

Abstract
The idiopathic inflammatory myopathies are a heterogeneous group of disorders characterised by diffuse muscle weakness and inflammation. A common immunopathogenic mechanism is the cytokine-driven infiltration of immune cells into the muscle tissue. Recent studies have further dissected the inflammatory cell types and associated cytokines involved in the immune-mediated myopathies and other chronic inflammatory and autoimmune disorders. In this review we outline the current knowledge of cytokine expression profiles and cellular sources in the major forms of inflammatory myopathy and detail the known mechanistic functions of these cytokines in the context of inflammatory myositis. Furthermore, we discuss how the application of this knowledge may lead to new therapeutic strategies for the treatment of the inflammatory myopathies, in particular for cases resistant to conventional forms of therapy.
Keywords: autoimmunity, autoinflammatory disease, cytokines, inflammation, neuroimmunology
Introduction
The idiopathic inflammatory myopathies are a heterogeneous group of autoimmune muscle disorders that have been classified into the following major types on the basis of their distinct clinical and pathological features and underlying immunopathogenic mechanisms: polymyositis (PM), dermatomyositis (DM), inclusion body myositis (IBM) and immune-mediated necrotizing myopathies (IMNM). Other subtypes include overlap syndromes and cancer-associated myositis 1,2 An autoimmune origin for these conditions is supported by immune cell-mediated myocytotoxicity, the presence of autoantibodies and over-expression of major histocompatibility complex (MHC) class I and II molecules in myositis tissue 3–6. However, there is still debate as to whether IBM is primarily autoimmune in origin or a degenerative myopathy with associated inflammatory features and an immune component.
A shared pathogenic mechanism in these diseases is the infiltration of muscle tissue by a variety of activated immune cells, a process that is heavily dependent upon the presence of multiple cytokines 7,8. Key cellular sources of these cytokines include regenerating myocytes within myositis muscle, cells from the adaptive immune response (CD4+ and CD8+ T cells, macrophages, dendritic cells and B cells) and cells from the innate immune system such as mast cells and gamma delta T cells 1,9–11.
The over-expression of tumour necrosis factor (TNF), interferon (IFN)-γ and interleukin (IL)-12 in the blood and muscle tissue of patients with various types of myositis has implicated the T helper type 1 (Th1) response as a key mediator of pathogenesis in these diseases 12. However, more recent studies detailing the presence of Th17-related cytokines such as IL-17, IL-22 and IL-6 have highlighted an alternative pathogenic mechanism and provided additional targets for therapy 13,14. In addition to their proinflammatory actions, these cytokines can also display anti-inflammatory properties and show duality of function depending on their concentrations, the local inflammatory milieu and the expression of co-stimulatory and adhesion molecules 15. In addition, a number of cytokines (such as IL-1α and IL-17) may also exert direct effects on the muscle tissue 13,16. These include the activation of signalling pathways such as nuclear factor (NF)-κB, further amplifying the inflammatory response through up-regulation of MHC-I expression and cytokine/chemokine production. Activation of NF-κB by proinflammatory cytokines can also have negative effects, inhibiting myocyte migration and differentiation, and may thereby impair muscle regeneration and repair 17.
In this review we will summarize the current understanding of key cellular sources of proinflammatory cytokines within myositis tissue and expression profiles among the different inflammatory myopathies. We also outline the multiple actions of these cytokines in the context of inflammatory myositis. Furthermore, we discuss the implications of these observations for the identification of novel therapeutic targets and development of new cytokine-based therapies for the treatment of resistant cases of IIM.
Immunopathogenesis of inflammatory myopathies
While the different inflammatory myopathies share a number of common characteristics, including muscle weakness and inflammation, each type has distinct clinical and pathological features 18. The underlying immunopathogenic mechanisms also differ, and the available evidence indicates that in PM and IBM a CD8+ T cell-mediated mechanism is primarily involved, whereas in DM and IMNM a humorally driven immune process is implicated 4,18–20. Although a number of autoantibodies have been identified (Table 1), their roles are uncertain and the specific antigenic targets of the immune response are still largely unknown, with the exception of a subgroup of cases of IMNM associated with antibodies to the signal recognition particle (SRP) and 3-hydroxymethyl glutaryl co-enzyme A reductase (HMGCR), which are thought to be involved in the pathogenesis of the myositis. In addition, there has been increasing recognition of the importance of non-immune mechanisms such as the possible contribution of MHC-I expression to muscle dysfunction and damage through the induction of endoplasmic stress and the unfolded protein response 32,33 It is also recognized that immature myogenic cells involved in muscle regeneration may activate Toll-like receptor (TLR) pathways leading to cytokine and chemokine production in addition to their role in antigen presentation, and may also be a target of the immune response 16,32,33.
