

Abstract
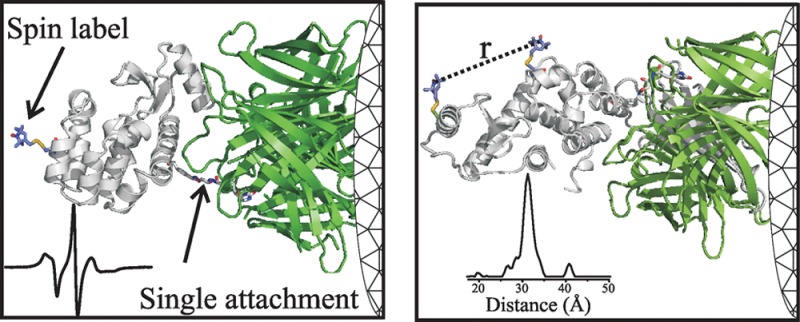
Proteins tethered to solid supports are of increasing interest in bioanalytical chemistry and protein science in general. However, the extent to which tethering modifies the energy landscape and dynamics of the protein is most often unknown because there are few biophysical methods that can determine secondary and tertiary structures and explore conformational equilibria and dynamics of a tethered protein with site-specific resolution. Site-directed spin labeling (SDSL) combined with electron paramagnetic resonance (EPR) offers a unique opportunity for this purpose. Here, we employ a general strategy using unnatural amino acids that enables efficient and site-specific tethering of a spin-labeled protein to a Sepharose solid support. Remarkably, EPR spectra of spin-labeled T4 lysozyme (T4L) reveal that a single site-specific attachment suppresses rotational motion of the protein sufficiently to allow interpretation of the spectral line shape in terms of protein internal dynamics. Importantly, line shape analysis and distance mapping using double electron–electron resonance reveal that internal dynamics, the tertiary fold, conformational equilibria, and ligand binding of the tethered proteins were similar to those in solution, in contrast to random attachment via native lysine residues. The results of this study set the stage for the development of an EPR-based flow system that will house soluble and membrane proteins immobilized site-specifically, thereby enabling facile screening of structural and dynamical effects of binding partners.
Tethering proteins to solid surfaces is fundamental for the development of biochips and optical biosensors for high-throughput analysis of molecular interactions based on surface plasmon resonance and bio-layer interferometry.1−4 In addition, tethering of spin-labeled proteins on a solid support suitable for EPR spectroscopy offers a number of potential advantages in site-directed spin labeling (SDSL) studies of protein structure and dynamics.5−10 For example, if a protein or complex can be rotationally immobilized on a solid support, then existing CW and time-domain methods in SDSL-EPR can be implemented to reveal internal dynamic modes of the protein on the nano- to microsecond time scale, free from contributions due to overall rotary diffusion,7,8,11 and for determination of interspin distances at room temperature using dipolar spectroscopy.7,8,11,12 Moreover, high local concentrations needed for time-domain applications such as saturation recovery13 can be achieved without concern for protein aggregation, and a continuous flow system could be designed that provides for EPR-based rapid screening of binding partners, including small ligands. In anticipation of this latter application, we refer to the general strategy using tethered proteins in SDSL as stationary-phase SDSL-EPR.
For these and other applications of tethered proteins aimed at investigating protein structure and function, it is important that the attachment to the surface not substantially alter the protein fold or conformational equilibria. SDSL can be used to determine the secondary and tertiary structures of a protein,14 to reveal the existence of conformational equilibria,15 to determine structural features of the substates involved,15,16 and to determine the time scale of exchange between them.7,13 Although other methods can be employed to determine many of these features for proteins in solution, SDSL-EPR may be unique in the capability to characterize proteins in solution and tethered to a solid support with equal ease. The main objectives of this work are to evaluate strategies for tethering spin-labeled proteins to a Sepharose matrix with respect to retention of solution-like structure, dynamics, and conformational equilibria and to determine if the potential advantages of tethering for SDSL can be realized. For SDSL applications, covalent attachment to CNBr-activated Sepharose via lysine residues has been previously employed.7,8 However, the nonselective nature of the attachment results in a heterogeneous preparation arising from the random orientation of protein molecules relative to the matrix and from differences in the number of linkages to the matrix.17 Consequently, each of the tethered molecules can potentially exhibit distinct structural, conformational, and hence functional properties. Indeed, it has been shown that enzyme activity and stability can be significantly altered by the number of attachment points to solid surfaces.18,19 Such molecular heterogeneity precludes quantitative analysis of structure-based changes in EPR spectra of tethered proteins due to contribution from species with distinct physical properties. Site-specific tethering strategies are expected to overcome the heterogeneous orientation problem, but questions remain as to the effect of attachment on the structure and conformation of the protein and the degree of rotational immobilization.
For site-specific tethering of a spin-labeled protein, it is necessary to have at least two orthogonal reactive groups in the protein: one selective for tethering and the other for introduction of the spin label. In the present study, we employ the biosynthetically incorporated unnatural amino acids (UAAs) p-azidophenylalanine (p-AzF) or p-acetylphenylalanine (p-AcF) for tethering to modified Sepharose together with a unique cysteine residue for introduction of a nitroxide spin label. The azidophenyl and acetylphenyl functionalities are not present in the 20 naturally occurring amino acids and enable the use of highly selective chemistry for tethering of proteins to solid matrices; indeed, p-AzF has already been employed for this purpose.20−22 T4 lysozyme (T4L) and variants thereof are used in this study as well-studied model systems to evaluate the immobilization strategy and to compare the status of the tethered protein with that in solution and that produced by the nonselective attachment to CNBr-activated Sepharose.
Remarkably, it is found that a single site-specific attachment is sufficient to suppress the rotational motion of the attached protein to a level beyond detection in the CW EPR line shape, a result similar to that found in earlier studies for a multipoint attachment using CNBr-Sepharose.7,8 Continuous wave (CW) EPR line shape analysis of a set of spin-labeled mutants and interspin distance mapping in doubly labeled proteins with double electron–electron resonance (DEER) reveal that the native secondary structure, tertiary fold, and backbone dynamics in the regions investigated are retained in the site-specifically tethered state. Most importantly, conformational equilibria in the native protein and in a ligand-binding cavity mutant are unperturbed in the tethered proteins. For the cavity mutant, ligand binding with a concomitant conformational shift previously observed in solution is also retained in the tethered protein. In contrast, tethering to CNBr-activated Sepharose is found to shift conformational equilibria and populate new states.
Experimental Procedures
Construction of T4L Mutants Containing Unnatural Amino Acids
The amber codon (TAG) was introduced to the T4L gene (pET11a vector) site-specifically using the QuikChange site-directed mutagenesis method. All mutants were introduced in the pseudo-WT background (WT*) that contains the mutations C54T and C97A. Mutations were verified by sequencing. Expression, purification, and spin labeling of the mutants were done as described in the Supporting Information.
Site-Specific Attachment of Proteins Bearing UAA to Modified Sepharose Beads
The general strategies for tethering to Sepharose are presented below in Results, and the detailed procedures for site-specific biotinylation of spin-labeled mutants bearing p-AcF or p-AzF and subsequent attachment to Sepharose derivatives are described in the Supporting Information.
