

Abstract
Background
Rapamycin, an inhibitor of mammalian target of rapamycin complex-1 (mTORC1), reduces platelet spreading, thrombus stability, and clot retraction. Despite an important role of mTORC1 in platelet function, little is known about how it is regulated. The objective of this study was to determine the signaling pathways that regulate mTORC1 in human platelets.
Methods
Mammalian target of rapamycin complex-1 activation was assessed by measuring the phosphorylation of its downstream substrate ribosomal S6 kinase 1 (p70S6K).
Results
Thrombin or the protein kinase C (PKC) activator phorbal 12-myristate 13-acetate stimulated activation of mTORC1 in a PKC-dependent, Akt-independent manner that correlated with phosphorylation of tuberin/tuberous sclerosis 2 (TSC2) (Ser939 and Thr1462). In contrast, insulin-like growth factor 1 (IGF-1)–stimulated TSC2 phosphorylation was completely dependent on phosphoinositide 3 kinase (PI3 kinase)/Akt but did not result in any detectable mTORC1 activation. Early (Ser939 and Thr1462) and late (Thr1462) TSC2 phosphorylation in response to thrombin were directly PKC dependent, whereas later TSC2 (Ser939) and p70S6K phosphorylation were largely dependent on paracrine signaling through P2Y12. PKC-mediated adenosine diphosphate (ADP) secretion was essential for thrombin-stimulated mTORC1 activation, as (i) ADP rescued p70S6K phosphorylation in the presence of a PKC inhibitor and (ii) P2Y12 antagonism prevented thrombin-mediated mTORC1 activation. Rescue of mTORC1 activation with exogenous ADP was completely dependent on the Src family kinases but independent of PI3 kinase/Akt. Interestingly, although inhibition of Src blocked the ADP rescue, it had little effect on thrombin-stimulated p70S6K phosphorylation under conditions where PKC was not inhibited.
Conclusion
These results demonstrate that thrombin activates the mTORC1 pathway in human platelets through PKC-mediated ADP secretion and subsequent activation of P2Y12, in a manner largely independent of the canonical PI3 kinase/Akt pathway.
Keywords: P2Y12 purinoceptor, platelets, protease-activated receptors, protein kinase C, target of rapamycin complex 1
Introduction
Rapamycin (Sirolimus) and its analogs (CCI-779, AP23573, and RAD001) are currently used in the clinic as immunosuppressants to prevent rejection of organ transplants, on drug-eluting stents to prevent restenosis after angioplasty, and as treatment for some forms of cancer. The target of rapamycin is the multiprotein complex mammalian target of rapamycin complex 1 (mTORC1), which is composed of the Ser/Thr kinase mTOR (FRAP), raptor (regulatory associated protein of mTOR), mLST8 (GβL), Ti1/Tel2 complex, and the negative regulators DEPTOR (Dep domain containing mTOR interacting protein) and PRAS40 (proline-rich Akt substrate 40) 1–3. Activation of this complex ultimately results in the phosphorylation of the mTOR substrates p70S6K (ribosomal S6 kinase 1) and 4E-BP1 (eukaryotic translation initiation factor 4E-binding protein 1), leading to regulation of growth processes such as transcription, protein synthesis, nutrient transport, and autophagy.
Mammalian target of rapamycin complex-1 integrates multiple inputs from mitogens and nutrient sensing, which converge predominantly on the TSC1 (hamartin/tuberous sclerosis 1)/TSC2/Rheb axis 4. Rheb is a small GTPase that relays upstream signals to modulate mTORC1 activity. Rheb is inhibited by the GAP activity of the TSC1-TSC2 complex, which promotes the turnover of GTP to GDP. The Rheb-GAP activity of TSC2 is in turn inhibited by Akt-dependent phosphorylation on Ser939 and Thr1462 in response to growth factor stimulation. Loss of TSC1-TSC2, such as in the cancer-prone syndrome tuberous sclerosis complex, results in Rheb becoming constitutively loaded with GTP, rendering the mTORC1 pathway active.
Historically, there has been little interest in the mTORC1 pathway in platelets. As these short-lived, anucleated cells do not possess a cell cycle and were generally considered to have little capacity (or requirement) for protein synthesis. Recently, interest in mTORC1 in platelets has been renewed for the following reasons. First, a series by Weyrich et al. 5–7 demonstrated that platelets can perform signal-dependent protein synthesis using a constitutive transcriptome. Further, rapamycin was found to inhibit synthesis of a subset of protein products in activated platelets 6. Second, rapamycin has been demonstrated to inhibit platelet activation, thrombin-mediated platelet clot retraction, and thrombus stability and remodeling 6,8,9. And, third, the observation was made that rapamycin-eluting stents may have antiplatelet properties, as platelets do not aggregate on the surfaces of such stents as they do in non-coated stents 10,11.
Despite the growing use of mTOR inhibitors in the clinic and their effects on platelet function, little is known of how mTORC1 activation in platelets is regulated. In this study, we aimed to elucidate the signaling pathways involved in the activation of mTORC1. We determined that (i) the mTORC1 regulator TSC2 is phosphorylated in platelets in response to agonist stimulation, but that phosphorylation of TSC2 alone is not sufficient for mTORC1 signaling and (ii) that mTORC1 activation by thrombin requires PKC-mediated ADP secretion and subsequent activation of P2Y12.
Materials and methods
Materials
pSer473 Akt, pThr202/Tyr204 ERK (extracellular signal-regulated kinase), pSer65 4EBP1, pSer9 GSK3β (glycogen synthase kinase 3), pThr389 p70S6K, pThr246 PRAS40, PKC phospho-motif (used for analysis of pleckstrin phosphorylation), pThr1462 TSC2, and pSer939 TSC2 antibodies were from Cell Signaling Technologies (New England Biolabs, Hitchin, UK). p70S6K (H-9) and p70S6K (C-18) were from Santa Cruz (Insight Biotechnology, Wembley, UK). FITC-conjugated PAC-1 antibody and PE-conjugated anti–P-selectin (CD62P) were from BD Bioscience (Oxford, UK). MK-2206 2HCl was from Selleckchem (Stratech, Newmarket, UK). AR-C 66096 tetrasodium salt, bisindolylmaleimide I (BIM I), H89, MRS 2279, PD98059, rapamycin, SL 327, SQ 22536, U0126, U46619, VU 0155069, VU 0364739 hydrochloride, and wortmannin were from Tocris (Avonmouth, UK). Protease activated receptor (PAR)1 activating peptide (SFLLRN-NH2) was from Bachem (Weil am Rhein, Germany). IGF-1 was from Immunological and Biochemical Test Systems (Binzwangen, Germany). Cross-linked collagen-related peptide (CRP-XL) was synthesized by Prof. Richard Farndale (Department of Biochemistry, University of Cambridge, UK). Microcystin-LR was from Axxora (Nottingham, UK). Enhanced chemiluminescent detection reagents were from GE Healthcare (Bucks, UK). Peroxidase-conjugated secondary antibodies were from Jackson Immunoresearch (Stratech, Newmarket, UK). NuPAGE SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) sample buffer was from Invitrogen (Paisley, UK). All other reagents were from Sigma (Poole, UK) unless otherwise indicated.
Platelet isolation
Blood was drawn into 4% trisodium citrate (1:9) from healthy drug-free volunteers in accordance with approved guidelines and with ethical approval granted by the local research ethics committee, University Hospitals Bristol NHS Foundation Trust (E5736). Written, informed consent was obtained in accordance with the Declaration of Helsinki. Platelets were isolated from whole blood as previously described 12 and resuspended at 4 × 108 mL−1 in modified HEPES-Tyrode buffer (145 mmol L−1 NaCl, 3 mmol L−1 KCl, 0.5 mmol L−1 Na2HPO4, 1 mmol L−1 MgSO4, 10 mmol L−1 HEPES, pH 7.2, 0.1% [w/v] d-glucose, 0.02 U mL−1 apyrase, and 10 μmol L−1 indomethacin).