Table 1.
Idiopathic inflammatory myopathies
| IM subtype | Muscle biopsy | Cellular infiltrate | Associated autoantibodies | References |
|---|---|---|---|---|
| Dermatomyositis | Perimysial and perivascular inflammation; perivascular atrophy; MAC deposition; MHC-I and II expression | CD4+ T cells; macrophages; B cells; plasma cells; BDCA-2 + plasmacytoid dendritic cells; CD8+ CD28−/− and CD4+ CD28n−/− T cells; mast cells; γ/Δ T cells | Anti-Mi 2 | 3,10,11,21–26 |
| Anti-TIF1y | ||||
| Anti-MDA5/anti-CADM-140 | ||||
| Anti-NXP-2 | ||||
| Anti-SAE | ||||
| Polymyositis | Endomysial inflammation; MHC-I and II expression | CD8+ T cells; macrophages; myeloid dendritic cells; CD68+ macrophages; BDCA-1 + myeloid dendritic cells; CD8+ CD28−/− and CD4+ CD28n−/− T cells; mast cells; γ/Δ T cells | Anti-tRNA synthetases (e.g. anti-Jo) | 3,10,11,21,25–27 |
| Anti-SRP | ||||
| Inclusion body myositis | Endomysial inflammation; rimmed vacuoles; protein aggregation, inclusions; MHC-I and II expression | CD8+ T cells; macrophages; myeloid dendritic cells; CD68+ macrophages; BDCA-1 + myeloid dendritic cells | Anti-cN1A | 3,21,25–27 |
| Immune-mediated necrotizing myopathies | Endomysial and perimysial inflammation; MAC, MHC-I expression | CD68+ macrophages; few CD4+, CD8+ T cells, B cells | Anti-HMGCR | 25,28–31 |
| Anti-SRP | ||||
| Anti-tRNA synthetases |
Adapted and modified from Dimachkie [25]. MAC = membrane attack complex; MHC = major histocompatibility complex; BDCA = blood dendritic cell antigen; Tif = and transcriptional intermediary factor; MDA = melanoma differentiation-associated protein; NXP = nuclear matrix protein; CADM = clinically amyopathic dermatomyositis; SAE = small ubiquitin-like modifier activating enzyme; SRP = signal recognition particle; HMGCR = 3-hydroxymethyl glutaryl co-enzyme A reductase.
Polymyositis and inclusion body myositis
In PM and IBM there is a mixed endomysial mononuclear inflammatory infiltrate comprising CD8+ T cells, macrophages and myeloid dendritic cells (Table 1). CD8+ cells surround and invade non-necrotic muscle fibres 34 and are thought to cause perforin-mediated cytotoxic injury as a result of the interaction between autoantigen-presenting MHC class I molecules on muscle fibres and co-stimulatory molecules on CD8+ cells 19. The invading mononuclear cells also include CD68+ macrophages and blood dendritic cell antigen (BDCA)-1+ myeloid dendritic cells, which are jointly thought to contribute to the cytotoxic injury and necrosis of muscle fibres 3,27. The infiltrating cells in polymyositis (and dermatomyositis) also include a population of apoptosis-resistant CD8+ and CD4+ T cells lacking the CD28 ligand (CD8+CD28−/−, CD4+CD28−/−), which are also thought to be cytotoxic effector cells 21. Studies employing T cell receptor spectratyping on muscle tissue and blood have shown that CD8+ T cells are clonally expanded in situ and persist over time and during relapses of disease in PM, while in IBM, which has a much more protracted clinical course, there is evidence that epitope spreading may occur with time 35–37. In IBM, in contrast to PM, there is increased expression of β-amyloid precursor protein (βAPP) and cell stress proteins and accumulation of amyloid proteins, and muscle fibres undergo progressive autophagic degeneration and atrophy 38. There is evidence from studies of muscle biopsies as well as in-vitro studies that this process may be secondary to the effects of proinflammatory cytokines such as IL-1β 39.
