arises from the molecules’ random fates, as their reverse transcription may abort at different sites due to differences in modification patterns and/or natural drop-off events. The many possible events yield cDNA fragments of varying lengths, thus contributing to variation in the counts of cDNAs of each possible length. It is worth noting that the likelihood that a molecule will give rise to a cDNA fragment of a certain length remains fixed under these settings, as it is fully determined by the reactivity profile and by RT's drop-off properties (see Materials and Methods). In other words, the distribution of fragment lengths is fixed, but finite samples from it display variation in fragment counts. One can think of these theoretical samples as representing technical replicates, i.e., multiple libraries, originating from the same RNA sample and modification experiment, which undergo sequencing and analysis separately.
Each simulation thus entailed finite sampling from a precomputed fragment-length distribution. Randomness in sample composition then propagated into variation in sample-based reactivity estimates. The complex relationship between these estimates and the observed fragment counts rendered direct assessment of estimation precision infeasible. It is common practice in such cases to resort to empirical assessment via resampling methods, where one repeats estimation multiple times from subsamples of the original data set. In our in silico study, we evaluated the true precision (under model assumptions) by repeating our workflow sufficiently many times, so as to faithfully reproduce the true distribution of the ML estimate. We utilized this approach to investigate the robustness of measurements and analysis under different experimental conditions.
Quantitative assessment of effects of controllable parameters
We sought to quantify the variation in reactivity estimates across a range of hit rates and data set sizes. Before we present our findings, we note that the lengths of variation intervals correlate with reactivity magnitudes, and in light of the range of SHAPE reactivities in a typical profile, it is challenging to visualize trends in these intervals across a profile. Instead, we depict the relative standard deviation (RSD) per nucleotide, that is, the ratio of estimated standard deviation (SD) to the true reactivity. We also filter out very small reactivities, as they are prone to zeroing out by our estimation method, which results in very large RSDs. Yet, in the context of an entire profile, these amount to minuscule fluctuations above zero that do not affect data interpretation. Finally, we note that we conducted simulations and observed similar results for several RNA sequences, but for coherence of exposition, in what follows we refer to a SHAPE profile of the P546 domain of the bI3 group I intron.
Effects of hit kinetics
Conducting structure mapping experiments routinely involves optimizing the reagent concentration. The optimum is often sequence- and system-specific, but a common aim is to balance between the adverse effects of too many and too few modifications (Low and Weeks 2010). This is because in molecules that carry multiple modifications, we detect only the one that is closest to the 3′ end (see Fig. 1). The loss of information from the 5′ region manifests itself in signal decay, which we correct for during analysis (Aviran et al. 2011a). Yet, high hit rates intensify signal decay and ultimately expedite signal loss, thereby shortening effective probing lengths. They might also introduce analysis-based inaccuracies due to substantial reliance on proper decay correction (Karabiber et al. 2013). Lowering the rate alleviates these concerns, but also decreases the signal-to-noise ratio (SNR), or signal quality, thus impacting analysis accuracy as well. The fact that both scenarios affect measurement precision, but in complex and subtle ways, motivated us to quantify their effects via simulations.
Figure 2 shows RSD values computed over a range of rates for the P546 RNA, along with its normalized SHAPE profile, where negligible reactivities were omitted from analysis (Fig. 2A). For ease of visualization, we divide the rates into three ranges, plotted separately in Figure 2B–D, and group the RSDs per nucleotide per each range group. Note that for comparison purposes, we plot the data for a hit rate of 2 in panels B and C (green bars). Some trends are immediately apparent from this comparison, most notably, a comprehensive increase in RSD with decreasing modification intensity, attributed to degradation in the obtained signal quality. One can also observe a threshold effect, where modification becomes so sparse such that it vastly degrades the quality (see panel D for rate 0.1 in pink). When scanning these trends across the sequence, we also note a change in pattern near the 5′ end. Specifically, reducing the rate from 3 to 2 (blue to green in panel B) results in decreased RSD (see sites 1–19), as opposed to increased RSD over the remaining sites. In other words, while increasing reagent concentration beyond two benefits measurements in one portion of the molecule, it trades off with the precision elsewhere. This observation captures the impact of severe signal decay on the SNR, as RT's drop-off process leaves very few fragments that can inform us of modifications at the 5′ region. The practical implication of this is that the molecule length for which high-quality data is effective is even shorter than the length for which signal is observed.
Nevertheless, if one aims to circumvent the signal decay problem by resorting to very low hit rates, then the obtained signal spans longer stretches and exhibits little decay, but it also results in overall poor quality (e.g., see high RSDs in the 5′ region in Fig. 2D). Importantly, when rates are low, the counts of fragments mapping to the 5′ region may be comparable to or sometimes even higher than their counterparts under higher rates, such that we indeed observe a longer and seemingly strong signal at the (+) channel (data not shown). But in fact, the SNR depends on the relative differences between counts at reactive versus unreactive sites, or alternatively, counts in the (+) versus (−) channels, which become negligible as fewer molecules are being modified. While frequent users of such probes are well aware of these tradeoffs and pitfalls, it is difficult to determine the quality and/or effective probing length based on eye inspection and/or acquired intuition alone.
More subtle observations from Figure 2 include inverse correlation between a site's normalized reactivity magnitude and its RSD. This raises the question whether observed variations have meaningful impact on the overall quality of the reconstructed profile or perhaps they amount to small absolute perturbations. We address it by overlaying the tenth and ninetieth percentiles of the simulated MLE distribution on the true normalized profile, as shown in Figure 3 for select hit rates. Note that absolute reactivities scale with the rate, and therefore, we consider variation around a fixed normalized profile. One can see from the figure that indeed, the large RSDs translate into fairly small profile perturbations, such that reactive/unreactive sites are well discriminated. It is also apparent that low reactivities cannot be determined accurately and often are indistinguishable from zero, but their range is also confined to strongly indicate structural constraints.
FIGURE 3.
