

Abstract
Epidemiologic studies have provided solid evidence for the neurotoxic effect of lead for decades of years. In view of the fact that children are more vulnerable to the neurotoxicity of lead, lead exposure has been an urgent public health concern for decades of years. The modes of action of lead neurotoxic effects include disturbance of neurotransmitter storage and release, damage of mitochondria, as well as induction of apoptosis in cerebrovascular endothelial cells, astroglia and oligodendroglia. Our studies here, from a novel point of view, demonstrates that lead specifically caused induction of COX-2, a well known inflammatory mediator in neurons and glia cells. Furthermore, we revealed that COX-2 was induced by lead in a transcription-dependent manner, which relayed on transcription factor NFAT, rather than AP-1 and NFκB, in glial cells. Considering the important functions of COX-2 in mediation of inflammation reaction and oxidative stress, our studies here provide a mechanistic insight into the understanding of lead-associated inflammatory neurotoxicity effect via activation of pro-inflammatory NFAT3/COX-2 axis.
Keywords: Lead, COX-2, NFAT, Neurotoxicity
1. Introduction
Lead is a naturally occurring heavy metal, and widely used in industries including batteries, welding and plastic manufacture (Ghareeb et al., 2009). Human intakes of lead at the occupational settings occur through the routes of ingestion, inhalation and dermal absorption, which can cause systemic disorders, such as cardiovascular, renal, immune, bone as well as nerve ailments (White et al., 2007). Lead is a well-known human neurotoxin. When the blood lead levels (PbB) exceed 40–60 μg/dL, various neurological disorders can occur, including encephalopathy and peripheral neuropathy (Spivey 2007; Murata et al., 2009). Notably, the developing nervous systems are more susceptible to toxicity of lead than the mature nerves so that children are at a higher risk to chronic lead exposure even at low levels (PbB below 10 μg/dL) (Lidsky and Schneider 2003).
The central nervous system comprise of two major types of cells, neurons and glial cells. Neurons receive sensory information at each synaptic terminal at the end of an axon, and send electrical signals over long distances within the body. Glial cells, including astrocytes, oligodendrocytes, and microglia, provide support and protection for neurons. They supply nutrients and oxygen to neurons, insulate one neuron from another, and destroy and remove the carcasses of dead neurons. Recently, a myriad of mounting evidence demonstrated that astrocytes are the type of brain cells that are responsible for sequestration of lead in brain tissue (Strużyńska et al., 2007). As a result, activation of astroglia is one of the modes of action underlying lead neurotoxicity, which may cause loss of the buffering function of astroglia. This leads to the death of neuronal cells by initiating the inflammatory events arising from the production of a wide range of cytokines and chemokines (Ricci et al., 2009).
The mechanisms underlying the neurotoxic effects of lead have been linked to excitotoxicity, alteration of neurotransmitter storage and release, induction of brain cell apoptosis, inflammation and oxidative stress (Lidsky and Schneider 2003). Particularly, lead-associated increase in inflammatory mediators has been reported in human population studies as well as experimental animal models and cell culture systems (Farkhondeh et al., 2013; Sirivarasai et al. 2013). For instance, Ghareeb et al. have reported that lead exposure in rats caused increases of inflammatory markers, NO and TNFα, coupled with a significant decrease of glutathione (GSH) levels and impairment of antioxidant activities of superoxide dismutase (SOD) and catalase (CAT) (Ghareeb et al., 2009).
Cyclooxygenase-2 (COX-2) is a source of inflammatory mediators and a multifunctional neuronal modulator. COX-2 catalyzes the conversion of free arachidonic acid (AA) to the intermediate prostaglandin H2 (PGH2), which is then transformed by a range of enzymes and nonenzymic mechanisms into the primary prostanoids, PGE2, PGF2α, PGD2, PGI2, and TXA2 and ROS are generated simultaneously (Vane et al., 1998). In peripheral tissues, COX-1 is the constitutively expressed form of cyclooxygenase, and COX-2 presents as an inducible form. In contrast, in normal brain COX-2 is the fundamental form exclusively expressed in neurons (Kaufmann et al., 1996; Breder et al., 1992; Yasojima et al., 1999). Immunoreactivity of COX-2 has been detected in the forebrain areas, including dentate gyrus granule cells, pyramidal cell neurons in the hippocampus, the piriform cortex, superficial cell layers of neocortex and the amygdala (Minghetti et al., 2004). Whereas, under pathologic conditions, such as hypoxia/ischemia and seizures, as well as in neurodegenerative diseases, including Alzheimer’s disease (AD), COX-2 over-expression has been associated with neurotoxicity (Minghetti et al., 2007). Thus, this reflects a role of COX-2 in neuroinflammation and neural cell death, which is reinforced by the therapeutic implications of COX-2 inhibitors and arachidonic acid shunting in alleviating brain injuries (Strauss 2008).
