

Abstract
Molecular approaches in ecotoxicology have greatly enhanced mechanistic understanding of the impact of aquatic pollutants in organisms. These methods+- have included high throughput Omics technologies, including quantitative proteomics methods such as 2D differential in-gel electrophoresis (DIGE) and isobaric tagging for relative and absolute quantitation (iTRAQ). These methods are becoming more widely used in ecotoxicology studies to identify and characterize protein bioindicators of adverse effects. In teleost fish, iTRAQ has been used successfully in different fish species (e.g. fathead minnow, goldfish, largemouth bass) and tissues (e.g. hypothalamus and liver) to quantify relative protein abundance. Of interest for ecotoxicology is that many proteins commonly utilized as bioindicators of toxicity or stress are quantifiable using iTRAQ on a larger scale, providing a global baseline of biological effect from which to assess additional changes in the proteome. This review highlights the successes to date for high throughput quantitative proteomics using DIGE and iTRAQ in aquatic toxicology. Current challenges for the iTRAQ method for biomarker discovery in fish are the high cost and the lack of complete annotated genomes for teleosts. However, the use of protein homology from teleost fishes in protein databases and the introduction of hybrid LTQ-FT (Linear ion trap –Fourier transform) mass spectrometers with high resolution, increased sensitivity, and high mass accuracy are able to improve significantly the protein identification rates. Despite these challenges, initial studies utilizing iTRAQ for ecotoxicoproteomics have exceeded expectations and it is anticipated that the use of non-gel based quantitative proteomics will increase for protein biomarker discovery and for characterization of chemical mode of action.
Keywords: ecotoxicoproteomics, LC MS/MS, fish database, quantitative proteomics
1. 2D gel electrophoresis as a biomarker discovery tool in aquatic toxicology
Two-dimensional polyacrylamide gel electrophoresis (2D PAGE) has been used since the late 1960’s as a powerful tool to separate proteins and to compare protein expression between controls and diseased or chemically-exposed biological samples. The method depends on exquisite separation of proteins by pI in the first dimension, followed by mass in the second dimension and is able to resolve up to 2,000 protein spots per gel. In earlier years (1970’s to mid 1990’s), proteins that were differentially expressed were identified by Edman Sequencing, a procedure that required at least 10 pmol of protein per spot if the protein was not blocked on the N-terminus. Blocked proteins could be identified by enzymatic digestion (usually with endoproteinase Lys C), followed by separation of the fragments and Edman Sequencing. The most significant innovation for proteomics was coupling mass spectrometry with 2D gels as a way to identify differentially expressed proteins (Lilley, 2001; Beranova-Giorgianni, 2003; Herbert, 2001).
The introduction of Differential in Gel Electrophoresis (DIGE) methods has increased the standard for this proteomics approach, allowing co-separation of a control set of proteins with proteins isolated from a treatment or disease (Tonge et al., 2001). The exquisite separation in the first dimension allows resolution of proteins that differ by a single positive or negative charge, making this method ideal for distinguishing protein isoforms and proteins that are post-translationally modified, in addition to quantifying protein expression changes.
While resolution of over 2,000 protein spots on large gels is considered excellent, there are some limitations to the method. Normally the spots that are visible on the gel include only those proteins that are relatively abundant in the preparation, have pI’s within the pH range of the IEF (normally pH 3 to 8) and that stain well by common protein stains. There are many proteins that are missed, including very basic or acidic proteins, very small or large proteins and proteins that are hydrophobic or of very low abundance (Beranova-Giorgianni, 2003; Lilley, 2001; Oh-Ishi, 2002).
In order to detect proteins that may be good biomarkers for chemical stress or disease but that may be present in lower concentrations, increased attention has focused on analysis of sub-proteomes. A sub-proteome is essentially the proteome of a sub-cellular compartment. Robust methods have been developed to fractionate sub-cellular compartments that rely on using a hyperosmotic buffer (at least containing 0.25 M sucrose) and differential centrifugation (Ankarcrona et al., 2002, Hogenboom et al., 2004, Jung et al., 2000, Kabbani et al., 2004, Nothwang et al., 2003). Nuclear, mitochondrial, membrane, cytoskeletal, and cytosolic subproteomic fractions are commonly separated by this technique which can then be further analyzed independently using DIGE.
The plasma proteome is an excellent bio-fluid that can be sampled for biomarkers, as has already been demonstrated for example with vitellogenin (Vtg) expression in male fish in response to estradiol (Sumpter and Jobling, 1995). The abundant proteins are readily accessible for identification and quantitation. However, the high concentrations of some proteins impede analysis of important biomarkers that are found in lower concentrations in the blood. Proteins in the plasma range in concentration over twelve orders of magnitude from the pg/ml level for cytokines to the mg/ml level for albumin and immunoglobulins and up to 20 mg/ml Vtg (normally found only in female fish plasma at the peak of reproduction but this can be as high at 100 mg/ml in plasma of male fish that have been treated with estrogens). For plasma biomarker discovery, it is essential to reduce the concentrations of the overly abundant proteins. Commercially available kits take advantage of affinity resins for albumin and up to 12 other high concentration proteins, and have been found to be fairly successful, at least in mammalian studies, and possibly to a lower extent in aquatic toxicology; however, there are several commercially available non-specific resins which can be used for aquatic species. While these resins reduce concentrations of the most abundant proteins, they sometimes can also remove other proteins that bind to the resins or albumin-bound proteins resulting in incomplete proteomic analysis (Mehta et al., 2003; Ahmed, et al., 2003).
With 2D gel electrophoresis, the quantitation is performed on the gel by densitometry. Some very elegant programs have been developed that compare protein patterns among gels, even allowing one to stretch a gel in either dimension to allow perfect matching. Gel spots corresponding to proteins that are identified as differentially expressed are then excised out of the gel, washed, dried and then reconstituted with trypsin to digest the proteins. Protein fragments elute out of the gel pieces and can be analyzed directly by mass spectrometry.
2D gel electrophoresis followed by identification of proteins by mass spectrometry has been used extensively in ecotoxicology (reviewed by Dowling and Sheehan, 2006; Iguchi et al., 2007; Monsinjon and Knigge, 2007; Sanchez et al., 2011). Among the first studies in this area was one in which zebrafish embryos were exposed to two concentrations of nonylphenol and E2 (1 ppm and 0.1 ppm) and analyzed by 2D gel electrophoresis (Shrader et al., 2003). Protein expression patterns were compared to identify proteins with chemical-dependent altered expression. Altered proteins were not identified, but the patterns were reproducible. Since then many studies have been conducted by other groups interested in identifying contaminant-specific protein expression patterns, but identification of the proteins continues to be problematic, especially since the species used are not fully sequenced or annotated. But progress in this area continues and several recent studies have shown excellent results (Costa et al. 2010; Fang et al., 2010; Li et al., 2010; Dorts et al., 2011; Forné et al., 2011; Karim et al., 2011). It is noted that this list is not exhaustive and only represent some of the aquatic ecotoxicology studies utilizing gel-based proteomics.