EPR Spectroscopy
CW EPR spectra of spin-labeled proteins were recorded at X-band in a Bruker ELEXYS 580 spectrometer at 295 K either in the indicated buffer, in buffer containing 30% w/w sucrose, or tethered to modified Sepharose beads as described in López et al.8 Protein concentrations were in the range of 100–500 μM. Samples on beads were loaded into glass capillaries (0.60 i.d. × 0.84 o.d.; VitroCom Inc., NJ) via capillary action, and the ends were sealed using a nonparamagnetic sealant (X-Sealant, Bruker). The beads were allowed to settle to the bottom of the capillary prior to data collection. DEER spectroscopy at Q-band was done according to standard procedures;23 details are provided in the Supporting Information.
Halothane Binding to the Immobilized T4L 128R1/121A/133A Mutant
For the halothane titration experiments, a saturated solution of halothane in buffer (20.6 mM) was diluted to the appropriate concentration and added to the tethered protein. The EPR spectra were recorded immediately.
Results
General Strategy for Tethering Protein Mutants Bearing UAAs
A p-AzF unnatural amino acid in a protein reacts with strained cyclooctynes at neutral pH and ambient temperature to yield a 1,3 triazole linkage (Cu-free click chemistry).24 This chemistry is exploited here for site-specific tethering of spin-labeled proteins. The azide group can be partially reduced to an amine during bacterial expression and by reducing agents such as DTT25,26 and TCEP27 that are often used to maintain cysteine residues in a reduced state (Supporting Information Methods and Figure S1). Thus, the purification procedure for the T4L mutants bearing p-AzF was adapted accordingly to miminize reduction of the azide during purification (Supporting Information Methods).
The p-AzF amino acid was employed for tethering in most experiments, but p-AcF was also investigated for comparative purposes in particular cases. The p-AcF amino acid reacts selectively with hydroxylamines to yield an oxime linkage, but it requires acidic conditions and elevated temperatures (pH 4; 37 °C),28 which can be incompatible with many proteins. However, effective catalysts for the reaction have recently been reported that allow facile coupling at neutral pH and ambient temperature.29 Considering the chemical reduction experienced in the p-AzF, the use of such catalysts may make p-AcF the amino acid of choice in future studies.
The UAAs were introduced at sites on a T4L surface distal to that where a cysteine mutation was engineered for spin labeling to ensure that direct contact of the spin label with the surface of the matrix did not occur (Figure 1). The proteins containing p-AzF were tethered via direct covalent attachment to Sepharose 4B beads functionalized with two different derivatives of dibenzylcyclooctyne (DBCO) differing by the length of the linker to the Sepharose (schemes 1 and 2, Figure 2). The attachment reaction was efficient and specific, with 80–100% of the protein being tethered in the mutant containing p-AzF, whereas only ≈10% was nonspecifically bound in the WT* protein (Figure S2A). The maximum protein loading observed on the DBCO beads was ∼13 mg/mL, corresponding to ≈700 μM. The high effective concentration of protein enabled EPR data collection with a high S/N ratio.
Figure 1.
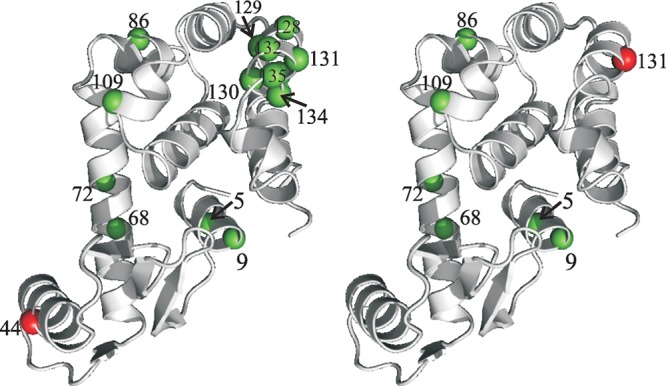
Sites where the UAA and cysteine mutations were introduced in T4L. The left- and right-hand panels show the two UAA sites employed for most studies (red spheres) together with the associated cysteine sites for spin labeling (green spheres).
Figure 2.

Tethering strategies employed in this study. For direct covalent attachment, proteins containing p-AzF were covalently attached to Sepharose beads functionalized with DBCO containing a 10-atom linker (scheme 1) or a 23-atom linker (scheme 2) as described in the Experimental Procedures and Supporting Information. For attachment mediated by biotin–streptavidin (schemes 3 and 4), proteins containing p-AcF or p-AzF were biotinylated and subsequently bound to Sepharose modified with streptavidin.
An alternative tethering strategy that relies on noncovalent tethering via bioaffinity using the biotin/streptavidin platform was evaluated (scheme 3, Figure 2). For proteins containing p-AcF, only the bioaffinity method was employed (scheme 4, Figure 2). Although many other bioaffinity strategies can be used, the biotin/streptavidin platform was selected due to the high affinity of streptavidin for biotin (Kd ∼ 10–15 M)30 and the stability of the interaction even under conditions normally associated with protein denaturation.31,32 For attachment using the biotin/streptavidin method, mutants bearing p-AzF or p-AcF amino acids were biotinylated with a biotin derivative containing either a DBCO or hydroxylamine functional group, respectively (schemes 3 and 4, Figure 2), and subsequently incubated with commercial streptavidin–Sepharose beads. For the examples tested, the biotinylation of the UAA-containing protein was quantitative, as evaluated with mass spectrometry (Figure S2B). For all biotinylated mutants, the attachment to the streptavidin beads was efficient and specific, i.e., ≈90% or greater of the biotinylated protein was retained, whereas no more than 5% was retained for a nonbiotinylated analogue (Figure S2A). The maximum binding capacity to the commercially available streptavidin beads used here was ∼10 mg/mL, corresponding to a protein concentration of ∼540 μM.
Rotational Immobilization of T4L Tethered to Sepharose Beads
The extent of protein immobilization due to tethering can be investigated with a spin label that is rigidly attached to the protein; in this case, the EPR spectrum will reflect purely rotational diffusion of the entire protein. One example is the side chain RX that cross-links pairs of cysteine residues at positions i and i ± 4 in helices or at appropriately spaced positions in β sheets7 (Figure 3, upper panel). Figure 3 (lower panel) shows EPR spectra for RX linking cysteine residues 5 and 9 in T4L (T4L 5RX9) under the indicated conditions.
Figure 3.
