

Abstract
Background
The ability of the spliceosomal small nuclear RNA U1 (U1snRNA) to rescue pre-mRNA splicing impaired by mutations makes it an attractive therapeutic molecule. Coagulation factor deficiencies due to splicing mutations are relatively frequent and could therefore benefit from this strategy. However, the effects of U1snRNAs in vivo remain unknown.
Objectives
To assess the rescue of the F7 c.859+5G>A splicing mutation (FVII+5A), causing severe human factor VII (hFVII) deficiency, by the modified U1snRNA+5a (U1+5a) in a murine model.
Methods
Mice expressing the human F7 c.859+5G>A mutant were generated following liver-directed expression by plasmid or recombinant adeno-associated viral (AAV) vector administration. The rescue of the splice-site defective pre-mRNA by U1+5a was monitored in liver and plasma through hFVII-specific assays.
Results
Injection of plasmids encoding the U1+5a rescued plasma hFVII levels, which increased from undetectable to ∼8.5% of those obtained with the wild-type hFVII plasmid control. To assess long-term effects, mice were injected with low and high doses of two AAV vectors encoding the FVII+5A splice site mutant as template to be corrected by U1+5a. This strategy resulted in hFVII plasma levels of 3.9 ± 0.8 or 23.3 ± 5.1 ng mL−1 in a dose-dependent manner, corresponding in patients to circulating FVII levels of ∼1–4.5% of normal. Moreover, in both experimental models, we also detected correctly spliced hFVII transcripts and hFVII-positive cells in liver cells.
Conclusions
Here we provide the first in vivo proof-of-principle of the rescue of the expression of a splicing-defective F7 mutant by U1snRNAs, thus highlighting their therapeutic potential in coagulation disorders.
Keywords: factor VII deficiency, genetic diseases, mouse, mutation, RNA splicing, U1 snRNA
Introduction
Protein replacement therapy has significantly improved the quality of life of patients with inherited coagulation factor deficiencies 1. However, there are still significant practical, technical and economical limitations that support research into alternative therapeutic strategies.
The U1 small nuclear RNA (U1snRNA), the ribonucleic acid component of the U1 ribonucleoprotein that mediates recognition of the donor splice site (5′ss) in the earliest splicing step 2, is emerging as an attractive therapeutic molecule for human diseases caused by splicing mutations, often associated (> 15%) with severe forms 3,4. It has been demonstrated that U1snRNAs with increased complementarity to mutated 5′ss are able to redirect the spliceosome assembly and rescue splicing in various cellular models of human genetic disease 5–10, including coagulation factor deficiencies 11–14. Recently, we have also demonstrated that modified U1snRNAs, designed to improve the definition of the 5′ss, can also restore splicing impaired by mutations at the acceptor splice site 14. However, the U1snRNAs strategy has not been explored in vivo, thus preventing the assessment of its potential therapeutic effect.
Mutations at splice sites are relatively frequent in patients with coagulation factor VII (FVII) deficiency (∼17%, and 8% at 5′ss) 13,15, as well as among patients with X-linked bleeding diseases such as FIX deficiency (∼14%, and 7% at 5′ss) (http://www.hgmd.cf.ac.uk/ac/index.php and http://www.factorix.org). Notably, the prevalence of these mutations further increases among patients with severe diseases (< 1% of normal factor levels). Here we sought to investigate the in vivo U1snRNA-mediated rescue of gene expression impaired by 5′ss splicing mutations in a model of human FVII deficiency caused by the F7 c.859+5G>A (NM_000131.4) mutation 16,17. We have previously shown this mutation to be efficiently rescued in vitro by the modified U1snRNA+5a (U1+5a) molecule 11,12. It is worth noting that intervention at the pre-mRNA level permits the restoration of gene expression while maintaining gene regulation in physiological tissues, and overcomes limitations related to delivery of large genes, such as the F8 gene, via vector-mediated approaches. In these inherited bleeding diseases the approach using U1snRNA can be particularly beneficial because even a modest increase of functional protein levels would result in improvement of the disease phenotype 18.
Materials and methods
Generation of vectors
Plasmids expressing human FVII (hFVII) wild-type (pFVIIwt) or harbouring the c.859+5G>A splicing mutation (pFVII+5A) were generated by cloning the wild-type or the mutated hFVII splicing-competent cassette 12 into the pAAV-hAAT backbone 19 using the ClaI and XhoI sites (Fig. 1). The hAAT promoter contains the human α1-antitrypsin (hAAT) promoter with four copies of the human ApoE enhancer and a synthetic intron 20,21. Plasmid pU1wt harbouring the wild-type U1snRNA cassette (U1wt) and pU1+5a were created by cloning the corresponding cassette 11 and its own promoter into a promoterless pAAV backbone through a BamHI cloning site. The U1+5a differs from the U1wt at positions +4 and +5 and contains a C and a U, respectively.
Figure 1.