One very interesting study by Berg et al., (2010) examined the effects of methyl mercury (MeHg) in brain of Atlantic cod. Cod were treated by intraperitoneal injection with two concentrations of MeHg (0.5 and 2 mg/Kg), divided into two injections administered one week apart. The total exposure was for two weeks. Differentially regulated proteins were analyzed by 2D gel electrophoresis. Seventy one different protein spots were altered by the treatment of which the authors were able to identify 40 by MALDI TOF MS and MS/MS. Some of the identified proteins belonged to groups already associated with MeHg targeting in mammals such as proteins involved in mitochondrial dysfunction, oxidative stress, calcium homeostasis and cellular cytoskeleton structure. However, they also identified novel proteins involved in the synthesis of neurotransmitters, the function of neurotransmitter receptors, among others (Berg et al, 2010). This study benefited from the exceptional care that was used to perform the 2D gel analysis and quantification and established protein identification methods that employed high resolution MS/MS.
New methods that take advantage of specific stains (e.g. Diamond Pro Q to identify phosphorylated proteins and Emerald Pro Q for glycosylated proteins) have also enabled researchers to answer specific questions in toxicology (Steinberg et al., 2001). Novel approaches such as the integration of 2D gels and immunostaining, as developed for redox proteomics (Sheehan et al., 2010) have led to using functional changes in understanding the effects of reactive oxygen species in environmental samples. Redox proteomics was coined to describe the method that uses knowledge from expected protein modifications caused by exposure to contaminants which cause oxidative stress. This approach was used to measure changes in proteins that were susceptible to reactive oxygen species (ROS) such as carbonylated and ubiquitined proteins (Chora et al, 2010). Proteins that are targeted by ROS are post-translationally modified by carbonylation and then by ubiquitin (to clear the protein from the cell by the proteasome). These proteins make suitable early biomarkers for oxidative stress.
2. iTRAQ as a biomarker discovery tool in aquatic toxicology
Proteomics has seen significant advances in recent years due to increases in resolution and scan speed of mass spectrometers as well as improved capabilities in spectral analysis algorithms. Recently, the potential of applying non-gel based proteomics methods to study fish physiology and ecotoxicology has been recognized (Martyniuk and Denslow, 2009; Forné et al., 2010, Sanchez et al., 2011). An important consideration for the non-gel based methods is quantitation. Over the past few years, several innovative methods have been suggested, all with their specific advantages and disadvantages. The various methods for protein quantitation in the context of aquatic toxicology have been previously summarized in Martyniuk and Denslow (2009) and will not be covered in detail here. Briefly, these non-gel based methods include spectral counting and isotope-coded affinity tags (ICAT) but there are limitations when using these methods for protein quantitation. In regards to spectral counting, quantitation depends on the number of spectra generated for a protein and small proteins may be less represented due to low numbers of tryptic peptides. In addition, proteins that are low in arginine and lysine residues or have them spaced far apart may not be adequately detected with this approach. A limitation to ICAT is that this method only labels cysteine containing peptides using isotope chemistry and peptides that do not contain a cysteine group will be missed in the analysis.
Another novel approach for quantifying changes in protein abundance termed isobaric tagging for relative and absolute quantitation (iTRAQ® Applied Biosystems, AB Sciex, Foster City, CA) (Ross et al., 2004) has recently become a powerful protein biomarker discovery tool in human disease research (Martin et al., 2008; Siu et al., 2009) and shows good promise in ecotoxicology. The iTRAQ method is based upon tagging the N-terminus of peptides generated from tryptic protein digests. N-terminal modification allows for a greater number of peptides to be quantified compared to other methods such as ICAT (i.e. limited by available cysteines). Amine labeled peptides from different samples will have a unique isobaric tag consisting of a reporter and a mass balance to ensure precursor ions enter the collision cell of the mass spectrometer simultaneously. The mass tags are cleaved during MS/MS and each reporter tag will yield low discriminating MS/MS signatures. Relative intensity of reporter tags yield information on relative abundance of proteins in each sample. Therefore, measurement of the intensity of reporter ions allows for relative quantification of the peptides.
Perhaps the most significant advantage of iTRAQ over more traditional proteomics approaches is that up to eight different samples can be simultaneously labeled and analyzed. This feature facilitates biological replicates, time course analysis, and/or dose responses, each an important parameter in aquatic toxicology studies. Overall, iTRAQ reduces the time and effort needed to study the proteome of multiple samples compared to gel-based methods.
There are also some inherent limitations of the non gel based proteomics methods such as iTRAQ that include (1) increased LC separation and MS run time due to larger amounts of peptides, (2) intensive sample separation before LC-MS/MS with pre-fractionation of proteins or peptides. Therefore, 2D- chromatography, in the form of strong cation exchange and reversed-phase chromatography, is usually needed with multiple peptide fractions collected for the analysis. Lastly, (3) proteins that are not readily soluble in buffers will not be processed and detected. It is noted that gel-based methods also have limitations that include size and pH restrictions for protein separation that lead to difficulties in protein resolution (reviewed in Martyniuk and Denslow, 2009). Despite these methodological limitations, complementary proteomics methods to investigate protein changes, for example using both iTRAQ and 2D gel electrophoresis in ecotoxicology studies, will only serve to improve the range of protein characterization as well as increase knowledge about the mode of action of aquatic contaminants found in the environment.
In a study comparing three quantitative proteomics methods (DIGE, iTRAQ, and ICAT), Wu et al. (2006) investigated a simplified protein mixture (6 proteins) and a more complex protein mixture of lysates from HCT-116 and HCT- 116 p53 −/− cells. The three methods were comparable in identifying and quantifying protein changes in simplified protein mixtures but only identified one common protein in the complex mixture, suggesting that protein characteristics (solubility, size, charge) can be more or less compatible with a specific method. One reason for the difference in protein detection can be attributed to isoelectric points or molecular weights being outside the ranges of the IPG strip used in 2D gel electrophoresis, as described above.
3. Quantitative proteomics for hepatic proteins in fish
The iTRAQ approach is poised to be a high-throughput biomarker discovery tool for proteomics studies using aquatic vertebrates (Domanski and Helbing, 2007; Martyniuk and Denslow, 2009; Serrano et al., 2010) and this method has been used with success in teleost fish, even though full genomic sequence information is lacking. Despite limitations in database content and annotation, the iTRAQ method can be successful at quantifying a significant number of proteins in teleosts. In female fathead minnows (Pimephales promelas, FHMs), a combination of two-dimensional liquid chromatography (2D LC) followed by tandem MS/MS on a quadrupole time of flight mass spectrometer (QSTAR XL, AB Sciex) identified a number of proteins in the liver that were responsive to androgen and anti-androgen waterborne treatments (Martyniuk et al. 2009). The experiment identified 293 proteins in the liver with high quality peptide data, of which 98 proteins were quantified. The study identified proteins that are potentially regulated through androgen receptor signaling, such as betaine homocysteine methyl-transferase (Bhmt), endozepine (Dbi), and fatty acid binding protein 1b (Fabp1b). The peptide coverage of proteins was variable in the FHM liver, and ranged from 4-8% for proteins such as calreticulin (Calr) and up to 81% and 87% for liver fatty acid binding protein 10 (Fabp10) and ubiquitin C (Ubc) respectively. Therefore, there can be tremendous variation in protein coverage in fish using mass spectrometry based approaches.