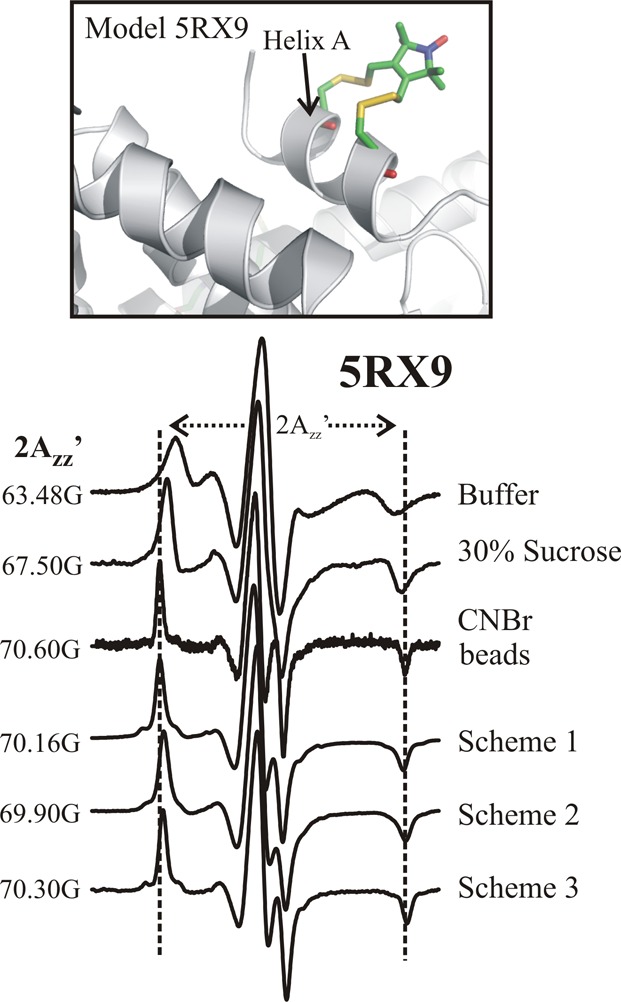
Effect of tethering on protein rotational motion. (Top) Ribbon model of 5RX9 based on a crystal structure.7 (Bottom) EPR spectra of 5RX9 recorded under the indicated conditions. The vertical dashed lines define the effective hyperfine splitting (2Azz′) of T4L 5RX9 tethered on CNBr-Sepharose; the hyperfine splitting values for the spectra are given. The site-specifically tethered proteins were attached via residue 131p-AzF. The spectra for T4L 5RX9 in buffer, 30% sucrose, and nonselectively attached to CNBr-activated Sepharose via native lysine residues have been previously published and are reproduced here for reference.7
The spectrum for T4L 5RX9 on CNBr has a powder line shape characteristic of a highly ordered nitroxide. A simulation of the spectrum was found to be consistent with a rapid (correlation time, τ ≈ 2 ns) but highly constrained (order parameter, S ≈ 0.9) motion.7 Because the motion is highly constrained relative to the protein, rotational diffusion of the entire protein modulates the motion of 5RX9 in a manner that is directly revealed in the CW EPR spectrum. A convenient measure of the nitroxide motion in the slow motional regime is the overall effective hyperfine splitting, 2Azz′ (Figure 3); 2Azz′ increases with decreasing mobility. For reference, the value of 2Azz′ for T4L 5RX9 in the complete absence of protein rotational diffusion is 70.8 ± 0.1 G at 295 K.7 As shown in Figure 3, 2Azz′ for the protein covalently attached to CNBr-activated Sepharose is close to this value; this is not surprising considering that multiple attachment points, which would strongly constrain protein motion, are likely.18,33 Thus, the rotational motion of T4L 5RX9 on CNBr is effectively frozen on the X-band EPR time scale. For comparison, the spectra in buffer and 30% sucrose have 2Azz′ of 63.5 and 67.5 G, corresponding to T4L rotational correlation times of τR ≈ 9 and 35 ns, respectively, consistent with predictions from the Stokes–Einstein equation.7
Figure 3 also shows representative data sets for T4L 5RX9/131p-AzF tethered to Sepharose via schemes 1 and 3; as is evident, the spectra have the characteristic powder lineshapes, and the 2Azz′ values are only slightly less (0.3 and 0.4 G, respectively) than that for multipoint attachment using CNBr-activated Sepharose. Similar results were obtained for attachment using 44p-AzF (Figure S3A). To investigate the effect of linker length between the protein and the matrix in the covalent attachment method, T4L 5RX9/131p-AzF was attached using a Sepharose derivative with an additional 16 atoms in a hydrophilic linker (Scheme 2). Remarkably, the 2Azz′ decreased only slightly (∼0.3 G) compared to that for the protein attached with the much shorter linker of Scheme 1 (Figure 3).
Thus, T4L 5RX9 tethered site selectively to Sepharose by a single attachment site leads to a high degree of immobilization, close to that for CNBr attachment. A similar conclusion was reached using another strongly immobilized nitroxide side chain (R8) that is introduced using a single cysteine residue (Figure S4).
Effect of Tethering on Local Protein Structure and Dynamics
The R1 nitroxide side chain is the most commonly used in SDSL. Unlike the internally constrained RX side chain, R1 at solvent-exposed sites in helices has rapid internal motion about the two terminal bonds adjacent to the nitroxide ring (Figure 4A).34 These internal modes dominate the EPR spectrum, and rotational diffusion of the protein has little contribution for T4L in 30% sucrose.8 Thus, the EPR spectrum encodes only information on internal local interactions and dynamics in the protein that modulate the motion of the nitroxide ring. As a result, it has been found that the CW spectral lineshapes of R1 faithfully report on both secondary and tertiary structures in stable proteins.35−37 Moreover, the rates and order of R1 motion at solvent-exposed sites on helices serve as metrics for nanosecond time scale backbone dynamics.15,38 Thus, R1 is well-suited to assess the effect of attachment on the structure and flexibility of T4L.
Figure 4.
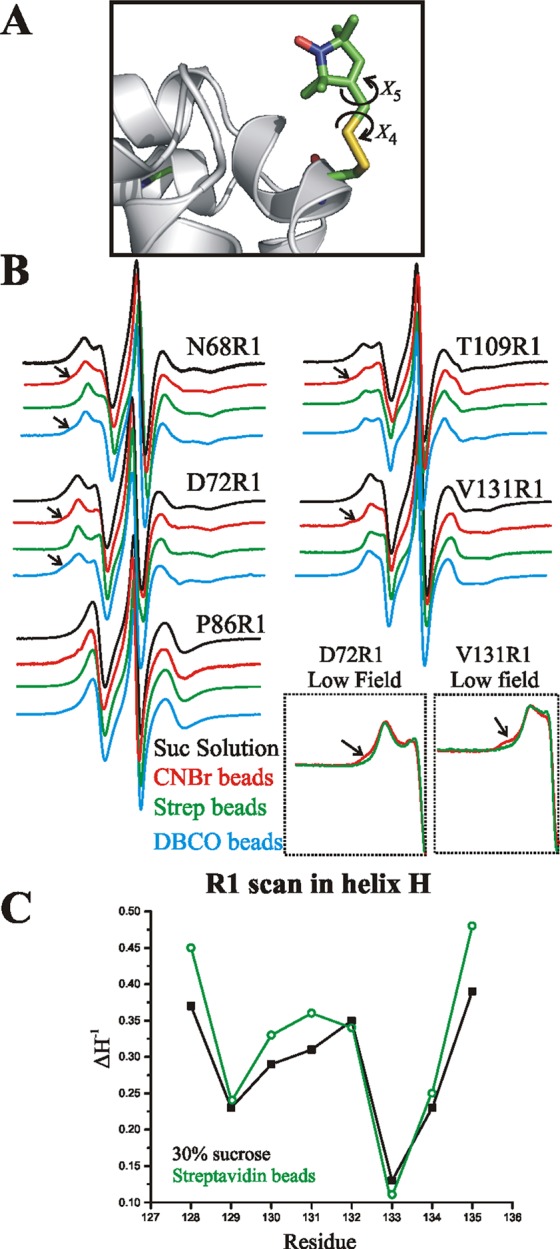
Monitoring structure and flexibility of T4L site-specifically attached to a solid support with CW lineshape analysis. (A) Model of R1 side chain in a helix showing points of internal flexibility about the last two dihedral angles (X4/X5; see text). (B) EPR spectra of the indicated sites recorded in 30% w/w sucrose (black), tethered to CNBr-Sepharose (red), or tethered site-specifically to streptavidin–Sepharose by scheme 3 (green) or by covalent attachment according to scheme 1 (cyan). The arrows identify new relatively immobile states observed after attachment. Insets: The low field lines of the spectra of 72R1 and 131R1 tethered to streptavidin and CNBr-Sepharose are magnified to reveal more clearly the spectral component corresponding to an immobile nitroxide. T4L 68R1 and 72R1 were tethered via site 131p-AzF, 86R1 via 44p-AzF, and 109R1 via 68p-AzF. (C) Plot of the inverse central line width (ΔH–1) along the sequence 128–135 in helix H; the proteins were tethered via 44p-AcF.