Dermatomyositis
In DM the inflammatory infiltrate comprises primarily CD4+ T cells, macrophages and small numbers of B cells and plasma cells, and is mainly perivascular and perimysial in distribution 22. In addition, BDCA-2+ plasmacytoid dendritic cells, which secrete type 1 IFNs, are present in the perimysium and endomysium (Table 1) 40. The immune response is thought to target the endothelium of capillaries and small blood vessels leading to activation of the complement pathway and deposition of C5b-9 membrane attack complexes, with resulting depletion of capillaries and muscle ischaemia 3,4. Deposition of immunoglobulins on intramuscular capillaries is postulated to activate the complement cascade, triggering the production of proinflammatory cytokines and chemokines which cause increased expression of adhesion molecules on endothelial cells and further recruitment of immune cells 3. Although antibodies reacting with a number of ubiquitous autoantigens have been identified in DM 20,23 endothelial cell-specific antibodies have not, as yet, been reported (Table 1).
Immune-mediated necrotizing myopathies
The IMNMs are a heterogeneous group of myopathies which, as a group, are characterized by a relative paucity or even absence of inflammatory changes in muscle tissue 41. However, in some cases CD68+ macrophages are prominent in the endomysium and perimysium, and small numbers of CD4+ and CD8+ T cells and B cells may also be present (Table 1) 28. In addition, there is diffuse expression of MHC-I antigens in muscle fibres, particularly in cases associated with statin therapy 29 or antibodies to 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGCR) 30,31, signal recognition particle (SRP) or tRNA aminoacyl synthetases 28,41. This finding is in keeping with an immune-mediated process in which muscle fibres participate in antigen presentation to the immune system. The complement pathway has also been implicated in IMNM, as shown by the presence of the membrane attack complex on muscle fibres in cases associated with anti-SRP or anti-synthetase antibodies 28 and evidence of complement-dependent antibody-mediated cytotoxicity in cases with anti-SRP antibodies 42.
Myositis-specific autoantibodies
Circulating antibodies to a number of ubiquitous autoantigens occur with variable frequencies in PM, DM and overlap syndromes and have more recently also been identified in IBM and IMNM (Table 1) 22,23,39–41. The most prevalent is the group of anti-synthetases, including anti-Jo-1 (anti-histidyl tRNA synthetase), which is present in about 20% of cases of PM and DM and is a marker for the anti-synthetase syndrome 29,30. In DM, antibodies to Mi-2, which is a component of the nucleosome remodelling deacetylase complex, have a high specificity especially for the adult form of the disease 42,43, and a number of other autoantibodies targeting melanoma differentiation-associated protein 5 (MDA-5), nuclear matrix protein 2 (NXP-2), small ubiquitin-like modifier activating enzyme (SAE) 1/2 and transcriptional intermediary factor 1 (TIF-1)α/γ have specificities for different subgroups of DM cases 22. There is increasing recognition that in addition to being potential biomarkers for different subgroups of inflammatory myopathy, some myositis-specific antibodies such as anti-Jo-1 and anti-Mi-2 may also have a role in the induction and maintenance of the autoimmune process as a result of over-expression of the target antigens in regenerating muscle fibres as part of the repair process in muscle 23,43,44.
Current concepts in cytokine functioning
Cytokines are small secreted proteins that regulate the immune response and play a major role in T cell differentiation and specialization and modulation of immune cell function. The release of cytokines from immune cells can be induced directly by immunoglobulin- or complement-receptor-mediated signalling or by the activation of a wide variety of cellular receptors by pathogen components. One family of such receptors, the Toll-like receptors (TLRs), play a crucial role in mediating inflammation by inducing cytokine release upon engagement of the receptor by an appropriate ligand, a process that is normally tightly regulated by a number of regulatory factors including microRNAs (miRNAs) 45,46. In muscle, activation of the TLR-3 pathway in human myoblasts induces production of IFN-β, which has also been shown to be expressed in myositis muscle tissue in immature muscle fibres which may be a target of the immune response 46.