For researchers interested in using proteomics in ecotoxicology, it is important to note that a number of well characterized bioindicators for general and oxidative stress are readily detectable in fish tissues by LC MS/MS and include stress-related proteins such as superoxide dismutase (Sod1), glutathione-S-transferases (Gst), catalase (Cat), glutathione peroxidases (Gxp), and heat shock proteins (Hsp). Many stress and detoxification proteins identified in the FHM liver have also been identified in livers of other teleost fish using high throughput proteomics, irrespective of the protein isolation and separation methods. Wang and colleagues (2007) separated hepatic proteins from zebrafish (Danio rerio) into cytosolic, nuclear, membrane, and cytoskeletal components via differential centrifugation and performed tryptic digests (buffer, methanol, SDS), and a microwave-assisted acid hydrolysate to increase coverage of the proteome. Using a QTOF Premier mass spectrometer, proteins such as epoxide hydrolase (Ephx), Sod, Hsp, and Vtg were readily identified. These proteins are relatively abundant in liver and conserved across teleosts families, which facilitate identification of increased numbers of peptide-protein assignments using databases that are not fish species specific. For example, when comparing the amino acid sequences of catalase for teleosts from different families of fish (zebrafish, Cyprinidae; barred knifejaw, Oplegnathidae; cobia, Rachycentridae; Atlantic salmon, Salmonidae; mangrove rivulus, Rivulidae), there is greater than 83% amino acid similarity across species from the different families. As pointed out by Wang et al (2007), the presence of well characterized biomarkers in the fish hepatic proteome can provide researchers with an internal standard to validate exposures and correlate protein responses to stress and metabolism of toxicants.
iTRAQ can also provide quantitative data for a number of classical, well characterized biomarkers in aquatic toxicology in addition to providing novel information on potential novel protein biomarkers for aquatic pollutants such as androgenic compounds. Using iTRAQ quantitation, Vtg was found to increase in abundance after treatment with flutamide, an anti-androgenic pharmaceutical (Martyniuk et al., 2009), corroborating earlier studies that demonstrate increases in plasma Vtg in teleosts after treatment with model anti-androgens (Jensen et al, 2004).
A new aquatic toxicology study investigated the proteome response of the marine diatom Thalassiosira pseudonana after benzo(a)pyrene (BaP) exposures with an iTRAQ-8 plex quantification using a QSTAR in a nanoLC-ESI-MS/MS analysis (Carvalho and Lettieri, 2011). The study identified a total of 308 different proteins in three biological replicates that were sensitive to BaP. Proteins affected by BaP included those involved in protein synthesis (ribosomes), metabolism, stress and regulation of protein folding such as heat shock protein/chaperone and Hsp70. The strength of the iTRAQ approach is that many utilized protein bioindicators can be quantified at once, providing a wealth of information that can be used to assess adverse effects in aquatic organisms.
4. Database limitations and strategies for teleost fish
In contrary to the high throughput DNA sequencers used for de novo sequencing of complete genomes or transcriptomes, the data acquired for protein identification by mass spectrometers becomes meaningful only after database searching, making proteomics largely dependent of the content of protein databases. When using LC-MS/MS, peptides are separated from complex sample mixtures. In the first level of the analysis, all the peptides are detected as an MS scan and in a second level, each isolated peptide is dissociated into fragments by collision with an inert gas and the MS/MS spectra are collected. Correlating the MS and MS/MS spectra with the candidate peptide is performed by a database search engine using the peptide mass from the first round and the short stretches of amino acid sequence obtained for each peptide from the second round. There are several database search engines (e.g. Mascot, Sequest and X!Tandem) available that compare an MS/MS fragment ion spectrum against theoretical fragmentation patterns derived from a user-defined protein database, and assign scores based on the quality of the match. This involves (i) simulation of the enzyme specific cleavage, (ii) specific fragmentation, and (iii) calculation of the mass values expected (Sadygov et al, 2004). Then, the identified peptide sequences are assembled into one or more proteins from the database. There are some protein families that contain identical blocks of sequences in particular domains of the protein. Often these similar sequence blocks are used to assign proteins to a family. If these blocks end up within a tryptic peptide, it may be difficult to assign the peptide to a specific protein. This problem is termed protein inference. Peptides that can be assigned to more than one protein in a family are not used for protein identification and quantification. To produce results that are meaningful, the source databases should be comprehensive, non-redundant, updated, and well annotated.
The quality and confidence of protein identification is also controlled in the case of shotgun proteomic experiments by assessing a false positive discovery rate (FDR). The FDR is calculated by using a randomized database also referred to as a decoy database. It is produced by reversing or randomizing the original protein sequences (Hidgon et al, 2007). The FDR can be then estimated after searching together the protein sequence database and its randomized version by comparing the score of the randomized versus nonrandomized matches. The number of matches to random sequences is used to estimate the FDR threshold.
Protein identification using mass spectrometry becomes a challenge when the proteins are from an organism in which the genome is not yet sequenced, as is the case for many teleost fish. Peptide de novo sequencing, (i.e. sequencing without assistance of a linear sequence database) using MS/MS spectrum can assist in determining amino acid tags. New techniques such as the use of labeling, specific proteases, and alternative ionization and dissociation techniques, as well as the introduction of new bioinformatics tools (Seidler et al, 2010) have improved the specificity of peptide identification. However, this is more commonly applied to specific proteins than to large-scale proteomics projects.
The main problem for identifying proteins for teleost fishes is the relative paucity of available protein sequence data. The NCBI nr database (non redundant sequences from GenBank CDS translations, Protein Data Bank (PDB), Swiss-Prot, Protein Information Resource (PIR), and Protein Research Foundation (PRF) which is commonly used for protein identification only contains a limited number of protein sequences for teleost fishes, ranging from only a few hundred for fathead minnow (Pimephales promelas) to several tens of thousands for zebrafish (Danio rerio) (Figure 1). However, in the past year, the number of protein sequences for teleosts in NCBI nr database has increased from 2 to 20 times, depending on the species, and this is largely due to the completion and annotation of genomics projects such as for the zebrafish and Atlantic salmon (Figure 1). The growing increase in teleost protein entries in databases is critical for the increase of protein identification rates based on protein homology for any proteomics study in teleost fishes. The increase in data entries for teleosts between 2008 and 2010 led to a net increase of 11% of the proteins identified for the FHM study mentioned above when the same raw data (Martyniuk et al, 2010a,b) was analyzed two years later (Appendix 1). The comparison of the two searches also revealed that a total of 29 % of the proteins identified were newly assigned proteins, derived from new MS/MS spectra assigned to new sequences in the database or from a different peptide reassignment because of changes in pre-existing entries (accession numbers removed or with updated sequences). However, it is important to note that results obtained from the use of other teleost species to identify proteins for a specific fish need to be used carefully. Peptides that are highly conserved among species are likely to fit within conserved blocks in protein families. The same peptide may match multiple proteins, including protein isoforms with different activities. This can lead to ambiguities in determining the identity and abundance of proteins. Others strategies to overcome these limitations include using available nucleotide sequences in EST databases (sequenced genomes or cDNA of teleost fish transcriptomics studies). Often, however, nucleotide sequences are not publicly available or are not adequately annotated. The separate search of multiple databases, protein and nucleotide, increases the number of proteins identified but also increases the analysis complexity. This approach requires additional computational analysis to merge the different identified proteins and it is important to remain alert for issues of protein inference, as described above. Existing database search engines still have to be improved to offer solutions to these problems.