Animal models have suggested that chronic lead exposure causes potential proinflammatory effect in CNS in immature rat brain, which might be achieved through activation of glial cells (Strużyńska et al., 2007). Activated glial cells generate an inflammatory reaction in the brain by producing cytokines. Developmental lead exposure has been shown to promote changes in inter-region gene expression of pro-inflammatory effectors, such as IL-1β, IL-18, IL-33, caspase 1 and NOS2 (Kasten-Jolly et al., 2012). However, the induction and role played by COX-2 in lead neurotoxicity is still unexplored. Therefore, in the present study, we investigated the COX-2 induction by lead in C6 rat glioma cell line; BV2 murine microglia cell line; primary cortical neuronal cultures; neural stem cells (NSCs); and RBE4 cell line (a well established in vitro model of the blood-brain barrier). Our data indicated that lead specifically caused COX-2 induction in C6 and BV12 glia cells, primary cortical neuronal cultures, and NSCs, with mild inductions in RBE4 (the cerebrovascular endothelial) cells. Further mechanistic investigation showed that the transcription factor, NFAT, rather than AP-1 and NFκB, was involved in lead-associated COX-2 induction in glial cells.
2. Results
2.1. Lead induced COX-2 expression in glia BV12 and C6 cells, primary cortical neuronal cultures and NSCs, but not in RBE4 cells
We first used a wide range doses of lead to treat various brain cell lines to determine whether and which type of brain cells were responsive to lead for COX-2 induction. As shown in Fig. 1A and C, COX-2 was remarkably induced by lead in BV2 cells at all the doses tested, ranging from 25 μM to 100 μM. Time course studies showed that the initial induction of COX-2 could be observed as early as 6 h and reached the peak induction by 24 h tested (Fig. 1B). Consistently, a dose- and time-dependent induction of COX-2 was observed in C6 cells following lead treatment (Fig. 1C and D). To examine the effect of lead in neurons, we isolated primary cortical neurons from embryonic rats. As shown in Fig. 1E, treatment of lead for 12 h induced COX-2 expression at various doses. In addition, we used neural stem cells (NSCs) derived from human induced pluripotent stem cells (iPSCs) to check COX-2 induction. As shown in Fig. 1F, lead treatment in iPSCs did not induce COX-2 expression, while incorporation of lead into the conditioned medium during the differentiation process from iPSCs to NSCs resulted in robust COX-2 induction (Fig. 1G). Oct4 was used as pluripotency marker to indicate the success of the induction. It suggested that brain cells were responsive to COX-2 induction by lead. Comparatively, RBE4 cells had higher basal level of COX-2 and lead treatment increased COX-2 expression only modestly at doses greater than 50 μM for 12 h (Fig. 1H).
Fig. 1.
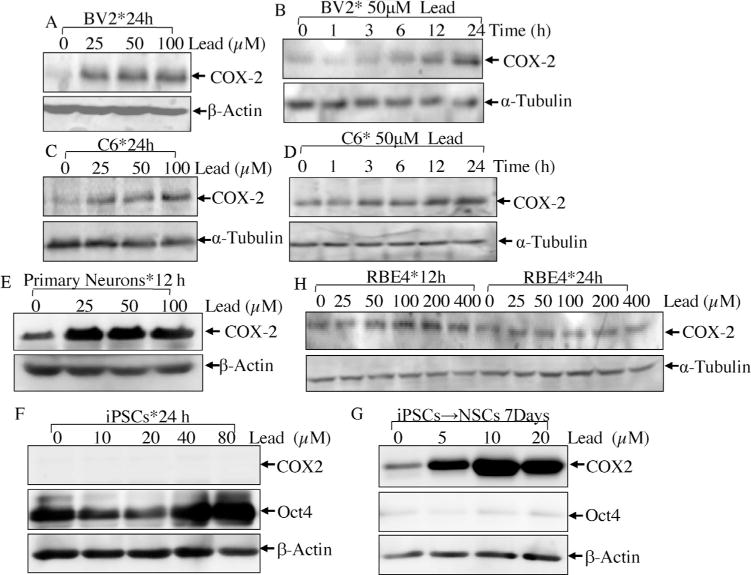
The effects of lead exposure on COX-2 protein induction in various types of brain cells.
BV2 murine microglia cell line(Aand B), C6rat glioma cell line(C and D), primarycortical neurons (E), iPSCs (F) and NSCs (G) were treated with the different doses of lead (25–400 μM) for various time points as indicated. The inductions of COX-2 were determined by Western blotting assay.
2.2. Lead induced COX-2 expression at transcriptional level
In order to exploit the molecular mechanism of COX-2 induction by lead, we first determined cox-2 mRNA levels by RT-PCR. The results showed that consistent with protein induction, cox-2 mRNA was elevated by lead treatment in BV2 cells in a similar dose and time responsive manner as that of protein (Fig. 2A and B). Namely, lead treatment of 25 μM led to obvious induction of cox-2 mRNA in BV12 cells (Fig. 2A), and the time window of cox-2 mRNA induction ranged from 3 h to 24 h (Fig. 2B). Comparable change of cox-2 mRNA was also detected in C6 cells in both dose-response and time-course assays (Fig. 2C). To further determine whether lead activated cox-2 transcription, cox-2 promoter-drive luciferase reporter was transfected into C6 cells stably. As shown in Fig. 2E, lead increased cox-2 promoter transactivation in a dose-dependent manner. The induction fold of cox-2 transcription was 1.48 ±0.08 for 12 h lead treatment, and 1.72±0.13 for 24 h treatment (Fig. 2F). It suggested that lead might increase cox-2 transcription in gliacytes.