Flow cytometry
Two-color analysis of the kinetics of platelet activation was conducted with the FITC-conjugated PAC-1 antibody to assess integrin αIIbβ3 activation and PE-conjugated anti-P-selectin (CD62P) to assess α-granule secretion as previously described 13. Aliquots of platelets (2 × 107 mL−1) were treated with vehicle (0.2% DMSO) or rapamycin (200 nmol L−1) for 15 min before stimulation with thrombin (0.2 U mL−1) at time ‘zero.’ At different time points, a fixed volume of platelets was removed and incubated with FITC–PAC-1 and PE–anti–P-selectin for 30 s before the addition of 1% formaldehyde. Samples were analyzed on a BD LSR II (BD Bioscience) using FACSDiva software (BD Bioscience, Oxford, UK), and a total of 10 000 platelet events per sample were collected. Data were analyzed using Flowing Version 1.6 software (Cell Imaging Core, Turku Centre for Biotechnology, Turku, Finland). Data are expressed as a percentage of maximal fluorescence in vehicle conditions.
Clot retraction
Clot retraction studies were performed in aggregometer tubes. Assays were started by adding platelets (3 × 108 mL−1) in the presence of 1 mg mL−1 fibrinogen and 1 mmol L−1 CaCl2 to thrombin (final concentration 1 U mL−1). Platelets were treated with vehicle (0.2% DMSO) or rapamycin for 15 min before starting the assay. Clot retraction was observed for 120 min and the extent of clot retraction recorded by digital camera and by the amount of clear liquid that could be removed at the end of the assay. Data are expressed as a percentage of the amount of fluid removed from the thrombin-treated vehicle control.
Protein extraction
Platelets were treated with vehicle (0.2% DMSO) or compound for 15 min and stimulated as indicated. Platelet suspensions were either lysed directly in 4× NuPAGE sample buffer (whole cell lysate) or extracted with an equal volume of ice-cold RIPA buffer (50 mmol L−1 HEPES, pH 7.4, 400 mmol L−1 NaCl, 2 mmol L−1 EDTA, 2% [v/v] IGEPAL CA-630, 1% [w/v] sodium deoxycholate, 0.2% [w/v] SDS, 40 mmol L−1 sodium β-glycerol phosphate, 20 mmol L−1 sodium pyrophosphate, 2 mmol L−1 benzamidine, 2 μmol L−1 microcystin-LR, 10 mmol L−1 sodium orthovanadate, and 2 μg mL−1 each of pepstatin, antipain, and leupeptin) for immunoprecipitation of p70S6K.
Immunoprecipitation and immunoblotting
p70S6K was immunoprecipitated from RIPA lysates as previously described 14. Immunoprecipitates and whole cell lysates were analyzed by SDS-PAGE/Western blotting using 7% Tris-glycine and 8% Bis-Tris gels, respectively, as previously described 15,16. For quantification of immunoblotting; densitometry was performed using ImageJ software (NIH, Bethesda, MD, USA). Data are expressed as a percentage of the maximal signal obtained in the vehicle control (mean ± SE).
Statistics
All data are presented as the mean ± SE of at least three independent observations. Data presented with statistical analysis were tested using a one-way anova or two-way anova as appropiate with either Dunnett or Bonferroni post hoc tests.
Results
Activation of mTORC1 by thrombin is delayed and correlates with the effect of rapamycin on later platelet activation events
To confirm and extend previous observations for a role of mTORC1 in platelet function, we examined the effect of rapamycin on integrin αIIbβ3 activation, P-selectin expression, and clot retraction. In agreement with our previous study 14, demonstrating that rapamycin is unable to inhibit PAR-mediated platelet aggregation, we observed that rapamycin had no significant effect on both early thrombin-induced integrin αIIbβ3 activation, as measured by PAC-1 binding (Fig. 1A) or P-selectin expression (Fig. 1B). In contrast, we saw a small decrease in later integrin αIIbβ3 activation (Fig. 1A, 20 min) and almost full blockade of clot retraction (Fig. 1C), suggesting that mTORC1 is mainly involved in later platelet functional events.
Figure 1.

Activation of mTORC1 by thrombin is delayed and correlates with the effect of rapamycin on later platelet activation events. Washed platelets were preincubated with either vehicle (0.2% DMSO) or rapamycin (200 nmol L−1) for 15 min. For platelet activation kinetic assays platelets were stimulated with 0.2 U mL−1 thrombin for indicated times before staining with FITC–PAC-1 and PE–anti–P-selectin. Samples were fixed in 1% formaldehyde and analyzed by FACS analysis. Results are expressed as mean ± SE of percentage maximal fluorescence of vehicle-treated platelets. **P < 0.01, indicating a significant difference from vehicle at the matching time point (A, B). Clot retraction assays were started by adding platelets (3 × 108 mL−1) in the presence of 1 mg mL−1 fibrinogen and 1 mmol L−1 CaCl2 to thrombin (final concentration 1 U mL−1). Platelets were treated with vehicle (0.2% DMSO) or rapamycin for 15 min before starting the assay. Clot retraction was recorded by digital camera and by measurement of the amount of fluid that could be removed. Data are expressed as a percentage of the amount of fluid removed from the vehicle control. **P < 0.01, indicating a significant difference from vehicle (C). Washed platelets were incubated with either SFLLRN (5 μmol L−1), thrombin (0.2 U mL−1), CRP-XL (5 μg mL−1), or U46619 (1 μmol L−1) for 15 min (D) or thrombin (0.2 U mL−1) for the indicated times (E) before extraction. Phosphorylation of the indicated proteins was assessed by Western blotting of either whole cell lysates or immunoprecipitates (IP). Membranes were reprobed for α-tubulin to confirm equal loading.
A well-characterized signaling pathway leading to mTORC1 activation is the insulin/PI3 kinase pathway. Akt activation downstream of PI3 kinase leads to TSC2 phosphorylation on Ser939 and Thr1462, which suppresses the inhibitory effect of TSC2-GAP on Rheb, leading to increased mTORC1 activation 17,18. To investigate the involvement of this pathway in mTORC1 activation in platelets, we examined the phosphorylation of TSC2 in human platelets. Stimulation of platelets with various agonists including thrombin, CRP-XL, and the thromboxane A2 analog U46619 stimulated TSC2 phosphorylation on Ser939 and Thr1462 and concomitant phosphorylation of p70S6K on Thr389 (Fig. 1D). Thrombin stimulation further resulted in phosphorylation of Akt (Ser473) and its downstream substrate, the mTORC1 component PRAS40 (Thr246). Timecourse experiments established that stimulation with thrombin resulted in rapid phosphorylation of TSC2 on both Ser939 and Thr1462, reaching maximal phosphorylation at 15 min (Fig. 1E, Figure S1A). In contrast, phosphorylation of the mTORC1 substrates p70S6K on Thr389 and 4EBP1 on Ser65 was significantly delayed compared to TSC2 phosphorylation, reaching a peak after 15 min. PRAS40 phosphorylation increased in parallel with activation of Akt, as monitored by increases in pSer473. These results demonstrate that thrombin stimulates TSC2 phosphorylation and activation of mTORC1. Furthermore, mTORC1 is likely to be involved in late rather than early thrombin-mediated platelet activation events due to the observations that (i) rapamycin has no effect on early integrin αIIbβ3 activation or α-granule secretion, (ii) thrombin-induced clot retraction is blocked by rapamycin, and (iii) phosphorylation of mTORC1 substrates by thrombin is delayed.