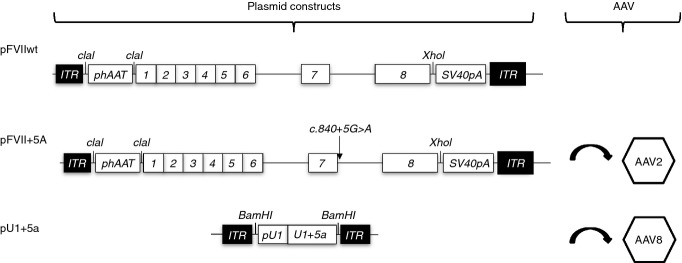
Experimental design and vectors. Schematic representation of the expression cassettes for the splicing-competent hFVII minigenes and for the modified U1+5a, which were used to create the pFVIIwt, pFVII+5A and pU1+5a plasmids, respectively. The splicing mutation under investigation (c.859+5G>A) and restriction sites used for cloning are indicated. Numbers indicate F7 exons. ITR, inverted terminal repeat; phAAT, chimeric promoter consisting of the human α1 antitrypsin promoter and human apolipoprotein E enhancer used for liver restricted expression of the transgene; SV40pA, polyadenylation site of SV40; pU1, endogenous promoter of the U1snRNA gene. The plasmids pFVII+5A and pU1+5a have been subsequently packaged into Adeno-Associated Virus (AAV) serotype 2 and 8, respectively.
Recombinant adeno-associated viral (AAV) vectors of serotype 2 (AAV2-FVII+5A) and serotype 8 (AAV8-U1+5a) were produced as previously described 22.
Animal procedures
All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at The Children's Hospital of Philadelphia. Eight-week-old male C57BL/6 mice were used for in vivo delivery of plasmids by hydrodynamic injection of DNA (2.5 mL phosphate buffer saline-PBS) or AAV vectors (0.2 mL in PBS/5% sorbitol) by tail vein injection. While plasmid DNA vectors were co-injected, the AAV vectors have been administrated in a sequential fashion, with the AAV8-U1+5A injected 2 weeks after the AAV2-FVII+5a. Blood samples were collected from the retro-orbital plexus into 3.8% sodium citrate.
Measurement of hFVII antigen, hFVII mRNA splicing profiles and liver enzymes
Human FVII antigen levels in mouse plasma were evaluated by a hFVII enzyme-linked immunosorbent assay (ELISA; Diagnostica Stago Inc., Asnieres-sur-Seine, France). To validate and optimize the assay, a standard curve was created by adding known amounts of human pooled normal plasma to mouse plasma. The sensitivity threshold of the assay in our hands was 0.25 ng mL−1 of hFVII. For the assay, mouse plasma samples were diluted 1 : 10 and 1 : 20.
To detect hFVII transcripts, total RNA was isolated from random sections of mouse liver using TRIZOL reagent (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's protocol. Two micrograms of total mouse liver RNA were retro-transcribed using SuperScript® III Reverse Transcriptase (Invitrogen) with random primers and subsequently PCR amplified 23 with hFVII-specific primers (h6F [5′-TAGAAAAAAGAAATGCCAGCAAACC-3′], h7F [5′-GTCCTGTTGTTGGTGAATGGAGCTCA-3′], h8R [5′-GCTGACCAATGAGAAGCGCACGAA-3′]). The PCRs were carried out in a total volume of 25 μL under the following conditions: preheating, 95 °C for 2 min followed by 40 cycles of 95 °C for 30 s, 60 °C for 20 s and 72 °C for 30 s.
Amplicons were resolved on a denaturing capillary electrophoresis system (Experion; BioRad Laboratories, Hercules, CA, USA).
Measurement of alanine aminotransferase (ALT) activity levels in plasma has been conducted through the commercially available kit from Teco Diagnostics (Anaheim, CA, USA).
Each sample has been evaluated in duplicate in each assay.
Immunohistochemical analysis
For immunofluorescence staining, livers were fixed overnight in 4% paraformaldeyhde and included in OCT embedding compound (Sakura Finetek, Leiden, the Netherlands) on a dry ice/methyl butane bath. Tissue sections (8 μm) were subjected to direct immunofluorescence staining. Briefly, sections were incubated with blocking buffer (10% fetal bovine serum [FBS]/0.1% Triton X-100/100 mm Glycine in PBS) at room temperature for 2 h. Sections were rinsed three times with PBS solution for 5 min each and then incubated with an unconjugated AffiniPure Fab Fragment Anti-Mouse IgG (H + L) solution (0.1 mg mL−1 antibody in 1%BSA in PBS) (Jackson Immunoresearch Laboratories, Baltimore, PA, USA) for 1 h at room temperature. Sections were rinsed three times with PBS solution (10 min each) and incubated overnight at 4 °C with a polyclonal sheep anti-human FVII FITC conjugated antibody (2 μg mL−1 in 1%FBS/PBS) (Affinity Biologicals, Ancaster, ON, Canada). Sections were mounted with mounting media containing DAPI to counter-stain nuclei and viewed with a NIKON Eclipse E800 microscope (Nikon Corporation Instrument Company, Shinagawa-ku, Tokyo, Japan) using a Plan APO 10 ×/0.75 objective and epifluorescent light (FITC HYQ filter, Nikon Corporation Instrument Company). Images were captured with a CoolSnap Pro camera and analyzed with Image Pro Plus software (Media Cybernetics, Bethesda, MD, USA).
Statistical analysis
Statistical differences among levels were evaluated by a Student's t-test with a P < 0.05 considered significant.
Results
We used two vector systems (viral and non-viral) for the assessment of the effect of U1+5a in rescuing the defective splice site F7 mutant harbouring the c.859+5G>A mutation (FVII+5A) as template (Fig. 1). Our model consists of the hepatocyte-restricted expression of the template pre-mRNA by using liver-specific promoter/enhancer elements. The availability of species-specific immunologic and nucleic acid amplification assays allows the evaluation of the hFVII expression without confounding effects with the endogenous murine FVII of the C57BL/6 mice (Fig. 2A, untreated cohort of mice exhibited undetectable levels in our hFVII ELISA). A series of previous attempts to use a liver-specific promoter for U1 expression were unsuccessful (D. Balestra and M. Pinotti, unpublished observations). Thus, we used the U1snRNA endogenous promoter in the experiments reported here.