Fig. 2.
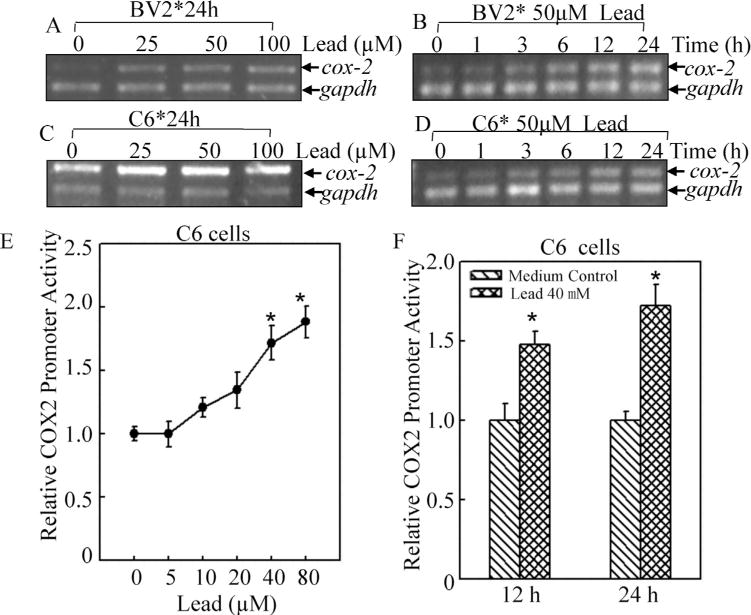
COX-2 transcription was induced by lead in BV2 and C6 cells.
The mRNA changes of cox-2 following lead treatment in BV2 cells (A and B) and C6 cells (C and D) were assessed by RT-PCR in both time and dose-dependent responses. (E and F), The transcription of cox-2 was evaluated by using cox-2 promoter-driven luciferase reporter in C6 cells. The symbol (*) indicates a significant difference between medium control and lead treatment (p<0.05). The value was showed as mean ± SD from three independent experiments.
2.3. The transcriptional regulation of cox-2 expression by lead was dependent on NFAT, but neither AP-1 nor NFκB
Analysis of the promoter regions of human and mouse cox-2 genes indicated that they both contain a canonical TATA-box and multiple transcription factor binding sites, including NFκB, AP-1, NFAT, NF-IL-6/CCAAT/enhancer-binding protein(C/EBP), and CREB, all of which regulate the expression of COX-2 in a cell type and stimuli dependent manner (Yan et al., 2006). Therefore, we checked the expressions of some of these transcription factors by Western Blotting. As shown in Fig. 3A, lead did not cause activation of c-Jun as indicated by marginal p-c-Jun S73 induction after lead treatment. In addition, changes of p-IKKα/β S176/180, p-IκBα S32/36 and total IκBα protein expression were observed after lead exposure (Fig. 3B). Expression of p-NFκB p65 S536 and p50 were not affected by lead treatment (Fig. 3B). While hypophosphorylation of NFAT was detected after lead exposure, indicating lead might cause NFAT activation because hyperphosphorylated NFAT was its inactivated form, which sequestered NFAT in the cytoplasm (Fig. 3C). When in its hypophosphorylation status, NFAT could translocate into nucleus and bind to DNA consensus sequences of NFAT-regulated gene, leading to the gene transcription. Next, we checked the transcriptional activities of AP-1, NFκB and NFAT by luciferase reporter assay to enforce what we observed from Western blotting assay. As expected, lead treatment increased NFAT transactivation activity, but neither AP-1 nor NFκB (Fig. 4A). The induction folds of NFAT activity were 1.50 ± 0.002 for 24 h and 2.58 ±0.014 for 48 h upon 40 μM of lead exposure (Fig. 4B). The results indicated that lead exposure specifically up-regulated NFAT transactivation, which might further contribute to COX-2 induction in glial cells. To further define the critical role of NFAT in COX-2 induction by lead, mutant luciferase reporter which harbor no NFAT responsive transcription factor binding sites was used (Ding et al., 2006). As shown in Fig. 4C and D, lead exposure induced significant wild-type COX-2 promoter transcription, while NFAT binding site mutations abolished the transcription of the cox-2 Promoter luciferase reporter in lead response. Our results demonstrated that the NFAT transactivation played a key role in cox-2 gene transcription and protein induction by lead in glia cells, which might provide a molecular basis for using of COX-2 inhibitor to overcome lead neurotoxicity.
Fig. 3.
