

Abstract
Aims
Chemotherapy-induced heart failure is increasingly recognized as a major clinical challenge. Cardiotoxicity of imatinib mesylate, a highly selective and effective anticancer drug belonging to the new class of tyrosine kinase inhibitors, is being reported in patients, some progressing to congestive heart failure. This represents an unanticipated challenge that could limit effective drug use. Understanding the mechanisms and risk factors of imatinib mesylate cardiotoxicity is crucial for prevention of cardiovascular complications in cancer patients.
Methods and results
We used genetically engineered mice and primary rat neonatal cardiomyocytes to analyse the action of imatinib on the heart. We found that treatment with imatinib (200 mg/kg/day for 5 weeks) leads to mitochondrial-dependent myocyte loss and cardiac dysfunction, as confirmed by electron microscopy, RNA analysis, and echocardiography. Imatinib cardiotoxicity was more severe in older mice, in part due to an age-dependent increase in oxidative stress. Mechanistically, depletion of the transcription factor GATA4 resulting in decreased levels of its prosurvival targets Bcl-2 and Bcl-XL was an underlying cause of imatinib toxicity. Consistent with this, GATA4 haploinsufficient mice were more susceptible to imatinib, and myocyte-specific up-regulation of GATA4 or Bcl-2 protected against drug-induced cardiotoxicity.
Conclusion
The results indicate that imatinib action on the heart targets cardiomyocytes and involves mitochondrial impairment and cell death that can be further aggravated by oxidative stress. This in turn offers a possible explanation for the current conflicting data regarding imatinib cardiotoxicity in cancer patients and suggests that cardiac monitoring of older patients receiving imatinib therapy may be especially warranted.
Keywords: Heart failure, Mitochondria, Chemotherapy, Cardiomyocytes, Cardioprotection
Introduction
Post-natal myocytes have limited regenerative capacity and their loss leads to cardiac remodelling that involves fibrosis and enlargement of the remaining myocytes.1 This process, which ultimately leads to heart failure, is often triggered by or accompanies ischaemic heart disease and is also increasingly observed in response to chemotherapy.2 In the case of anthracyclines, dysregulated mitochondrial biogenesis and increased myocyte death,3 which are sufficient to cause heart failure, underlie drug-induced heart failure.4
Imatinib mesylate (imatinib) was the first of a new generation of highly selective anticancer drugs that paved the way for several novel tyrosine kinase-targeted therapies. Imatinib targets the tyrosine kinase activity of the BCR–Abl fusion protein resulting from a chromosomal translocation, the Philadelphia chromosome which causes chronic myelogenous leukaemia (CML).5 The introduction of imatinib has revolutionized the treatment of CML patients, with an outstanding 70% cytogenic remission and initial minimal side effects, namely peripheral oedema and dyspnoea.6,7 Imatinib use is being extended to the treatment of other cancers, including gastrointestinal stromal tumours and prostate cancer.8–10
Imatinib therapy is often a life-long treatment; a decade after its introduction into the clinic, other side effects are emerging, notably the development of congestive heart failure (CHF).11–16 The finding that imatinib may be cardiotoxic was unexpected given the results of IRIS (International Randomized Study of Interferon versus STI571), where the overall incidence of CHF was ∼1% in both the imatinib and interferon arms.17 Other studies reported no cardiac toxicity in imatinib-treated patients.18 This discrepancy could be attributed to the lack of accurate and extensive cardiac function monitoring in most cancer clinical trials, and/or to variables that can affect cardiovascular parameters such as age, sex, or obesity. Of note, the median age in the IRIS study was 50 years, with a broad range from 19 to 70 years, while studies from registries reveal that the median age of CML patients at diagnosis is 60–65 years.19,20 It is also important to point out that cardiac toxicity could be latent, as seen with anthracyclines where cardiac dysfunction can occur years after the end of drug usage.2 Mechanistically, the latent effects could be the result of subclinical alterations of cardiac gene expression that ultimately impair the adaptive stress response of the heart.3 In the case of imatinib, no early changes in cardiac biomarkers have been analysed, but an earlier study suggested that imatinib-induced cardiac dysfunction might be due to mitochondrial alterations induced by c-Abl inhibition.12 At present, the effects of and the mechanisms underlying imatinib action on the heart remain incompletely understood. In the present work, we analysed the mechanism of imatinib action on the heart and investigated the impact of ageing on imatinib-induced cardiotoxicity. We found that imatinib induces cardiomyocyte loss via the activation of mitochondrial death pathways and that prevention of mitochondrial dysfunction through up-regulation of Bcl-2 protects against imatinib cardiotoxicity. Remarkably, clinical evidence of cardiotoxicity was found to be age dependent, and more overt in ageing mice. These results provide new insights into the mechanism of imatinib action on the heart and offer a possible explanation for the current controversy regarding imatinib-induced cardiotoxicity in cancer patients.
Methods
Cell cultures
Procedures with neonatal cardiomyocytes were as previously described.21 Imatinib mesylate (Gleevec®, Novartis) 100 mg tablets were dissolved in water and purified by successive centrifugation. Primary neonatal cardiomyocytes were treated with vehicle or imatinib at the indicated concentrations and times. Doxorubicin was used at 300 nM. The cloning and production of adeno-LacZ and adeno-GATA4 (G4) were previously reported.21
In vivo experiments
Mice were handled in accordance with institutional guidelines for animal care. Experiments were approved by the institutional Animal Care Committees and the investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1985). The Gata4+/− mice were described previously.3 Bcl-2-overexpressing mice were generated using an SV40 expression vector containing the human Bcl-2 cDNA under the control of the α-myosin heavy chain (MHC) promoter22 to direct expression specifically to cardiomyocytes. Human Bcl-2 expression was verified using northern and western blots. Treatment with imatinib mesylate (200 mg/kg/day) or vehicle was for 5 weeks, as suggested in published reports.12,23 Doxorubicin (Dox) treatment was a single i.p. injection of 15 mg/kg as previously described.3 M-mode echocardiography was performed using two Visual-Sonics VEVO 770 and VEVO 2100 systems and a 30 MHz linear array transducer, on mice lightly anaesthetized using 2% isofluorane and 80 mL/min of 100% oxygen, as described by Aries et al.,3 and echocardiographic indices were calculated as described by Yang et al.24 Heart failure was defined as an EF <45%.
Immunohistochemistry and terminal deoxynucleotidyltransferase-mediated dUTP end labelling
Immunohistochemical studies were performed as described.25,26 The ANF antibody [T4014, RGG-9103 (dilution 1:1000)] was purchased from Peninsula Laboratories (San Carlos, CA, USA). Homemade GATA4 antibody was used at a dilution of 1:1000. Nitrotyrosine antibody was used at a dilution of 1:400 (Abcam, UK). A Zeiss AxioImager.A2 light microscope (Objectives: Zeiss EC Plan-Neufluar; Camera: Zeiss AxioCam MRC) was used for image acquisition. Terminal deoxynucleotidyltransferase-mediated dUTP end labelling (TUNEL) assays were carried out using an Apoptag kit according to the manufacturer’s instructions (Intergen, Purchase, NY, USA).