Thrombin-mediated TSC2 Ser939 and TSC2 Thr1462 phosphorylation and activation of mTORC1 is largely Akt independent
Inhibitors of PI3 kinase (wortmannin) or Akt (MK-2206) reduced thrombin-stimulated TSC2 phosphorylation to between 20% and 40% of maximal phosphorylation (Fig. 2A). Interestingly, p70S6K phosphorylation was only partially inhibited by wortmannin and was unaffected by MK-2206 under conditions where phosphorylation of Akt and its downstream substrate PRAS40 were completely absent (Fig. 2A). In contrast, the generic PKC inhibitor BIM I blocked both TSC2 and p70S6K phosphorylation. Rapamycin blocked p70S6K phosphorylation, confirming that it is an mTORC1 substrate. In view of TSC2 phosphorylation being rapid and sustained, we determined the effects of PI3 kinase and PKC inhibition on early (1 min) and late (15 min) TSC2 phosphorylation (Fig. 2B). Wortmannin only partially inhibited early Ser939 TSC2 phosphorylation and failed to inhibit early Thr1462 TSC2 phosphorylation, a result that was mirrored by the P2Y12 antagonist AR-C 66096. In contrast, BIM I ablated both early and late phosphorylation. The reduction of late TSC2 phosphorylation by wortmannin and AR-C 66096 was also observed in pleckstrin and extracellular signal-regulated kinase (ERK) phosphorylation (Fig. 2B, Figure S1B, C). Together, these results demonstrate that thrombin stimulates TSC2 phosphorylation and activation of mTORC1 in a PKC-dependent manner that is in part independent of the PI3 kinase/Akt pathway.
Figure 2.
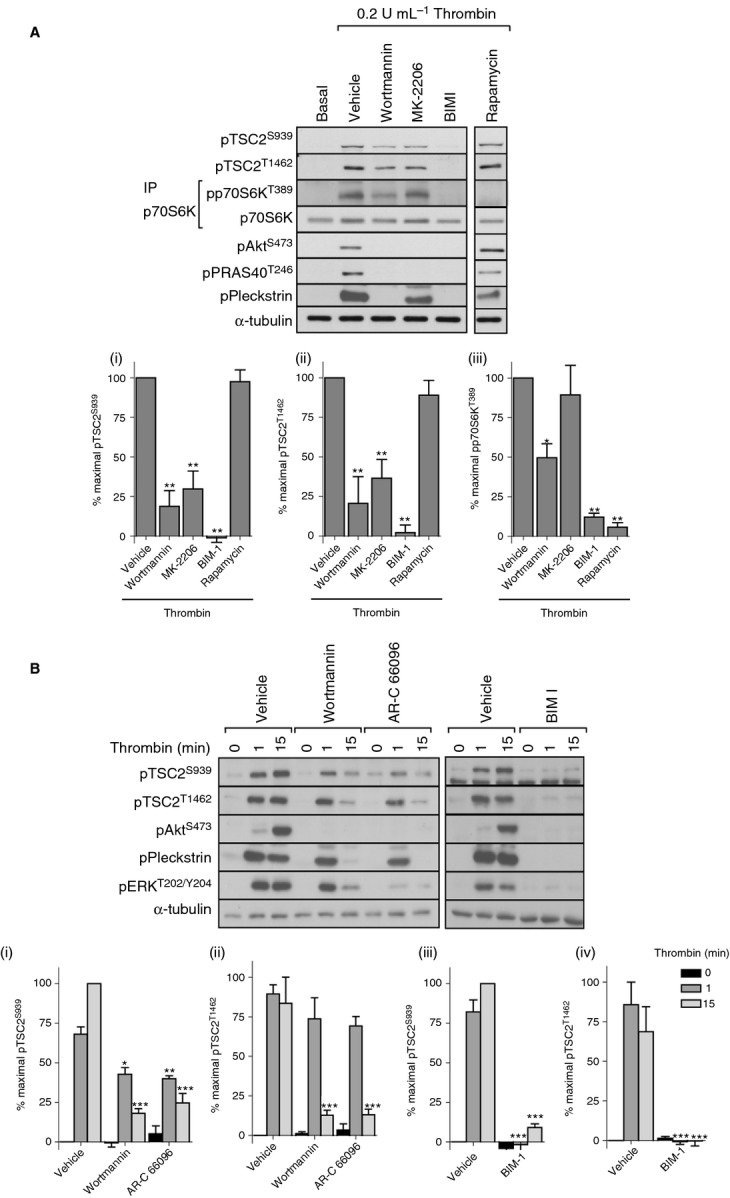
Akt-independent, PKC-dependent phosphorylation of p70S6K. Washed platelets were incubated with vehicle (0.2% DMSO), wortmannin (100 nmol L−1), MK-2206 (1 μmol L−1), BIM I (10 μmol L−1), or rapamycin (200 nmol L−1) for 15 min before stimulation with thrombin (0.2 U mL−1) for 15 min (A) or with vehicle (0.2% DMSO), wortmannin (100 nmol L−1), AR-C 66096 (1 μmol L−1), or BIM I (10 μmol L−1) for 15 min before stimulation with thrombin (0.2 U mL−1) for 1 or 15 min (B). Phosphorylation of the indicated proteins was assessed by western blotting of either whole cell lysates or immunoprecipitates (IP). Membranes were reprobed for α-tubulin to confirm equal loading. The bar graphs depict quantification of pTSC2 at Ser939 and Thr1462 (ratio phosphorylated/loading control) and pp70S6K at Thr389 (ratio phosphorylated/total) expressed as a percentage of the maximal signal induced by thrombin in control conditions. *P < 0.05, **P < 0.01, ***P < 0.001, indicating a significant difference from vehicle control (A) or between vehicle control and inhibitor-treated samples at the matching time point (B). Results are expressed as mean ± SE.
IGF-1 stimulates Akt-dependent TSC2 Ser939 and TSC2 Thr1462 phosphorylation but does not activate mTORC1
The mitogen IGF-1 is a classic agonist of the PI3 kinase/Akt pathway leading to activation of mTORC1 in many cell types 19,20. In platelets, IGF-1 stimulated transient phosphorylation of TSC2 on Ser939 and Thr1462, which closely followed phosphorylation of Akt Ser473 and the Akt substrate PRAS40 (Fig. 3A). Interestingly, despite robust TSC2 phosphorylation, we were unable to detect activation of mTORC1 by observation of either pThr389 p70S6K (Fig. 3A) or pSer65 4E-BP1 (data not shown), suggesting that TSC2 phosphorylation is not sufficient to activate mTORC1. In contrast to thrombin, IGF-1–stimulated TSC2 phosphorylation was completely dependent on PI3 kinase/Akt and ablated by both wortmannin and MK-2206 (Fig. 3B). These results demonstrate that IGF-1 stimulates Akt-dependent TSC2 phosphorylation but that this is insufficient to activate mTORC1 in human platelets.
Figure 3.
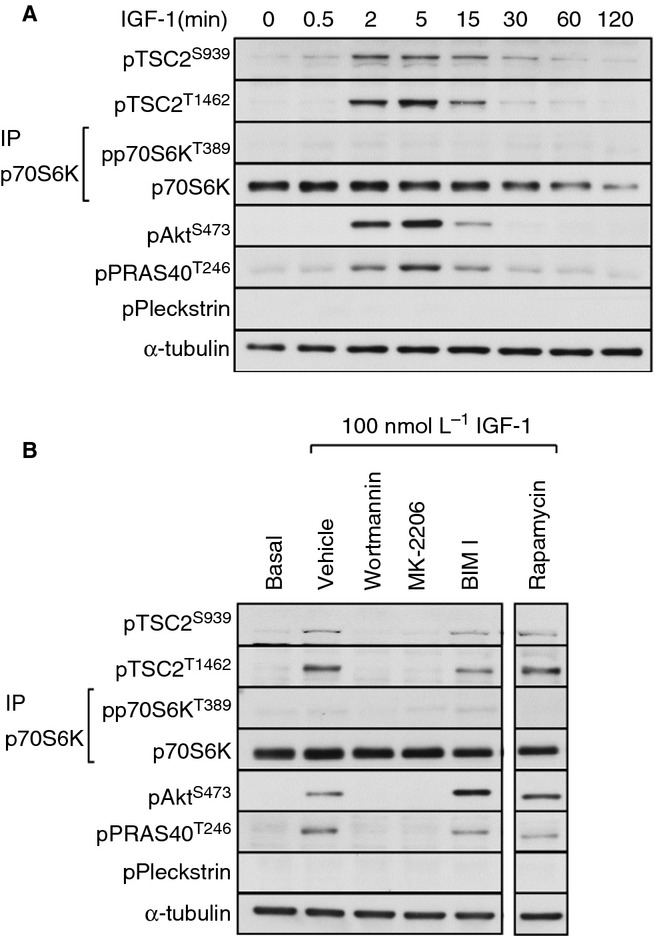
IGF-1–stimulated TSC2 phosphorylation is insufficient to activate mTORC1. Washed platelets were stimulated with IGF-1 (100 nmol L−1) for the indicated times (A) or incubated with vehicle (0.2% DMSO), wortmannin (100 nmol L−1), MK-2206 (1 μmol L−1), BIM I (10 μmol L−1), or rapamycin (200 nmol L−1) for 15 min before stimulation with IGF-1 (100 nmol L−1) for 15 min (B). Phosphorylation of the indicated proteins was assessed by Western blotting of either whole cell lysates or immunoprecipitates (IP). Membranes were reprobed for α-tubulin to confirm equal loading.