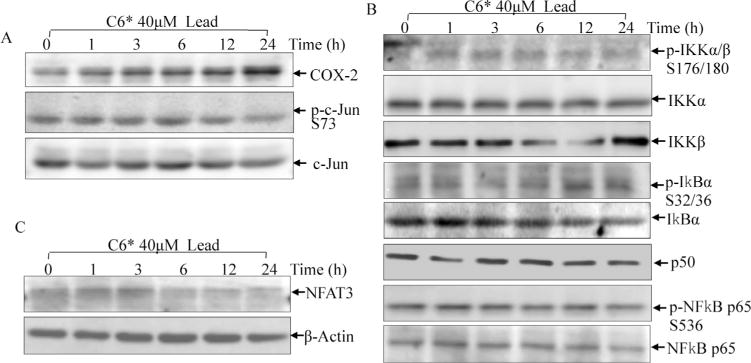
The effect of lead exposure on potential transcription factors that might involved in regulation of cox-2 transcription.
C6 cells were exposed to 40 μM lead for 1h, 3h, 6h, 12h and 24h. The cells were extracted and subjected to Western blotting assay. The induction of COX-2 and activation of AP-1 were detected using anti-COX-2 and anti-p-c-Jun S73 antibodies (A). The lead-associated activation of NFκB pathway was assessed using antibodies recognizing phospho- and total IKKα/β, IkBα, p65 and total p50 (B). Activation of NFAT3 was determined by checking the hypophosphorylated form of NFAT3 using anti-NFAT3 antibody (C).
Fig. 4.
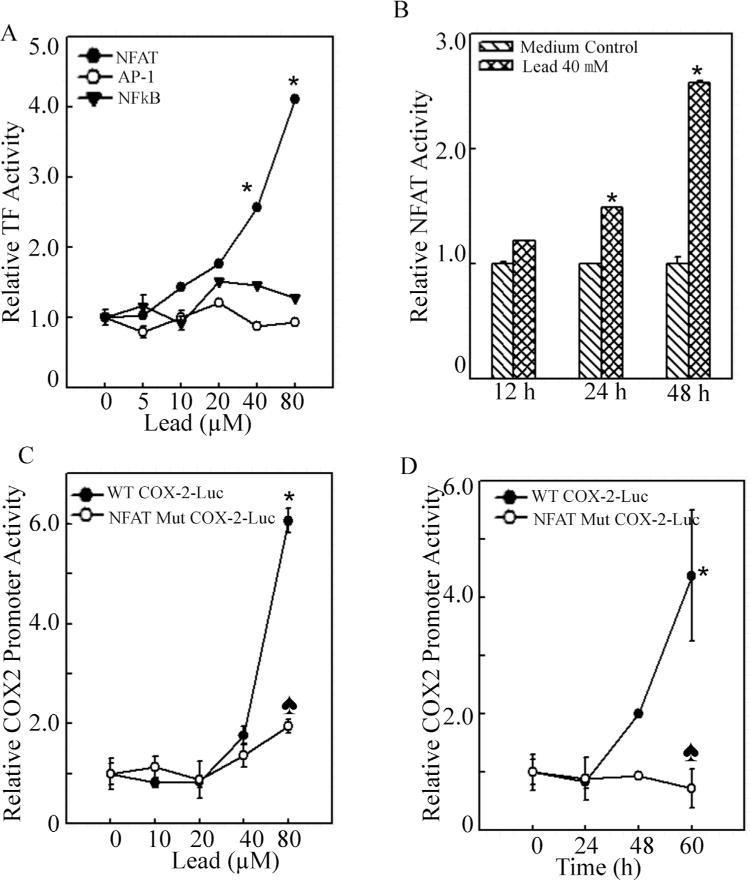
NFAT transactivation was crucial for cox-2 transcription following lead exposure.
(A), C6 cells were stably transfected with the AP-1, NFκB and NFAT luciferase reporter in combination with the pRL-TK vector (Promega) as an internal control. After the cells were treated with lead at the indicated doses, the luciferase activities were determined using a luminometer to determine transactivation activities of the individual transcription factor. The asterisk (*) indicates a significant increase compared with medium control (p < 0.05). (B), The C6 stable transfectants of NFAT luciferase reporter in combination with the pRL-TK vector were treated with 40 μM lead for 12 h, 24 h and 48 h. The transactivation of NFAT was determined using a luminometer. The asterisk (*) indicates a significant increase compared with medium control (p < 0.05). (C and D), The cox-2-luciferase reporter plasmid containing site-directed mutant of the NFAT binding sequence was used to validate the involvement of NFAT in induction of COX-2 by lead. The asterisk (*) indicates a significant increase compared with medium control (p < 0.05). The heart symbol (♠) indicates a significant decrease compared with that of wild type cox-2 promoter luciferase reporter (p<0.05).
3. Discussion
Our studies here, from a novel point of view, demonstrated that lead specifically caused induction of COX-2, a well known inflammatory mediator, in brain cells. Furthermore, we revealed that COX-2 was induced by lead in a transcription-dependent manner, which relayed on transcription factor NFAT, rather than AP-1 and NFκB in glial cells.