RNA and protein
Northern and western blots were performed as reported previously.21 The Bcl-2 antibody [Ab-2; PC-68 (dilution 1:1000)] was from Oncogene Research Products (San Diego, California). The anti GATA4 and GATA6 antibodies were previously described.21 Real-time PCR (qPCR) was carried out as described by Debrus et al.26 Oligonucleotide sequences are available on request.
Transmission electron microscopy
Mice hearts were perfused with KCl–phosphate-buffered saline (PBS-KCl) solution to stop the heart in diastole and wash the blood out, and then for 10 min with 2% glutaraldehyde in 0.1 M sodium cacodylate (pH 7.4) buffer. Left ventricles were dissected, finely sliced, and incubated in the same fixative until processing. A JEOL 1230 transmission electron microscope was used.
Statistical analysis
Data are reported as means ± standard error of the mean (SEM). A Student unpaired t-test was used to compare any two groups, while the one-way analysis of variance (ANOVA) test was used to compare multiple groups. In all cases, a P- value <0.05 was considered as an index of statistical significance.
Results
Imatinib mesylate negatively affects cardiac function and structure
To analyse the effect of imatinib on the heart, wild-type mice (150 days old, n = 6 per group) were treated for 5 weeks with vehicle or with 200 mg/kg/day of imatinib. Earlier studies have shown that this dose produces blood concentrations comparable with those seen in human.23 Cardiac function was assessed using echocardiography. Table 1 lists all the parameters measured and shows that no significant difference was noted in the body weights of the two groups. Imatinib induced a reduction in the mitral valve mean gradient probably due to impaired cardiac relaxation, characteristic of diastolic dysfunction (Supplementary material online, Figure S1A). In contrast, fractional shortening, reflective of systolic function, was not significantly changed upon imatinib treatment. Mice treated with imatinib showed a reduced LV posterior wall thickness (LVPW) (Table ; Supplementary material online, Figure S1B and C), which was further confirmed by histological examination of Masson trichrome-stained tissue sections (Supplementary material online, Figure S1D). As a reduction in LV wall thickness may be indicative of myocyte loss, TUNEL assays were performed on heart sections to assess cell death. As shown in the representative Figure S1E in the Supplementary material online, imatinib treatment induced a three-fold increase in TUNEL-positive nuclei (percentage labelled nuclei vs. total cardiomyocyte nuclei) in treated mice as compared with the vehicle-treated mice (1.05 ± 0.08% vs. 0.300 ± 0.01%, P < 0.02).
Table 1.
Echocardiographic indices in the various mice groups studied
| Group | IVS (mm) | LVID (mm) | LVPW (mm) | HR (b.p.m.) | FS (%) | LV mass/BW (mg/g) | Age (days) |
|---|---|---|---|---|---|---|---|
| Young | 0.62 ± 0.03 | 4.29 ± 0.09 | 0.65 ± 0.02 | 442.5 ± 7.6 | 28.8 ± 1.3 | 3.31 ± 0.18 | 118.1 ± 3.6 |
| Young + I | 0.60 ± 0.04 | 4.47 ± 0.09 | 0.58 ± 0.02* | 449.5 ± 8.0 | 28.2 ± 1.4 | 3.13 ± 0.19* | 142.2 ± 3.8 |
| G4+/− | 0.76 ± 0.01 | 4.29 ± 0.05 | 0.70 ± 0.04 | 428.0 ± 34.0 | 29.5 ± 2.2 | 2.80 ± 0.10 | 145.0 ± 0.0 |
| G4+/− + I | 0.70 ± 0.04** | 4.46 ± 0.16 | 0.63 ± 0.02* | 396.0 ± 27.0 | 28.6 ± 2.4 | 2.60 ± 0.20** | 167.0 ± 11.0 |
| Bcl-2 | 0.61 ± 0.05 | 4.00 ± 0.15 | 0.66 ± 0.04 | 432.0 ± 5.0 | 30.2 ± 2.9 | 3.10 ± 0.00 | 122.0 ± 5.0 |
| Bcl-2 + I | 0.65 ± 0.03 | 3.94 ± 0.09 | 0.63 ± 0.03 | 441.0 ± 11.0 | 30.9 ± 0.9 | 3.10 ± 0.20 | 122.0 ± 5.0 |
| Old | 0.75 ± 0.06 | 4.40 ± 0.13 | 0.78 ± 0.07 | 484.0 ± 15.1 | 30.0 ± 0.8 | 3.41 ± 0.41 | 449.8 ± 14.9 |
| Old + I | 0.69 ± 0.03 | 4.63 ± 0.09* | 0.70 ± 0.06 | 453.2 ± 18.1 | 21.3 ± 0.8* | 3.18 ± 0.23 | 480.1 ± 15.1 |
| Old G4+/− | 0.79 ± 0.06 | 4.11 ± 0.19 | 1.01 ± 0.18 | 409.0 ± 29.0 | 24.3 ± 3.8 | 3.60 ± 0.80 | 441.0 ± 2.0 |
| Old G4+/− + I | 0.77 ± 0.06 | 5.33 ± 0.52* | 0.78 ± 0.12 | 422.0 ± 20.0 | 17.8 ± 2.6*,** | 5.00 ± 1.50 | 441.0 ± 1.0 |
BW, body weight; FS, fractional shortening; G4, GATA4; HR, heart rate; I, imatinib; IVS, interventricular septum (diastole); LVID, left ventricular interdimension (diastole); LVPW, left ventricular posterior wall (diastole).
Two-way analysis of variance was used to test significance with the genotype as the first variable and treatment as the second variable.
*Significance of treatment (P < 0.05);
**significance of genotype (P < 0.05).
Imatinib mesylate cardiotoxicity
WMaharsy et al.