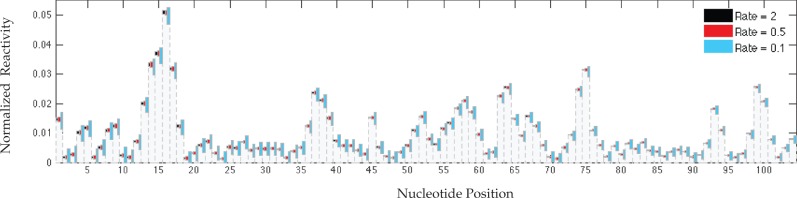
MLE variation at different SHAPE-Seq hit kinetics. Normalized target reactivities are shown in gray bars, with colored boxes representing variation around them. Box boundaries mark tenth and ninetieth percentiles of the empirical MLE distribution.
Effects of sequencing coverage
The recent coupling of digital sequencing with structure probing not only facilitated multiplexing and increased throughputs, but also opened the door to more predictable experiment design through precise control over the volume of collected data. Previously, this was nontrivial, as CE platforms generate analog signals from which relative, but not absolute, quantities are determined. Several factors commonly affect the choice of sequencing volume, including platform availability (e.g., desktop versus large-scale machines), data processing cost, multiplexing capacity, and data quality. Next, we elucidate the dependence of measurement precision on the number of analyzed fragments, to shed light on tradeoffs associated with these factors and on relations with reagent concentration.
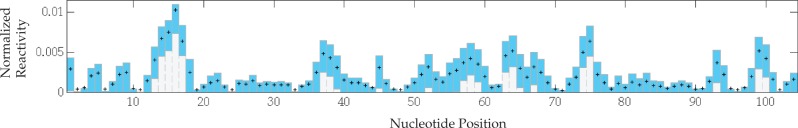
Figure 4A shows RSDs obtained at rates 2 and 1, after a 10-fold reduction in the number of reads. It illustrates the same trends as in Figure 2, but with considerably larger variations. These are even more pronounced for rates <1 (data not shown), and obviously, for lower read numbers. Figure 4B illustrates the overall degradation in quality of reconstruction for rates 0.5 and 0.1, with 10% of the reads. Again, effects are exacerbated at lower depths (data not shown). While it is expected that measurements under low hit kinetics are more susceptible to reductions in data size, Figure 4 shows that variation can be significant under high hit kinetics as well, especially at the 5′ region. In such cases, collecting fewer sequences might further shorten the effective probing length and may be suitable only for very short RNAs. These results also demonstrate that one can compensate for effects of low-hit kinetics by collecting more data. This is particularly important in multiplexed settings, since less structured RNAs tend to be more reactive than highly structured ones, and may thereby attract more reagent molecules. In well-controlled conditions, sequencing deeper or populating the sample with more low-reactivity RNAs could bias the coverage toward them. However, such strategies apply predominantly to cell-free studies, where sample manipulation at the laboratory is common, but are irrelevant when RNA material is limited or sample composition cannot be easily altered.
FIGURE 4.
MLE variation following a 10-fold reduction in SHAPE-Seq sequencing volume from 4 × 106 to 4 × 105 reads. (A) RSD values at high hit kinetics. (B) Tenth and ninetieth percentiles of empirically assessed MLE distribution at low-hit kinetics.
Analysis of transcriptome-wide mapping via random primer extension
Length limitations inherent in detection via primer extension are apparent from our analysis and widely appreciated. Traditionally, long molecules were probed with multiple primers, carefully designed to anneal at intermediate locations (Wilkinson et al. 2008), a labor-intensive effort that also precludes de novo characterization. With the advent of NGS, techniques such as RNA-Seq leveraged multiple distinct hexamer primers capable of random pervasive annealing to enable transcriptome-wide (TW) studies. Alternatively, transcripts are fragmented into random templates that are ligated to adapter sequences, where primers are designed to bind.
SHAPE-Seq and similar assays which rely on single primer extension (SPE) set the foundation to more advanced protocols that detect modifications via random primer extension (RPE) along with capabilities to probe structure in vivo (Ding et al. 2014; Rouskin et al. 2014; Talkish et al. 2014). Implementation and nuances differ between methods, but here we attempt to provide the broadest assessment of RPE-based strategies, because RPE may be coupled to a range of probes and is applicable in diverse conditions, and as such it opens up many more possibilities. For example, in vivo mapping was obtained with dimethyl sulfate (DMS), but a SHAPE-NAI probe has similar functionality (Spitale et al. 2013), whereas other probes enhance structural characterization at in vitro or near-in vivo conditions (Kielpinski and Vinther 2014; Wan et al. 2014).
A useful property of RPE is that it circumvents 3′ directionality bias. Ideally, all modifications are equally amenable to detection, as a primer could drop, for example, in between the two modifications cartooned in Figure 1. Our SPE analysis warrants revisiting then, for balancing between too many and too few modifications may no longer be relevant. Furthermore, RPE spreads the reads (i.e., their 3′ end site) across a molecule, thereby redistributing the amount of information allocated per site. Intuitively, it improves signal quality near the 5′ end at the expense of reducing it near the SPE site, while obviating reliance on signal correction methods.
In this work, we avoid detailed SPE versus RPE comparisons, since we view them as geared toward distinct endeavors, e.g., molecular engineering (Qi and Arkin 2014) versus genome-scale studies (Mortimer et al. 2014), respectively. Instead, we extended our model and analysis to capture key additional features of RPE and TW mapping data and to highlight new complexities and tradeoffs in design and informatics. An evident new challenge is that multiplexing is no longer easily manipulable. Biasing coverage toward select transcripts becomes nontrivial and furthermore, one now faces natural variation in abundances ranging over several orders of magnitude (Mortazavi et al. 2008). Consequent variation in effective coverage per RNA is a clear cause of SNR and performance differences, which, to date, has been circumvented with low-throughput targeted experiments (Kwok et al. 2013). Before discussing additional layers of complexity, we introduce two new design parameters: primer rate and fragment length range. Importantly, priming and fragmentation are equivalent from a modeling perspective (see Materials and Methods), thus primer rate stands for the average density of RT start sites within either setting. Prior to sequencing, fragments are size-selected to obtain a library of fragments that are within an admissible range.
Unlike the SPE case, the analysis below ties measurement quality to three pivotal factors, or design decisions, rather than directly to parameters. Moreover, it reveals how entangled these factors and decisions are. While a simple model suffices to render key performance determinants, it fails to capture additional real-world intricacies of ribonomic big data. Thus, we complement our in silico analysis with a qualitative discussion that elucidates finer details as well as conveys difficulties in their comprehensive treatment by simplistic data analysis schemes. To simplify exposition, we discuss primarily the primer-based DMS approach in Ding et al. (2014)—a natural extension of SPE. For the most part, results carry over to fragment-based methods (Rouskin et al. 2014; Talkish et al. 2014), but otherwise we specifically address them.