Figure 1.
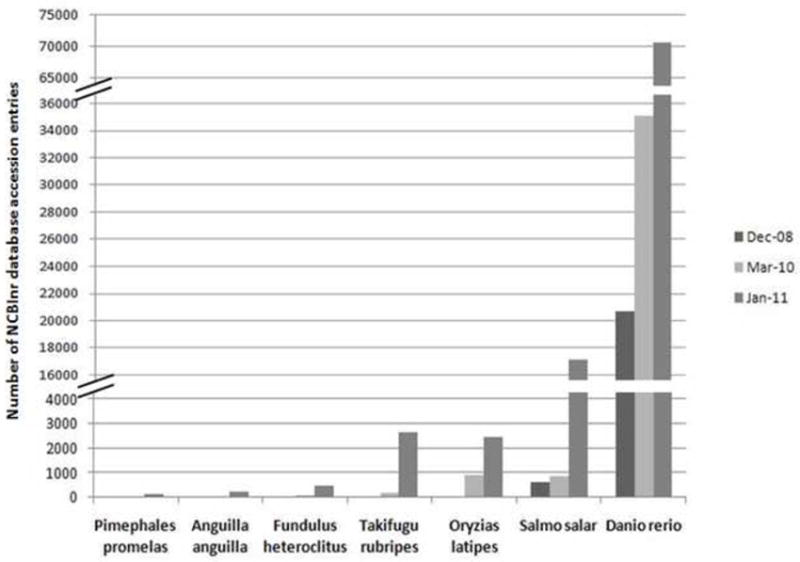
Number of accession entries available in NCBI nr database for several model teleost fish species (Pimephales promelas, Anguilla anguilla, Fundulus heteroclitus, Takifugu rubripes, Oryzias latipes, Salmo salar, Danio rerio) in ecotoxicology and genetics (as of December 2008- January 2011). There has been an increase of approximately 2-20 fold in protein information in all species over the past two years in the NCBI nr database.
Quadrupole time of flight configurations such as the QSTAR and quadrupole ion trap such as LTQ (ThermoScientific) instruments are commonly used in large-scale proteomics studies, although the specifications in terms of mass accuracy, resolution and dynamic range affect the quality of mass measurement. The LTQ has the advantage of being more sensitive with its higher ion capacity and scan rates while QSTAR shows higher mass accuracy and resolution. With the introduction of hybrid systems such as the LTQ-FT and the Orbitrap LC-mass spectrometers combining high resolution and high mass accuracy and faster scan rate, the amount and quality of data acquired has been greatly improved and the identification rates have increased significantly for teleost fish. In a study of zebrafish brain, Singh et al, (2010) were able to identify 8475 proteins with an LTQ-FT mass spectrometer by using sophisticated protein pre-fractionation techniques by gel electrophoresis. A point to note is that the zebrafish genome is completely sequenced and this facilitates more protein identifications. More challenging is identifying proteins for non model species for which only limited DNA sequences exists. But even for a species such as fathead minnow, the use of an LTQ-FT system in conjunction with the most recent protein databases increased the number of proteins identified in the liver by approximately 4-5 times more compared to a QSTAR (Martyniuk et al., unpublished).
5. Limitations of the iTRAQ approach in fish tissues
While a strength of the iTRAQ method is that in a single experiment all of the samples that are being compared are mixed after labeling and chromatographed and separated together, it can be difficult to repeat the entire experiment again and get the same results (Gan et al, 2007). While most of the same proteins are identified in replicate experiments, some proteins are only identified in one of the replicate experiments (Table 1). This problem is largely due to the sensitivity and duty cycle of the mass spectrometer as well as protein abundance and complexity within each sample (i.e. biological variation in proteomic response) and database limitations as outlined above. Therefore, in some cases, peptides from a protein may only be detected in one iTRAQ experiment. In the study by Martyniuk et al., (2009), of the 98 proteins quantified in FHM liver, peptides from 18 proteins were detected only in a single experiment, peptides from 18 proteins were detected in 2/3 iTRAQ experiments, and peptides from 19 proteins were detected in all three iTRAQ experiments (Table 1). The proteins identified in only a single iTRAQ experiment included nucleolin (Ncl), enoyl coenzyme A hydratase 1 (Ech1), and Fabp1B while those identified in all three experiments in the liver included phosphoglycerate mutase 1 (Pgam1) and triosephosphate isomerase 1b (Tpi1b). It is expected that the most abundant proteins will be more often detected in consecutive iTRAQ experiments.
Table 1.
Number of proteins quantified and differentially expressed in teleost tissues during independent iTRAQ sample analysis. Each experiment consisted of three iTRAQ experiments (n=3 biological replicates) using LC MS/MS (QSTAR) and ProteinPilot™ Software (Applied Biosystems) for data analysis. The number of fractions analyzed for each independent iTRAQ experiment ranged between 8-11. The ratios under the iTRAQ column refers to the number of independent experiments a protein was detected. For example, 3/3 means that the protein was identified in all three iTRAQ experiments.
| Teleost Species | Tissue | Chemical Treatment | Proteins Quantified | Proteins Altered (p<0.10) | iTRAQ 1 in 3 | iTRAQ 2 in 3 | iTRAQ 3 in 3 | References |
|---|---|---|---|---|---|---|---|---|
| Fathead minnow (Pimephales promelas) | Liver | 17β-trenbolone/flutamide | 98 | 55 | 18 | 18 | 19 | Martyniuk et al. 2009 |
| Fathead minnow (Pimephales promelas) | Telencephalon | 17α-ethinylestradiol | 77 | 26 | 5 | 5 | 16 | Martyniuk et al. 2010a |
| Goldfish (Carassius auratus) | Hypothalamus | Dopamine receptor agonists (LY 171555 and SKF 38393) | 222 | 70 | 30 | 26 | 14 | Popesku et al. 2010 |
| Largemouth bass (Micropterus salmoides) | Hypothalamus | Dieldrin | 90 | 23 | 6 | 4 | 13 | Martyniuk et al. 2010b |
In another example, in the FHM telencephalon, there were 77 proteins that were quantified using LC MS/MS (Martyniuk et al., 2010a). Peptides from 5 proteins were detected in a single experiment, peptides from 5 proteins were detected in 2/3 iTRAQ experiments, and peptides from 16 proteins were detected in all three iTRAQ experiments. The proteins identified in only a single iTRAQ experiment included myelin protein zero (Mpz) and malate dehydrogenase 1a, NAD (soluble) (Mdh1a) while those identified in all three experiments in the telencephalon included 14 kDa apolipoprotein and lactate dehydrogenase B4 (Ldhb). Despite this limitation, the number of peptides used in the quantification of a protein can be quite impressive. In the FHM liver, translationally-controlled tumor protein was quantified using 120 high quality spectra (Martyniuk et al., 2009) while in the brain, golli-mbp isoform 1 and calmodulin were quantified using 128 and 136 spectra respectively (Martyniuk et al., 2010a), increasing confidence in relative protein abundance changes after experimental treatments.