Imatinib-induced cardiotoxicity is age dependent
Next, we investigated the impact of ageing on the severity of imatinib-induced cardiotoxicity. Old (450-day-old) and young (150-day-old) mice (n = 8 per group) were treated with imatinib and their cardiac parameters were measured 0, 2, and 5 weeks into the treatment (Table ). While the younger mice hearts showed no change in fractional shortening (contractility) and LV volume (chamber size), older mice hearts started to show a reduced contractility (fractional shortening) and an increased chamber size (LV volume) as early as 2 weeks into the treatment (Figure 1A and B). Not surprisingly, at the end of the 5 weeks of imatinib administration, fractional shortening and LV volume were significantly changed in these mice (Figure 1B). Trichrome staining revealed increased myocyte loss and fibrosis in the left ventricles of treated old mice; younger mice heart sections had little if any increase in fibrosis (Figure 1C). Consistent with the differential effects of imatinib on the two age groups, differential changes in gene expression were observed in old vs. young imatinib-treated mice (Supplementary material online, Figure S2). Real-time PCR analysis performed on cDNA samples obtained from mice ventricles showed similar down-regulation of certain genes such as βMHC and Bcl-XL in both treated groups. Other genes were differentially regulated in older mice. For example, the prosurvival transcription factor GATA4 and important markers of contractility such as ion channels KV4.2 and SERCA2A were down-regulated specifically in the treated older mice. Interestingly, mitochondrial integrity markers, such as PGC-1 and CYTB mRNA levels, were reduced only in the ventricles of older treated mice. Moreover, profibrotic genes, COL3A and CTGF, were up-regulated in old but not in young mice treated with imatinib. These changes in gene expression are consistent with an exaggerated cardiac stress response and remodelling of older imatinib-treated mice. This could be due to impaired homeostatic mechanisms predisposing ageing cardiomyocytes to drug-induced cardiac injury. A closer look at gene expression differences between untreated young and old mice (Supplementary material online, Figure S3) revealed a basal up-regulation of stress response genes such as βMHC, SERCA2A, and CYTB in the older population. Moreover, pro-death genes such as Beclin1, an autophagy marker, and BAX, as well as the hypoxia-responsive HIF1α gene were also increased in older mice.
Figure 1.
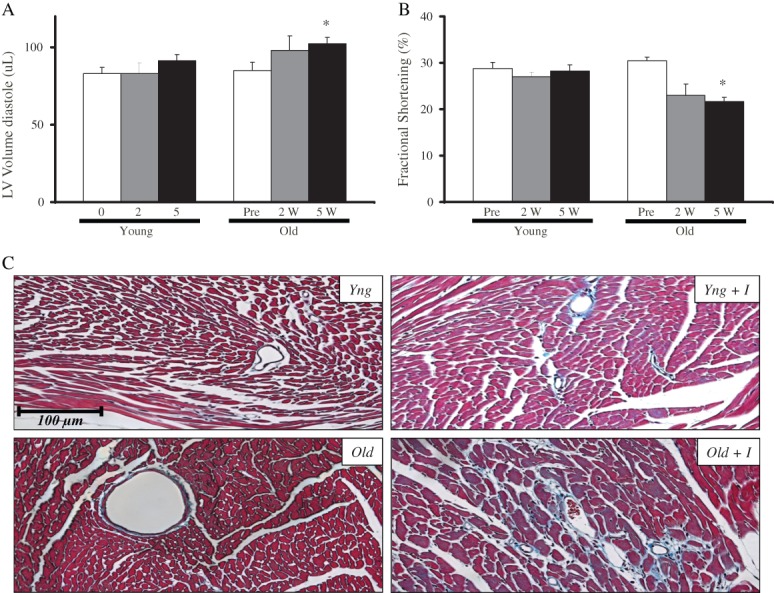
Imatinib-induced cardiotoxicity is age dependent. Echocardiographic data showing (A) changes in left ventricular chamber diastolic volume in young (Yng, 150 days old) and old (Old, 450 days old) mice treated with imatinib (200 mg/kg/day) at 0, 2 and 5 weeks of treatment; day 0 = no treatment, and (B) changes in fractional shortening in the same groups. The data shown are the mean ± SEM of n = 8 per group. (C) Trichrome-stained left ventricle sections from young and old mice treated with vehicle or imatinib (200 mg/kg/day) for 5 weeks. Note excessive remodelling in old treated hearts.
Transmission electron microscopy (TEM) was performed on mice heart tissues to determine the degree of tissue injury and the mechanisms of cell death. TEM analysis revealed the presence of extensive mitochondrial abnormalities, such as swelling, in imatinib-treated mice (Figure 2A and B). Abnormal mitochondria were also more abundant in older vs. younger untreated mice ventricles (Figure 2B). In the imatinib-treated older mice hearts, the basally stressed mitochondria became extremely disorganized, with some even losing their cristae (Figure 2A). Interestingly, abnormal nuclei and autophagic structures were common in imatinib-treated tissues (Figure 2C), suggesting the activation of necrotic, apoptotic, and autophagic cell death mechanisms, in agreement with previous findings reported by Herman et al.16 Necrotic nuclei were detected in all imatinib-treated mice, but were found at significantly higher levels in older vs. younger mice receiving imatinib (Figure 2D). Although autophagic lysosomal structures were more abundant in untreated old vs. young ventricles, their levels were comparable in both treated groups, suggesting increased sensitivity of younger hearts to the autophagic response (Figure 2E). Together, the ultrastructural changes observed are consistent with the gene expression analysis; they confirm that imatinib induces mitochondrial damage and mitochondrial-dependent cell death; they also suggest that the higher sensitivity of older mice to the drug may be due to the increased basal level of mitochondrial stress.
Figure 2.

Light microscopy and transmission electron microscopy (TEM) of imatinib-treated mouse ventricles. (A–D) TEM: (A) ×4000 magnification revealing mitochondrial disarray in the left ventricles of imatinib-treated mice, especially in the older animals (arrows). (B) Graph showing the quantification of disrupted mitochondria per square micrometre. (C) ×2000 magnification showing a representative normal nucleus from non-treated mice ventricles compared with swollen–apoptotic and fragmented–necrotic nuclei, as well as autophagy structures (asterisks) in imatinib-treated hearts. (D) Graph showing the quantification of necrotic nuclei per 100 µm2. (E) Quantification of autophagic/lysosomal vacuoles per 100 µm2. *P < 0.05.
Oxidative stress could be a major player in mitochondrial damage, and previously Herman et al. reported an increase in peroxynitrite, known to be a powerful oxidant, in imatinib-treated rat ventricles.16 Immunohistochemistry on sections from mice hearts detected no nitrotyrosine staining in either control or imatinib-treated young animals. However, a nitrotyrosine-positive reaction was observed in old non-treated left ventricles and increased significantly in imatinib-treated littermates (Supplementary material online, Figure S4F).
Imatinib-induced death is exacerbated in hydrogen peroxide-treated primary neonatal cardiomyocytes
To confirm whether the observed myocyte death was a direct effect of the drug on myocytes, we used primary neonatal cardiomyocytes cultures to carry out a time course and dose–response analysis. TUNEL assays performed on ventricular myocytes showed that imatinib concentrations as low as 0.5 µM were able to induce cell death significantly 18 h post-treatment (Figure 3A). Furthermore, the time course study revealed that the effects of imatinib (5 µM) on cell survival can be observed as early as 3 h post-treatment with a two-fold increase in TUNEL-positive cells (Figure 3B). These effects were comparable with the well-known effects of another cancer agent, Dox, on cardiomyocyte apoptosis (Figure 3C). We also tested whether the increased oxidative stress seen exclusively in older mice ventricles and known to be a hallmark of ageing hearts27,28 can reproduce the effects of ageing on imatinib-induced cardiotoxicity. Co-treatment of primary neonatal cardiac myocytes with H2O2 and imatinib led to a higher increase in TUNEL-positive cardiomyocytes than treatment with either H2O2 or imatinib alone (Figure 3D). Together, the data indicate that imatinib treatment has direct effects on cardiomyocytes which are further exacerbated by oxidative stress.