(1) Ratio of hit to primer rates. To consider the ratio's effects, it is helpful to draw an analogy to SPE. In SPE, we essentially fix the primer density at 1 per the RNA length, and when changing hit rates we in fact modulate the ratio. Dynamics generally carry over to RPE, with two deviations: No stochasticity in priming location prevails in SPE; and RT stops due to primer encounters are unique to RPE. At this point, we note that our understanding of standard NGS protocols, integrated into our model, is that no strand displacement takes place at such encounters, and that RT aborts. Yet, similar models can accommodate nonstandard RT steps. Furthermore, our modeling assumption aligns with fragment-based approaches, where RT drops off at a template's end—the analog of a priming site, thus extending the scope of analysis.
While fragment dynamics in SPE and RPE are not identical, the ratio presents a similar tradeoff, namely, decreased SNR due to background noise versus increased 3′ directionality bias (see Materials and Methods for formal analysis). For example, under small ratios, RPE features frequent consecutive primers, preventing RT from reaching adducts (see Fig. 5B, inset). Primer encounters have two undesired outcomes: (1) background noise and (2) economic inefficiency due to high proportion of noninformative, but sequenced, cDNA (see Fig. 5B versus Fig. 5C,D). A straightforward way to reduce these encounters is to sparsely modify and prime, in which case long interprimer distances allow for significant natural RT drop off in between primers—yet another source of background. In fragment-based protocols, template-end background can be discerned and removed prior to sequencing via fragment selection (Rouskin et al 2014; Talkish et al. 2014). Yet, this trades economic inefficiency with experimental one, as the fraction of informative, modification-based, fragments remains small. As we discuss below, when biological material is limited, both remedies might be infeasible. High hit-to-primer rate ratios, in contrast, give rise to frequent consecutive adducts with no intermediary primer—a source of signal decay and information loss when reactivities are not uniform (see Fig. 5C, in particular highlighted regions).
FIGURE 5.

Read distribution in RPE experiment at different ratios of hit-to-primer rates and cDNA length ranges. (A) Normalized reactivity profile for a fictive RNA obtained by replicating the P546 SHAPE profile five times. Model-derived fractions of reads that reach each site at hit rates 0.003 (B) and 0.05 (C,D), with primer rate fixed at 0.01 and length range upper bounded at 500 nt. Signal decay intensifies when lower size cutoff of 25 nt is applied (D). Highlighted windows depict regions of signal decay.
The analogy to SPE aims to recapitulate our previous points that avoiding signal decay and thereby reliance on nontrivial data correction does not necessarily translate into better-quality data, and that quantitative evaluation of design choices and informatics pipelines is beneficial. Notably, resemblance to SPE dynamics increases once high-pass cDNA filtering is introduced via lower size cutoff, as it imposes a fixed blind window downstream from each modification, which intensifies signal decay (see Fig. 5D, highlighted regions). For example, highly reactive sites located downstream from a modification and within this window frequently display modifications that “shadow” it (see Fig. 5D, inset). As we increase the hit rate, the more severe this directionality bias is. For a window size w, we can think of it as similar to positioning a primer w nucleotides downstream, which means the decay spans a window's size and can be difficult to spot for short windows. Other effects of fragment filtering are discussed next.
(2) Fragment lower-size cutoff. Size-selection throws away potentially valuable information. While normally undesired, it is common practice when data entail complexities or ambiguities which are nontrivial to resolve. Mining information from NGS readouts has been an ongoing challenge in TW studies, mainly because read lengths are such that alignment to multiple genomic locations is common (Trapnell et al. 2010). Despite consistent increases in read lengths, the generation of short cDNAs is inherent to existing mapping methods, and is even more pervasive in experiment than in control. Ambiguous cDNA alignments then give rise to uncertainty with respect to a cDNA's true origin, translating into noisy hit counts per site.
A straightforward remedy is to discard all ambiguously aligned reads, but that decreases total counts, or signal strength, and might leave some regions unmapped. One may also increase the size cutoff point as a means to reduce uncertainty in counts, but that too leaves us with less usable information. Either measure reduces both noise and signal power, making the composite effect on SNR difficult to predict. Furthermore, the extent of ambiguity in alignments is system- and reference-dependent. For example, transcriptomes often consist of multiple gene isoforms with substantial sequence overlap, that are absent from the matching genome reference. Isoform-level studies are then more prone to this issue than gene-level ones. At the same time, the extent of ambiguity is design-dependent, as it is tightly linked to the shape of the cDNA length distribution. When cDNAs are relatively short, larger fractions of them trigger uncertainty in comparison to data sets comprising of longer fragments. Length distributions largely depend on the sum of the hit and primer rates, which sets the interprimer/adduct distances (see Materials and Methods for detail). Sparse dynamics would then be more robust to this issue, but as we discuss next, they pose other critical challenges.
(3) Sum of hit and primer rates. The total frequency of priming and modification events determines key features of the cDNA length distribution, e.g., its mean and variance. As mentioned, one can circumvent some confounding issues by targeting low hit and primer rates (i.e., sparse dynamics). Sparse dynamics yield fewer reads per molecule—a problematic outcome when biological material is limited to a degree that a “sequence deeper” brute force solution is infeasible. Taking the wide variation in RNA abundances into consideration, design also greatly depends on the transcripts of interest.
Material limitations are analogous to limiting coverage per transcript. To get a sense of current capabilities and associated data quality, we revisit our SPE analysis with coverage anticipated based on recent work. For example, extended Figure 2 in Ding et al. (2014) shows maximal coverage close to 100 reads on average per site, obtained for a minute fraction of the RNAs from libraries of tens of millions of reads. For the P546 RNA, this amounts to an order of total 104 reads, whereas Figure 4 depicts an order of magnitude deeper coverage (4 × 105). If we allocate an average of 100 reads to a transcript of length 775 nt (profile shown in Fig. 5) and set hit and primer rates to 0.003 per site, our model predicts variation as shown in Figure 6, where lower rates or coverage display further degradation (data not shown). Critically, reported coverage-per-transcript ranges over five orders of magnitude, with merely a quarter of them featuring at least one read per site on average, yielding about 102 or more reads in the P546 example. Unfortunately, the need for deep coverage has not been assessed quantitatively, albeit highlighted qualitatively in Talkish et al. (2014). Notably, crude preliminary assessment does not require sophisticated models. Instead, one can bootstrap the data for preliminary quality measures, for example, by using the NGS-based approach introduced in Aviran et al. (2011a). Yet, prior in silico design is still useful. For example, we showed that the read-per-molecule yield also depends on the size cutoff, with sparse dynamics affording higher fractions of retained fragments, thus linking this factor to another design choice.