As is common in proteomics, abundant proteins are more frequently detected by mass spectrometry. Based on protein data in fish collected with a QSTAR (AB Sciex) mass spectrometer with information-dependent acquisition, the iTRAQ approach quantified peptides between 33-70% of the time in biological replicates (n=3, 8-11 fractions/iTRAQ experiment) (i.e. the protein is detected in multiple iTRAQ experiments). Increased reproducibility in the replicates would be obtained by examining more salt fractions, increasing the duration of the gradients (>2h) for reverse phase separation, and using an instrument with higher sensitivity such as a hybrid LTQ-FT or Orbitrap. Measuring and quantifying individual proteome responses will improve with time as increasingly more iTRAQ samples can be analyzed in high throughput approaches for a toxicological experiment.
The confidence in protein abundance changes increases with the numbers of unique peptides used in the quantitation. In experiments with teleost fishes, the number of unique peptides used in quantitation can be very good. For example, the minimum number of high quality peptide data for quantitation can be as low as 3 for hepatic proteins such as 60S ribosomal protein L28 and electron-transfer-flavoprotein, beta polypeptide (Etfb) but >100 peptides across fractions for proteins such as Fabp10 in the fish liver and golli-myelin basic protein isoform 1 (Mbp) in the neuroendocrine brain. As an example, in the LMB hypothalamus, multiple peptides were detected for Eno1, a glycolytic enzyme that is highly expressed in the CNS and functions to catalyze the inter-conversion of 2-phosphoglycerate and phosphoenolpyruvate. When examining spectral intensity of reporter tags, each tag (and peptide) was significantly more abundant in dieldrin-treated LMB (label 116) compared to control LMB (label 117) (Figure 2). This example demonstrates the consistency that can be observed in reporter tags across multiple peptides from a fish protein.
Figure 2.
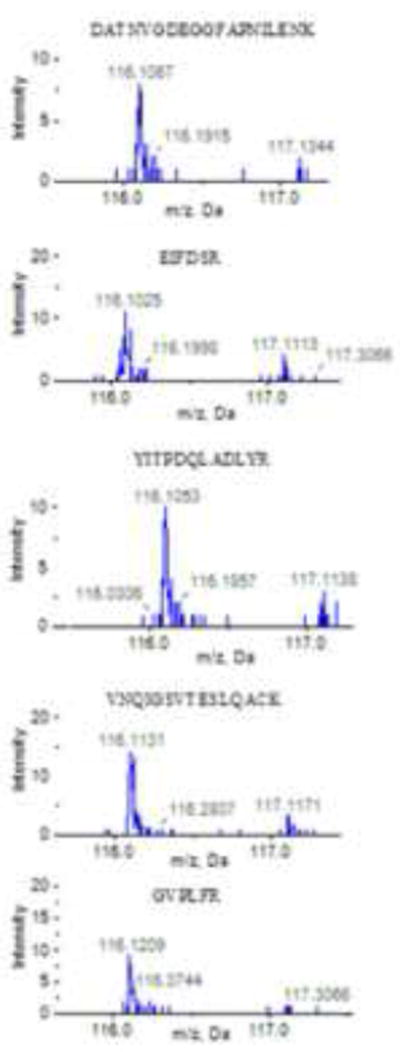
Comparisons for relative spectral intensity in an iTRAQ experiment for reporter ions originating from multiple peptides from Eno1. Shown is the spectral intensity for iTRAQ isobaric tag 116 m/z Da (dieldrin-treated) and tag 117 m/z Da (control). Note that tag 116 is consistently higher in intensity than tag 117, indicating that Eno1 is up-regulated in the dieldrin-treated LMB hypothalamus compared to control.
Currently, high throughput proteomics is costly. For good proteomic coverage and a sufficient dataset of peptides from which to quantify proteins, it is recommended that a minimum of three to four iTRAQ labeling experiments be conducted per condition. More experiments must be conducted to obtain good estimates on individual biological variation. The first separation is usually via strong cation chromatography. It is recommended that at least 8 to 10 fractions be collected for the second separation by reverse phase. Undoubtedly, with the use of the new LTQ-FT systems, the power of detection will be significantly greater.
6. Applying proteomics to study the fish ovarian proteome
There are a number of environmental pollutants that directly impact the reproductive system in fish. The effect can occur at any point along the hypothalamic-pituitary-gonadal axis and the ovary has been a significant focus of many transcriptomic studies in fish toxicology characterizing genomics responses to model compounds in this tissue to better identify EDC modes of action (Garcia-Reyero et al., 2009; Villeneuve et al., 2009). However, large scale toxicoproteomics studies in the fish ovary have been limited. Knoll-Gellida et al. (2006) investigated the protein complement of fully grown zebrafish follicles and identified a number of proteins using 1D-SDS-PAGE and 2D-PAGE analysis. The proteins identified included zona pellucida glycoproteins and Vtg isoforms (Vtg1 and Vtg). Vtg is a large protein that is highly abundant in the ovary because of active uptake into maturing follicles during sexual maturation. The researchers reported that the three most abundant categories for proteins were structural molecules (e.g. beta-actins and ribosomes), binding proteins and chaperones (e.g. heat shock proteins) and proteins with a catalytic activity (e.g. dehydrogenase or enolases).
In a recent study by Groh et al. (2011), multidimensional protein identification technology (MudPit) and a LTQ-Orbitrap XL identified 1,379 unique proteins is the zebrafish ovary. Interestingly the top five positions in the ovarian protein list (total peptide hits ranging between 3,901 and 14,593) were vitellogenins and this was > 95% of the peptides. Thus, proteomics studies in the teleostean ovary are challenging due to the abundance of Vtg. To delve deeper into the ovarian proteome, samples were additionally analyzed using a “Reject Mass List” containing the masses of Vtg proteins. This strategy worked very well and increased the number of protein identifications in the zebrafish ovary.