Figure 3.
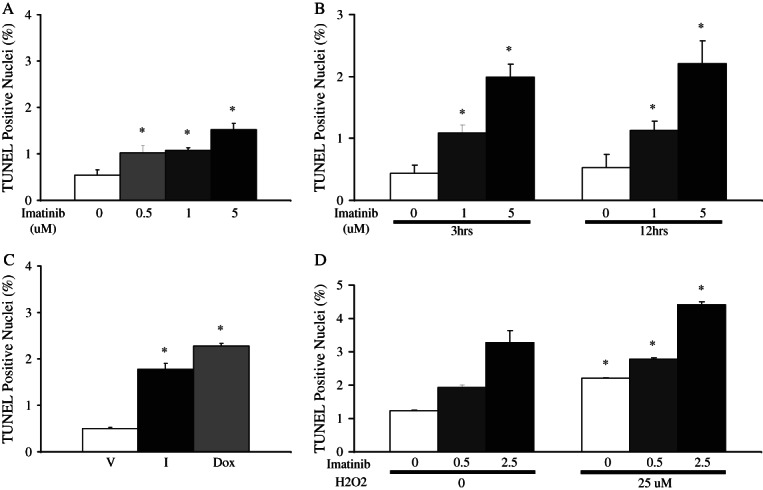
Imatinib acts directly on cardiomyocytes to induce cell death. The results shown were obtained in rat primary neonatal cardiomyocytes (CMCs) following treatment with imatinib (I) at the indicated doses and time. (A) Dose response at 18 h. (B) Time course. (C) Effects of imatinib- (5 mM) as compared with doxorubicin- (Dox) and vehicle- (V) treated cardiomyocytes on cell death. Treatments were for 18 h. (D) CMCs were treated with either vehicle or H2O2 (25 mM) for 24 h and then treated with different doses of imatinib (0, 0.5, and 2. 5 mM) for 18 h. In all cases, the data shown are the mean of three different experiments done, with 10 fields counted per experiment. *P < 0.05 vs. the respective control. TUNEL, terminal deoxynucleotidyltransferase-mediated dUTP end labelling.
GATA4 down-regulation is associated with Imatinib-induced cardiotoxicity
Transcriptome analysis revealed down-regulation of GATA4, an important regulator of cardiomyocyte survival.3 We analysed protein levels following imatinib treatment. Staining of histological sections with a specific GATA4 antibody indicated that GATA4 protein levels (brown nuclei) were markedly down-regulated in imatinib-treated mice vs. their control littermates (Figure 4A). This effect was also observed in cultured primary neonatal cardiomyocytes (Figure 4B) where GATA4 but not GATA6 down-regulation was evident following imatinib treatment (Figure 4B). Since decreased GATA4 levels sensitize cardiomyocytes to apoptosis and impair the heart adaptive response,3 we hypothesized that GATA4-haploinsufficient mice might be more sensitive to imatinib-induced cardiac toxicity. Wild-type and Gata4+/− mice (n = 7) were treated for 5 weeks with imatinib, and cardiac gene expression and function were assessed using qPCR and echocardiography. The QPCR analysis revealed a large increase (>40 fold, P < 0.05) in ANF mRNA levels in Gata4+/− mice treated with imatinib compared with a two-fold increase in the treated wild-type counterparts (Figure 4C), indicative of significant cardiac stress.29 The Bcl-XL mRNA level was significantly lower in untreated Gata4+/− hearts relative to their wild-type control littermates. Decreased levels of prosurvival and mitochondrial protective genes such as Bcl-XL could contribute to increased imatinib-induced cardiotoxicity in GATA4-haploinsufficient mice (Figure 4D). Indeed, echocardiography confirmed the higher sensitivity of Gata4+/− hearts to imatinib: whereas none of the young imatinib-treated wild-type mice had an EF <45% (suggestive of heart failure), a third of the similarly treated Gata4+/− littermates showed signs of heart failure with EFs of <45% (Figure 4E). Additionally, in the case of the older mice, all treated Gata4+/− mice were in heart failure compared with 60% of imatinib-treated age-matched wild-type mice (Figure 4F). In response to imatinib, the Gata4+/− mice showed a more significant increase in LV diameter compared with the wild-type mice, indicative of a maladaptive dilated cardiomyopathy response (Table ).
Figure 4.
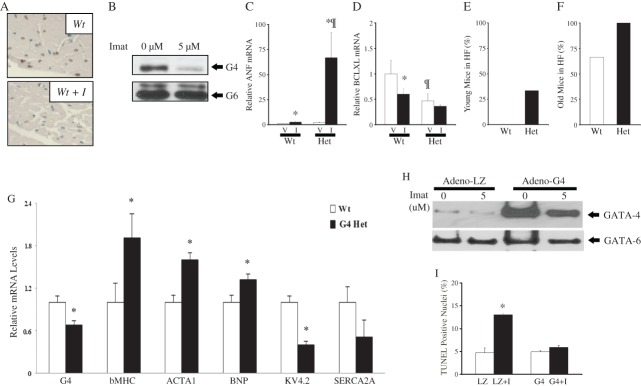
A role for GATA4 in imatinib-induced cardiotoxicity. (A) Histological sections stained with a GATA4 antibody and counterstained with methyl green reveal decreased GATA4 signal (brown nuclei) in ventricles from imatinib-treated mice. (B) Western blot analysis of GATA4 protein levels in primary neonatal cardiac myocytes treated with 5 mM imatinib for 18 h. Note the decrease in GATA4 but not in the related GATA6 protein. (C and D) Graph showing ANF and Bcl-XL mRNA levels in the ventricles of wild-type (Wt) or GATA4 heterozygous mice (Het) treated with vehicle (V) or imatinib (I) as obtained by real-time PCR (QPCR). (E and F) Increased incidence of heart failure (HF) post-imatinib treatment in Gata4+/− mice (Het) as compared with their wild-type (Wt) littermates (HF = EF <45%). (G) Graphs showing transcript changes using QPCR analysis on RNA from wild type (Wt) and Gata4+/− (Het) mice ventricles. (H) Primary neonatal cardiomyocytes were infected with adenoviruses expressing GATA4 or, as a control, LacZ (LZ). Western blot showing GATA4 and GATA6 levels in the indicated conditions. (I) Quantification of TUNEL (terminal deoxynucleotidyltransferase-mediated dUTP end labelling) assay. The data are the mean of triplicate plates with 10 fields counted per plate.