PKC-dependent ADP secretion is essential for thrombin-stimulated mTORC1 activity
The strong inhibitory effect of BIM I on thrombin-stimulated TSC2 and p70S6K phosphorylation suggests an important role for PKC in activation of the mTORC1 pathway. Diverse signaling pathways have been described that can directly or indirectly modulate mTORC1 activity 21–24. Of these, ERK, p90RSK and phospholipase D are activated downstream of PKC in human platelets 25,26. To evaluate the role of these pathways in PKC-mediated TSC2 phosphorylation and mTORC1 activation, we incubated platelets with pharmacological inhibitors of the MEK/ERK pathway and PLD. Inhibition of MEK (U0126 and PD98059) or PLD (VU 0155069 [PLD1] and VU 0364739 HCl [PLD2]) had no significant effect on TSC2 phosphorylation (Fig. 4A), demonstrating that ERK, RSK, and PLD do not contribute to thrombin-mediated TSC2 phosphorylation. Although U0126 resulted in a significant reduction in p70S6K phosphorylation, this was not observed with the structually unrelated compound PD98059, indicating that this effect of U0126 is likely to be off-target. This is supported by the finding that the MEK inhibitor SL 327 had no significant effect on p70S6K phosphorylation (data not shown). Both PLD inhibitors had no effect on p70S6K phosphorylation. Together, these results establish that ERK, p90RSK, and PLD do not contribute to thrombin-mediated mTORC1 activation. An alternative mechanism by which PKC can regulate platelet function is by contributing to platelet granule secretion 27, thereby releasing ADP, which further supports platelet function through the P2Y1 and P2Y12 receptors. The P2Y12 antagonist AR-C 66096, but not the P2Y1 antagonist MRS 2279, largely prevented thrombin-stimulated TSC2 and p70S6K phosphorylation (Fig. 4B). In contrast, blocking the other major amplification pathway, integrin αIIbβ3 activation with Abciximab or RGDS had no effect. The inhibitory effect of the PKC inhibitor on p70S6K phosphorylation was completely restored by the addition of ADP, whereas TSC2 phosphorylation was only partially rescued (Fig. 4C). Together, these results indicate that PKC contributes to TSC2 phosphorylation and activation of mTORC1 by promoting ADP secretion and subsequent activation of P2Y12.
Figure 4.

PKC-mediated ADP secretion and subsequent P2Y12 activation are essential for thrombin-induced p70S6K phosphorylation. Washed platelets were incubated with vehicle (0.2% DMSO), BIM I (10 μmol L−1), U0126 (10 μmol L−1), PD98059 (50 μmol L−1), VU 0155069 (10 μmol L−1), VU 0364739 HCl (10 μmol L−1), MRS 2279 (10 μmol L−1), AR-C 66096 (1 μmol L−1), abciximab (10 μg mL−1), or RGDS (1 mmol L−1) for 15 min before stimulation with thrombin (0.2 U mL−1) for 15 min (A, B). Washed platelets were incubated with vehicle (0.2% DMSO) or BIM I (10 μmol L−1) for 15 min before stimulation with thrombin (0.2 U mL−1), ADP (10 μmol L−1), or both for 15 min (C). Phosphorylation of the indicated proteins was assessed by Western blotting of either whole cell lysates or immunoprecipitates (IP). Membranes were reprobed for α-tubulin to confirm equal loading. The bar graphs depict quantification of pTSC2 at Ser939 and Thr1462 (ratio phosphorylated/loading control) and pp70S6K at Thr389 (ratio phosphorylated/total) expressed as a percentage of the maximal signal induced by thrombin in vehicle conditions. *P < 0.05, **P < 0.01, indicating a significant difference from vehicle control (A, B, C). In order to assess whether significant rescue of the effect of BIM-1 on thrombin stimulation was achieved, sample phosphorylation was compared to phosphorylation in thrombin + BIM-1–treated samples. †P < 0.05, ††P < 0.01, indicating a significant difference from thrombin + BIM-1–treated samples (C). Results are expressed as mean ± SE.
Phorbal 12-myristate 13-acetate stimulates TSC2 phosphorylation and activation of mTORC1 in a PKC-dependent, Akt-independent manner
To evaluate whether activation of PKC is sufficient for TSC2 phosphorylation and activation of mTORC1, we stimulated platelets with the PKC activator phorbal 12-myristate 13-acetate (PMA). Figure 5A demonstrates that PMA stimulated rapid TSC2 phosphorylation, which was sustained for 60 min, whereas p70S6K phosphorylation was much more delayed (as observed in response to thrombin in Fig. 2A). Although Akt Ser473 phosphorylation was detected on long exposures of the film, only very low levels of PRAS40 phosphorylation could be detected (Fig. 5A), suggesting that PMA is a weak activator of Akt. Indeed, inhibitor studies confirmed that PKC, and not the PI3 kinase/Akt pathway, was involved in PMA-mediated TSC2 phosphorylation and mTORC1 activity (Fig. 5B). Earlier, we demonstrate that P2Y12 was essential for thrombin-stimulated mTORC1 activity (Fig. 4B). PMA is a very poor platelet secretagogue and, unsurprisingly, antagonism of P2Y12 with AR-C 66096 had no effect on either TSC2 or p70S6K phosphorylation (Fig. 5B). This suggests that under non-physiological/PMA-stimulated conditions, active PKC is directly responsible for TSC2 and p70S6K phosphorylation and does not require ADP secretion.
Figure 5.