In order to first examine the influence of the different attachment strategies on the local backbone dynamics and tertiary fold, the spectra of 68R1, 72R1, 86R1, 109R1, and 131R1 were recorded in sucrose solution, bound nonspecifically to CNBr-activated Sepharose, and attached site-specifically with a p-AzF amino acid using direct attachment (scheme 1) or the streptavidin strategies (schemes 3 and 4). The EPR spectra are provided in Figure 4B.
The EPR spectra of all R1-labeled mutants attached via lysine residues to CNBr-Sepharose are essentially identical to those in sucrose solution except for the appearance of a new minor population of a relatively immobilized state of R1 in the former (arrows in Figure 4B). T4L contains 13 lysine residues distributed throughout the protein surface, some of which are in close proximity to the labeling sites (Figure S3B). Earlier work suggested that the immobilized component arises, at least partially, from a subset of proteins where the attachment places the nitroxide close to the surface of the matrix.8
This is confirmed here by a reduction in the immobile component upon replacing lysine residues at 84, 85, 124, and 147 that surround 131R1 by alanine and residue 135 by arginine (Figure S3B). Site-specific tethering by direct covalent attachment (scheme 1) also gives rise to a small immobilized component R1 at sites 68 and 72, each tethered via 131p-AzF. A possible origin of this component will be discussed below. Importantly, site-specific tethering mediated by streptavidin (scheme 3) gives EPR spectra that are very similar to those in sucrose solution, indicating that the structure of the protein near the R1 sites as well as the nanosecond backbone dynamics15,38 are similar in solution and on the Sepharose support. For the sites investigated, similar results were obtained by streptavidin-mediated tethering using p-AcF rather than p-AzF, suggesting that the two strategies are equivalent, as expected (Figure S3D).
To further explore the influence of solid-phase immobilization on secondary and tertiary structures, a continuous R1 scan was generated through helix H from residues 128–135. In previous studies, the variation of R1 mobility35,39 and solvent accessibility40 along this sequence were shown to be periodic in position, thus documenting the helical secondary structure and its orientation in the fold. Qualitatively, the EPR spectra of the tethered mutants are nearly identical in line shape to those in solution, with subtle differences likely due to the reduction in protein correlation time (Figure S3C). A plot of the inverse central line width (ΔH–1) as a semiquantitative measure of nitroxide mobility36 is shown in Figure 4C, which reveals the same periodic dependence on position as for the protein in solution. The maxima in the plot at 128, 131/132, and 135 identify the solvent-exposed surface of the helix where R1 has relatively high mobility; the minima at 129 and 133 correspond to the buried helix surface.
Effect of Tethering on Global Tertiary Structure
While the CW spectrum of R1 in a protein provides information on the local structure, measurement of interspin distances in doubly spin-labeled proteins using double electron–electron resonance (DEER) spectroscopy provides global information on the tertiary structure.41 To investigate the tertiary structure of tethered T4L, four R1 pairs were engineered, three of which contain the two R1 side chains within the C-terminal domain (i.e., 68R1/109R1, 68R1/131R1, and 109R1/131R1), whereas a 44R1/109R1 mutant monitored the relative positions of the N and C domains (Figures 5A and 6A).
Figure 5.
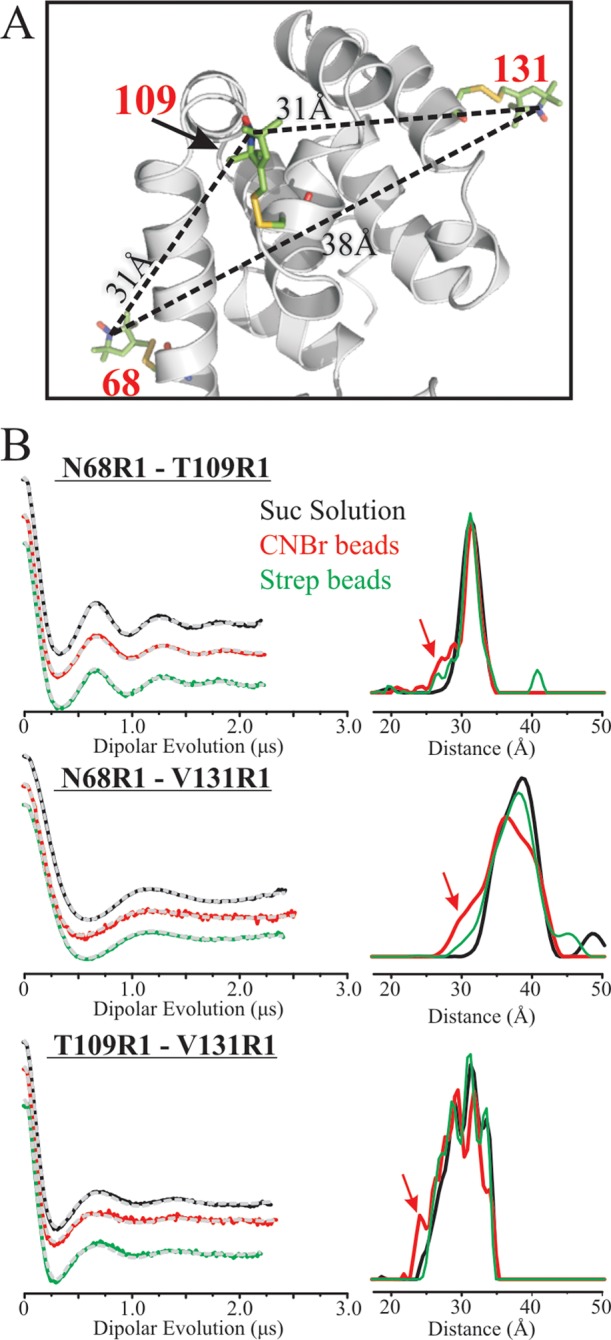
Interspin distance measurements within the C-terminal domain of T4L in solution and tethered to beads. (A) Ribbon diagram showing the distances measured along with the expected values based on modeling. (B) Background-corrected DEFs (left panel) and corresponding distance distributions (right panel) for the indicated R1 doubles in solution (black), tethered to CNBr-Sepharose (red), and tethered site-specifically via site 44p-AzF and scheme 3 (green) are shown. The dashed gray traces are the best fit of the DEF. The red arrows identify an increase in the distance probability due to random attachment compared to solution and site-specific attachment.