Lead is a naturally-occurring heavy metal, and can be found in small amount in the earth’s crust and soil (Laidlaw and Filippelli, 2008). Human exposure to lead occurs in the living environment and occupational settings through the routes of inhalation and ingestion (Spivey, 2007). US Environmental Protection Agency (EPA) sets 1.5 μg/m3 as the health-based national air quality standard for lead. After lead was banned from usages in gasoline, paint and food cans, the average adult blood lead levels dropped markedly from 15 μg/dL to 1–2 μg/dL so that workplace exposure became the major source of human lead exposure (Spivey, 2007). Lead usually presents as a dust or fume in the air, and is inhaled through the lung in the workplace, therefore, the National Institute of Occupational Safety and Health (NIOSH) sets 100 μg/m3 as the recommended exposure limit (REL) for lead in the air. It is also regulated that when the worker PbB exceeds 50 μg/dL, immediate removal from exposure is required to prevent various clinical symptoms of lead intoxication, such as central nervous system effects and peripheral neuropathy (Spivey, 2007; Murata et al., 2009). For reference, the concentration of lead used in this study was between 500 μg/dL and 2000 μg/dL.
Depending on the variety of cell models and experimental procedures, different doses of lead have been used in cell culture system in either chronic or acute exposure. For instance, incubation of lead at the concentrations of 0.25–1.0 μM (5.2–20.7 μg/dL) for 7–21 days resulted in an obvious reduction in functions of primary cultures of rat astrocytes (Lidsky and Schneider, 2003). Our published studies showed that short-term treatment of lead (within 48 h) at 50 μM (1035.2 μg/dL) activated the primary cultured rat microglia (Liu et al., 2012). To further reveal the molecular basis for lead activation of microglia, we used acute lead exposure here since COX-2 is an immediate early response gene, and during our observance periods (within 24 h), no severe cell death was observed. The major dose range of lead we used in the current studies was from 25 to 100 μM (517.6–2070.4 μg/dL). During iPSCs to NSCs differentiation process, the cells were cultured in the presence of lead for 7 days at 5, 10 and 20 μM (103.5, 207.0 and 414.1 μg/dL). For REB4, the cerebrovascular endothelial cells, which were tolerant to lead exposure, the lead treatment doses were 25–400 μM (517.6–8281.6 μg/dL), and only marginal induction of COX-2 was observed compared with those in glial cells and neurons, indicating that glial cells and neurons were responsive to COX-2 induction by lead.
Our current results of RT-PCR and cox-2 promoter-driven luciferase reporter assay indicated that lead induced COX-2 expression at transcriptional level. COX-2 has been shown to be regulated by transcription factors of NF-κB, AP-1 and NFAT upon exposure to heavy metals, such as nickel and arsenite (Ding et al., 2006; Zhang et al., 2006; Cai et al., 2011; Ouyang et al., 2007; Zuo et al., 2012). Therefore we compared their transactivations following lead treatment and found only NFAT activity was up-regulated by lead. Next we mutated the NFAT binding sites on cox-2 promoter and found it abolished the induction of cox-2 transcription, therefore we concluded that lead induced COX-2 transcription in a NFAT-dependent manner. NFAT is a master transcription factor that controls expressions of a variety genes involved in inflammation, such as iNOS, COX-2 and TNFα (Yan et al., 2006; Li et al., 2006; Obasanjo-Blackshire et al., 2006; Ke et al., 2006). Of the four members of NFAT family, NFAT3 is the most abundant form in the nervous system, and involves in neural development and axon growth as well as pathologic features (Vihma et al., 2008; Nguyen and Giovanni, 2008). The activity of NFAT3 is tightly regulated by its phosphorylation status. When NFAT3 is hyper-phosphorylated, it sequesters in cytoplasm; while its dephosphorylation executed by calcium-related phosphatase, calcineurin, causes its translocation to nucleus, where NFAT3 interacts with the promoters of genes containing its DNA binding consensus and regulates gene transcription (Hogan et al., 2003).
Calcium signals in nervous systems are the fundamental mechanisms underlying physiological functions of learning and memory, whereas disruption of these process may lead to declines of cognition (Oliveira and Bading, 2011). The calcium signals are regulated by several factors, including ion influx/efflux through voltage or transmembrane-dependent ion channels, storage/release of internal calcium reservoir mitochondria and the endoplasmic reticulum (ER), as well as sequestration by calcium-binding proteins (Oliveira and Bading, 2011). Lead is known to substitute calcium in several tissues including nervous systems so that lead can block the uptake of calcium by brain mitochondria or ER, increasing the concentration of ionic calcium in the cytosol (Goldstein, 1977). Calmodulin belongs to EF-hand superfamily which can bind to calcium. The activation of calmodulin bycalcium binding leads to displacement of the auto-inhibiting domain and exposes the calcineurin active site; so that the activated calcineurin subsequently dephosphorylates the cytoplasmic NFAT proteins, leading to NFAT nuclear translocation and activation (Smith, 2009). Therefore, our current studies indicated that lead increase NFAT3 activity which might be achieved through up-regulating intracellular calcium concentration by displacing calcium from mitochondria or ER uptake.