These results indicate that decreased GATA4 levels worsen imatinib cardiotoxicity and suggest that inhibition of GATA4 by imatinib may be one of the mechanisms underlying its cardiac toxicity. As shown in Figure 4G, gene expression profiling revealed a basal up-regulation of stress response genes in untreated young Gata4+/− hearts including βMHC, ACTA1, and BNP, and a decrease in GATA4-responsive genes such as KV4.2 and SERCA2A that were found to be inhibited by imatinib. The up-regulation of nitrotyrosine in Gata4+/− ventricles further confirmed our hypothesis that reduced GATA4 levels hypersensitize mice hearts to imatinib-induced mitochondrial stress (Supplementary material online, Figure S4). To confirm a role for GATA4 in imatinib-induced cardiotoxicity, we infected primary neonatal cardiomyocyte cultures with adenoviruses expressing GATA4 protein, or a LacZ control. Treatment of these cultures with imatinib decreased GATA4 levels in LacZ-infected cells but not in adeno-GATA4-infected cells in which GATA4 levels were increased three-fold over control cells (Figure 4H). Preventing GATA4 depletion blunted the imatinib-dependent increase in cell death as measured by TUNEL assays (Figure 4I). Together the results indicate that GATA4 is an important target in imatinib-induced cardiotoxicity and that restoring GATA4 can be beneficial.
Bcl-2 protein overexpression in mice hearts is protective
The cardioprotective effects of GATA4 are mediated, in part, by the prosurvival Bcl-2 and Bcl-XL proteins which inhibit apoptosis by counterbalancing the effects of the proapoptotic proteins (e.g. BAX and BAD), preventing mitochondrial uncoupling and cytochrome c release.30–32 By decreasing Bcl-2 and Bcl-X levels, imatinib treatment alters the ratio of pro-/antiapoptotic proteins which is critical to mitochondrial integrity and function. We tested whether overcoming decreases in GATA4 target prosurvival genes can prevent imatinib cardiotoxicity. Transgenic mice overexpressing human Bcl-2 under the control of the cardiomyocyte-specific α-MHC promoter were used (Figure 5A) and the levels of Bcl-2 were confirmed via northern and western blots (Figure 5B). Echocardiography showed that imatinib induced reductions in the LV mass to body weight ratio and the mitral valve gradient was attenuated in the Bcl-2 transgenic mice as compared with their treated wild-type littermates (Table ; Figure 5C). The ANF marker of cardiac stress was up-regulated in the ventricles of imatinib-treated wild-type mice but not in those from similarly treated Bcl-2 transgenics (Figure 5D). TUNEL assays performed on mice heart sections showed a five-fold increase in apoptotic nuclei in wild-type mice treated with imatinib but no detectable change in the number of TUNEL-positive nuclei in imatinib-treated Bcl-2 mice (Figure5E). As control, a group of wild-type and Bcl-2 transgenic mice were treated with Dox which is known to induce cardiomyocyte apoptosis and cardiac dysfunction.3 Imatinib-induced apoptosis was comparable with that induced by Dox, and Bcl-2 up-regulation in cardiomyocytes was protective against both treatments. Finally, left ventricles from both the non-treated and treated mice stained negative for nitrotyrosine (Supplementary material online, Figure S4).
Figure 5.
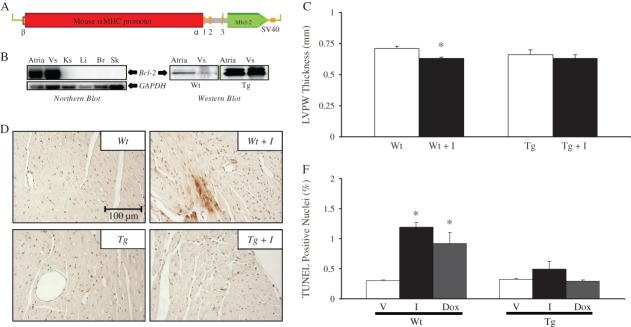
Myocardial up-regulation of Bcl-2 prevents imatinib cardiotoxicity. (A) Cardiomyocyte-specific Bcl-2 overexpression in transgenic mice using the α-myosin heavy chain (MHC) promoter. (B) Expression of Bcl-2 was confirmed using northern (left panel) and western blots (right panel). Vs, ventricles; Ks, kidneys; Li, liver; Br, brain; Sk, skeletal muscle. (C) Left ventricular posterior wall thickness (LVPW) obtained by echocardiographic analysis in treated wild-type (Wt) and Bcl-2 transgenics (Tg) relative to their respective vehicle-treated controls. Data are the mean ± SEM of n = 4–6. (D) Histological sections from mice from the four different groups stained with ANF antibody and counterstained with methyl green. Note the cytoplasmic presence of the ANF stress marker (brown) in the ventricles of Wt imatinib-treated but not Bcl-2 mice. (E) Percentage of apoptotic nuclei as detected by TUNEL (terminal deoxynucleotidyltransferase-mediated dUTP end labelling) assays in Wt and Bcl-2 (Tg) mice treated with vehicle (V), imatinib (I), or doxorubicin (Dox). The data shown are the mean from four different mice per group.
Together, the data presented indicate that imatinib directly and profoundly affects cardiomyocyte survival and biogenesis, and suggest that ageing is a risk factor in imatinib-induced cardiotoxicity.
Discussion
Chemotherapy-induced cardiotoxicity is a major impediment to the effective use of several anticancer drugs and has been most extensively studied in the case of anthracyclines.33–35 Drug-induced cardiotoxicity ranges from acute effects on ion channels—resulting in mild to life-threatening arrhythmias—to latent irreversible cardiomyopathies and heart failure. Because of the latter, undesirable cardiac effects of drugs often emerge after several years in clinical use. Over the past few years, accumulating reports are revealing unexpected cardiotoxicity of the new generation of antineoplastic ‘designer drugs’ belonging to the class of receptor tyrosine kinase inhibitors.11–14,36,37 Imatinib mesylate was the first of such highly specific anticancer drugs to be approved by the Food and Drug Administration (FDA)11 after results from the IRIS clinical study confirmed its potency in the treatment of CML patients with minimal side effects. Conflicting reports of imatinib-induced cardiotoxicity may be due to the modest or short-term cardiac monitoring associated with human anticancer clinical studies. Our study also suggests that variability in patients’ age and length of treatment may account for the differing levels of cardiac dysfunction observed.
Unlike the case of anthracyclines, few studies have analysed the effects of imatinib on the heart in well-defined experimental settings. In the present work, we investigated the mechanism(s) of imatinib action on the heart and the impact of age on cardiac response to imatinib treatment. Our results both in vivo and in vitro in cultured cardiomyocytes are consistent with a deleterious effect of imatinib on the heart that is dose, time, and age dependent. They also suggest that diastolic dysfunction may be an early manifestation of imatinib cardiotoxicity that could ultimately lead to heart failure. Moreover, reduced LV mass and increased cell death suggest that imatinib affects myocyte cell survival, an irreversible effect that could cause delayed cardiac manifestations as in the case of anthracycline cardiotoxicity. This is especially noteworthy given that imatinib treatment is life long and that cardiac damage can be cumulative, with evidence of heart failure appearing with the long-term therapy.