Figure 2.

U1+5a-mediated rescue of hFVII expression by hydrodynamic injection. (A) hFVII levels in plasma (mean and 95% confidence intervals) at 48 h upon hydrodynamic injection in mice (four mice per group) of two doses of pFVII plasmids (1 or 2 μg g−1 mouse body weight; 1× or 2×, respectively) without or with a corresponding 1.5× molar excess of pU1+5a. The negative control (no plasmid injection) is represented by mice injected with PBS only. Inset: hFVII levels in plasma (mean and 95% confidence intervals) at 48 h upon hydrodynamic injection in mice (five mice per group) of 2 μg g−1 mouse body weight of pFVII plasmids with a corresponding 1.5× molar excess of pU1wt. (B) Analysis of hFVII mRNA forms in mouse livers. The aberrant (a) and normal (n) transcripts are depicted on the right. Arrows indicate primers used for RT-PCR. Amplicons were resolved on a denaturing capillary electrophoresis system. Histograms report the quantification of transcripts, expressed as percentage (mean and 95% confidence intervals) of total hFVII forms, in liver mRNA from mice injected with pFVIIwt or pFVII+5A, without or with a 1.5× molar excess of pU1+5a. M, size marker; bp, base pairs. (C) Representative examples of liver sections stained with a species-specific anti-hFVII antibody (green). Mice were injected with 2 μg g−1 mouse body weight of plasmids pFVIIwt or pFVII+5A, without or with a 1.5× molar excess of pU1+5a. Images are taken at 100× magnification. Scale bar, 50 μm.
Rescue of human FVII expression by transient expression of U1+5a
Hydrodynamic injection of two different doses of plasmid pFVIIwt (1 or 2 μg g−1 of mouse body weight), used as a positive control, resulted in a dose-dependent increase of hFVII antigen levels in mouse plasma, reaching ∼2 μg mL−1 at 48 h post-injection. These findings were further supported by data on hFVII mRNA and protein in the liver. Specifically, mice injected with pFVIIwt displayed only the normally spliced transcripts (first column, Fig. 2B) and immunohistochemical analysis of their livers revealed frequent hFVII-positive cells (Fig. 2C).
Injection of pFVII+5A alone at both doses tested (1 or 2 μg g−1 of mouse body weight) showed no detectable hFVII antigen levels (Fig. 2A), as expected. Analysis of hFVII mRNA in the liver showed that the recipient mice generated almost exclusively the aberrant mRNA form, having the intronic 37 bp repeat inclusion (Fig. 2B), with minimally detectable levels of correct transcripts (mean ± standard deviation, 2 ± 1.5% of total hFVII transcripts; 95% CI, −0.7 to 6.5). Furthermore, we could not detect any hFVII expressing liver cells by immunofluorescence staining (Fig. 2C). These findings suggest that the splicing change in the hFVII mutant template prevents the hFVII biosynthesis and secretion in mice, thus mimicking findings in humans homozygous for the F7 c.859+5G>A mutation.
To evaluate the U1-mediated rescue of hFVII expression impaired by the c.859+5G>A mutation, we injected the pFVII+5A plasmid at either 1 or 2 μg g−1 of mouse body weight with a corresponding molar excess (1.5 fold) of pU1+5a (Fig. 1). Forty-eight hours post-injection, a significant increase (P < 0.05) of circulating hFVII levels was detected (50.6 ± 16.0 ng mL−1, 95% CI 9–91, or 178 ± 126 ng mL−1, 95% CI −21 to 379, for each plasmid amount, respectively, Fig. 2A). These values correspond to 5% and 8.5% of the expression levels of pFVIIwt at low and high dose cohorts, respectively (Fig. 2A). Moreover, co-expression of the U1+5a resulted in rescue of hFVII splicing (26 ± 10% of total transcripts, 95% CI −0.04 to 52) in the liver (Fig. 2B, last column). Corroborating these findings, immunohistochemical analysis of livers from mice co-injected with pFVII+5A and pU1+5a showed an appreciable number of hFVII-positive staining cells that were absent in livers of mice injected with pFVII+5A alone (Fig. 2C). Injection of pU1+5a alone in C57Bl/6 showed no effect on circulating hFVII, as expected (Fig. 2A).
To confirm the specificity of the U1+5a towards the defective splice site, we injected mice with the pFVIIwt (2 μg g−1 of mouse body weight) and a molar excess of pU1+5a (Fig. 2A). This resulted in circulating hFVII levels of 1780 ± 391 ng mL−1 (95% CI 767–2713), comparable (P = 0.31) to those measured in mice injected with pFVIIwt alone (2146 ± 466 ng mL−1, 95% CI 988–3304). Furthermore, we assessed the ability of the U1wt to rescue the hFVII+5A mutant (Fig. 2A, inset). Injection of the high dose of pFVII+5A (2 μg g−1) with a molar excess of pU1wt produced minimal hFVII levels in mouse plasma (13 ± 8 ng mL−1, 95% CI 10–22), which was significantly lower (P < 0.05) compared with what we observed using pU1+5a (178 ± 126 ng mL−1). Furthermore, injection of pU1wt together with pFVIIwt resulted in similar hFVII levels compared with mice receiving only pFVIIwt (2584 ± 728, CI 1680–3488, vs. 2146 ± 466 ng mL−1, respectively, P = 0.4). Thus, the U1wt is not effective in rescuing the splice defect in vivo, in accordance with data obtained in cellular models 11,12.