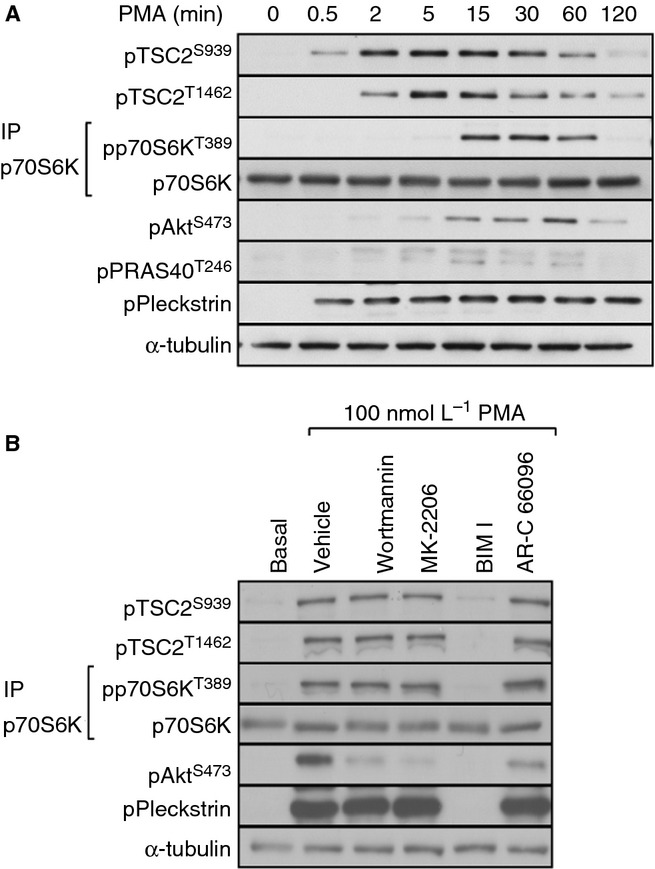
PMA stimulates TSC2 and p70SK phosphorylation independently of PI3 kinase/Akt. Washed platelets were stimulated with PMA (100 nmol L−1) for the indicated times (A) or incubated with vehicle (0.2% DMSO), wortmannin (100 nmol L−1), MK-2206 (1 μmol L−1), BIM I (10 μmol L−1), or AR-C 66096 (1 μmol L−1) for 15 min before stimulation with PMA (100 nmol L−1) for 15 min (B). Blots were performed on whole cell lysates with the exception of p70S6K which was immunoprecipitated from RIPA lysates. Membranes were reprobed for α-tubulin as a loading control.
P2Y12 contributes to mTORC1 activity through a PI3 kinase–independent pathway
The principal signaling pathways activated downstream of the P2Y12 receptor are Gαi-mediated inhibition of adenylate cyclase activity leading to protein kinase A (PKA) inhibition and activation of both Src kinases and PI3 kinase 28,29. In addition, diacylglycerol (DAG) kinase and potassium channels have been reported to be regulated downstream of P2Y12 29,30. Pharmacological mimetics of these effect (inhibition of adenylate cyclase with SQ 22536, PKA inhibition with H89, or blocking DAG kinase with R59949) did not restore activation of mTORC1 in thrombin-stimulated platelets in the presence of BIM I (Fig. 6A). We next investigated whether the rescue by ADP could be mimicked by mediators that activate Gαz and/or the PI3 kinase pathway, respectively 16,31. Although epinephrine and IGF-1 partially rescued BIM-mediated inhibition of Akt Ser473 phosphorylation, they failed to significantly reverse the effect of BIM on p70S6K phosphorylation (Fig. 6B). Interestingly, there appeared to be a trend for epinephrine to induce some restoration of the BIM-mediated inhibition of p70S6K phosphorylation; however, this did not translate into full rescue and did not reach significance. Conversely, epinephrine induced a significant partial rescue of TSC2 phosphorylation at both Ser939 and Thr1462. Thus, activation of Gαz or the PI3 kinase pathway alone is insufficient to fully rescue BIM-mediated inhibition of mTORC1 activity. In contrast, we found that the Src kinase inhibitor SU6656 blocked ADP-mediated rescue of p70S6K phosphorylation (Fig. 7A). Src therefore contributes to p70S6K phosphorylation downstream of ADP under conditions where PKC activity is compromised. Interestingly, SU6656 had little effect on thrombin-stimulated p70S6K phosphorylation and did not further reduce p70S6K phosphorylation in the presence of wortmannin (Fig. 7B), suggesting that PKC can bypass the requirement for Src downstream of P2Y12. Taken together, these results demonstrate that thrombin stimulates mTORC1 activity through PKC-mediated secretion of ADP and subsequent stimulation of P2Y12. Under conditions where PKC activity is absent, thrombin stimulates mTORC1 activity through ADP-mediated activation of the Src family of kinases.
Figure 6.
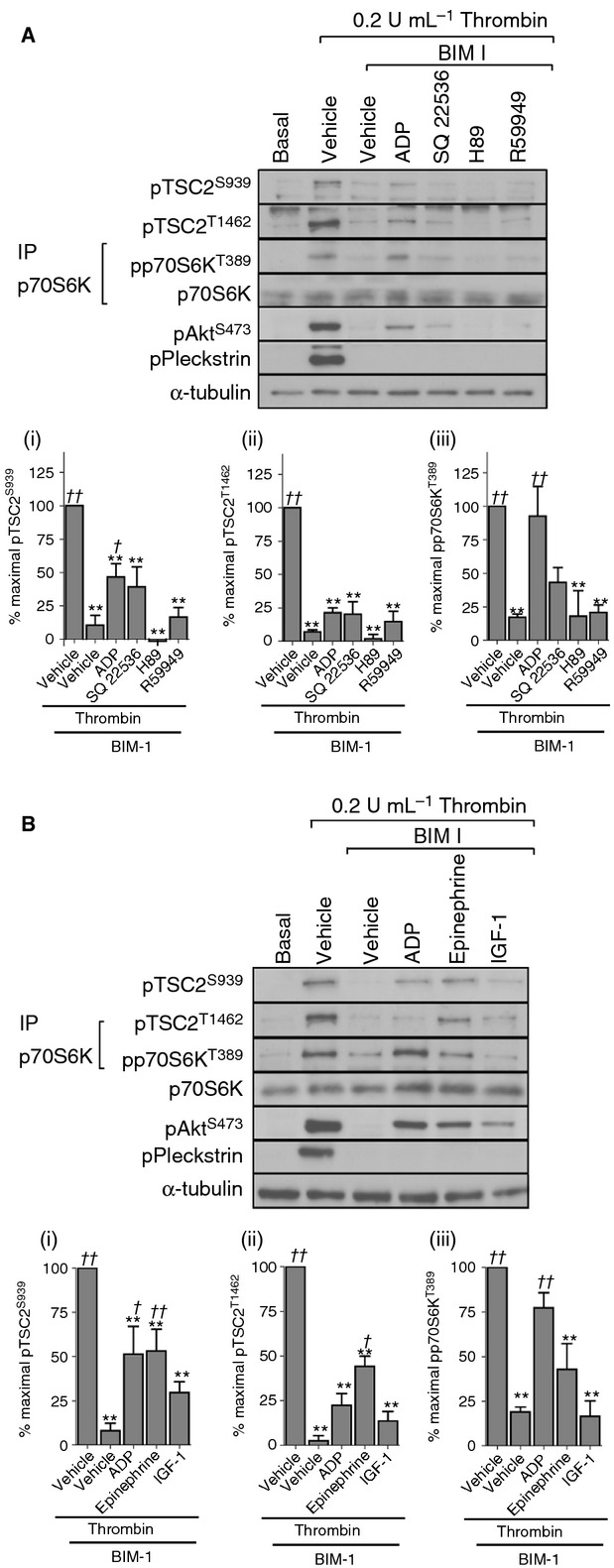
P2Y12 contributes to mTORC1 activity through a PI3 kinase–independent pathway. Washed platelets were incubated with vehicle (0.2% DMSO) or BIM I (10 μmol L−1) in the absence/presence of SQ 22536 (100 μmol L−1), H89 (10 μmol L−1), or R59949 (1 μmol L−1) for 15 min before stimulation with thrombin (0.2 U mL−1) (A) or thrombin (0.2 U mL−1) and ADP (10 μmol L−1), epinephrine (10 μmol L−1), or IGF-1(100 nmol L−1) for 15 min (B). Blots were performed on whole cell lysates with the exception of p70S6K, which was immunoprecipitated from RIPA lysates. Membranes were reprobed for α-tubulin as a loading control. The bar graphs depict quantification of pTSC2 at Ser939 and Thr1462 (ratio phosphorylated/loading control) and pp70S6K at Thr389 (ratio phosphorylated/total) expressed as a percentage of the maximal signal induced by thrombin in control conditions. **P < 0.01, indicating a significant difference from vehicle control(A, B). In order to assess whether significant rescue of the effect of BIM-1 on thrombin stimulation was achieved sample phosphorylation was compared to phosphorylation in thrombin + BIM-1–treated samples. †P < 0.05, ††P < 0.01, indicating a significant difference from thrombin + BIM-1–treated samples (A, B). Results are expressed as mean ± SE.