Gene expression analysis and high resolution TEM indicate that imatinib affects mitochondrial structure and that ageing exacerbates imatinib-induced cardiac tissue damage. Heart function and structure severely deteriorated in older mice compared with their younger counterparts and the alterations were accompanied by differential changes in gene expression, including up-regulation of the cardiac stress marker ANF and the profibrotic genes COL3A and CTGF. Up-regulated expression of several stress response genes in older untreated mice may explain their heightened susceptibility to imatinib-induced injury. The increased vacuolation and extensive mitochondrial swelling and disintegration seen by TEM in older hearts together with the increased levels of nitrotyrosine support the hypothesis that oxidative stress, a hallmark of ageing hearts, underlies their hypersensitivity to imatinib.
Our data generally support and extend the findings of Kerkela et al.12 In their study, Kerkela et al. suggested that inhibition of cardiomyocyte c-Abl and activation of the JunK pathway are the mechanisms of imatinib cardiotoxicity. It should be noted that this pathway was examined after 20–24 h treatment at a time when over half the cardiomyocytes were TUNEL positive, possibly reflecting an advanced stage of imatinib action. Others have shown that c-Abl suppression is neither cytotoxic to cardiomyocytes nor does it induce endoplasmic reticulum stress.38 In our study, we examined earlier time points after treatment with <5% of TUNEL-positive cardiomyocytes. Under these conditions, inhibition of the transcription factor GATA4 was evident, and preventing it rescued imatinib cardiotoxicity. GATA4 has emerged as a critical regulator of cardiomyocyte survival and the adaptive stress response of the adult heart.3,39 Our data uncover a role for the transcription factor GATA4 in imatinib cardiotoxicity. GATA4-haploinsufficient mice were more susceptible to imatinib cardiotoxicity with significantly increased incidence (reaching almost 100% of treated older mice) of heart failure and dilated cardiomyopathy. Previously, we found that the widely used class of anticancer drugs anthracyclines also decrease cardiomyocyte GATA4, and restoring GATA4 prevents drug-induced cardiotoxicity.3 It is therefore noteworthy that two chemically distinct anticancer drugs converge on GATA4 to alter cardiomyocyte survival and biogenesis.
Altogether these results show that imatinib-induced cardiotoxicity involves mitochondrial impairment and cell death that can be further worsened by increased oxidative stress as occurs in ageing myocytes. This offers a plausible explanation for the conflicting reports regarding imatinib cardiotoxicity in cancer patients and advocates for cardiac follow-up of CML patients receiving imatinib treatment. Finally, the fact that imatinib use has been extended to the treatment of patients with prostate cancer,40 a disease of older male subjects, coupled to a recent report about an elderly patient developing decompensated heart failure upon imatinib treatment,41 underscores the need for closer cardiac monitoring of older patients during and after imatinib therapy.
Acknowledgments
We thank Dr John Veinot for advice on electron microscopy, Janie Beauregard and Chantal Lefebvre for technical support, and Hélène Touchette for editorial assistance. We are grateful to the transgenic, histology, and pathology core facilities at IRCM, at the University of Ottawa, and at the University of Ottawa Heart Institute. We thank all members of the Nemer lab for helpful discussions throughout the study.
Funding
The Canadian Institutes of Health Research (CIHR).
Conflict of interest: none declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Imatinib induces cardiotoxicity in mice. A–C Echocardiographic analysis of heart function. A. Mitral valve (MV) mean gradient in vehicle (Wt) and imatinib-(Wt + I) treated mice. B. Fractional shortening (FS). C. LV posterior wall (LVPW) thickness in the same mice. The data shown are the mean of measurements obtained from 4–6 different mice (150 days old) per group. *P < 0.05 vs. the respective control. D. Quantification of LV wall thickness in Wt and Wt + I mice obtained via ImageJ® software analysis of Masson trichrome-stained heart Sections. A representative image is shown on the right. E. Representative TUNEL images with arrows indicating brown-stained apoptotic nuclei. Note the significant increase of TUNEL-positive nuclei in imatinib-treated heart sections.
Figure S2. Impaired regulation of stress response and mitochondrial biogenesis genes in older imatinib-treated mice. Graphs showing transcript changes using QPCR analysis and RNA from young (Y) or old (O) mice ventricles. Genes depicted in the bottom row correspond to known GATA4 targets. Note how changes in old animals treated with imatinib (0 + I) are indicative of mitochondrial dysregulation (BCL2, PGCla, and CYTB) fibrosis (COL3A and CTGF), and cardiac dysfunction (ANF, SERCA2A, and KV4.2). The data shown are the mean ± SEM of n = 6 and are corrected to S16 (used as internal control); values obtained from the non-treated ventricular samples were assigned an arbitrary value of 1. *P < 0.05.
Figure S3. Graphs showing QPCR results for some markers of mitochondrial and oxidative stress genes in untreated young vs. old hearts. Values obtained from the non-treated young ventricular samples were assigned an arbitrary value of 1. *P < 0.05.
Figure S4. Nitrotyrosine staining in ventricle sections from the different study groups. (+I = treatment with imatinib). Positive nitrotyrosine labelling indicative of oxidative stress was mostly observed in old treated ventricular sections. Positive nitrotyrosine labelling was also detected in untreated Gata4+/− ventricular sections (G4+/−) but not in imatinib-treated Bcl-2 transgenic mice (Tg).