Figure 6.
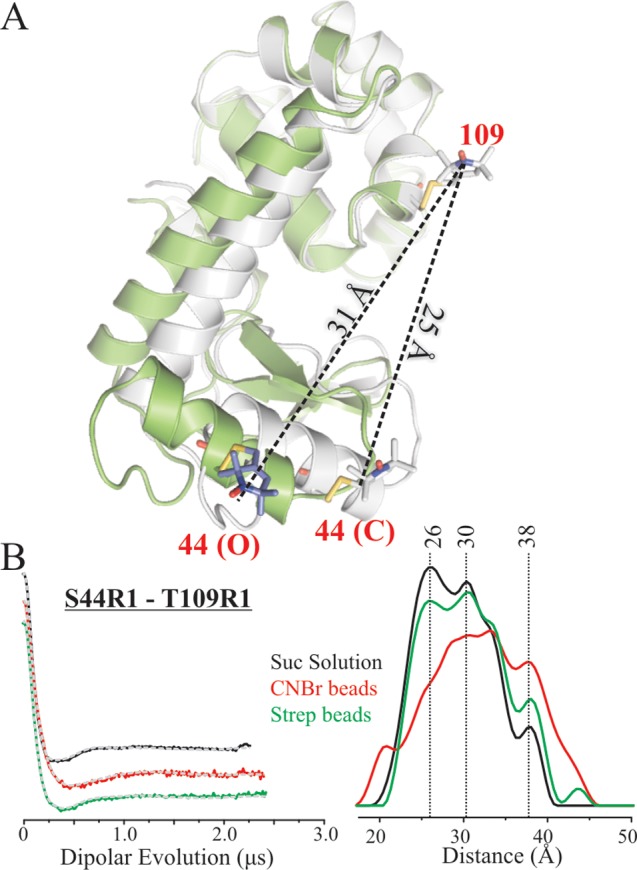
Interdomain distance measurements of T4L in solution and tethered to beads. (A) Ribbon models of the closed (gray, PDB 3lzm) and open states (green, PDB 150L) of T4L aligned with the C-terminal domain (residues 83–164); expected interspin distances for 109R1/44R1 in the two states are indicated. (B) Background-corrected DEFs (left panel) and corresponding distance distributions (right panel) in solution (black), tethered to CNBr-Sepharose (red), and tethered site-specifically via 131p-AzF and scheme 3 (green) are shown. The dashed gray traces are the best fit of the DEFs.
The background-corrected dipolar evolution functions (DEF) and corresponding distance distributions for 68R1/109R1, 68R1/131R1, and 109R1/131R1 tethered using scheme 3 are shown in Figure 5B (green traces); for the pairs tested, essentially identical distance distributions were obtained using the direct covalent coupling via scheme 1 (Figure S5). For comparison, data for the three pairs in solution and tethered nonspecifically to CNBr-Sepharose are shown in black and red, respectively.
The most probable distances for the proteins tethered site-specifically are within 0.5 Å of those in solution and are in excellent agreement with distances predicted by modeling (Figure 5A). Moreover, the distance distributions show only subtle differences; the three populations in 109R1/131R1 may be due to rotamers of R1. Thus, the interspin distance measurements show unequivocally that the tertiary fold of the protein in the C-terminal domain is retained after site-specific attachment via either scheme 1 or 3. In contrast, the distance distributions of the three mutants tethered to CNBr-Sepharose show increased populations of minor states observed in the site-specific attachment (see red arrows in Figure 5B) as well as population shifts among the major states of 109R1/131R1; for 68R1/131R1, the distribution mode shifts by ∼2.5 Å.
The 44R1/109R1 double is of special interest because it measures the distance between the two domains of T4L. The active site of the enzyme is located between the two domains, and solution SDSL studies42 have shown that there is an equilibrium between open and closed conformations related by a hinge-bending motion that results in changes in the distance and relative angle between the two domains. Thus, the interspin distance of 44R1/109R1 serves as a metric to monitor conformational properties of the protein with respect to the hinge-bending motion.
DEER data for 44R1/109R1 are shown in Figure 6B. In all cases, the widths of the distributions observed for 44R1/109R1 are broader than those of Figure 5, consistent with the structural heterogeneity expected between the domains. In solution, populations at 26, 30, and 38 Å are resolved, the first two of which correspond reasonably well with distances of 25 and 31 Å, corresponding to the crystallographic closed and open states, respectively. The distance distribution of the protein tethered site-specifically is very similar, with only subtle changes in the relative probability of the populations (Figures 6 and S5). On the other hand, tethering via CNBr-Sepharose leads to significant changes in the distance distribution profile relative to that for the solution state; the most probable distance is ≈7 Å longer than that observed in solution, and new populations are evident. Apparently, the nonspecific, multipoint, and heterogeneous attachment via lysine residues modulates the conformational properties of T4L.
Monitoring Ligand Binding to a Tethered T4L Cavity Mutant
With a judiciously placed spin label, the EPR spectrum can serve to monitor ligand binding via associated shifts in conformational equilibria.23 When the protein is tethered to a solid support, the surrounding solution can be readily changed, thereby allowing for facile screening of potential ligands with SDSL-EPR on the same protein sample. This application requires that the tethering does not substantially alter the conformational or ligand-binding equilibria of the protein. The data in Figure 6 already indicate that site-specific tethering does not alter a known conformational equilibrium, i.e., the T4L hinge bending.
To investigate the influence of tethering on ligand-binding equilibria, the cavity mutant T4L 121A/133A was selected.43 A recent SDSL-EPR study showed that this mutant exists in a conformational equilibrium between at least two states of similar free energy, one of which binds halothane and other organic ligands, thereby shifting the equilibrium.23 Figure 7A shows a model of the mutant and the location of 128R1 used to monitor the conformational equilibrium. Figure 7B shows the EPR spectra of T4L 128R1/121A/133A in the absence (apo) and presence (holo) of halothane (10 mM) both in solution and tethered site-specifically. For the latter, a complete set of spectra for halothane concentrations from 0 to 20 mM is provided in Figure S6. The spectra in solution are reproduced from López et al.,23 where it was shown that each spectrum consists of two components, i and m, arising from distinct protein conformations, and that the effect of ligand binding was to shift the conformational equilibrium toward the m component. The structural origins of the spectral components are discussed in López et al.23 The spectra and spectral changes upon halothane addition are remarkably similar for the protein in solution and tethered to Sepharose, demonstrating that site-specific tethering did not substantially alter either the protein conformational equilibrium or the ligand binding and concomitant shift in equilibrium; the small differences that do exist are attributable to the reduction in correlation time for the tethered protein relative to that in sucrose solution (e.g., Figure 3).