Figure 7.
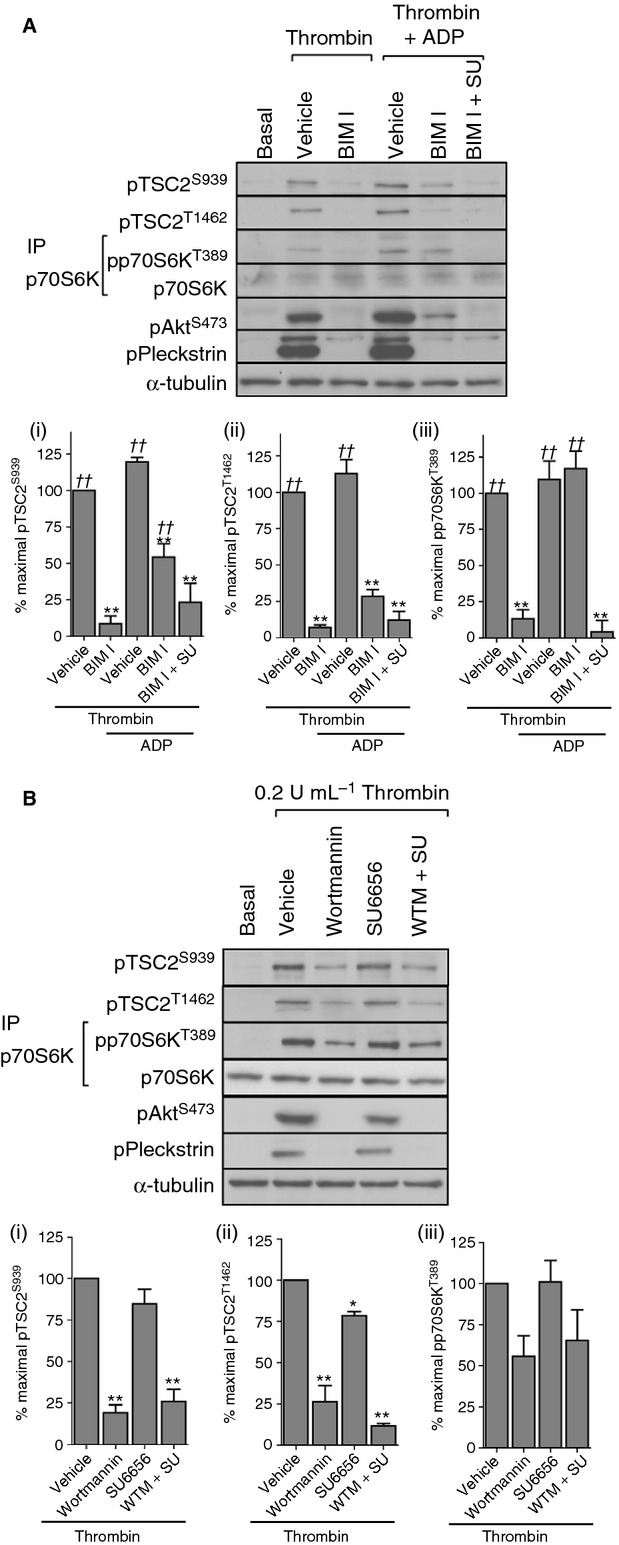
Involvement of Src in P2Y12-mediated mTORC1 activation under conditions of PKC inhibition. Washed platelets were incubated with vehicle (0.2% DMSO) or BIM I (10 μmol L−1) in the absence/presence of SU 6656 (20 μmol L−1) for 15 min before stimulation with thrombin (0.2 U mL−1) or thrombin (0.2 U mL−1) and ADP (10 μmol L−1) (A). Washed platelets were incubated with vehicle (0.2% DMSO) or wortmannin (100 nmol L−1) and/or SU6656 for 15 min before stimulation with thrombin (0.2 U mL−1) for 15 min (B). Blots were performed on whole cell lysates with the exception of p70S6K, which was immunoprecipitated from RIPA lysates. Membranes were reprobed for α-tubulin as a loading control. The bar graphs depict quantification of pTSC2 at Ser939 and Thr1462 (ratio phosphorylated/loading control) and pp70S6K at Thr389 (ratio phosphorylated/total) expressed as a percentage of the maximal signal induced by thrombin in control conditions. *P < 0.05, **P < 0.01, indicating a significant difference vehicle control (A, B). In order to assess whether significant rescue of the effect of BIM-1 on thrombin stimulation was achieved, sample phosphorylation was compared to phosphorylation in thrombin + BIM-1–treated samples. ††P < 0.01, indicating a significant difference from thrombin + BIM-1–treated samples (A). Results are expressed as mean ± SE.
Discussion
Despite growing evidence that activation of mTORC1 can alter platelet function, little is known about how this signaling complex is regulated. In other cell types, it is well characterized that growth factor–mediated activation of PI3 kinase and subsequent phosphorylation of Akt leads to phosphorylation of TSC on Ser939 and Thr1462, reducing its GAP activity on Rheb and ultimately resulting in the activation of mTORC1 and phosphorylation of its downstream substrates 32. Here, we demonstrate for the first time that in platelets, TSC2 and mTORC1 substrates are phosphorylated in response to a wide range of agonists. Further, we find that the regulation of TSC2 and mTORC1 is highly complex and differs between platelet stimuli. We found that TSC2 is phosphorylated at Ser939 and Thr1462 in response to physiological platelet agonists, the growth factor IGF-1, and the PKC activator PMA. Phosphorylation of TSC2 mediated by IGF-1 is solely dependent on PI3 kinase/Akt; conversely, phosphorylation induced by PMA is independent from PI3 kinase/Akt, despite weak Akt activation under these conditions. Regulation by the physiological platelet agonist thrombin is more complex. We found that early (1 min) phosphorylation of TSC2 was completely blocked by the PKC inhibitor BIM I, not rescued by the addition of exogenous ADP (data not shown), and only partially inhibited by loss of PI3 kinase or P2Y12 signaling (Fig. 2B). Whereas late (15 min) phosphorylation of TSC2, although completely blocked by the PKC inhibitor BIM I, was partially rescued by the addition of exogenous ADP (Fig. 4C). Further, loss of PI3 kinase/Akt or P2Y12 signaling resulted in attenuation of late TSC2 phosphorylation. Interestingly, under these conditions, both wortmannin and AR-C 66096 reduced phosphorylation of the PKC substrate pleckstrin (Figs 2B and 4B), suggesting an intricate interplay between the PI3 kinase pathway and PKC activation. Indeed, both P2Y12 and PI3 kinase have been implicated in sustaining pleckstrin phosphorylation 30,33,34. Thus, early TSC2 phosphorylation occurs through PAR-mediated Gαq activation of PKC, whereas sustained phosphorylation requires concomitant P2Y12 -mediated GαI activation of PI3 kinase/PKC signaling (Fig. 8). Our initial experiments showed that loss of PI3 kinase signaling resulted in a partial loss in thrombin-stimulated p70S6K phosphorylation. In contrast, inhibition of PKC prevented p70S6K phosphorylation by blocking PKC-mediated secretion of ADP and subsequent P2Y12 activation. However, ADP alone is unable to induce phosphorylation of p70S6K, suggesting that PAR and P2Y12 are necessary but individually insufficient to activate mTORC1. Several signaling pathways have been classically demonstrated to be activated downstream of P2Y12, but we ruled out roles for adenylyl cyclase, PKA, and DAG kinase. Further, activators of PI3 kinase such as IGF-1 and epinephrine failed to match the level of rescue induced by ADP, suggesting that rescue of PI3 kinase activation pathways alone does not lead to a concurrent rescue of mTORC1 activation. As recent reports suggest that Src family members are activated in ADP stimulated platelets 28,35, we also studied the effect of Src kinase inhibitors. We found that SU6656 completely prevented ADP-mediated rescue of p70S6K phosphorylation, demonstrating that Src kinases play an essential role in thrombin-stimulated mTORC1 under conditions where PKC is blocked. Interestingly, a recent study described a role for Src in mTORC1 activation in cells expressing hyperactive Src 36, supporting the idea that Src can promote mTORC1 activation under certain conditions. Despite the importance of P2Y12-mediated activation of Src kinases in the ADP-mediated rescue of mTORC1 activity, thrombin-mediated mTORC1 activity was surprisingly unaffected by SU6656 and SU6656 did not further reduce p70S6K phosphorylation in the presence of wortmannin. This is an interesting finding, as it demonstrates clear redundancy between signaling pathways that mediate mTORC1 activity, with a role for Src kinases only becoming apparent only under conditions where PKC activity is impaired. Together, these results show that thrombin is likely to stimulate mTORC1 activation through synergy between PKC and a P2Y12-stimulated pathway other than PI3 kinase/Src.