Considering the important functions of COX-2 in mediation of inflammation reaction and oxidative stress, our studies here provide one more piece of evidence for the involvement of pro-inflammatory NFAT3/COX-2 axis in lead-associated neurotoxicity.
4. Materials and methods
4.1. Cell culture
PC12, BV2, C6 and RBE4 as well as their stable transfectants were maintained at 37 °C in 5% CO2 incubator with Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, and 25 μg/mL of gentamicin. The cultures were dissociated with trypsin and transferred to new 75 cm2 culture flasks (Fisher, Pittsburgh, PA) twice a week. FBS was purchased from Nova-Tech (Grand Island, NE). The cells were cultured in 0.1% FBS for 24 h before lead treatment which was also carried out in 0.1% FBS.
4.2. Primary neuronal culture
Rat primary cortical neuronal cultures were kindly provided by Dr. Huaye Zhang and Dr. Zhiping Pang (Child Health Institute of New Jersey, Dept. of Neuroscience and Cell Biology, UMDNJ-Robert Wood Johnson Medical School) according to the protocol as published previously (Kim et al., 2011). Briefly, cerebral cortex tissues were dissected from embryonic day 19 rat embryos, trypsinized, and triturated through a glass Pasteur pipette. Dissociated neurons were plated in 6-well culture plates pre-coated with 1% (w/v) poly-L-lysine at the density of 1 × 106 cells. Cultures were grown in Neurobasal medium (Invitrogen) supplemented with B27 (Invitrogen) and 2 mM GlutaMAX (Invitrogen).
4.3. Constructs and transfection
The COX-2 and NFAT luciferase reporter plasmid were constructed as described previously (Subbaramaiah et al., 2001; Rincón et al., 1997). The p-AP-1-Luc and NFκB-Luc reporter plasmid was purchased from Stratagene (LaJolla, CA). The COX-2-luciferase reporter plasmid containing site-directed mutant of the NFAT binding sequence was reported in our previous publication (Ding et al., 2006). They were stably transfected into C6 cells individually.
4.4. Reverse transcription polymerase chain reaction (RT-PCR)
Total RNA was extracted from the cells using Trizol reagent (Invitrogen, Carlsbad, California). Total cDNAs were synthesized by ThermoScript™ RT-PCR system (Invitrogen). The mRNA amount presented in the cells was measured by semiquantitative RT-PCR. The primers for murine cox-2 were 5′-TCC TCC TGG AAC ATG GAC TC-3′ and 5′-GCT CGG CTT CCA GTA TTG AG-3′, for murine gapdh were 5′-TGC AGT GGC AAA GTG GAG ATT-3′ and 5′-TTT TGG CTC CAC CCT TCA AGT-3′. The PCR products were separated on 2% agarose gels, stained with EB, and scanned for the images under UV light.
4.5. Luciferase reporter assay
C6 cells were transfected with the indicated luciferase reporter in combination with the pRL-TK vector (Promega) as an internal control. The luciferase activities were determined using a luminometer (Wallac 1420 Victor 2 multilabel counter system) as described in previous studies (Song et al., 2007).
4.6. Western blotting assay
The cells were washed twice with ice-cold PBS and collected with the cell lysis buffer (10 mM Tris-HCl, pH 7.4, 1% SDS, and 1 mM Na3VO4). The cell extracts were sonicated, denatured by heating at 100 °C for 5 min, and quantified with a Dc protein assay kit (Bio-Rad Hercules, CA). Equal aliquots of cell extracts were separated on SDS-polyacrylamide gels. The proteins were then transferred to PVDF membranes (Bio-Rad Hercules, CA), blocked, and probed with one of the antibodies against COX-2 (Cayman Chemical Co.), p-c-Jun S73, p-IKKα/β S176/180, p-IκBα S32/36, p-NFκB p65 S536, total c-Jun IκBα and IKKα/β (Cell Signaling Technology, Beverly, MA), NFAT3, p65, and p50 (Santa Cruz Biotechnology, CA, USA), or β-Actin (Sigma). Primary antibody-bound proteins were detected by using an alkaline phosphatase-linked secondary antibody and an ECF Western blotting system (Amersham, Piscataway, NJ).
4.7. Statistical analysis
The significance of the difference between the treated and untreated groups was determined with the Wilcoxon rank sum test. The results are expressed as mean ± S.D.
Acknowledgments
We thank Dr. Huaye Zhang, Dr. Zhiping Pang and Dr. Yasunari Seita (Child Health Institute of New Jersey, Dept. of Neuroscience and Cell Biology, UMDNJ-Robert Wood Johnson Medical School) for the generous providing of rat primary cortical neuronal cultures and iPSCs. This work was supported partially by grants from NBRPC2012CB525004, NSFC81229002, NIH/NCICA177665 and CA112557, as well as NIH/NIEHS ES000260.
Footnotes
Conflicts of interest
The authors declare that there are no conflicts of interest.