For future studies using quantitative proteomics in fish ovarian tissues, strategies to deplete high abundance proteins, specifically Vtg, will serve to increase the proteome coverage. Strategies similar to immunodepletion columns for high abundant proteins found in human plasma can be employed where proteins such as serum albumin, immunoglobins, alpha-2-macroglobulin, fibrinogen, and Apoa1 and Apoa2 can comprise up to 85% of the serum protein content (Li et al., 2005). The implementation of immunodepletion has been successful in iTRAQ experiments for biomarker discovery in human plasma (Song et al., 2008). However, it is pointed out that these methods have been successful in mammalian tissues and have not been adequately demonstrated in fish tissues for quantitative proteomics.
7. Future of quantitative proteomics in aquatic toxicology
Applications for quantitative proteomics will increase in ecotoxicology for biomarker discovery and mechanistic understanding of an aquatic toxicant’s MOA. With the exponential increase in genomic information from next generation sequencing for non-model fish species, improved proteomics databases will increase peptide-protein identifications in fish. In the studies outlined here, between 40-60% of all spectra were assigned to a known protein, leaving approximately 50% of spectra unidentified. As databases improve, proteomic data files can be re-searched and analyzed to obtain new quantitative information about the fish proteome. Another approach that will potentially aid in the peptide-protein assignments is de novo sequencing that will reduce the number of false positives in peptide-protein identification.
A final consideration for experimental designs is to consider the weak correlation between gene and protein expression changes in tissues. It is not well understood how the transcriptome relates to the proteome in fish and data are scarce in the literature (Martyniuk and Denslow et al., 2009). This is important since both genes and proteins should be included in environmental monitoring. To improve predictive ability in environmental risk assessment, efforts are focused on integration of molecular and physiological data (i.e. systems toxicology approach). This overall framework in aquatic toxicology must include some knowledge of the complex relationship between gene expression, protein abundance, and metabolites (Martyniuk and Denslow, 2009; Helbing et al., 2010; Ekman et al., 2011, Garcia-Reyero and Perkins, 2011; Katsiadaki et al., 2010). Although there is much to be learned, there have been significant advances in moving towards these goals in the past few years in order to adapt Omics approaches to field based environmental studies.
Supplementary Material
Highlights.
We review recent advances for proteomics in aquatic toxicology.
iTRAQ is a powerful tool for biomarker discovery but there are challenges.
Fish ovarian proteome can be studied but there are special considerations.
Fish databases have improved and can be re-searched with spectra to gain new information.
Acknowledgments
We regret not being able to cite all proteomics studies in this review, but have highlighted just a few examples to illustrate how proteomics is being utilized to solve environmental problems. We would like to thank the current and former members of the Proteomics Research Group (C. Diaz, S. Chen, and S. McClung) at the University of Florida for their support and expertise in adapting the iTRAQ method to teleost fish. We would also like to acknowledge and thank D. Barber for his expertise and advice in quantitative proteomics. This research was funded by a Canada Research Chair and NSERC Discovery Grant to CJM and NIH RO1 ES015449 grant and EPA STAR grant (R831848) to NDD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmed N, Barker G, Oliva K, Garfin D, Talmadge K, Georgiou H, Quinn M, Rice G. An approach to remove albumin for the proteomic analysis of low abundance biomarkers in human serum. Proteomics. 2003;3:1980–1987. doi: 10.1002/pmic.200300465. [DOI] [PubMed] [Google Scholar]
- Ankarcrona M, Hultenby K. Presenilin-1 is located in rat mitochondria. Biochem Biophys Res Commun. 2003;295:766–770. doi: 10.1016/s0006-291x(02)00735-0. [DOI] [PubMed] [Google Scholar]
- Beranova-Giorgianni S. Proteome analysis by two-dimensional gel electrophoresis and mass spectrometry: strengths and limitations. Trac-Trend Anal Chem. 2003;5:273–281. [Google Scholar]
- Berg K, Puntervoll P, Valdersnes S, Goksøyr A. Responses in the brain proteome of Atlantic cod (Gadus morhua) exposed to methylmercury. Aquat Toxicol. 2010;100:51–65. doi: 10.1016/j.aquatox.2010.07.008. [DOI] [PubMed] [Google Scholar]
- Carvalho RN, Lettieri T. Proteomic analysis of the marine diatom Thalassiosira pseudonana upon exposure to benzo(a)pyrene. BMC Genomics. 2011;12:159. doi: 10.1186/1471-2164-12-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chora S, McDonagh B, Sheehan D, Starita-Geribaldi M, Roméo M, Bebianno MJ. Ubiquitination and carbonylation of proteins in the clam Ruditapes decussatus, exposed to nonylphenol using redox proteomics. Chemosphere. 2010;81:1212–1217. doi: 10.1016/j.chemosphere.2010.09.038. [DOI] [PubMed] [Google Scholar]
- Costa PM, Chicano-Gálvez E, López Barea J, Del Valls TA, Costa MH. Alterations to proteome and tissue recovery responses in fish liver caused by a short-term combination treatment with cadmium and benzo[a]pyrene. Environ Pollut. 2010;158:3338–3346. doi: 10.1016/j.envpol.2010.07.030. [DOI] [PubMed] [Google Scholar]
- Dorts J, Kestemont P, Marchand PA, D’Hollander W, Thézenas ML, Raes M, Silvestre F. Ecotoxicoproteomics in gills of the sentinel fish species, Cottus gobio, exposed to perfluorooctane sulfonate (PFOS) Aquat Toxicol. 2011;103:1–8. doi: 10.1016/j.aquatox.2011.01.015. [DOI] [PubMed] [Google Scholar]
- Domanski D, Helbing CC. Analysis of the Rana catesbeiana tadpole tail fin proteome and phosphoproteome during T3-induced apoptosis: identification of a novel type I keratin. BMC Dev Biol. 2007;7:94. doi: 10.1186/1471-213X-7-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling VA, Sheehan D. Proteomics as a route to identification of toxicity targets in environmental toxicology. Proteomics. 2006;6:5597–5604. doi: 10.1002/pmic.200600274. [DOI] [PubMed] [Google Scholar]