Figure 8.

Diagram demonstrating how TSC2 and mTORC1 are regulated in thrombin-activated human platelets. Early (1 min) TSC2 phosphorylation is regulated by PKC, whereas late (15 min) is regulated downstream of P2Y12 by PI3 kinase and PKC. mTORC1 activation requires concomitant PAR and P2Y12 signaling and either PKC or Src activation.
A well-established pathway of growth factor–mediated mTORC1 activity is Akt-mediated phosphorylation of the negative mTORC1 regulator PRAS40 at Thr246, resulting in its dissociation from mTORC1 and Akt-mediated inactivation of Rheb-GAP TSC2 37. Interestingly, our results demonstrate that the PI3 kinase/Akt/PRAS40 pathway is redundant in mTORC1 activation in human platelets. This is supported by the following findings: (i) although inhibition of Akt did result in the loss of thrombin-mediated PRAS40 phosphorylation, p70S6K phosphorylation was unaffected, (ii) PMA-stimulated phosphorylation of p70S6K occurred in the apparent absence of significant PRAS40 phosphorylation, and (iii) IGF-1 stimulated phosphorylation of Akt and PRAS40 without concomitant p70S6K phosphorylation. The role of TSC2 phosphorylation in mTORC1 activation is more controversial. The inability of IGF-1 to activate mTORC1 despite phosphorylating Akt/TSC2 suggests that TSC2 phosphorylation is not sufficient for mTORC1 activation. However, we cannot rule out that (i) TSC2 phosphorylation must exceed a critical threshold to permit mTORC1 activation, something that Akt alone cannot achieve, (ii) PKC phosphorylates additional residues on TSC2 that are important in mTORC1 activation, (iii) Akt and PKC phosphorylate different pools of TSC2, of which only the PKC pool couples to mTORC1, and (iv) PKC provides an additional signal that is also required for mTORC1 activation. The observations that (i) thrombin-induced TSC2 phosphorylation was partially inhibited by loss of Akt signaling without loss of p70S6K phosphorylation and (ii) that p70S6K phosphorylation was fully rescued by the addition of ADP on BIM I–treated platelets without full rescue of TSC2 phosphorylation, which suggests that TSC2 may be dispensable for thrombin-mediated mTORC1 activation. Although controversial, there is a precedent for this in the literature with activation of mTORC1 by amino acids and noxious stimuli requiring Rheb, but not TSC2 38, and activation of mTORC1 in glioma cells by AMOG (adhesion molecule on glial cells) being independent of both Rheb and TSC2 39. However, we clearly cannot rule out the possibility that lower levels of TSC2 phosphorylation are still sufficient to maximally support thrombin-stimulated mTORC1 activity. Indeed, without the possibility to use a pharmacological approach to target TSC2 in platelets, we cannot exclude its role in mTORC1 activation in platelets.
In conclusion, we demonstrate that TSC2 is activated downstream of PKC and Akt and that Akt/PRAS40 signaling is dispensable for mTORC1 activation in human platelets. We also propose a novel model of thrombin-mediated mTORC1 activation in which PKC and P2Y12 play a key role. Interestingly, there is clear redundancy in this system as the role for Src kinases only becomes apparent under conditions where PKC activity is impaired.
Addendum
S. F. Moore designed, analyzed, performed experiments, and wrote the manuscript; R. W. Hunter designed, analyzed, performed experiments, and edited the manuscript; I. Hers supervised the project, designed experiments, and wrote the manuscript.
Acknowledgments
We thank the healthy blood donors within the Medical Sciences Building, University of Bristol, for their generous donations. We also wish to acknowledge the assistance of Dr Andrew Herman, director of the Flow Cytometry Facility at the University of Bristol. This work was supported by the British Heart Foundation (grants PG/08/056/25325, PG/10/100/28658).
Disclosure of Conflict of Interest
The authors state that they have no conflicts of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Quantification of the kinetics of TSC2 phosphorylation and inhibition of pleckstrin and EBK phosphorylation by PI3 kinase and P2Y12 inhibitors.
References
- 1.Kaizuka T, Hara T, Oshiro N, Kikkawa U, Yonezawa K, Takehana K, Iemura S, Natsume T, Mizushima N. Tti1 and Tel2 are critical factors in mammalian target of rapamycin complex assembly. J Biol Chem. 2010;285:20109–16. doi: 10.1074/jbc.M110.121699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS, Sabatini DM. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137:873–86. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9:316–23. doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- 4.Avruch J, Hara K, Lin Y, Liu M, Long X, Ortiz-Vega S, Yonezawa K. Insulin and amino-acid regulation of mTOR signaling and kinase activity through the Rheb GTPase. Oncogene. 2006;25:6361–72. doi: 10.1038/sj.onc.1209882. [DOI] [PubMed] [Google Scholar]
- 5.Weyrich AS, Dixon DA, Pabla R, Elstad MR, McIntyre TM, Prescott SM, Zimmerman GA. Signal-dependent translation of a regulatory protein, Bcl-3, in activated human platelets. Proc Natl Acad Sci USA. 1998;95:5556–61. doi: 10.1073/pnas.95.10.5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weyrich AS, Denis MM, Schwertz H, Tolley ND, Foulks J, Spencer E, Kraiss LW, Albertine KH, McIntyre TM, Zimmerman GA. mTOR-dependent synthesis of Bcl-3 controls the retraction of fibrin clots by activated human platelets. Blood. 2007;109:1975–83. doi: 10.1182/blood-2006-08-042192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zimmerman GA, Weyrich AS. Signal-dependent protein synthesis by activated platelets: new pathways to altered phenotype and function. Arterioscler Thromb Vasc Biol. 2008;28:s17–24. doi: 10.1161/ATVBAHA.107.160218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aslan JE, Tormoen GW, Loren CP, Pang J, McCarty OJ. S6K1 and mTOR regulate Rac1-driven platelet activation and aggregation. Blood. 2011;118:3129–36. doi: 10.1182/blood-2011-02-331579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.López E, Berna-Erro A, Bermejo N, Brull JM, Martinez R, Garcia Pino G, Alvarado R, Salido GM, Rosado JA, Cubero JJ, Redondo PC. Long-term mTOR inhibitors administration evokes altered calcium homeostasis and platelet dysfunction in kidney transplant patients. J Cell Mol Med. 2013;17:636–47. doi: 10.1111/jcmm.12044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aslan JE, McCarty OJ. Regulation of the mTOR-Rac1 axis in platelet function. Small GTPases. 2012;3:67–70. doi: 10.4161/sgtp.19137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pan CJ, Tang JJ, Weng YJ, Wang J, Huang N. Preparation and characterization of rapamycin-loaded PLGA coating stent. J Mater Sci Mater Med. 2007;18:2193–8. doi: 10.1007/s10856-007-3075-9. [DOI] [PubMed] [Google Scholar]
- 12.Hunter R, Hers I. Insulin/IGF-1 hybrid receptor expression on human platelets; consequences for the effect of insulin on platelet function. J Thromb Haemost. 2009;7:2123–30. doi: 10.1111/j.1538-7836.2009.03637.x. [DOI] [PubMed] [Google Scholar]