References
- 1.van Berlo JH, Maillet M, Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest. 2013;123:37–45. doi: 10.1172/JCI62839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ewer MS, Ewer SM. Cardiotoxicity of anticancer treatments: what the cardiologist needs to know. Nat Rev Cardiol. 2010;7:564–575. doi: 10.1038/nrcardio.2010.121. [DOI] [PubMed] [Google Scholar]
- 3.Aries A, Paradis P, Lefebvre C, Schwartz RJ, Nemer M. Essential role of GATA-4 in cell survival and drug-induced cardiotoxicity. Proc Natl Acad Sci USA. 2004;101:6975–6980. doi: 10.1073/pnas.0401833101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whelan RS, Kaplinskiy V, Kitsis RN. Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol. 2010;72:19–44. doi: 10.1146/annurev.physiol.010908.163111. [DOI] [PubMed] [Google Scholar]
- 5.Goldman JM, Melo JV. Chronic myeloid leukemia—advances in biology and new approaches to treatment. N Engl J Med. 2003;349:1451–1464. doi: 10.1056/NEJMra020777. [DOI] [PubMed] [Google Scholar]
- 6.Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005;105:2640–2653. doi: 10.1182/blood-2004-08-3097. [DOI] [PubMed] [Google Scholar]
- 7.Kantarjian HM, Shah NP, Cortes JE, Baccarani M, Agarwal MB, Undurraga MS, Wang J, Ipina JJ, Kim DW, Ogura M, Pavlovsky C, Junghanss C, Milone JH, Nicolini FE, Robak T, Van DJ, Vellenga E, Bradley-Garelik MB, Zhu C, Hochhaus A. Dasatinib or imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: 2-year follow-up from a randomized phase 3 trial (DASISION) Blood. 2012;119:1123–1129. doi: 10.1182/blood-2011-08-376087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim SJ, Uehara H, Yazici S, Busby JE, Nakamura T, He J, Maya M, Logothetis C, Mathew P, Wang X, Do KA, Fan D, Fidler IJ. Targeting platelet-derived growth factor receptor on endothelial cells of multidrug-resistant prostate cancer. J Natl Cancer Inst. 2006;98:783–793. doi: 10.1093/jnci/djj211. [DOI] [PubMed] [Google Scholar]
- 9.Nabhan C, Villines D, Valdez TV, Tolzien K, Lestingi TM, Bitran JD, Christner SM, Egorin MJ, Beumer JH. Phase I study investigating the safety and feasibility of combining imatinib mesylate (Gleevec) with sorafenib in patients with refractory castration-resistant prostate cancer. Br J Cancer. 2012;107:592–597. doi: 10.1038/bjc.2012.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Le CA, Ray-Coquard I, Bui BN, Adenis A, Rios M, Bertucci F, Duffaud F, Chevreau C, Cupissol D, Cioffi A, Emile JF, Chabaud S, Perol D, Blay JY. Discontinuation of imatinib in patients with advanced gastrointestinal stromal tumours after 3 years of treatment: an open-label multicentre randomised phase 3 trial. Lancet Oncol. 2010;11:942–949. doi: 10.1016/S1470-2045(10)70222-9. [DOI] [PubMed] [Google Scholar]
- 11.Force T, Krause DS, Van Etten RA. Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat Rev Cancer. 2007;7:332–344. doi: 10.1038/nrc2106. [DOI] [PubMed] [Google Scholar]
- 12.Kerkela R, Grazette L, Yacobi R, Iliescu C, Patten R, Beahm C, Walters B, Shevtsov S, Pesant S, Clubb FJ, Rosenzweig A, Salomon RN, Van Etten RA, Alroy J, Durand JB, Force T. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat Med. 2006;12:908–916. doi: 10.1038/nm1446. [DOI] [PubMed] [Google Scholar]
- 13.Demetri GD. Structural reengineering of imatinib to decrease cardiac risk in cancer therapy. J Clin Invest. 2007;117:3650–3653. doi: 10.1172/JCI34252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toubert ME, Vercellino L, Faugeron I, Lussato D, Hindie E, Bousquet G. Fatal heart failure after a 26-month combination of tyrosine kinase inhibitors in a papillary thyroid cancer. Thyroid. 2011;21:451–454. doi: 10.1089/thy.2010.0270. [DOI] [PubMed] [Google Scholar]
- 15.Sarszegi Z, Bognar E, Gaszner B, Konyi A, Gallyas F, Jr, Sumegi B, Berente Z. BGP-15, a PARP-inhibitor, prevents imatinib-induced cardiotoxicity by activating Akt and suppressing JNK and p38 MAP kinases. Mol Cell Biochem. 2012;365:129–137. doi: 10.1007/s11010-012-1252-8. [DOI] [PubMed] [Google Scholar]
- 16.Herman EH, Knapton A, Rosen E, Thompson K, Rosenzweig B, Estis J, Agee S, Lu QA, Todd JA, Lipshultz S, Hasinoff B, Zhang J. A multifaceted evaluation of imatinib-induced cardiotoxicity in the rat. Toxicol Pathol. 2011;39:1091–1106. doi: 10.1177/0192623311419524. [DOI] [PubMed] [Google Scholar]
- 17.Hahn EA, Glendenning GA, Sorensen MV, Hudgens SA, Druker BJ, Guilhot F, Larson RA, O’Brien SG, Dobrez DG, Hensley ML, Cella D. Quality of life in patients with newly diagnosed chronic phase chronic myeloid leukemia on imatinib versus interferon alfa plus low-dose cytarabine: results from the IRIS Study. J Clin Oncol. 2003;21:2138–2146. doi: 10.1200/JCO.2003.12.154. [DOI] [PubMed] [Google Scholar]
- 18.Wolf A, Couttet P, Dong M, Grenet O, Heron M, Junker U, Laengle U, Ledieu D, Marrer E, Nussher A, Persohn E, Pognan F, Riviere GJ, Roth DR, Trendelenburg C, Tsao J, Roman D. Imatinib does not induce cardiotoxicity at clinically relevant concentrations in preclinical studies. Leuk Res. 2010;34:1180–1188. doi: 10.1016/j.leukres.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 19.Rohrbacher M, Berger U, Hochhaus A, Metzgeroth G, Adam K, Lahaye T, Saussele S, Muller MC, Hasford J, Heimpel H, Hehlmann R. Clinical trials underestimate the age of chronic myeloid leukemia (CML) patients. Incidence and median age of Ph/BCR-ABL-positive CML and other chronic myeloproliferative disorders in a representative area in Germany. Leukemia. 2009;23:602–604. doi: 10.1038/leu.2008.245. [DOI] [PubMed] [Google Scholar]
- 20.Rousselot P, Cony-Makhoul P, Nicolini F, Mahon FX, Berthou C, Rea D, Reiffers J, Bornand A, Saint-Jean O, Guilhot J, Guilhot F. Long-term safety and efficacy of imatinib mesylate (Gleevec®) in elderly patients with chronic phase chronic myelogenous leukemia: results of the AFR04 study. Am J Hematol. 2013;88:1–4. doi: 10.1002/ajh.23330. [DOI] [PubMed] [Google Scholar]
- 21.Charron F, Paradis P, Bronchain O, Nemer G, Nemer M. Cooperative interaction between GATA-4 and GATA-6 regulates myocardial gene expression. Mol Cell Biol. 1999;19:4355–4365. doi: 10.1128/mcb.19.6.4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paradis P, Dali-Youcef N, Paradis FW, Thibault G, Nemer M. Overexpression of angiotensin II type 1 receptor in cardiomyocytes induces cardiac hypertrophy and remodeling. Proc Natl Acad Sci USA. 2000;97:931–936. doi: 10.1073/pnas.97.2.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff NC, Ilaria RL., Jr Establishment of a murine model for therapy-treated chronic myelogenous leukemia using the tyrosine kinase inhibitor STI571. Blood. 2001;98:2808–2816. doi: 10.1182/blood.v98.9.2808. [DOI] [PubMed] [Google Scholar]