- Ekman DR, Villeneuve DL, Teng Q, Ralston-Hooper KJ, Martinović-Weigelt D, Kahl MD, Jensen KM, Durhan EJ, Makynen EA, Ankley GT, Collette TW. Use of gene expression, biochemical and metabolite profiles to enhance exposure and effects assessment of the model androgen 17β-trenbolone in fish. Environ Toxicol Chem. 2011;30:319–329. doi: 10.1002/etc.406. [DOI] [PubMed] [Google Scholar]
- Fang Y, Gao X, Zha J, Ning B, Li X, Gao Z, Chao F. Identification ofdifferential hepatic proteins in rare minnow (Gobiocypris rarus) exposed to pentachlorophenol (PCP) by proteomic analysis. Toxicol Lett. 2010;199:69–79. doi: 10.1016/j.toxlet.2010.08.008. [DOI] [PubMed] [Google Scholar]
- Forné I, Abián J, Cerdà J. Fish proteome analysis: model organisms and non-sequenced species. Proteomics. 2010;10:858–872. doi: 10.1002/pmic.200900609. [DOI] [PubMed] [Google Scholar]
- Forné I, Castellana B, Marín-Juez R, Cerdà J, Abián J, Planas JV. Transcriptional and proteomic profiling of flatfish (Solea senegalensis) spermatogenesis. Proteomics. 2011;11:2195–2211. doi: 10.1002/pmic.201000296. [DOI] [PubMed] [Google Scholar]
- Gan CS, Chong PK, Pham TK, Wright PC. Technical, Experimental, and Biological Variations in Isobaric Tags for Relative and Absolute Quantitation (iTRAQ) J Proteome Res. 2007;6:821–827. doi: 10.1021/pr060474i. [DOI] [PubMed] [Google Scholar]
- Garcia-Reyero N, Villeneuve DL, Kroll KJ, Liu L, Orlando EF, Watanabe KH, Sepúlveda MS, Ankley GT, Denslow ND. Expression signatures for a model androgen and antiandrogen in the fathead minnow (Pimephales promelas) ovary. Environ Sci Technol. 2009;43:2614–2619. doi: 10.1021/es8024484. [DOI] [PubMed] [Google Scholar]
- Garcia-Reyero N, Perkins EJ. Systems biology: leading the revolution in ecotoxicology. Environ Toxicol Chem. 2011;30:265–273. doi: 10.1002/etc.401. [DOI] [PubMed] [Google Scholar]
- Groh KJ, Nesatyy VJ, Segner H, Eggen RI, Suter MJ. Global proteomics analysis of testis and ovary in adult zebrafish (Danio rerio) Fish Physiol Biochem. 2011;37:619–647. doi: 10.1007/s10695-010-9464-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbing CC, Maher SK, Han J, Gunderson MP, Borchers C. Peering into molecular mechanisms of action with frogSCOPE. Gen Comp Endocrinol. 2010;168:190–198. doi: 10.1016/j.ygcen.2010.01.012. [DOI] [PubMed] [Google Scholar]
- Herbert BR, Harry JL, Packer NH, Gooley AA, Pederson SK, Williams KL. What place for polyacrylamide in proteomics? Trends Biotechnol. 2001;19(10):S3–9. doi: 10.1016/S0167-7799(01)01796-6. [DOI] [PubMed] [Google Scholar]
- Hidgon R, Hogan JM, van Belle G, Kolker E. Experiment-specific estimation of peptide identification probabilities using a randomized database. OMICS. 2007;11:351–366. doi: 10.1089/omi.2007.0040. [DOI] [PubMed] [Google Scholar]
- Hogenboom S, Tuyp JJM, Espeel M, Koster J, Wanders RJA, Waterham HR. Human mevalonate pyrophosphate decarboxylase is localized in the cytosol. Mol Genet Metab. 2004;81:216–224. doi: 10.1016/j.ymgme.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Iguchi T, Watanabe H, Katsu Y. Toxicogenomics and ecotoxicogenomics for studying endocrine disruption and basic biology. Gen Comp Endocrinol. 2007;153:25–29. doi: 10.1016/j.ygcen.2007.01.013. [DOI] [PubMed] [Google Scholar]
- Jiang L, He L, Fountoulakis M. Comparison of protein precipitation methods for sample preparation prior to proteomic analysis. J Chromatogr A. 2004;1023:317–320. doi: 10.1016/j.chroma.2003.10.029. [DOI] [PubMed] [Google Scholar]
- Jensen KM, Kahl MD, Makynen EA, Korte JJ, Leino RL, Butterworth BC, Ankley GT. Characterization of responses to the antiandrogen flutamide in a short-term reproduction assay with the fathead minnow. Aquat Toxicol. 2004;70:99–110. doi: 10.1016/j.aquatox.2004.06.012. [DOI] [PubMed] [Google Scholar]
- Jung E, Hoogland C, Chiappe D, Sanchez J, Hochstrasser DF. The establishment of a human liver nuclei two-dimensional electrophoresis reference map. Electrophoresis. 2000;21:3483–3487. doi: 10.1002/1522-2683(20001001)21:16<3483::AID-ELPS3483>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Kabbani N, Jeromin A, Levenson R. Dynamin-2 associated with the dopamine receptor signalplex and regulates internalization of activated D2 receptors. Cell Signal. 2004;16:497–503. doi: 10.1016/j.cellsig.2003.09.011. [DOI] [PubMed] [Google Scholar]
- Katsiadaki I, Williams TD, Ball JS, Bean TP, Sanders MB, Wu H, Santos EM, Brown MM, Baker P, Ortega F, Falciani F, Craft JA, Tyler CR, Viant MR, Chipman JK. Hepatic transcriptomic and metabolomic responses in the Stickleback (Gasterosteus aculeatus) exposed to ethinylestradiol. Aquat Toxicol. 2010;97:174–187. doi: 10.1016/j.aquatox.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Karim M, Puiseux-Dao S, Edery M. Toxins and stress in fish: Proteomic analyses and response network. Toxicon. 2011;57:959–969. doi: 10.1016/j.toxicon.2011.03.018. [DOI] [PubMed] [Google Scholar]
- Knoll-Gellida A, André M, Gattegno T, Forgue J, Admon A, Babin PJ. Molecular phenotype of zebrafish ovarian follicle by serial analysis of gene expression and proteomic profiling, and comparison with the transcriptomes of other animals. BMC Genomics. 2006;7:46. doi: 10.1186/1471-2164-7-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Gong Y, Wang Y, Wu S, Cai Y, He P, Lu Z, Ying W, Zhang Y, Jiao L, He H, Zhang Z, He F, Zhao X, Qian X. Comparison of alternative analytical techniques for the characterisation of the human serum proteome in HUPO Plasma Proteome Project. Proteomics. 2005;5:3423–3441. doi: 10.1002/pmic.200401226. [DOI] [PubMed] [Google Scholar]
- Li P, Hulak M, Rodina M, Sulc M, Li ZH, Linhart O. Comparative protein profiles: potential molecular markers from spermatozoa of Acipenseriformes (Chondrostei, Pisces) Comp Biochem Physiol Part D Genomics Proteomics. 2010;5:302–307. doi: 10.1016/j.cbd.2010.08.003. [DOI] [PubMed] [Google Scholar]
- Lilley KS, Razzaq A, Dupree P. Two-dimensional gel electrophoresis: recent advances in sample preparation, detection and quantitation. Curr Opin Chem Biol. 2001;6:46–50. doi: 10.1016/s1367-5931(01)00275-7. [DOI] [PubMed] [Google Scholar]