FIGURE 6.
Variation in reactivities reconstructed by the scheme in Ding et al. (2014) and computed from 100,000 RPE simulations of 77.5 × 103 reads. Box boundaries mark tenth and ninetieth percentiles of the empirical distribution; plus signs mark target normalized SHAPE reactivities in the fictive 775 nt-long RNA depicted in Figure 5. Hit and primer rates are 0.003 per site, and shown is a middle window of reactivities to circumvent end-effects.
Key principles of judicious design are well-captured by our model, but numerous other confounding factors are beyond its scope, some unique to structure mapping and others widely prevalent in functional genomics NGS assays. Interestingly, our experience is that some issues can be addressed by model-based statistical approaches, which typically treat most reads as valuable information and include them in analysis (Trapnell et al. 2010; Roberts et al. 2011). In what follows, we touch briefly upon factors we have become aware of while working in this field.
(1) Nonuniform priming. Analysis of RNA-Seq data reveals systematic biases in cDNA generation, attributed to hexamer binding or fragmentation (Roberts et al. 2011). These biases introduce local signal amplitude changes, which might alter the relativity among inferred reactivities. Distortion may be more pronounced when a narrow range of fragment sizes is selected (e.g., 25–45-nt fragments in Rouskin et al. 2014), in which case the information per site originates from a short stretch of RT start sites. In other words, a narrow range localizes a perturbation's effect whereas a wide range smoothens it out. Note, in passing, that some published analyses alleviate these discrepancies by comparing normalized counts between control and experiment, with normalization accounting for transcript abundance and possibly length (Ding et al. 2014; Talkish et al. 2014). Local count normalization over 50–200-nt windows is carried out in Rouskin et al. (2014) to remedy fragmentation-specific artifacts at the 3′ end. Such heuristic could have somewhat compensated for nonuniformity, if normalization had spanned a similar window size (i.e., 45–25 = 20 nt). Instead, boosted fragmentation at a site would result in attenuation of all reactivities in a window of, say, 200 nt, whereas counts are effectively enriched within 25–45-nt upstream of that site. This not only leaves local perturbations in place, but also generates further imbalance in relativity in between normalization windows.
(2) Multiple alignments and transcript abundances. Statistical uncertainty due to multiple alignments is intricately related to another confounding factor—unknown RNA abundances. Knowledge of relative abundances often implies that certain alignments are more probable than others, and this way, it can inform alignments, counts, and reactivities. For example, a subset of reads mapping to two isoforms would be split differently if the isoforms are equally or differentially expressed. In RNA-Seq, statistical methods resolve such ambiguities jointly with quantification of abundances, read error rates, and biases (Roberts et al. 2011), but one must keep in mind that mapping assays introduce additional complexity in the form of unknown reactivities.
(3) Fragment upper size cutoff and ambiguous RT stops. A useful property of RPE is that no sequence information is needed a priori. But there is also no notion of full-length RNA template with well-defined ends, which makes it impossible to discern by sequencing alone between fragments arising from modification and those resulting from RT runs through template ends or bound primers. Current fragment-based methods (Rouskin et al. 2014; Talkish et al. 2014) approach this ambiguity experimentally by filtering all fragments of the latter type. From an informatics standpoint, Rouskin et al.’s approach is more brute force, as it discards more than just full-template copies, but nonetheless, both protocols throw away potentially valuable information. For example, if signal decay prevails, its correction relies on the number of successful elongations past a site (see Equation 4 in Materials and Methods), a quantity whose recovery may suffer bias due to missing information. It is interesting to note that this issue becomes negligible under sufficiently sparse conditions, because RT's imperfect processivity limits achievable cDNA lengths and chances to run through template ends or primers. This appears to be the case in Ding et al. (2014), although their approach may potentially account for primer encounters under different conditions through integration of (+) and (−) data into the reactivity estimates.
(4) Protein–RNA interactions. A fundamental difference between in vitro and in vivo probing is the absence/presence of protein–RNA interactions (PRI), many of which are yet to be revealed. PRI can trigger structural rearrangements, and indeed, recent studies reveal global measurement differences between conditions (Kwok et al. 2013; Rouskin et al. 2014). Yet, observed changes may also be attributed to protein protection from modification by way of solvent inaccessibility (Kwok et al. 2013), yielding low reactivities. PRI thus give rise to ambiguity, as one cannot readily discern between structurally constrained regions and protein-bound ones from weak signal alone. This has been a long-standing challenge, but with recent breakthroughs and anticipated wealth of data, it becomes a critical barrier and possibly a primary bottleneck to accurate interpretation of in vivo data and their power to improve structure prediction. Clearly, this is unique to these emerging techniques, and more so, increasing the information content of these probes via statistics or deeper coverage does not seem plausible. We anticipate that progress will be achieved through integration and joint analysis of complementary assays.
(5) Background noise. A (−) channel controls for RT's imperfect processivity, which generally features nonuniform, possibly structure-dependent, rates, with occasional spikes. Given the nonwhite nature of this noise, it is standard practice to integrate it at nucleotide resolution into SPE analysis. In RPE, this is also warranted and obtained in Ding et al. (2014) and Talkish et al. (2014) through comparisons of (−) and (+) readouts. There are several points one should keep in mind when integrating background. First, its magnitude depends on experimental conditions, which can be probe-dependent (e.g., DMS versus SHAPE), as well as on fragment lengths. Second, it is important to retain the same RNA structure in (−) and (+). This is problematic when randomly fragmenting, as each fragment adopts its own structure prior to the RT step. Since short fragments are quicker to denature when heated, they are advantageous for noise reduction (Rouskin et al. 2014). Third, when signal decay prevails, it is also present in the (−) channel, albeit more moderately (Aviran et al. 2011a). Decay can then become significant upstream of spikes or of sites with high noise levels.