Collectively, these data suggest that the rescue of the defective splice site mutation in the F7 gene by transient expression of U1+5a is feasible and specific towards the mutant template, thus providing the first evidence of the efficacy of this strategy in vivo.
Sustained expression of FVII by AAV-U15a
To investigate whether U1+5a-mediated correction of the hFVII splicing mutation could be sustained over time we used a strategy based on AAV vectors. The FVII+5A and the U1+5a expression cassettes (Fig. 1) were packaged into AAV serotype 2 (AAV2-FVII+5A) and serotype 8 (AAV8-U1 + 5a), respectively. These serotypes were chosen because of their tropism for the liver following peripheral intravascular delivery. In a preliminary study, AAV2-FVII+5A was delivered at doses of 1.2 × 1012 vector genomes (vg) per mouse and during the 5-week period, no expression of circulating hFVII was detected (Fig. 3A). In the rescue mode, at day 0, mice (n = 4) received AAV2-FVII+5A at doses of 1.2 × 1012 or 6.0 × 1012 vg per mouse and, 2 weeks later, the AAV8-U1+5a was administered at a fixed dose of 1.2 × 1011 vg per mouse (Fig. 3A). The initial injection of AAV2-FVII+5A resulted in no increased levels of circulating FVII for the first 2 weeks, as expected (Fig. 3A). However, upon injection of AAV8-U1+5a, there was a progressive increase in plasma hFVII antigen levels in a dose-dependent manner (P < 0.02), reaching 5.5 ± 0.7 ng mL−1 (95% CI 3.7–7.2) and 8.7 ± 1.9 ng mL−1 (95% CI 6.9–10.4) during the 6-week follow-up (Fig. 3A). Conversely, using a fixed dose of the AAV2-FVII+5A (1.2 × 1012 vg per mouse, template) we increased the AAV8-U1+5a dose by 5-fold (6.0 × 1011 vg per mouse). The U1+5a-mediated correction effect was directly related to the AAV8-U1+5a dose, as measured by circulating hFVII levels of 3.9 ± 0.8 ng mL−1 (95% CI 1.8–5.8) with 1.2 × 1011 vg per mouse or 23.3 ± 5.1 ng mL−1 (95% CI 10.6–35.9) with 6 × 1011 vg per mouse at 2 weeks post-injection, but with the highest AAV8-U1+5a dose the mice did not survive beyond this time-point (see below). These findings were further supported by the detection of the correctly spliced hFVII transcripts (4 ± 0.5% or 16 ± 3% of the total transcripts; Fig. 3B) and of hFVII-positive staining cells in the liver (Fig. 3C). Altogether these findings showed that there is a dose-related effect on the rescue of the hFVII splice site mutant by increasing the expression of the therapeutic gene (U1+5a) or of the template (FVII+5A). In humans, the range of FVII levels in plasma is 350–500 ng mL−1. Therefore, if the sustained hFVII levels obtained in mice (8.7 ± 1.9 ng mL−1) are translated into F7 c.859+5G>A homozygous patients, it would achieve the ∼2% of normal, which could ameliorate the severe bleeding phenotype 18.
Figure 3.

U1+5a-mediated rescue of hFVII expression by AAV delivery. (A) Plasma hFVII antigen levels (mean and 95% confidence intervals) in mice (four mice per group) injected with the AAV2-FVII+5A and, 2 weeks later (arrow), with the AAV8-U1+5a viral vectors at the doses reported in the table. *Denotes the last time-point for this group of mice (n = 7). Inset: scheme of the sequential injection protocol. (B) Analysis of hFVII mRNA forms in mouse liver. The aberrant (a) and normal (n) transcripts are depicted on the right. Arrows indicate primers used for RT-PCR. Electrophoretic separation of amplicons and analysis was as in Fig. 1. Mice were injected with 1.2 × 1012 vector genomes (vg) per mouse of AAV2-FVII+5A and 6 × 1011 or 1.2 × 1011 vg per mouse of AAV8-U1+5a. M, size marker; bp, base pairs. (C) Representative examples of liver sections from mice treated as indicated. Cells were stained with a species-specific anti-hFVII antibody (green) and cell nuclei (blue) with DAPI. Stained sections were captured as indicated in Fig. 1. Scale bar, 50 μm.
Over-expression of U1+5a is associated with hepatocellular toxicity
In the experimental design we choose AAV8 for the expression of U1+5a because of the superior liver transduction efficiency compared with AAV2 24; thus a more robust correction of the defective splice site could be achieved. We carried out a pilot dose-response experiment using AAV8-U1+5a vector at doses ranging from 1.2 × 1011 to 2.4 × 1012 vg per mouse (n = 2–4 per dose) while keeping the AAV2-FVII+5a dose of 1.2 × 1012 vg per mouse. A Kaplan–Meier survival curve demonstrated premature mortality in mice injected with AAV8-U1+5a doses of 6.0 × 1011 vg per mouse compared with the lower dose cohort, but at the highest doses all mice died by day 20 (Fig. 4A). There was a direct correlation between dose and liver toxicity as determined by increased levels of ALT over time in mice injected with 6.0 × 1011 vg per mouse or higher, whereas in the low-dose cohort the ALT levels remained similar to the baseline values (Fig. 4B). Notably, the mice injected with 1.2 or 6.0 × 1012 vg per mouse of AAV2-FVII+5A and 1.2 × 1011 AAV8-U1+5a, resulting in sustained levels of hFVII (Fig. 3A), exhibited no change in the levels of ALT. Unfortunately, in mice injected with the two highest AAV8-U1+5a doses, due to liver failure, no detectable hFVII levels were observed (data not shown). Thus, these data support our choice of the two low doses of U1+5a for the rescue experiments (Fig. 3A). These studies showed that a dose of AAV8-U1+5a of 1.2 × 1011 vg per mouse was the safest because it was not associated with increased mortality or liver toxicity, thus allowing long-term correction of the defective splice site and sustained expression of hFVII (Fig. 3A).