- 13.Moore SF, Hunter RW, Harper MT, Savage JS, Siddiq S, Westbury SK, Poole AW, Mumford AD, Hers I. Dysfunction of the PI3 kinase/Rap1/integrin α(IIb)β(3) pathway underlies ex vivo platelet hypoactivity in essential thrombocythemia. Blood. 2013;121:1209–19. doi: 10.1182/blood-2012-05-431288. [DOI] [PubMed] [Google Scholar]
- 14.Moore SF, Hunter RW, Hers I. mTORC2 protein-mediated protein kinase B (Akt) serine 473 phosphorylation is not required for Akt1 activity in human platelets. J Biol Chem. 2011;286:24553–60. doi: 10.1074/jbc.M110.202341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hunter RW, Mackintosh C, Hers I. Protein kinase C-mediated phosphorylation and activation of PDE3A regulate cAMP levels in human platelets. J Biol Chem. 2009;284:12339–48. doi: 10.1074/jbc.M807536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hers I. Insulin-like growth factor-1 potentiates platelet activation via the IRS/PI3Kalpha pathway. Blood. 2007;110:4243–52. doi: 10.1182/blood-2006-10-050633. [DOI] [PubMed] [Google Scholar]
- 17.Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412:179–90. doi: 10.1042/BJ20080281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cai SL, Tee AR, Short JD, Bergeron JM, Kim J, Shen J, Guo R, Johnson CL, Kiguchi K, Walker CL. Activity of TSC2 is inhibited by AKT-mediated phosphorylation and membrane partitioning. J Cell Biol. 2006;173:279–89. doi: 10.1083/jcb.200507119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuemmerle JF. IGF-I elicits growth of human intestinal smooth muscle cells by activation of PI3K, PDK-1, and p70S6 kinase. Am J Physiol Gastrointest Liver Physiol. 2003;284:G411–22. doi: 10.1152/ajpgi.00310.2002. [DOI] [PubMed] [Google Scholar]
- 20.Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–5. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 21.Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science. 2001;294:1942–5. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- 22.Kam Y, Exton JH. Role of phospholipase D1 in the regulation of mTOR activity by lysophosphatidic acid. FASEB J. 2004;18:311–9. doi: 10.1096/fj.03-0731com. [DOI] [PubMed] [Google Scholar]
- 23.Lehman N, Ledford B, Di Fulvio M, Frondorf K, McPhail LC, Gomez-Cambronero J. Phospholipase D2-derived phosphatidic acid binds to and activates ribosomal p70 S6 kinase independently of mTOR. FASEB J. 2007;21:1075–87. doi: 10.1096/fj.06-6652com. [DOI] [PubMed] [Google Scholar]
- 24.Iijima Y, Laser M, Shiraishi H, Willey CD, Sundaravadivel B, Xu L, McDermott PJ, Kuppuswamy D. c-Raf/MEK/ERK pathway controls protein kinase C-mediated p70S6K activation in adult cardiac muscle cells. J Biol Chem. 2002;277:23065–75. doi: 10.1074/jbc.M200328200. [DOI] [PubMed] [Google Scholar]
- 25.Nadal-Wollbold F, Pawlowski M, Lévy-Toledano S, Berrou E, Rosa JP, Bryckaert M. Platelet ERK2 activation by thrombin is dependent on calcium and conventional protein kinases C but not Raf-1 or B-Raf. FEBS Lett. 2002;531:475–82. doi: 10.1016/s0014-5793(02)03587-1. [DOI] [PubMed] [Google Scholar]
- 26.Martinson EA, Scheible S, Greinacher A, Presek P. Platelet phospholipase D is activated by protein kinase C via an integrin alpha IIb beta 3-independent mechanism. Biochem J. 1995;310(Pt 2):623–8. doi: 10.1042/bj3100623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Konopatskaya O, Gilio K, Harper M, Zhao Y, Cosemans J, Karim Z, Whiteheart S, Molkentin J, Verkade P, Watson S, Heemskerk J, Poole A. PKCalpha regulates platelet granule secretion and thrombus formation in mice. J Clin Invest. 2009;119:399–407. doi: 10.1172/JCI34665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xiang B, Zhang G, Stefanini L, Bergmeier W, Gartner TK, Whiteheart SW, Li Z. The Src family kinases and protein kinase C synergize to mediate Gq-dependent platelet activation. J Biol Chem. 2012;287:41277–87. doi: 10.1074/jbc.M112.393124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shankar H, Kahner BN, Prabhakar J, Lakhani P, Kim S, Kunapuli SP. G-protein-gated inwardly rectifying potassium channels regulate ADP-induced cPLA2 activity in platelets through Src family kinases. Blood. 2006;108:3027–34. doi: 10.1182/blood-2006-03-010330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guidetti GF, Lova P, Bernardi B, Campus F, Baldanzi G, Graziani A, Balduini C, Torti M. The Gi-coupled P2Y12 receptor regulates diacylglycerol-mediated signaling in human platelets. J Biol Chem. 2008;283:28795–805. doi: 10.1074/jbc.M801588200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang J, Wu J, Kowalska MA, Dalvi A, Prevost N, O'Brien PJ, Manning D, Poncz M, Lucki I, Blendy JA, Brass LF. Loss of signaling through the G protein, Gz, results in abnormal platelet activation and altered responses to psychoactive drugs. Proc Natl Acad Sci USA. 2000;97:9984–9. doi: 10.1073/pnas.180194597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol. 2003;13:1259–68. doi: 10.1016/s0960-9822(03)00506-2. [DOI] [PubMed] [Google Scholar]
- 33.Wu CC, Wu SY, Liao CY, Teng CM, Wu YC, Kuo SC. The roles and mechanisms of PAR4 and P2Y12/phosphatidylinositol 3-kinase pathway in maintaining thrombin-induced platelet aggregation. Br J Pharmacol. 2010;161:643–58. doi: 10.1111/j.1476-5381.2010.00921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang J, Falck JR, Reddy KK, Abrams CS, Zhao W, Rittenhouse SE. Phosphatidylinositol (3,4,5)-trisphosphate stimulates phosphorylation of pleckstrin in human platelets. J Biol Chem. 1995;270:22807–10. doi: 10.1074/jbc.270.39.22807. [DOI] [PubMed] [Google Scholar]
- 35.Kim S, Jin J, Kunapuli SP. Relative contribution of G-protein-coupled pathways to protease-activated receptor-mediated Akt phosphorylation in platelets. Blood. 2006;107:947–54. doi: 10.1182/blood-2005-07-3040. [DOI] [PubMed] [Google Scholar]
- 36.Vojtechová M, Turecková J, Kucerová D, Sloncová E, Vachtenheim J, Tuhácková Z. Regulation of mTORC1 signaling by Src kinase activity is Akt1-independent in RSV-transformed cells. Neoplasia. 2008;10:99–107. doi: 10.1593/neo.07905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25:903–15. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 38.Smith EM, Finn SG, Tee AR, Browne GJ, Proud CG. The tuberous sclerosis protein TSC2 is not required for the regulation of the mammalian target of rapamycin by amino acids and certain cellular stresses. J Biol Chem. 2005;280:18717–27. doi: 10.1074/jbc.M414499200. [DOI] [PubMed] [Google Scholar]
- 39.Scheidenhelm DK, Cresswell J, Haipek CA, Fleming TP, Mercer RW, Gutmann DH. Akt-dependent cell size regulation by the adhesion molecule on glia occurs independently of phosphatidylinositol 3-kinase and Rheb signaling. Mol Cell Biol. 2005;25:3151–62. doi: 10.1128/MCB.25.8.3151-3162.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Quantification of the kinetics of TSC2 phosphorylation and inhibition of pleckstrin and EBK phosphorylation by PI3 kinase and P2Y12 inhibitors.