(6) Missing information near transcript ends. Coverage levels decline gradually toward the 3′ end due to shortening of regions accessible for hexamer priming. The longer the cDNA fragments are (on average), the more pervasive the associated SNR degradation is, rendering sparse conditions less ideal for short transcript studies. Near the 5′ end, information is lost when attempting to discriminate between fragmentation and modification by way of two size-selection rounds (Rouskin et al. 2014), leaving an unmapped stretch matching the length gap between rounds.
(7) Comparative analysis. Our SPE analysis illustrates the role of profile normalization in facilitating comparisons. Commonly used normalization schemes bridge varying signal intensities (Low and Weeks 2010) and may successfully accommodate variation in coverage-per-transcript. However, we anticipate unprecedented diversity of structural profiles, encompassing a range of lengths, probes, and conditions, which would require thoughtful comparisons. A comprehensive framework is currently lacking, along with standardization of analysis routines, such that the entire process of analysis followed by normalization is meaningful.
Software availability
The computational tools developed for this study are freely available at http://www.bme.ucdavis.edu/aviranlab/sms_software/.
DISCUSSION
We presented novel informatics methodology for assessing the precision and reproducibility of measurements obtained from an emerging class of assays that leverage NGS to dramatically enhance the throughput, scope, and efficiency of structural RNA studies. From a data analysis standpoint, NGS is also transformative by virtue of delivering digital readouts, as compared with previous readout of analog dye intensities. This new wealth of digital information provides opportunities to improve experiment design and reproducibility. In the case of structure mapping assays, we can now determine the number of collected reads and directly link it to measured quantities via computer simulations. Yet, measurements suffer from complex dependencies on reagent concentrations and on fragment size selection. Integration of mathematical models into simulations allows linkage of these experimental parameters to measurements as well as automation of data analysis (Aviran et al. 2011a). These new capabilities motivated us to use model-based simulations to elucidate effects of controllable parameters on data quality.
While our work provides platform and conceptual framework for quantitative evaluation of these effects, its main contribution is in rendering the complexity of input–output relationships. Furthermore, our results highlight the difficulty in accurately determining them by intuition or visual data inspection. In SPE setting, we showed that factors such as reactivity magnitude and probing length modulate the SNR, and that the gradual quality degradation trend as hit rates decrease may reverse at some point. However, such events are case-specific and may not be readily detected. Similarly, it is difficult to infer an effective probing length for obtaining high-quality data through observation of a signal's strength. The advent of RPE shifts the scale of experiments and introduces additional parameters and confounding factors, bringing complexities to levels that warrant dedicated big data infrastructure for computer-aided design. Finally, one must keep in mind that tradeoffs are RNA-specific, and in and of itself, this justifies careful evaluation. The workflow we developed is useful for this purpose and will aid new users of these transformative technologies in gaining the intuition required for experiment design.
At the core of our work is a model of SHAPE-Seq and similar chemistries. While modeling is what facilitates such study, it may also constrain its applicability as long as a model has not been thoroughly validated. In modeling SHAPE, we made two assumptions: (1) site-wise independence of measured features and (2) Poisson reaction dynamics. While the latter is standard in modeling biochemical or low-incidence reactions (Aviran et al. 2011b), the former is not yet fully established, likely because these methods gained popularity only recently. We thus anticipate that ongoing data collection will trigger much needed data-driven modeling (see, e.g., Bindewald et al. 2011; Sükösd et al. 2013), which we can then reiterate to refine the model and improve its predictive power. There is also need to assess the degree of other noise and bias sources, for example, those incurred in NGS library preparation, although progress is being made in overcoming these issues (Jayaprakash et al. 2011; Shiroguchi et al. 2012; Ding et al. 2014). With the emergence of TW assays, additional modeling questions arise: (1) Does cDNA synthesis proceed through encounters with primers via strand displacement or does RT abort? and (2) Does modification interfere with primer binding by preclusion or biasing? Answers may be protocol-dependent (e.g., the choice of RT and reagent) and would alter the model and data properties, particularly the differences between the distributions in control and experiment, from which reactivities are derived (data not shown). A more overarching question concerns the new capacity for in vivo studies—do bound proteins interfere with the probing chemistry, for example, by protecting sites from modification (Kwok et al. 2013)? If yes, then how do they alter a structural signature and how can a model account for that? Nevertheless, we emphasize that the conceptual analysis framework we presented is generic in that it is not tied to any protocol and can be readily adapted to other experimental choices.
The field of nucleic acid structure probing is rapidly evolving, with the maturation of recent techniques and the emergence of more complex ones that enhance scope to in vivo and TW studies. We believe that these advances should be accompanied by matching progress and refinement in informatics infrastructure, to aid in accelerating their optimization and adoption by the research community and to improve their robustness and fidelity. Alternatively, clever new experiments may resolve numerous issues, with the recent SHAPE-MaP (Siegfried et al. 2014) establishing exciting progress in this direction. In SHAPE-MaP, modified sites are encoded by incorporation of noncomplementary nucleotides in cDNA synthesis, where detection by sequencing amounts to careful and elaborate alignment and mismatch identification. Two additional libraries are needed to control for background and for sequence context effects on adduct detection likelihood. This new experimental paradigm eliminates the directionality inherent in the reviewed methods, thus vastly simplifying analysis by reducing it to site-by-site inference. This in turn eliminates some key issues we discussed, in particular those involving the relationship between priming and modification dynamics. SHAPE-MaP signal appears to exhibit dependencies on the hit rate, RT mutation rates (natural and adduct-induced), sequencing errors (which are platform-specific), alignment and read selection strategies, and coverage. Some of these dependencies may not be trivial, and it could be valuable to use our conceptual framework to gain more insight into this promising technique. Furthermore, the simplification of data analysis suggests that in-house in silico optimization of such experiments may be readily feasible for experimentalists.
The anticipated stream of ribonomic big data also highlights the importance of data-informed computational structure analysis, and indeed, much recent progress has been made in this domain (see, e.g., Deigan et al. 2008; Quarrier et al. 2010; Ding et al. 2012; Hajdin et al. 2013; Eddy 2014). It is of interest to quantify effects of data variation on structure prediction, for example, by concatenating such algorithms to our workflow, and further identifying which ones are more robust with respect to technical data variation.