Figure 4.
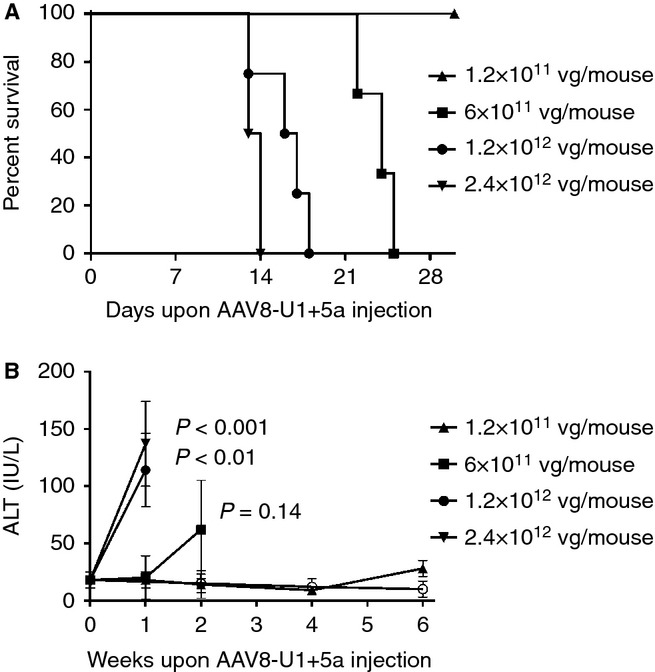
Kaplan–Meier survival curves (A) and ALT levels (B) in relationship with the AAV8-U1+5a dose injected. Mice (n = 2–4 per group) were injected with AAV vector by the intravascular route at day 0. The vector doses per mouse are indicated. The open circle in panel B indicates mice injected with 6 × 1012 vg per mouse of AAV2-FVII+5A and 1.2 × 1011 vg per mouse of AAV8-U1+5a. The P values refer to statistical differences from a given ALT value and the baseline values. ALT, alanine aminotransferase enzyme.
Discussion
Gene replacement and the correction of disease-causing mutations are the most attractive strategies for the treatment of genetic diseases. Currently, clinical studies are focused on gene replacement of the mutated gene. RNA-targeted strategies have been also developed for human diseases. To date, modified U1snRNAs have been successfully exploited in vivo to mask splicing regulatory elements and induce skipping of defective exons 25. However, this strategy is not suitable for the correction of the most common inherited bleeding disorders. U1snRNA-based therapy, by redirecting the spliceosome assembly to the mutated splicing junction and restoring proper exon inclusion, represents a correction strategy able to produce full-length functional proteins. There are several advantages of such an approach for bleeding disorders: (i) there is a relatively high frequency of splice site mutations 3,4,26,27, (ii) correction is independent of the size of the target gene, while (iii) maintaining the endogenous regulatory elements for gene expression. Moreover, the corrected protein product can be measured in plasma over time. However, the efficacy of the U1snRNA approach in vivo for inherited bleeding disorders as well as for other human genetic diseases has not been demonstrated. Based on our encouraging results from the rescue of FVII expression impaired by a donor splice site mutation in cellular models 11,12, we evaluated the U1snRNA efficacy in mice in a quantitative manner.
Homozygosity for the c.859+5G>A mutation in the F7 gene 16,17 is associated with clinically severe FVII deficiency. It represents a prototypical example of a point mutation affecting the 5′ss and inducing aberrant splicing, and we have previously demonstrated its effective correction by the modified U1snRNA U1+5a in cellular models 11,12. Specifically, the in vitro studies indicated that the presence of the modified 5′ tail of the U1+5a with increased complementarity to the mutated F7 5′ss is fundamental for the correction effect 11. The absence of mouse models of FVII deficiency for splicing mutations, and the perinatal lethal phenotype of F7 knock-out mice 28, prompted us to generate a novel in vivo model by hepatocyte-restricted expression of the mutated hFVII splicing-competent cassette harboring the F7 c.859+5G>A mutation (FVII+5A, template). The rationale behind this strategy is the possibility of evaluating the hFVII transcripts and the hFVII protein levels in mice by exploiting hFVII-specific assays. For the delivery of the F7 mutant expression cassette we chose two vector systems, non-viral vector (plasmid DNA) and transduction by AAV vectors, to evaluate transient and prolonged hFVII expression, respectively. Although these vectors do not integrate into the hepatocyte genome, they guarantee the liver-specific transcription of the hFVII pre-RNA template (FVII+5A), the key target of the proposed U1snRNA-mediated correction strategy. Expression of the mutated cassette by both delivery systems did not result in detectable circulating hFVII levels in mouse plasma, thus mirroring the coagulation phenotype observed in the F7 c.859+5G>A homozygotes 16,17. Moreover, investigation of hFVII mRNA in mouse liver revealed aberrantly spliced forms due to the usage of the cryptic 5′ss in the intron, and trace levels of the correct form, thus recapitulating the pathological mechanisms dissected by studies in cellular models 11. Altogether these data provided us with a mouse model for the in vivo evaluation of U1+5a efficacy at different levels, from the mRNA to the protein in liver and into the circulation.