Transparency document
The Transparency document associated with this article can be found in the online version.
References
- Abdul H, Furman J, Sama M, Mathis D, Norris C. NFATs and Alzheimer’s Disease. Mol Cell Pharmacol. 2010;2(1):7–14. [PMC free article] [PubMed] [Google Scholar]
- Breder C, et al. Distribution and characterization of cyclooxygenase immunoreactivityin the ovine brain. J Comp Neurol. 1992;322(3):409–438. doi: 10.1002/cne.903220309. [DOI] [PubMed] [Google Scholar]
- Cai T, et al. A Cross-Talk Between NFAT and NF-κB Pathways is Crucial for Nickel- Induced COX-2 Expression in Beas-2B Cells. Current Cancer Drug Targets. 2011;11(5):548–559. doi: 10.2174/156800911795656001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, et al. Cyclooxygenase-2 induction by arsenite is through a nuclear factor of activated T-cell-dependent pathway and plays an antiapoptotic role in beas-2B cells. J Biol Chem. 2006;281(34):24405–24413. doi: 10.1074/jbc.M600751200. [DOI] [PubMed] [Google Scholar]
- Farkhondeh T, Boskabady M, Koohi M, Sadeghi-Hashjin G, Moin M. The effect oflead exposure on selected blood inflammatory biomarkers in guinea pigs. Cardiovasc. Hematol Disord Drug Targets. 2013;13(1):45–49. doi: 10.2174/1871529x11313010005. [DOI] [PubMed] [Google Scholar]
- Goldstein GW. Lead encephalopathy: the significance of lead inhibition of calcium uptake by brain mitochondria. Brain Res. 1977;136(1):185–188. doi: 10.1016/0006-8993(77)90145-7. [DOI] [PubMed] [Google Scholar]
- Ghareeb DA, Hussien HM, Khalil AA, El-Saadani MA, Ali AN. Toxic effects of leadexposure on the brain of rats: Involvement of oxidative stress, inflammation, acetylcholinesterase, and the beneficial role of flaxseed extract. Toxicol Environ Chem. 2009;92(1):187–195. [Google Scholar]
- Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes & Development. 2003;17(18):2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- Ke Q, et al. Essential role of ROS-mediated NFAT activation in TNF-α induction by crystalline silica exposure. Am J Physiol – Lung Cell Mol Physiol. 2006;291(2):L257–L264. doi: 10.1152/ajplung.00007.2006. [DOI] [PubMed] [Google Scholar]
- Kim JY, Oh MH, Bernard LP, Macara IG, Zhang H. The RhoG/ELMO1/Dock180 signaling module is required for spine morphogenesis in hippocampal neurons. J Biol Chem. 2011;286(43):37615–37624. doi: 10.1074/jbc.M111.268029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann WE, Worley PF, Pegg J, Bremer M, Isakson P. COX-2, a synaptically induced enzyme, is expressed by excitatory neurons at postsynaptic sites in rat cerebral cortex. Proc Natl Acad Sci. 1996;3(6):2317–2321. doi: 10.1073/pnas.93.6.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasten-Jolly J, Pabello N, Bolivar VJ, Lawrence DA. Developmental lead effects on behavior and brain gene expression in male and female BALB/cAnNTac mice. Neurotoxicology. 2012;33(5):1005–1020. doi: 10.1016/j.neuro.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lidsky TI, Schneider JS. Lead neurotoxicity in children: basic mechanisms and clinical correlates. Brain. 2003;126(1):5–19. doi: 10.1093/brain/awg014. [DOI] [PubMed] [Google Scholar]
- Laidlaw MAS, Filippelli GM. Resuspension of urban soils as a persistent source of lead poisoning in children: a review and new direct ions. Appl Geochem. 2008;23(8):2021–2039. [Google Scholar]
- Liu MC, et al. Involvement of microglia activation in the lead induced long-term potentiation impairment. PLoS ONE. 2012;7(8):e43924. doi: 10.1371/journal.pone.0043924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Song L, Zhang D, Wei L, Huang C. Knockdown of NFAT3 blocked TPA-induced COX-2 and iNOS expression, and enhanced cell transformation in Cl41 cells. J Cell Biochem. 2006;99(4):1010–1020. doi: 10.1002/jcb.20834. [DOI] [PubMed] [Google Scholar]
- Murata K, Iwata T, Dakeishi M, Karita K. Lead toxicity: does the critical level of lead resulting in adverse effects differ between adults and children? J Occup Health. 2009;1(1):1–12. doi: 10.1539/joh.k8003. [DOI] [PubMed] [Google Scholar]
- Minghetti L. Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J Neuropathol Exp Neurol. 2004;3(9):901–910. doi: 10.1093/jnen/63.9.901. [DOI] [PubMed] [Google Scholar]