Figure 7.
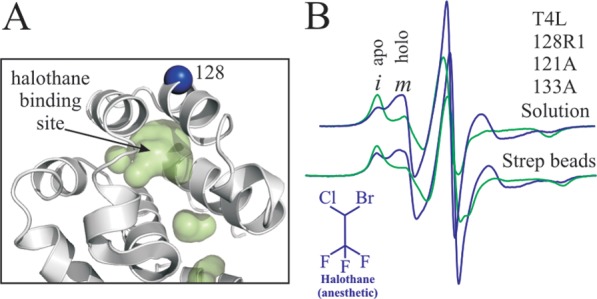
Ligand binding to T4L 121A/131A tethered site-specifically via site 44p-AzF and scheme 3. (A) Ribbon diagram showing the structure of the T4L mutant 121A/133A (PDB 251L), the internal ligand binding cavity as a green surface, and the location of the 128R1 site used to detect ligand binding. (B) EPR spectra of 128R1/121A/133A in the apo state (green) and with 10 mM halothane (blue) in solution (top panel) and site-specifically tethered (bottom panel).
Discussion
Although nonspecific, covalent attachment of proteins to commercially available CNBr-activated Sepharose is convenient, the nonuniform orientation of the tethered proteins leads to some fraction of the population in which the spin label interacts directly with the matrix, giving rise to immobilized states of the nitroxides that complicate the interpretation of the EPR spectra in terms of protein structure. Moreover, the potential of multipoint attachment to distort the structure make this method unattractive for conformational analysis. These effects are illustrated by the data of Figures 4–6, and this method of tethering was not investigated further.
To overcome the above problems, the two basic strategies for site-specific tethering to Sepharose shown in Figure 2 were evaluated. The direct covalent attachment of T4L (scheme 1) resulted in the appearance of a spectral component corresponding to an immobilized state of R1 at certain sites (Figure 4B), much like the case for CNBr attachment. The origin of this component is likely to be R1–matrix interaction due to the short linker. Such interaction will depend on the relative orientation of the surfaces containing the linker and R1 and will not appear for every site, as observed. For T4L tethering mediated by streptavidin/biotin, the streptavidin molecule provides a rigid spacer of at least 40 Å between the spin-labeled protein and the surface of the bead, so direct interaction with the surface is not possible. This effect, together with the extremely high stability of streptavidin, and the small Kd of the biotin/streptavidin association (≈10–15 M) make the streptavidin/biotin-mediated coupling the method of choice for most applications. For the few examples compared, tethering via either p-AzF or p-AcF mediated by streptavidin/biotin was equivalent in terms of the metrics employed.
A single-site tether of T4L to Sepharose, either by direct covalent attachment with a short linker or mediated by streptavidin/biotin, is apparently sufficient to rotationally immobilize the protein on the CW EPR time scale (Figure 3), thus allowing the EPR spectra of R1 side chains to be interpreted in terms of internal protein dynamics on the nanosecond time scale. Presumably, the single-site tether pays the entropic cost of localization, thus allowing multiple weak enthalpically driven interactions with the matrix (or streptavidin) that lead to rotational immobilization. Future studies will be needed to determine whether immobilization extends to the micro- to millisecond time domain. If so, the methods developed here will enable application of CW saturation transfer44 and pulsed EPR methods7 to directly monitor internal modes of protein motion in this slow motional regime. The high effective concentrations (≈700 μM) that can be achieved without aggregation will provide high signal-to-noise data with these methods and enable real-time recording of transient changes initiated, for example, by pressure or temperature-jump methods designed to measure the kinetics of conformational exchange.
SDSL-EPR offers a unique opportunity to map sequence-specific secondary structure and both local and global tertiary structure of tethered proteins for comparison with their solution counterparts to assess the effect of attachment. The EPR lineshapes of R1 spectra in a protein are fingerprints of the local secondary structure and tertiary fold.35 For example, the periodic dependence of R1 dynamics along the sequence 128–135 in T4L in solution reveals a helical structure of orientation within the fold, as predicted for helix H in the crystal structure (Figure 4).35 The same periodicity and phase of R1 dynamics are found for the protein site-specifically tethered, clearly showing that helix H structure and interactions with the surrounding protein are unperturbed by the streptavidin-mediated tether (Figures 3C and S3). In addition to structural information, the EPR lineshapes of R1 at solvent-exposed sites on helices also reveal the amplitude of local backbone dynamics on the nanosecond time scale.11,15,38 The near identity of such spectra for helix H in solution and in the tethered state (Figure S3D) confirms that backbone dynamics are unperturbed in the tethered state. Although complete R1 scans were not done throughout the structure, other helices in the fold (C, D, F) were sampled by individual sites. In each case, the spectral line shapes are consistent with an ordered helical structure and are very similar to the corresponding spectra in solution (Figure 4). These data provide strong evidence that the tertiary fold and backbone dynamics of the C-terminal domain are retained in the tethered protein.
The above conclusion is quantitatively supported by the narrow and very similar distance distributions between R1 pairs within the C terminal domain for both the solution and tethered states of T4L (Figure 5); in each case, the distances are in excellent agreement with the single conformation of the WT T4L crystal structure (PDB 3LZM). On the other hand, the broad multimodal distance distributions observed for an interdomain pair in solution is consistent with the existence of a conformational ensemble, the members of which are related by bending about an interdomain hinge that was identified in crystal structures from different space groups.45 The fact that the same distribution is found for the tethered protein indicates that this dynamic conformational equilibrium between substates of a similar free energy is unperturbed (Figure 6). A different conformational equilibrium, previously identified in the ligand-binding cavity mutant T4L 121A/133A,23 gives a similar result. As shown in Figure 7, the tethered mutant retains not only the conformational equilibrium observed in solution but also the striking conformational selection due to ligand binding (halothane). Collectively, the data make a convincing argument that single and site-specific tethering has little effect on secondary or tertiary structure, backbone dynamics, or conformational equilibria.