In the transient model using plasmid DNA, the co-expression of the U1+5a was associated with the detection of correctly-spliced transcripts in mouse liver, thus indicating the ability of U1+5a to redirect usage of the mutated 5′ss in vivo by the spliceosome machinery. This observation was paralleled by the synthesis of hFVII, as indicated by hFVII-positive hepatocytes, and most importantly, by the increase of circulating hFVII levels, the final goal of the U1+5a-mediated correction strategy. In addition, in this model we demonstrated that U1+5a is specific for the splice site mutant, because no increase in hFVII levels was obtained by replacing the template with a hFVII-wt expressing plasmid. Moreover, over-expression of the U1wt had very modest effects on the FVII+5A mutant, further indicating the necessity of the engineered U1snRNA 5′ tail to mediate a significant correction effect in vivo.
In the context of AAV vectors, the rescue was prolonged over time, as indicated by the low but appreciable levels of plasma hFVII up to 6 weeks post-injection of the lowest AAV8-U1+5a dose. Clearly, prior to utilizing U1snRNA in a clinical application, further improvements will be necessary to enhance the correction efficacy. Towards that goal, our data show that increasing the template for the U1+5a (higher AAV2-FVII+5A dose) led to increased hFVII plasma levels. In our model, the use of AAV2 for the expression of the template is typically restricted to only 20–30% of hepatocytes. Thus, it is anticipated that in FVII-deficient patients, where all liver cells express the mutated pre-mRNA, targeting by U1-5a will likely result in a more robust increase of hFVII levels. It is worth noting that even this seemingly minimal increase in FVII levels could be associated with an improved phenotype, as seen in hemophilia patients on prophylaxis with protein replacement (aimed at levels > 1% of normal) as well as from emerging data from an early clinical trial on AAV liver gene transfer in subjects with severe hemophilia B 29.
A rational strategy to enhance the extent of rescue of the splicing-defective hFVII allele is to increase the expression of U1+5a. However, our data show that there is a dose-dependent effect that limits the safety of this proposal. As the dose of the AAV8 encoding U1+5a increased by 5-fold the dose associated with long-term expression, it resulted in a significantly increased hFVII expression at 2 weeks but did not reach plateau levels due to liver toxicity. AAV8-U1+5a administered at even higher doses was associated with premature mortality. Early studies on RNA interference technology also encountered liver toxicity upon high doses of AAV8 expressing short hairpin RNA (shRNA) 30. Remarkably, these findings prompted the development of shRNA/microRNA technologies that are well tolerated.
Clearly, the hepatocellular toxicity that we observed from U1+5a over-expression will need to be sufficiently and extensively addressed prior to a clinical application using our approach. Current ongoing studies will attempt to define the underlying mechanism(s) by which U1+5a induced liver toxicity at high AAV8 doses. It must be pointed out that the U1+5a targets the donor splice site sequence of F7 intron 7. The mammalian donor splice site sequences, apart from the invariant dinucleotide +1G and +2T, can significantly differ from the consensus sequence at the other positions, particularly in alternatively spliced exons. Thus, it is possible that the U1+5a, differing from the normal U1snRNA at the two non-conserved positions +4 and +5, interacts directly with other 5′ss and elicits negative effects on the fine regulation of critical alternatively spliced exons. Similar results have been reported in spinal muscular atrophy 31, where a U1snRNAs complementary to the 5′ss of SMN2 exon7 showed some degree of cellular toxicity.
Towards the goal of overcoming the off-target effects and potential toxicity of U1snRNA, we have recently developed Exon Specific U1snRNA (ExSpeU1), targeting non-conserved intronic sequences downstream of the 5′ss and still able to restore exon definition impaired by different mutations in cellular models of hemophilia B 14. This approach was not feasible for the F7 c.859+5G>A mutation due to the presence in the F7 IVS7 of six highly conserved 37 bp repeated sequences 32, which prevents the identification of unique targets for ExSpeU1. Further optimization on the AAV8-U1+5a vector such as modification in the promoter and vector serotype combined with the use of ExSpeU1 in a mouse model expressing the target pre-mRNA in the majority of (all) hepatocytes will allow the full evaluation of the efficacy as well as the safety of the U1 snRNA-based correction strategy.
In conclusion, we have provided the first proof-of-concept that exploiting the U1snRNA pathway results in splice site correction and increased circulating levels of a therapeutic protein in a mouse model. Therefore, these data support further studies exploiting U1snRNAs as novel therapeutic strategies for coagulation factor and other genetic disorders.
Addendum
D. Balestra and A. Faella performed the experiments in the animal models and evaluated the expression of hFVII in mice; N. Cavallari created the plasmid constructs; P. Margaritis designed experiments in animals and wrote the manuscript; F. Pagani analyzed data and revised the manuscript; M. Pinotti, V. R. Arruda and F. Bernardi conceived the study and designed research, analyzed and interpreted data and wrote the manuscript.
Acknowledgments
The financial support of Telethon-Italy (GGP09183) (D. Balestra, N. Cavallari, F. Pagani and M. Pinotti) and AIFA (AIFA 2008 – Bando per le malattie rare – Progetto RF-null-2008-1235892) (F. Bernardi and M. Pinotti) is gratefully acknowledged. We thank the Research Vector Core of the Center for Cell and Molecular Therapies at The Children's Hospital of Philadelphia for the production of AAV vectors.