- Minghetti L. Role of COX-2 in inflammatory and degenerative brain diseases. Sub-Cell Biochem. 2007;2:127–141. doi: 10.1007/1-4020-5688-5_5. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Di Giovanni S. NFAT signaling in neural development and axon growth. Int J Dev Neurosci. 2008;26(2):141–145. doi: 10.1016/j.ijdevneu.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obasanjo-Blackshire K, et al. Calcineurin regulates NFAT-dependent iNOS expression and protection of cardiomyocytes: Co-operation with Src tyrosine kinase. Cardiovas Res. 2006;1(4):672–683. doi: 10.1016/j.cardiores.2006.05.026. [DOI] [PubMed] [Google Scholar]
- Ouyang W, Zhang D, Ma Q, Li J, Huang C. Cyclooxygenase-2 induction by arsenite through the IKKβ/NFκB pathway exerts an antiapoptotic effect in mouse epidermal Cl41 cells. Environ Health Perspect. 2007;115(4):513–518. doi: 10.1289/ehp.9588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira AMM, Bading H. Calcium signaling in cognition and aging-dependentcognitive decline. Biofactors. 2011;37(3):168–174. doi: 10.1002/biof.148. [DOI] [PubMed] [Google Scholar]
- Ricci G, Volpi L, Pasquali L, Petrozzi L, Siciliano G. AstrocyteKneuron interactions in neurological disorders. J Biol Phys. 2009;35(4):317–336. doi: 10.1007/s10867-009-9157-9. (in English) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rincón M, Flavell RA. Transcription mediated by NFAT is highly inducible in effector CD4+ T helper 2 (Th2) cells but not in Th1cells. Mol Cell Biol. 1997;7(3):1522–1534. doi: 10.1128/mcb.17.3.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spivey A. The weight of lead: effects add up in adults. Environ Health Perspect. 2007;15(1):A30–A36. doi: 10.1289/ehp.115-a30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strużyńska L, Dąbrowska-Bouta B, Koza K, Sulkowski G. Inflammationlike glial response in lead-exposed immature rat brain. Toxicol Sci. 2007;95(1):156–162. doi: 10.1093/toxsci/kfl134. [DOI] [PubMed] [Google Scholar]
- Sirivarasai J, et al. Association between inflammatory marker, environmental leadexposure, and glutathione S-transferase gene. BioMed Res Int. 2013;2013:6. doi: 10.1155/2013/474963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss KI. Antiinflammatory and neuroprotective actions of COX2 inhibitors in theinjured brain. Brain, Behav Immun. 2008;22(3):285–298. doi: 10.1016/j.bbi.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith H. Calcineurin as a nociceptor modulator. Pain Physician. 2009;12(4):E309–E318. [PubMed] [Google Scholar]
- Subbaramaiah K, Bulic P, Lin Y, Dannenberg AJ, Pasco DS. Development and use of a gene promoter-based screen to identify novel inhibitors of Cyclooxygenase-2 transcription. J Biomol Screen. 2001;6(2):101–110. doi: 10.1177/108705710100600206. [DOI] [PubMed] [Google Scholar]
- Song L, et al. p85{alpha} Acts as a novel signal transducer for mediation of cellular apoptotic response to UV radiation. Mol Cell Biol. 2007;27(7):2713–2731. doi: 10.1128/MCB.00657-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vane JR, Bakhle YS, Botting RM. Cyclooxygenses1 and 2. Ann Rev Pharmacol Toxicol. 1998;38(1):97–120. doi: 10.1146/annurev.pharmtox.38.1.97. [DOI] [PubMed] [Google Scholar]
- Vihma H, Pruunsild P, Timmusk T. Alternative splicing and expression of human and mouse NFAT genes. Genomics. 2008;92(5):279–291. doi: 10.1016/j.ygeno.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White LD, et al. New and evolving concepts in the neurotoxicology of lead. Toxicol Appl Pharmacol. 2007;225(1):1–27. doi: 10.1016/j.taap.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Yasojima K, Schwab C, McGeer EG, McGeer PL. Distribution of cyclooxygenase-1 and cyclooxygenase-2 mRNAs and proteins in human brain and peripheral organs. Brain Res. 1999;830(2):226–236. doi: 10.1016/s0006-8993(99)01389-x. [DOI] [PubMed] [Google Scholar]
- Yan Y, et al. NFAT3 is specifically required for TNF-α-induced cyclooxygenase-2(COX-2) expression and transformation of Cl41 cells. J Cell Sci. 2006;119(14):2985–2994. doi: 10.1242/jcs.03014. [DOI] [PubMed] [Google Scholar]
- Zhang D, et al. JNK1, but not JNK2, is required for COX-2 induction by nickelcompounds. Carcinogenesis. 2006;28(4):883–891. doi: 10.1093/carcin/bgl186. [DOI] [PubMed] [Google Scholar]
- Zuo Z, Ouyang W, Li J, Costa M, Huang C. Cyclooxygenase-2 (COX-2) mediates arsenite inhibition of UVB-induced cellular apoptosis in mouse epidermal Cl41 cells. Curr. Cancer Drug Targets. 2012;12(6):607–616. doi: 10.2174/156800912801784802. [DOI] [PMC free article] [PubMed] [Google Scholar]