Traditionally, cytokines were divided into classes on the basis of their known function; that is, proinflammatory versus anti-inflammatory/regulatory 15. However, increasing evidence demonstrates that a number of proinflammatory cytokines can have both stimulatory and inhibitory effects on immune cells, a phenomenon dependent upon the concentration of cytokine present, the local immune milieu and the influence of other interacting cells and molecules 15,47. A key example of this is transforming growth factor (TGF)-β, previously only considered to be an immune suppressor cytokine because of its role in the development and differentiation of regulatory T cells (Tregs). The recent identification of Th17 cells has also implicated TGF-β in proinflammatory responses due to its role in Th17 differentiation and function 48. Interestingly, recent studies have shown that the presence of pathogens or inflammatory mediators such as IL-6 and IL-1 can induce conversion of Tregs to Th17 cells and a switch in cytokine profiles. Furthermore, a number of studies have demonstrated that in the presence of IL-12 and/or TNF-α Th17 cells can be converted into non-classic Th1 cells which secrete both IFN-γ and IL-17. In comparison, differentiated Th1 and Th2 cells are stably expressed and prevented from converting to alternative T effector phenotypes associated with the reciprocal lineage. Differentiation of T-helper-cell subsets is controlled by lineage-specifying transcription factors that bind to regulatory elements in genes encoding cytokines and other transcription factors. Acetylation and methylation of histone molecules at the promoter and enhancer region of genes dictates the expression of genes associated with one T helper effector phenotype and the suppression of those associated with other lineages 49.
Nearly two decades ago, patients with rheumatoid arthritis were treated successfully with the first monoclonal antibody targeting TNF-α 50. Since then, intensive research efforts have been focused on key cytokines such as IL-1, IL-6, IL-12 and IL-17 and the targeting of these molecules therapeutically in a variety of inflammatory and autoimmune diseases 15,48. It is being increasingly recognized that cytokines do not act in isolation, but function within a milieu of multiple cytokines with the ability to synergize with or antagonize one another's functional capacity 47,51. For example, in-vitro studies examining the combined effects of TNF-α, IL-1β and IL-17 demonstrated enhanced activation of inflammatory signalling pathways in comparison to the individual cytokines alone. Furthermore, a number of pathways not activated by the individual cytokines alone were activated in the presence of the combined cytokines 52,53. This highlights the importance of investigating combination therapies or developing therapeutic strategies that will modulate multiple inflammatory pathways in the treatment of chronic inflammatory and autoimmune diseases 47,54.
Key cytokines in myositis
Over-expression of cytokines within DM, PM, IBM and IMNM muscle tissue is thought to contribute to a number of common immunopathogenic mechanisms in the inflammatory myopathies: co-stimulation, immune cell activation and transmigration of inflammatory cells into the muscle fibres (Fig. 1). Furthermore, cytokines can act directly on the muscle fibres, leading to the synthesis of soluble proinflammatory mediators, thus contributing to the persistence of the inflammatory response 19. The study of cytokine expression within myositis muscle began in 1986 with the identification of IL-2 and the IFNs α, β and γ in PM tissue 7.
Fig. 1.
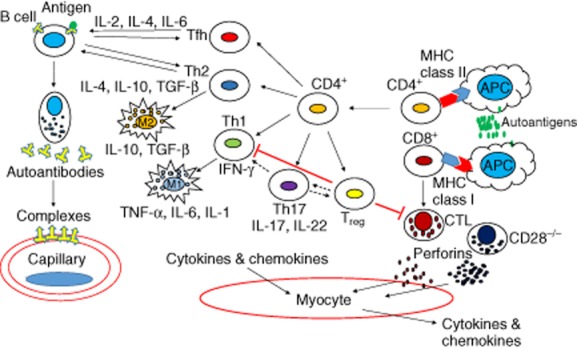
Immune effector cells and associated cytokines in myositis. CD4+ and CD8+ T cells are activated by autoantigen-expressing antigen-presenting cells (APCs). Activated CD4+ T cells differentiate into the various T helper effector cells. T helper type 1 (Th1) and Th17 cells secrete cytokines that mediate muscle damage and inflammation and the activation of additional immune cells. Th2 and Tfh cells modulate B cell function and differentiation into antibody producing plasma cells leading to complement mediated capillary damage. The presence of regulatory T cells (Treg) reduces inflammation and tissue damage by inhibiting CD4+ and CD8+ effector T cells. A degree of plasticity can also occur in these cell types Th17 → Th1 and Th17 ↔ Treg. MHC = major histocompatibility complex; CTL = cytotoxic T lymphocyte; CD28−/− = CD28 null T lymphocyte; M1 = type 1 macrophage (proinflammatory); M2 = type 2 macrophage (anti-inflammatory). Adapted and modified from Rayavarapu et al. 9
Since then, extensive research has characterized a wide variety of cytokines expressed in the different inflammatory myopathies (Table 2) and their functional effects. Cytokine profiling has been carried out both in muscle tissue and skin, in addition to blood and serum analysis for systemic expression.