Disclosure of Conflict of Interest
M. Pinotti, F. Pagani and F. Bernardi are founders of the start-up company Raresplice. The remaining authors declare no competing financial interests.
References
- 1.Batorova A, High KA, Gringeri A. Special lectures in haemophilia management. Haemophilia. 2010;16(Suppl. 5):22–8. doi: 10.1111/j.1365-2516.2010.02289.x. [DOI] [PubMed] [Google Scholar]
- 2.Horowitz DS, Krainer AR. Mechanisms for selecting 5′ splice sites in mammalian pre-mRNA splicing. Trends Genet. 1994;10:100–6. doi: 10.1016/0168-9525(94)90233-x. [DOI] [PubMed] [Google Scholar]
- 3.Faustino NA, Cooper TA. Pre-mRNA splicing and human disease. Gene Dev. 2003;17:419–37. doi: 10.1101/gad.1048803. [DOI] [PubMed] [Google Scholar]
- 4.Buratti E, Chivers M, Kralovicova J, Romano M, Baralle M, Krainer AR, Vorechovsky I. Aberrant 5′ splice sites in human disease genes: mutation pattern, nucleotide structure and comparison of computational tools that predict their utilization. Nucleic Acids Res. 2007;35:4250–63. doi: 10.1093/nar/gkm402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baralle M, Baralle D, De Conti L, Mattocks C, Whittaker J, Knezevich A, Ffrench-Constant C, Baralle FE. Identification of a mutation that perturbs NF1 agene splicing using genomic DNA samples and a minigene assay. J Med Genet. 2003;40:220–2. doi: 10.1136/jmg.40.3.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Susani L, Pangrazio A, Sobacchi C, Taranta A, Mortier G, Savarirayan R, Villa A, Orchard P, Vezzoni P, Albertini A, Frattini A, Pagani F. TCIRG1-dependent recessive osteopetrosis: mutation analysis, functional identification of the splicing defects, and in vitro rescue by U1 snRNA. Hum Mutat. 2004;24:225–35. doi: 10.1002/humu.20076. [DOI] [PubMed] [Google Scholar]
- 7.Tanner G, Glaus E, Barthelmes D, Ader M, Fleischhauer J, Pagani F, Berger W, Neidhardt J. Therapeutic strategy to rescue mutation-induced exon skipping in rhodopsin by adaptation of U1 snRNA. Hum Mutat. 2009;30:255–63. doi: 10.1002/humu.20861. [DOI] [PubMed] [Google Scholar]
- 8.Hartmann L, Neveling K, Borkens S, Schneider H, Freund M, Grassman E, Theiss S, Wawer A, Burdach S, Auerbach AD, Schindler D, Hanenberg H, Schaal H. Correct mRNA processing at a mutant TT splice donor in FANCC ameliorates the clinical phenotype in patients and is enhanced by delivery of suppressor U1 snRNAs. Am J Hum Genet. 2010;87:480–93. doi: 10.1016/j.ajhg.2010.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmid F, Glaus E, Barthelmes D, Fliegauf M, Gaspar H, Nurnberg G, Nurnberg P, Omran H, Berger W, Neidhardt J. U1 snRNA-mediated gene therapeutic correction of splice defects caused by an exceptionally mild BBS mutation. Hum Mutat. 2011;32:815–24. doi: 10.1002/humu.21509. [DOI] [PubMed] [Google Scholar]
- 10.Glaus E, Schmid F, Da Costa R, Berger W, Neidhardt J. Gene therapeutic approach using mutation-adapted U1 snRNA to correct a RPGR splice defect in patient-derived cells. Mol Ther. 2011;19:936–41. doi: 10.1038/mt.2011.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pinotti M, Rizzotto L, Balestra D, Lewandowska MA, Cavallari N, Marchetti G, Bernardi F, Pagani F. U1-snRNA-mediated rescue of mRNA processing in severe factor VII deficiency. Blood. 2008;111:2681–4. doi: 10.1182/blood-2007-10-117440. [DOI] [PubMed] [Google Scholar]
- 12.Pinotti M, Balestra D, Rizzotto L, Maestri I, Pagani F, Bernardi F. Rescue of coagulation factor VII function by the U1 + 5A snRNA. Blood. 2009;113:6461–4. doi: 10.1182/blood-2009-03-207613. [DOI] [PubMed] [Google Scholar]
- 13.Pinotti M, Bernardi F, Dal Mas A, Pagani F. RNA-based therapeutic approaches for coagulation factor deficiencies. J Thromb Haemost. 2011;9:2143–52. doi: 10.1111/j.1538-7836.2011.04481.x. [DOI] [PubMed] [Google Scholar]
- 14.Fernandez Alanis E, Pinotti M, Dal Mas A, Balestra D, Cavallari N, Rogalska ME, Bernardi F, Pagani F. An exon-specific U1 small nuclear RNA (snRNA) strategy to correct splicing defects. Hum Mol Genet. 2012;21:2389–98. doi: 10.1093/hmg/dds045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernardi F, Dolce A, Pinotti M, Shapiro AD, Santagostino E, Peyvandi F, Batorova A, Lapecorella M, Schved JF, Ingerslev J, Mariani G. Major differences in bleeding symptoms between factor VII deficiency and hemophilia B. J Thromb Haemost. 2009;7:774–9. doi: 10.1111/j.1538-7836.2009.03329.x. [DOI] [PubMed] [Google Scholar]
- 16.Bernardi F, Patracchini P, Gemmati D, Ferrati M, Arcieri P, Papacchini M, Redaelli R, Baudo F, Mariani G, Marchetti G. Molecular analysis of factor VII deficiency in Italy: a frequent mutation (FVII Lazio) in a repeated intronic region. Hum Genet. 1993;92:446–50. doi: 10.1007/BF00216448. [DOI] [PubMed] [Google Scholar]