Summary and Conclusions
The main conclusion of general interest is that the tertiary structure, backbone dynamics, and conformational equilibria of T4L derivatives attached site-selectively to a Sepharose matrix are unperturbed relative to the protein in solution for the sites investigated. The spin-labeled sites were chosen distal to the attachment point, and, while it is likely that backbone dynamics and perhaps local structure are influenced in close proximity to that point, the data clearly show that the global structure and conformational equilibria as well as the free energy of ligand binding are retained in the tethered state. Although here it is shown that T4 lysozyme retains solution structure and conformational equilibria upon site-specific tethering, like any strategy that requires mutagenesis, the introduction of unnatural amino acids and subsequent tethering to a solid support could potentially perturb the local structure and dynamics in other proteins. Judicious selection of the attachment sites far from functional regions and appropriate functional assays should be performed in any protein of interest to assess the effect of mutagenesis and subsequent attachment on the native properties
A general advantage of the strategy employed here is the broad applicability, particularly for proteins expressed in Escherichia coli, where site-specific incorporation of unnatural amino acids for attachment is relatively simple and produces quantitative yields of functionalized proteins.28,46 For SDSL-EPR, the technology offers important advantages demonstrated here, namely, the rotational immobilization of proteins on the EPR time scale and the ability to achieve high local concentrations (≈1 mM) without aggregation. Thus, tethering, in principle, enables the investigation of marginally stable states of proteins, such as molten globules, that aggregate at concentrations needed for spectroscopic study and for studies of unfolding/folding mechanisms under reversible conditions. Site-specific tethering of spin-labeled proteins is also an enabling technology for an EPR-based continuous flow system that could be employed for structure-based screening of ligand binding using the full complement of SDSL-EPR methods; this application is currently under investigation.
Acknowledgments
We thank Dr. Peter G. Schultz for providing the plasmids pSUPAR-p-AcF and pEVOL-p-AzF used for incorporation of UAA in E. coli. We would like to thank Liming Tan for providing assistance in expression, purification, and tethering of T4 lysozyme mutants 72p-AcF/131C and T4L 72C/131p-AcF. We thank Dr. Zhongyu Yang, Michael T. Lerch, and Kevin Eden for careful reading of the manuscript.
Glossary
Abbreviations
- SDSL
site-directed spin labeling
- EPR
electron paramagnetic resonance
- DEER
double electron–electron resonance
- CW
continuous wave
- CNBr
cyanogen bromide
- UAA
unnatural amino acids
- p-AzF
para-azidophenylalanine
- p-AcF
para-acetylphenylalanine
- DEF
dipolar evolution function
Supporting Information Available
Full experimental procedures; data showing reduction of p-AzF with DTT and TCEP; EPR spectra of R1, R8, and RX labeled T4L; MALDI-TOF data; DEER data of doubly labeled T4L mutants tethered to DBCO-Sepharose; and EPR spectra of 128R1/121A/133A tethered to streptavidin beads showing complete titration of halothane. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† (M.R.F.) Genzyme Corporation, 500 Kendall Street, Cambridge, Massachusetts 02138, United States.
This work was supported by National Institutes of Health Grant 5R01 EY005216, Jules Stein Eye Institute Training Grant 2T32EY007026-36A1, and a Jules Stein Professor Endowment.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hämäläinen M. D.; Markgren P.-O.; Schaal W.; Karlén A.; Classon B.; Vrang L.; Samuelsson B.; Hallberg A.; Danielson U. H. (2000) Characterization of a set of HIV-1 protease inhibitors using binding kinetics data from a biosensor-based screen. J. Biomol. Screening 5, 353–359. [DOI] [PubMed] [Google Scholar]
- Karlsson R.; Kullman-Magnusson M.; Hämäläinen M. D.; Remaeus A.; Andersson K.; Borg P.; Gyzander E.; Deinum J. (2000) Biosensor analysis of drug–target interactions: direct and competitive binding assays for investigation of interactions between thrombin and thrombin inhibitors. Anal. Biochem. 278, 1–13. [DOI] [PubMed] [Google Scholar]
- Cooper M. A. (2002) Optical biosensors in drug discovery. Nat. Rev. Drug Discovery 1, 515–528. [DOI] [PubMed] [Google Scholar]
- Abdiche Y.; Malashock D.; Pinkerton A.; Pons J. (2008) Determining kinetics and affinities of protein interactions using a parallel real-time label-free biosensor, the Octet. Anal. Biochem. 377, 209–217. [DOI] [PubMed] [Google Scholar]
- Jacobsen K.; Oga S.; Hubbell W. L.; Risse T. (2005) Determination of the orientation of T4 lysozyme vectorially bound to a planar-supported lipid bilayer using site-directed spin labeling. Biophys. J. 88, 4351–4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen K.; Risse T. (2008) On the origin of the polar order of T4 lysozyme on planar model surfaces. J. Phys. Chem. B 112, 967–972. [DOI] [PubMed] [Google Scholar]
- Fleissner M. R.; Bridges M. D.; Brooks E. K.; Cascio D.; Kalai T.; Hideg K.; Hubbell W. L. (2011) Structure and dynamics of a conformationally constrained nitroxide side chain and applications in EPR spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 108, 16241–16246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López C. J.; Fleissner M. R.; Guo Z.; Kusnetzow A. K.; Hubbell W. L. (2009) Osmolyte perturbation reveals conformational equilibria in spin-labeled proteins. Protein Sci. 18, 1637–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim C. B.; Zhang Z.; Thomas D. D. (2007) Synthesis of TOAC spin-labeled proteins and reconstitution in lipid membranes. Nat. Protoc. 2, 42–49. [DOI] [PubMed] [Google Scholar]
- Regan M. C.; Horanyi P. S.; Pryor E. E. Jr.; Sarver J. L.; Cafiso D. S.; Bushweller J. H. (2013) Structural and dynamic studies of the transcription factor ERG reveal DNA binding is allosterically autoinhibited. Proc. Natl. Acad. Sci. U.S.A. 110, 13374–13379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbell W. L.; Lopez C. J.; Altenbach C.; Yang Z. (2013) Technological advances in site-directed spin labeling of proteins. Curr. Opin. Struct. Biol. 23, 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.; Liu Y.; Borbat P.; Zweier J. L.; Freed J. H.; Hubbell W. L. (2012) Pulsed ESR dipolar spectroscopy for distance measurements in immobilized spin labeled proteins in liquid solution. J. Am. Chem. Soc. 134, 9950–9952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges M. D.; Hideg K.; Hubbell W. L. (2010) Resolving conformational and rotameric exchange in spin-labeled proteins using saturation recovery EPR. Appl. Magn. Reson. 37, 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbell W. L.; Gross A.; Langen R.; Lietzow M. A. (1998) Recent advances in site-directed spin labeling of proteins. Curr. Opin. Struct. Biol. 8, 649–656. [DOI] [PubMed] [Google Scholar]