- Martin B, Brenneman R, Becker KG, Gucek M, Cole RN, Maudsley S. iTRAQ analysis of complex proteome alterations in 3xTgAD Alzheimer’s mice: understanding the interface between physiology and disease. PLoS One. 2008;3:e2750. doi: 10.1371/journal.pone.0002750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martyniuk CJ, Alvarez S, McClung S, Villeneuve DL, Ankley GT, Denslow ND. Quantitative proteomic profiles of androgen receptor signaling in the liver of fathead minnows (Pimephales promelas) J Proteome Res. 2009;8:2186–2200. doi: 10.1021/pr800627n. [DOI] [PubMed] [Google Scholar]
- Martyniuk CJ, Denslow ND. Towards functional genomics in fish using quantitative proteomics. Gen Comp Endocrinol. 2009;164:135–141. doi: 10.1016/j.ygcen.2009.01.023. [DOI] [PubMed] [Google Scholar]
- Martyniuk CJ, Kroll KJ, Doperalski NJ, Barber DS, Denslow ND. Environmentally relevant exposure to 17alpha-ethinylestradiol affects the telencephalic proteome of male fathead minnows. Aquat Toxicol. 2010a;98:344–353. doi: 10.1016/j.aquatox.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martyniuk CJ, Kroll KJ, Doperalski NJ, Barber DS, Denslow ND. Genomic and proteomic responses to environmentally relevant exposures to dieldrin: indicators of neurodegeneration? Toxicol Sci. 2010b;117:190–199. doi: 10.1093/toxsci/kfq192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta AI, Ross S, Lowenthal MS, Fusaro V, Fishman DA, Petricoin EF, Liotta LA. Biomarker amplification by serum carrier protein binding. Dis Markers. 2003;19:1–10. doi: 10.1155/2003/104879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsinjon T, Knigge T. Proteomic applications in ecotoxicology. Proteomics. 2007;7:2997–3009. doi: 10.1002/pmic.200700101. [DOI] [PubMed] [Google Scholar]
- Nothwang HG, Becker M, Ociepka K, Friauf E. Protein analysis in the rat auditory brainstem by two-dimensional electrophoresis and mass spectrometry. Mol Brain Res. 2003;116:59–69. doi: 10.1016/s0169-328x(03)00234-1. [DOI] [PubMed] [Google Scholar]
- Oh-Ishi M, Maeda T. Separation techniques for high-molecular-mass proteins. J Chromatogr B. 2002;771:49–66. doi: 10.1016/s1570-0232(02)00112-5. [DOI] [PubMed] [Google Scholar]
- Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Pillai S, Dey S, Daniels S, Martin S, Barlet-Jones M, He F, Jacobson A, Pappin DJ. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- Sadygov RG, Cociorva D, Yates JR., III Large-scale database searching using tandem mass spectra: Looking up the answer in the back of the book. Nat Methods. 2004;1:195–202. doi: 10.1038/nmeth725. [DOI] [PubMed] [Google Scholar]
- Sanchez BC, Ralston-Hooper K, Sepúlveda MS. Review of recent proteomic applications in aquatic toxicology. Environ Toxicol Chem. 2011;30:274–282. doi: 10.1002/etc.402. [DOI] [PubMed] [Google Scholar]
- Seidler J, Zinn N, Boehm ME, Lehmann WD. De novo sequencing of peptides by MS/MS. Proteomics. 2010;10:634–649. doi: 10.1002/pmic.200900459. [DOI] [PubMed] [Google Scholar]
- Serrano J, Higgins L, Witthuhn BA, Anderson LB, Markowski T, Holcombe GW, Kosian PA, Korte JJ, Tietge JE, Degitz SJ. In vivo assessment and potential diagnosis of xenobiotics that perturb the thyroid pathway: Proteomic analysis of Xenopus laevis brain tissue following exposure to model T4 inhibitors. Comp Biochem Physiol Part D Genomics Proteomics. 2010;5:138–150. doi: 10.1016/j.cbd.2010.03.007. [DOI] [PubMed] [Google Scholar]
- Shrader EA, Henry TR, Greeley MS, Jr, Bradley BP. Proteomics in zebrafish exposed to endocrine disrupting chemicals. Ecotoxicology. 2003;12:485–488. doi: 10.1023/b:ectx.0000003034.69538.eb. [DOI] [PubMed] [Google Scholar]
- Singh SK, Rakesh KS, Ramamoorthy K, Saradhi AVP, Idris MM. Proteome profile of zebrafish brain based on Gel LC-ESI MS/MS analysis. J Proteomics Bioinformatics. 2010;3:135–142. [Google Scholar]
- Siu KW, DeSouza LV, Scorilas A, Romaschin AD, Honey RJ, Stewart R, Pace K, Youssef Y, Chow TF, Yousef GM. Differential protein expressions in renal cell carcinoma: new biomarker discovery by mass spectrometry. J Proteome Res. 2009;8:3797–3807. doi: 10.1021/pr800389e. [DOI] [PubMed] [Google Scholar]
- Sheehan D, McDonagh B, Bárcena JA. Redox proteomics. Expert Rev Proteomics. 2010;7:1–4. doi: 10.1586/epr.09.98. [DOI] [PubMed] [Google Scholar]
- Song X, Bandow J, Sherman J, Baker JD, Brown PW, McDowell MT, Molloy MP. iTRAQ experimental design for plasma biomarker discovery. J Proteome Res. 2008;7:2952–2958. doi: 10.1021/pr800072x. [DOI] [PubMed] [Google Scholar]
- Steinberg TH, Pretty, On Top K, Berggren KN, Kemper C, Jones L, Diwu Z, Haugland RP, Patton WF. Rapid and simple single nanogram detection of glycoproteins in polyacrylamide gels and on electroblots. Proteomics. 2001;1:841–855. doi: 10.1002/1615-9861(200107)1:7<841::AID-PROT841>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Sumpter JP, Jobling S. Vitellogenesis as a biomarker for estrogenic contamination of the aquatic environment. Environ Health Perspect. 1995;103(Suppl 7):173–178. doi: 10.1289/ehp.95103s7173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonge R, Shaw J, Middleton B, Rowlinson R, Rayner S, Young J, Pognan F, Hawkins E, Currie I, Davison M. Validation and development of fluorescence two-dimensional differential gel electrophoresis proteomics technology. Proteomics. 2001;1:377–396. doi: 10.1002/1615-9861(200103)1:3<377::AID-PROT377>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Wang N, Mackenzie L, De Souza AG, Zhong H, Goss G, Li L. Proteome profile of cytosolic component of zebrafish liver generated by LC-ESI MS/MS combined with trypsin digestion and microwave-assisted acid hydrolysis. J Proteome Res. 2007;6:263–272. doi: 10.1021/pr060367o. [DOI] [PubMed] [Google Scholar]
- Wu WW, Wang G, Baek SJ, Shen RF. Comparative study of three proteomic quantitative methods, DIGE, cICAT, and iTRAQ, using 2D gel- or LC-MALDI TOF/TOF. J Proteome Res. 2006;5:651–658. doi: 10.1021/pr050405o. [DOI] [PubMed] [Google Scholar]
- Villeneuve L, Wang RL, Bencic DC, Biales AD, Martinović D, Lazorchak JM, Toth G, Ankley GT. Altered gene expression in the brain and ovaries of zebrafish (Danio rerio) exposed to the aromatase inhibitor fadrozole: microarray analysis and hypothesis generation. Environ Toxicol Chem. 2009;28:1767–1782. doi: 10.1897/08-653.1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.