- 23.Yang XP, Liu YH, Rhaleb NE, Kurihara N, Kim HE, Carretero OA. Echocardiographic assessment of cardiac function in conscious and anesthetized mice. Am J Physiol. 1999;277:H1967–H1974. doi: 10.1152/ajpheart.1999.277.5.H1967. [DOI] [PubMed] [Google Scholar]
- 24.Nemer G, Nemer M. Cooperative interaction between GATA-5 and NF-ATc regulates endothelial–endocardial differentiation of cardiogenic cells. Development. 2002;129:4045–4055. doi: 10.1242/dev.129.17.4045. [DOI] [PubMed] [Google Scholar]
- 25.Debrus S, Rahbani L, Marttila M, Delorme B, Paradis S, Nemer M. The zinc finger-only protein Zfp260 is a novel cardiac regulator and a nuclear effector of α1-adrenergic signaling. Mol Cell Biol. 2005;25:8669–8682. doi: 10.1128/MCB.25.19.8669-8682.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raju R, Jian B, Hubbard W, Chaudry I. The mitoscriptome in aging and disease. Aging Dis. 2011;2:174–180. [PMC free article] [PubMed] [Google Scholar]
- 27.Bratic A, Larsson NG. The role of mitochondria in aging. J Clin Invest. 2013;123:951–957. doi: 10.1172/JCI64125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hayek S, Nemer M. Cardiac natriuretic peptides: from basic discovery to clinical practice. Cardiovasc Ther. 2010;29:362–376. doi: 10.1111/j.1755-5922.2010.00152.x. [DOI] [PubMed] [Google Scholar]
- 29.Gallagher JM, Komati H, Roy E, Nemer M, Latinkic BV. Dissociation of cardiogenic and postnatal myocardial activities of GATA4. Mol Cell Biol. 2012;32:2214–2223. doi: 10.1128/MCB.00218-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 31.Gupta S, Kass GE, Szegezdi E, Joseph B. The mitochondrial death pathway: a promising therapeutic target in diseases. J Cell Mol Med. 2009;13:1004–1033. doi: 10.1111/j.1582-4934.2009.00697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suter TM, Ewer MS. Cancer drugs and the heart: importance and management. Eur Heart J. 2013;34:1102–1111. doi: 10.1093/eurheartj/ehs181. [DOI] [PubMed] [Google Scholar]
- 33.Eschenhagen T, Force T, Ewer MS, De Keulenaer GW, Suter TM, Anker SD, Avkiran M, de AE, Balligand JL, Brutsaert DL, Condorelli G, Hansen A, Heymans S, Hill JA, Hirsch E, Hilfiker-Kleiner D, Janssens S, de JS NeubauerG, Pieske B, Ponikowski P, Pirmohamed M, Rauchhaus M, Sawyer D, Sugden PH, Wojta J, Zannad F, Shah AM. Cardiovascular side effects of cancer therapies: a position statement from the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. 2011;13:1–10. doi: 10.1093/eurjhf/hfq213. [DOI] [PubMed] [Google Scholar]
- 34.Steingart RM, Bakris GL, Chen HX, Chen MH, Force T, Ivy SP, Leier CV, Liu G, Lenihan D, Lindenfeld J, Maitland ML, Remick SC, Tang WH. Management of cardiac toxicity in patients receiving vascular endothelial growth factor signaling pathway inhibitors. Am Heart J. 2012;163:156–163. doi: 10.1016/j.ahj.2011.10.018. [DOI] [PubMed] [Google Scholar]
- 35.Turrisi G, Montagnani F, Grotti S, Marinozzi C, Bolognese L, Fiorentini G. Congestive heart failure during imatinib mesylate treatment. Int J Cardiol. 2010;145:148–150. doi: 10.1016/j.ijcard.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 36.Chu TF, Rupnick MA, Kerkela R, Dallabrida SM, Zurakowski D, Nguyen L, Woulfe K, Pravda E, Cassiola F, Desai J, George S, Morgan JA, Harris DM, Ismail NS, Chen JH, Schoen FJ, Van den Abbeele AD, Demetri GD, Force T, Chen MH. Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet. 2007;370:2011–2019. doi: 10.1016/S0140-6736(07)61865-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu W, Lu S, McAlpine I, Jamieson JD, Lee DU, Marroquin LD, Heyen JR, Jessen BA. Mechanistic investigation of imatinib-induced cardiac toxicity and the involvement of c-Abl kinase. Toxicol Sci. 2012;129:188–199. doi: 10.1093/toxsci/kfs192. [DOI] [PubMed] [Google Scholar]
- 38.Oka T, Maillet M, Watt AJ, Schwartz RJ, Aronow BJ, Duncan SA, Molkentin JD. Cardiac-specific deletion of Gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ Res. 2006;98:837–845. doi: 10.1161/01.RES.0000215985.18538.c4. [DOI] [PubMed] [Google Scholar]
- 39.Xu Y, Fang F, Sun Y, St Clair DK, St Clair WH. RelB-dependent differential radiosensitization effect of STI571 on prostate cancer cells. Mol Cancer Ther. 2010;9:803–812. doi: 10.1158/1535-7163.MCT-09-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ran HH, Zhang R, Lu XC, Yang B, Fan H, Zhu HL. Imatinib-induced decompensated heart failure in an elderly patient with chronic myeloid leukemia: case report and literature review. J Geriatr Cardiol. 2012;9:411–414. doi: 10.3724/SP.J.1263.2012.05251. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Imatinib induces cardiotoxicity in mice. A–C Echocardiographic analysis of heart function. A. Mitral valve (MV) mean gradient in vehicle (Wt) and imatinib-(Wt + I) treated mice. B. Fractional shortening (FS). C. LV posterior wall (LVPW) thickness in the same mice. The data shown are the mean of measurements obtained from 4–6 different mice (150 days old) per group. *P < 0.05 vs. the respective control. D. Quantification of LV wall thickness in Wt and Wt + I mice obtained via ImageJ® software analysis of Masson trichrome-stained heart Sections. A representative image is shown on the right. E. Representative TUNEL images with arrows indicating brown-stained apoptotic nuclei. Note the significant increase of TUNEL-positive nuclei in imatinib-treated heart sections.
Figure S2. Impaired regulation of stress response and mitochondrial biogenesis genes in older imatinib-treated mice. Graphs showing transcript changes using QPCR analysis and RNA from young (Y) or old (O) mice ventricles. Genes depicted in the bottom row correspond to known GATA4 targets. Note how changes in old animals treated with imatinib (0 + I) are indicative of mitochondrial dysregulation (BCL2, PGCla, and CYTB) fibrosis (COL3A and CTGF), and cardiac dysfunction (ANF, SERCA2A, and KV4.2). The data shown are the mean ± SEM of n = 6 and are corrected to S16 (used as internal control); values obtained from the non-treated ventricular samples were assigned an arbitrary value of 1. *P < 0.05.
Figure S3. Graphs showing QPCR results for some markers of mitochondrial and oxidative stress genes in untreated young vs. old hearts. Values obtained from the non-treated young ventricular samples were assigned an arbitrary value of 1. *P < 0.05.
Figure S4. Nitrotyrosine staining in ventricle sections from the different study groups. (+I = treatment with imatinib). Positive nitrotyrosine labelling indicative of oxidative stress was mostly observed in old treated ventricular sections. Positive nitrotyrosine labelling was also detected in untreated Gata4+/− ventricular sections (G4+/−) but not in imatinib-treated Bcl-2 transgenic mice (Tg).