- 17.Pinotti M, Toso R, Redaelli R, Berrettini M, Marchetti G, Bernardi F. Molecular mechanisms of FVII deficiency: expression of mutations clustered in the IVS7 donor splice site of factor VII gene. Blood. 1998;92:1646–51. [PubMed] [Google Scholar]
- 18.Pollak E, High KA. Genetic disorders of coagulation. In: Warrell D, Cox T, Firth J, Benz E, editors. Oxford Textbook of Medicine. 4. Vol. 3. Oxford: Oxford University Press; 2003. pp. 757–67. [Google Scholar]
- 19.Margaritis P, Roy E, Aljamali MN, Downey HD, Giger U, Zhou S, Merricks E, Dillow A, Ezban M, Nichols TC, High KA. Successful treatment of canine hemophilia by continuous expression of canine FVIIa. Blood. 2009;113:3682–9. doi: 10.1182/blood-2008-07-168377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okuyama T, Huber RM, Bowling W, Pearline R, Kennedy SC, Flye MW, Ponder KP. Liver-directed gene therapy: a retroviral vector with a complete LTR and the ApoE enhancer-alpha 1-antitrypsin promoter dramatically increases expression of human alpha 1-antitrypsin in vivo. Hum Gene Ther. 1996;7:637–45. doi: 10.1089/hum.1996.7.5-637. [DOI] [PubMed] [Google Scholar]
- 21.Herzog RW, Yang EY, Couto LB, Hagstrom JN, Elwell D, Fields PA, Burton M, Bellinger DA, Read MS, Brinkhous KM, Podsakoff GM, Nichols TC, Kurtzman GJ, High KA. Long-term correction of canine hemophilia B by gene transfer of blood coagulation factor IX mediated by adeno-associated viral vector. Nat Med. 1999;5:56–63. doi: 10.1038/4743. [DOI] [PubMed] [Google Scholar]
- 22.Matsushita T, Elliger S, Elliger C, Podsakoff G, Villarreal L, Kurtzman GJ, Iwaki Y, Colosi P. Adeno-associated virus vectors can be efficiently produced without helper virus. Gene Ther. 1998;5:938–45. doi: 10.1038/sj.gt.3300680. [DOI] [PubMed] [Google Scholar]
- 23.Bertolucci C, Cavallari N, Colognesi I, Aguzzi J, Chen Z, Caruso P, Foa A, Tosini G, Bernardi F, Pinotti M. Evidence for an overlapping role of CLOCK and NPAS2 transcription factors in liver circadian oscillators. Mol Cell Biol. 2008;28:3070–5. doi: 10.1128/MCB.01931-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao G, Alvira M, Wang L, Calcedo R, Johnston J, Wilson J. (2002) Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci USA. 2002;99:11854–9. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Denti MA, Rosa A, D'Antona G, Sthandier O, De Angelis FG, Nicoletti C, Allocca M, Pansarasa O, Parente V, Musaro A, Auricchio A, Bottinelli R, Bozzoni I. Body-wide gene therapy of Duchenne muscular dystrophy in the mdx mouse model. Proc Natl Acad Sci USA. 2006;103:3758–63. doi: 10.1073/pnas.0508917103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krawczak M, Reiss J, Cooper DN. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet. 1992;90:41–54. doi: 10.1007/BF00210743. [DOI] [PubMed] [Google Scholar]
- 27.Krawczak M, Thomas NS, Hundrieser B, Mort M, Wittig M, Hampe J, Cooper DN. Single base-pair substitutions in exon-intron junctions of human genes: nature, distribution, and consequences for mRNA splicing. Hum Mutat. 2007;28:150–8. doi: 10.1002/humu.20400. [DOI] [PubMed] [Google Scholar]
- 28.Rosen ED, Chan JC, Idusogie E, Clotman F, Vlasuk G, Luther T, Jalbert LR, Albrecht S, Zhong L, Lissens A, Schoonjans L, Moons L, Collen D, Castellino FJ, Carmeliet P. Mice lacking factor VII develop normally but suffer fatal perinatal bleeding. Nature. 1997;390:290–4. doi: 10.1038/36862. [DOI] [PubMed] [Google Scholar]
- 29.Nathwani AC, Tuddenham EG, Rangarajan S, Rosales C, McIntosh J, Linch DC, Chowdary P, Riddell A, Pie AJ, Harrington C, O'Beirne J, Smith K, Pasi J, Glader B, Rustagi P, Ng CY, Kay MA, Zhou J, Spence Y, Morton CL, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365:2357–65. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR, Marion P, Salazar F, Kay MA. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537–41. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 31.Nlend Nlend R, Meyer K, Schümperli D. Repair of pre-mRNA splicing: prospects for a therapy for spinal muscular atrophy. RNA Biol. 2010;7:430–40. doi: 10.4161/rna.7.4.12206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O'Hara PJ, Grant FJ, Haldeman BA, Gray CL, Insley MY, Hagen FS, Murray MJ. Nucleotide sequence of the gene coding for human factor VII, a vitamin K-dependent protein participating in blood coagulation. Proc Natl Acad Sci USA. 1987;84:5158–62. doi: 10.1073/pnas.84.15.5158. [DOI] [PMC free article] [PubMed] [Google Scholar]