MATERIALS AND METHODS
Model of SHAPE/DMS chemistry
Since the principles of SHAPE, DMS, and other chemistries are similar from a modeling perspective (Weeks 2010), a SHAPE model is representative of several techniques. We consider an RNA sequence whose nucleotides (or sites) are numbered 1–n by their distance from the 3′ end, where a cDNA primer binds to initiate its extension by RT. In the (+) channel of a SHAPE experiment, the RNA is treated with an electrophile that reacts with conformationally flexible nucleotides to form 2′-O-adducts (see Fig. 1). Each molecule may be exposed to varying numbers of electrophile molecules, where each exposure may result in a site's modification (i.e., adduct formation). We model the number of times an RNA molecule reacts with electrophile molecules as a Poisson process of unknown hit rate c > 0, that is,
 |
The site of adduct formation is determined by a probability distribution, denoted Θ = (θ1,…,θn). One can think of this formulation as expressing a competition between n sites over an electrophile molecule, where θk is site k’s relative attraction power. We call Θ the normalized relative SHAPE reactivity profile, or in short, normalized profile, and we use it as a baseline for comparison of measurements taken across varying experimental conditions.
In our model, the number of modifications at site k is also Poisson-distributed, with hit rate rk = cθk ≥ 0, i.e., we have
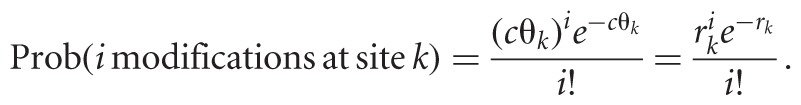 |
We therefore also consider the SHAPE reactivity profile R = (r1,…, rn), which we estimate from sequencing data. R is a scaled version of the normalized profile Θ (R = c Θ), hence the rk’s do not form a probability distribution but rather sum to the hit rate . Scaling by c implies that R lumps the modification intensity, or hit kinetics, into it while Θ is invariant to c. In practice, this means that changes in reagent concentration modulate R but not Θ, motivating us to use Θ for comparisons across modification conditions. In a control experiment, called (−) channel, the primary source of sequencing data is RT's imperfect processivity, resulting in its dropping off during transcription, potentially at varying rates across the molecule. We define the drop-off propensity at site k, γk, to be the conditional probability that transcription terminates at site k, given that RT has reached this site. The parameters Γ = (γ1,…,γn), 0 ≤ γk ≤ 1 ∀k, characterize RT's natural drop off and are unknown and thus estimated jointly with R from data.
Statistical inference for SHAPE-Seq data
The products of the (+) and (−) channels are cDNA fragments of varying lengths, whose one end maps to the priming site at the 3′ end (see Fig. 1). We call a fragment of length k, mapping to sites 0 to k − 1, a k-fragment (1 ≤ k ≤ n + 1). When quantifying fragments via NGS, we summarize the data by k-fragment counts, where (X1, …, Xn+1) and (Y1, …, Yn+1) are counts from the (+) and (−) channels, respectively. In previous work (Aviran et al. 2011a,b), we used this model to derive probabilities of observing each potential outcome as functions of R and Γ:
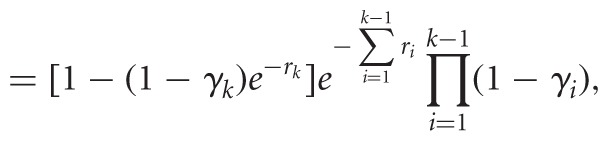 |
(1) |
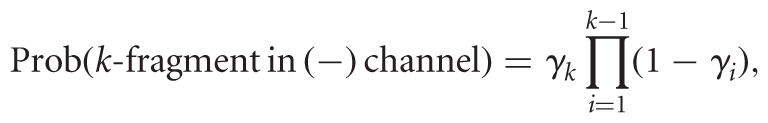 |
(2) |
(1 ≤ k ≤ n + 1), where we set γn+1 = 1 and rn+1 = ∞. We used Equations 1 and 2 to formulate the likelihood of observing the data, which we maximized to find the R and Γ values that best explain them. This approach, known as maximum-likelihood estimation (MLE), provides reactivity estimates:
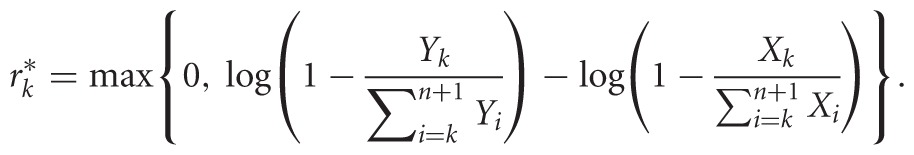 |
(3) |
We further showed in Aviran et al. (2011a) that these are often well approximated by
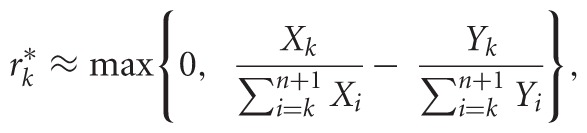 |
(4) |
where a correction factor accounts for all RT termination events observed at or upstream of site k (recall that sites are numbered from 3′ to 5′, to reflect fragment lengths). Equation 4 is more intuitive and also applies to capillary-based signal correction (Aviran et al. 2011b), as recently implemented in analysis platforms (Karabiber et al. 2013).
Models of random primer extension (RPE)
RPE diversifies the data, introducing variable start sites, i.e., a (j,k)-fragment now maps to sites j to k—1 in the RNA, with varying j and k. We introduce n parameters, Δ = (δ1,…,δn), 0 ≤ δk ≤ 1, which capture priming or cleavage affinities. The expressions below pertain to random priming, but with slight adaptation they would model fragmentation. Here, δj is the probability that a hexamer binds sites j to j + 5. Three factors trigger RT stops: natural drop off, modification, or bound primer upstream of j + 5. In the (−) channel, only two factors take effect, yielding
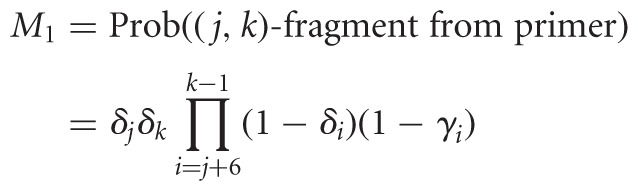 |
and
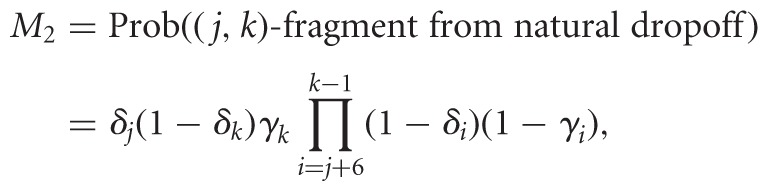 |
where Prob((j,k)-fragment in (−) channel) = M1 + M2. This is the probability of priming at j, not priming or dropping off anywhere between j + 6 and k − 1, and dropping off at k either naturally or due to a primer.