- López C. J.; Oga S.; Hubbell W. L. (2012) Mapping molecular flexibility of proteins with site-directed spin labeling: a case study of myoglobin. Biochemistry 51, 6568–6583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerch M. T.; Yang Z.; Brooks E. K.; Hubbell W. L. (2014) Mapping protein conformational heterogeneity under pressure with site-directed spin labeling and double electron–electron resonance. Proc. Natl. Acad. Sci. U.S.A. 111, 1201–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pye E. K.; Chance B. (1976) Investigation of the physical properties of immobilized enzymes. Methods Enzymol. 44, 357–372. [DOI] [PubMed] [Google Scholar]
- Koch-Schmidt A. C.; Mosbach K. (1977) Studies on conformation of soluble and immobilized enzymes using differential scanning calorimetry. 2. Specific activity and thermal stability of enzymes bound weakly and strongly to Sepharose CL 4B. Biochemistry 16, 2105–2109. [DOI] [PubMed] [Google Scholar]
- Pedroche J.; del Mar Yust M.; Mateo C.; Fernández-Lafuente R.; Girón-Calle J.; Alaiz M.; Vioque J.; Guisán J. M.; Millán F. (2007) Effect of the support and experimental conditions in the intensity of the multipoint covalent attachment of proteins on glyoxyl-agarose supports: correlation between enzyme–support linkages and thermal stability. Enzyme Microb. Technol. 40, 1160–1166. [Google Scholar]
- Yoshimura S. H.; Khan S.; Ohno S.; Yokogawa T.; Nishikawa K.; Hosoya T.; Maruyama H.; Nakayama Y.; Takeyasu K. (2012) Site-specific attachment of a protein to a carbon nanotube end without loss of protein function. Bioconjugate Chem. 23, 1488–1493. [DOI] [PubMed] [Google Scholar]
- Seo M. H.; Han J.; Jin Z.; Lee D. W.; Park H. S.; Kim H. S. (2011) Controlled and oriented immobilization of protein by site-specific incorporation of unnatural amino acid. Anal. Chem. 83, 2841–2845. [DOI] [PubMed] [Google Scholar]
- Camarero J. A. (2008) Recent developments in the site-specific immobilization of proteins onto solid supports. Pept. Sci. 90, 450–458. [DOI] [PubMed] [Google Scholar]
- López C. J.; Yang Z.; Altenbach C.; Hubbell W. L. (2013) Conformational selection and adaptation to ligand binding in T4 lysozyme cavity mutants. Proc. Natl. Acad. Sci. U.S.A. 110, 4306–4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sletten E. M.; Bertozzi C. R. (2011) From mechanism to mouse: a tale of two bioorthogonal reactions. Acc. Chem. Res. 44, 666–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartwright I. L.; H D.; Armstrong V. W. (1976) The reaction between thiols and 8-azidoadenosine derivatives. Nucleic Acid Res. 3, 2331–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staros J. V.; Bayley H.; Standring D. N.; Knowles J. R. (1978) Reduction of aryl azides by thiols: implications for the use of photoaffinity reagents. Biochem. Biophys. Res. Commun. 80, 568–572. [DOI] [PubMed] [Google Scholar]
- Faucher A.-M.; Grand-Maître C. (2003) Tris(2-carboxyethyl)phosphine (TCEP) for the reduction of sulfoxides, sulfonylchlorides, N-oxides, and azides. Synth. Commun. 33, 3503–3511. [Google Scholar]
- Fleissner M. R.; Brustad E. M.; Kalai T.; Altenbach C.; Cascio D.; Peters F. B.; Hideg K.; Peuker S.; Schultz P. G.; Hubbell W. L. (2009) Site-directed spin labeling of a genetically encoded unnatural amino acid. Proc. Natl. Acad. Sci. U.S.A. 106, 21637–21642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendeler M.; Grinberg L.; Wang X.; Dawson P. E.; Baca M. (2013) Enhanced catalysis of oxime-based bioconjugations by substituted anilines. Bioconjugate Chem. 25, 93–101. [DOI] [PubMed] [Google Scholar]
- Green N. M. (1975) Avidin. Adv. Protein Chem. 29, 85–133. [DOI] [PubMed] [Google Scholar]
- Lesaicherre M.-L.; Lue R. Y. P.; Chen G. Y. J.; Zhu Q.; Yao S. Q. (2002) Intein-mediated biotinylation of proteins and its application in a protein microarray. J. Am. Chem. Soc. 124, 8768–8769. [DOI] [PubMed] [Google Scholar]
- González M. n.; Argaraña C. E.; Fidelio G. D. (1999) Extremely high thermal stability of streptavidin and avidin upon biotin binding. Biomol. Eng. 16, 67–72. [DOI] [PubMed] [Google Scholar]
- Wilchek M.; Miron T. (2003) Oriented versus random protein immobilization. J. Biochemical Biophys. Methods 55, 67–70. [DOI] [PubMed] [Google Scholar]
- Columbus L.; Kalai T.; Jeko J.; Hideg K.; Hubbell W. L. (2001) Molecular motion of spin labeled side chains in alpha-helices: analysis by variation of side chain structure. Biochemistry 40, 3828–3846. [DOI] [PubMed] [Google Scholar]
- Mchaourab H. S.; Lietzow M. A.; Hideg K.; Hubbell W. L. (1996) Motion of spin-labeled side chains in T4 lysozyme. Correlation with protein structure and dynamics. Biochemistry 35, 7692–7704. [DOI] [PubMed] [Google Scholar]
- Columbus L.; Hubbell W. L. (2002) A new spin on protein dynamics. Trends Biochem. Sci. 27, 288–295. [DOI] [PubMed] [Google Scholar]
- Lietzow M. A.; Hubbell W. L. (2004) Motion of spin label side chains in cellular retinol-binding protein: correlation with structure and nearest-neighbor interactions in an antiparallel beta-sheet. Biochemistry 43, 3137–3151. [DOI] [PubMed] [Google Scholar]
- Columbus L.; Hubbell W. L. (2004) Mapping backbone dynamics in solution with site-directed spin labeling: GCN4-58 bZip free and bound to DNA. Biochemistry 43, 7273–7287. [DOI] [PubMed] [Google Scholar]
- Guo Z.; Cascio D.; Hideg K.; Kálái T.; Hubbell W. L. (2007) Structural determinants of nitroxide motion in spin-labeled proteins: tertiary contact and solvent-inaccessible sites in helix G of T4 lysozyme. Protein Sci. 16, 1069–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyka J.; Ilnicki J.; Altenbach C.; Hubbell W. L.; Froncisz W. (2005) Accessibility and dynamics of nitroxide side chains in T4 lysozyme measured by saturation recovery EPR. Biophys. J. 89, 2059–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeschke G.; Polyhach Y. (2007) Distance measurements on spin-labelled biomacromolecules by pulsed electron paramagnetic resonance. Phys. Chem. Chem. Phys. 9, 1895–1910. [DOI] [PubMed] [Google Scholar]
- Mchaourab H. S.; Oh K. J.; Fang C. J.; Hubbell W. L. (1997) Conformation of T4 lysozyme in solution. Hinge-bending motion and the substrate-induced conformational transition studied by site-directed spin labeling. Biochemistry 36, 307–316. [DOI] [PubMed] [Google Scholar]
- Xu J.; Baase W. A.; Baldwin E.; Matthews B. W. (1998) The response of T4 lysozyme to large-to-small substitutions within the core and its relation to the hydrophobic effect. Protein Sci. 7, 158–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moen R. J.; Thomas D. D.; Klein J. C. (2013) Conformationally trapping the actin-binding cleft of myosin with a bifunctional spin label. J. Biol. Chem. 288, 3016–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber H. R.; Matthews B. W. (1990) A mutant T4 lysozyme displays five different crystal conformations. Nature 348, 263–266. [DOI] [PubMed] [Google Scholar]
- Kálai T.; Fleissner M. R.; Jekő J.; Hubbell W. L.; Hideg K. (2011) Synthesis of new spin labels for Cu-free click conjugation. Tetrahedron Lett. 52, 2747–2749. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.