In the (+) channel, experimental considerations affect the model since modification takes place prior to hexamer binding and may or may not preclude it, or it may merely bias it. This is not yet well understood and may be probe-dependent. DMS, for example, interferes with the Watson–Crick base-pairing face of adenines and cytosines, whereas SHAPE targets the backbone. Relevance to fragmentation is also unclear since cleavage occurs between nucleotides. Furthermore, hydroxyl radical probes of solvent accessibility and tertiary structure do not face this issue as they substitute modification for cleavage (Kielpinski and Vinther 2014), but they naturally fit in our analysis framework. For these reasons, simulation results reflect an assumption that modifications do not impede binding, but we developed and implemented in software a model describing mutually exclusive events. The chances of events are as follows:
 |
and Prob((j,k)-fragment in (+) channel) = P1 + P2 + P3. Additionally, our software implements a reactivity reconstruction scheme described in Ding et al. (2014). Fragment-length range defaults to 25–500 nt.
Poisson-based dynamics. It is helpful to simplify analysis by modeling modification and priming as two independent Poisson processes, with rates λ1 and λ2 per nucleotide, respectively. Note that this imposes equal rates per site, that is, uniform priming and equal reactivities. Since Poisson-based waiting times are memoryless, given an adduct at site k, the chances that the next event will be an adduct or a primer are λ1/(λ1 + λ2), λ2/(λ1 + λ2), respectively; hence, dynamics are governed by λ1/λ2. A more realistic model allows varying rates per nucleotide, as modeled for SPE, thus breaking the symmetry among sites. This means that some sites are modified more frequently than others, and that low-reactivity sites are more likely to “see” a shadowing adduct downstream than highly reactive ones.
SHAPE data
To render simulations realistic, we used available SHAPE profiles, which we normalized and set as the true structural signatures to be fixed throughout simulations. For SPE, we focused on short RNAs, since RT's imperfect processivity results in loss of signal typically within a few hundreds of nucleotides, an effect that is expedited in high hit kinetics. For illustration purposes, we chose the 155-nt long P546 domain of the bI3 group I intron, quantified via SHAPE-CE (Deigan et al. 2008). It has an attractive property that it is well-balanced such that reactivities of various magnitudes are spread fairly evenly. Our simulations also rely on quantified RT natural drop-off likelihoods, and despite being determined during analysis, these auxiliary measures are not typically reported along with the reactivities. We therefore fixed γk’s to be within 0.005–0.01, an average drop-off probability range we calculated from SHAPE-Seq data (Mortimer et al. 2012). These values also align with SHAPE-CE estimates (Wilkinson et al. 2008). For RPE, we considered very long transcripts in order to faithfully emulate sparse reaction dynamics and realistic mRNA lengths and also to avoid end effects. We mimicked a long transcript through concatenation of multiple copies of a characterized short RNA.
Empirical MLE distribution
We empirically assessed the distribution of estimates per site by drawing N = 105 independent samples (with replacement) of 4 × 106 reads from the distributions in Equations 1–2 and running them through MLE. The large sample size was chosen to ensure that the sample variance, , , which we treat as if it were the true variance, would be narrowly distributed around the true value. Since each Binomial k-fragment distribution is nearly Gaussian for large read numbers, we first adjusted N such that is negligible at all sites, where μk and σk2 are the mean and variance of the Gaussian approximation at k. However, the distribution of estimates is not necessarily Gaussian, in which case the above calculation may not apply. To remedy a situation where the RSD is higher than that under the Gaussian assumption, we increased N by an additional two orders of magnitude, to obtain N = 105.
ACKNOWLEDGMENTS
We thank Yiliang Ding, Julius Lucks, Shujun Luo, Stefanie Mortimer, and Silvi Rouskin for many discussions and for clarifications on the methods they developed. This work is supported by National Institutes of Health (NIH) grants R00 HG006860 to S.A. and R01 HG006129 to L.P.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.043844.113.
REFERENCES
- Aviran S, Trapnell C, Lucks JB, Mortimere SA, Luo S, Schroth GP, Doudna JA, Arkin AP, Pachter L 2011a. Modeling and automation of sequencing-based characterization of RNA structure. Proc Natl Acad Sci 108: 11069–11074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aviran S, Lucks JB, Pachter L 2011b. RNA structure characterization from chemical mapping experiments. In Proceedings of the 49th Annual Allerton Conference on Communication, Control, and Computing, pp. 1743–1750, Monticello, IL. [Google Scholar]
- Bindewald E, Wendeler M, Legiewicz M, Bona MK, Wang Y, Pritt MJ, Le Grice SFJ, Shapiro BA 2011. Correlating SHAPE signatures with three-dimensional RNA structures. RNA 17: 1688–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YY, Jensen MC, Smolke CD 2010. Genetic control of mammalian T-cell proliferation with synthetic RNA regulatory systems. Proc Natl Acad Sci 107: 8531–8536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YJ, Liu P, Nielsen AAK, Brophy JAN, Clancy K, Peterson T, Voigt CA 2013. Characterization of 582 natural and synthetic terminators and quantification of their design constraints. Nat Methods 10: 659–664. [DOI] [PubMed] [Google Scholar]
- Deigan KE, Li TW, Mathews DH, Weeks KM 2008. Accurate SHAPE-directed RNA structure determination. Proc Natl Acad Sci 106: 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding F, Lavender CA, Weeks KM, Dokholyan NV 2012. Three-dimensional RNA structure refinement by hydroxyl radical probing. Nat Methods 9: 603–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Tang Y, Kwok CK, Zhang Y, Bevilacqua PC, Assmann SM 2014. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 505: 696–700. [DOI] [PubMed] [Google Scholar]
- Eddy SR 2014. Computational analysis of conserved RNA secondary structure in transcriptomes and genomes. Annu Rev Biophys 43: 433–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajdin CE, Bellaousov S, Huggins W, Leonard CW, Mathews DH, Weeks KM 2013. Accurate SHAPE-directed RNA secondary structure modeling, including pseudoknots. Proc Natl Acad Sci 110: 5498–5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaprakash AD, Jabado O, Brown BD, Sachidanandam R 2011. Identification and remediation of biases in the activity of RNA ligases in small-RNA deep sequencing. Nucleic Acids Res 39: e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karabiber F, McGinnis JL, Favorov OV, Weeks KM 2013. QuShape: rapid, accurate, and best-practices quantification of nucleic acid probing information, resolved by capillary electrophoresis. RNA 19: 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielpinski LJ, Vinther J 2014. Massive parallel-sequencing-based hydroxyl radical probing of RNA accessibility. Nucleic Acids Res 42: e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok CK, Ding Y, Tang Y, Assmann SM, Bevilacqua PC 2013. Determination of in vivo RNA structure in low-abundance transcripts. Nat Commun 4: 2971. [DOI] [PubMed] [Google Scholar]
- Low JT, Weeks KM 2010. SHAPE-directed RNA secondary structure prediction. Methods 52: 150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM 2013. RNA-guided human genome engineering via Cas9. Science 339: 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino EJ, Wilkinson KA, Coughlan JL, Weeks KM 2005. RNA structure analysis at single nucleotide resolution by selective 2′-hydroxyl acylation and primer extension (SHAPE). J Am Chem Soc 127: 4223–4231. [DOI] [PubMed] [Google Scholar]
- Mitra S, Shcherbakova IV, Altman RB, Brenowitz M, Laederach A 2008. High-throughput single-nucleotide structural mapping by capillary automated footprinting analysis. Nucleic Acids Res 36: e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5: 621–628. [DOI] [PubMed] [Google Scholar]
- Mortimer SA, Trapnell C, Aviran S, Pachter L, Lucks JB 2012. SHAPE-Seq: high throughput RNA structure analysis. Curr Protoc Chem Biol 4: 275–299. [DOI] [PubMed] [Google Scholar]
- Mortimer SA, Kidwell MA, Doudna JA 2014. Insights into RNA structure and function from genome-wide studies. Nat Rev Genet 15: 469–479. [DOI] [PubMed] [Google Scholar]
- Qi S, Arkin AP 2014. A versatile framework for microbial engineering using synthetic noncoding RNAs. Nat Rev 12: 341–354. [DOI] [PubMed] [Google Scholar]
- Quarrier S, Martin JS, Davis-Neulander L, Beauregard A, Laederach A 2010. Evaluation of the information content of RNA structure mapping data for secondary structure prediction. RNA 16: 1108–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A, Trapnell C, Donaghey J, Rinn JL, Pachter L 2011. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol 12: R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouskin S, Zubradt M, Washietl S, Kellis M, Weissman JS 2014. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature 505: 701–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seetin MG, Kladwang W, Bida JP, Das R 2014. Massively parallel RNA chemical mapping with a reduced bias MAP-seq protocol. Methods Mol Biol 1086: 95–117. [DOI] [PubMed] [Google Scholar]
- Sharp PA 2009. The centrality of RNA. Cell 136: 577–580. [DOI] [PubMed] [Google Scholar]
- Shiroguchi K, Jia TZ, Sims PA, Xie XS 2012. Digital RNA sequencing minimizes sequence-dependent bias and amplification noise with optimized single-molecule barcodes. Proc Natl Acad Sci 109: 1347–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegfried NA, Busan S, Rice GM, Nelson JAE, Weeks KM 2014. RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP). Nat Methods 11: 959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman IM, Li F, Gregory BD 2013. Genomic era analysis of RNA secondary structure and RNA-binding proteins reveal their significance to post-transcriptional regulation in plants. Plant Sci 205-206: 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitale RC, Crisalli P, Flynn RA, Torre EA, Kool ET, Chang HY 2013. RNA SHAPE analysis in living cells. Nat Chem Biol 9: 18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sükösd Z, Swenson MS, Kjems J, Heitsch CE 2013. Evaluating the accuracy of SHAPE-directed RNA secondary structure predictions. Nucleic Acids Res 41: 2807–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkish J, May G, Lin Y, Woolford JL, McManus CJ 2014. Mod-seq: high-throughput sequencing for chemical probing of RNA structure. RNA 20: 713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underwood JG, Uzilov AV, Katzman S, Onodera CS, Mainzer JE, Mathews DH, Lowe TD, Salama SR, Haussler D 2010. FragSeq: transcriptome-wide RNA structure probing using high-throughput sequencing. Nat Methods 7: 995–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicens Q, Gooding AR, Laederach A, Cech TR 2007. Local RNA structural changes induced by crystallization are revealed by SHAPE. RNA 13: 536–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y, Qu K, Ouyang Z, Chang HY 2013. Genome-wide mapping of RNA structure using nuclease digestion and high-throughput sequencing. Nat Protoc 8: 849–869. [DOI] [PubMed] [Google Scholar]
- Wan Y, Qu K, Zhang QC, Flynn RA, Manor O, Ouyang Z, Zhang J, Spitale RC, Snyder MP, Segal E, et al. 2014. Landscape and variation of RNA secondary structure across the human transcriptome. Nature 505: 706–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks KM 2010. Advances in RNA structure analysis by chemical probing. Curr Opin Struct Biol 20: 295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson KA, Gorelick RJ, Vasa SM, Guex N, Rein A, Mathews DH, Giddings MC, Weeks KM 2008. High-throughput SHAPE analysis reveals structures in HIV-1 genomic RNA strongly conserved across distinct biological states. PLoS Biol 6: e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q, Ryvkin P, Li F, Dragomir I, Valladares O, Yang J, Cao K, Wang LS, Gregory BD 2010. Genome-wide double-stranded RNA sequencing reveals the functional significance of base-paired RNAs in Arabidopsis. PLoS Genet 6: e1001141. [DOI] [PMC free article] [PubMed] [Google Scholar]