Table 2.
Sources of key cytokines in the inflammatory myopathies (IM)
| Cytokines | IM subtypes | Serum | Mononuclear cells | Endothelial cells | Muscle | ECM | Skin | References |
|---|---|---|---|---|---|---|---|---|
| Th1 | ||||||||
| IFN-γ | All | + | + | – | DM, PM, IBM | – | DM | 7,12,28,55–57 |
| IL-2 | All | + | DM, PM, IBM | – | DM, PM, IBM | – | DM | 7,12,28,55 |
| IL-12 | All | + | + | – | + | – | – | 7,12,28,55 |
| TNF-α | All | + | + | – | + | – | – | 7,12,39,55,58 |
| Th2 | – | |||||||
| IL-4 | All | + | DM | – | + | – | DM | 7,12,59,60 |
| IL-13 | All | + | – | – | – | – | DM | 7,12,59,60 |
| Th17 | – | |||||||
| IL-17 | All | + | + | – | DM, PM, IBM | – | DM | 12,13,60–62 |
| IL-22 | DM, PM | – | + | PM | + | – | – | 13,63,64 |
| IL-23 | DM, PM | – | + | – | + | – | – | 13,65 |
| IL-6 | All | + | DM, PM, IBM | – | DM, PM, IBM | – | – | 12,13,65 |
| TWEAK | DM, PM, IBM | – | DM, PM, IBM | – | DM, PM, IBM | – | – | 66,67 |
| Treg | ||||||||
| IL-10 | DM, PM, IBM | + | + | – | DM, PM, IBM | – | – | 7,12,46,68,69 |
| TGF-β | DM, PM, IBM | – | DM | DM, PM, IBM | DM, PM, IBM | DM | DM | 7,64,65,68–71 |
| IL-1 family | ||||||||
| IL-1α | DM, PM, IBM | – | DM, PM, IBM | DM, PM, IBM | DM, PM, IBM | – | – | 7,12,70 |
| IL-1β | All | All | DM, PM, IBM | – | DM, PM, IBM | – | – | 7,12,39,58,70 |
| Type I IFNs | ||||||||
| IFN-α | All | + | DM, PM, IBM | – | DM | – | DM | 1,7,12,40,64,72–75 |
| IFN-β | DM, PM | + | DM, PM, IBM | – | DM, PM | – | DM | 1,7,40,64,72,74,76 |
Adapted and modified from Figarella-Branger et al. 7. All refers to dermatomyositis (DM), polymyositis (PM), inclusion body myositis (IBM), immune-mediated necrotizing myopathies (IMNM). ECM = extracellular matrix; IL = interleukin; IFN = interferon; TGF = transforming growth factor; TNF = tumour necrosis factor; TWEAK = TNF-like weak inducer of apoptosis; Treg = regulatory T cell; Th1 = T helper type 1.
Muscle tissue
Investigation of cytokine expression in myositis began initially with immunohistochemical studies of muscle tissue. As a method for cytokine detection, immunohistochemistry is hampered by the low concentrations of cytokines within the tissue, their transient expression and issues of sensitivity and specificity 7. As a result, a number of research groups turned their focus to the examination of cytokine mRNA transcripts in muscle tissue 77. These studies showed strong expression of inflammatory cytokines in all myositis subtypes, with the majority suggesting a Th1 response 7,55. The application of gene expression array technology to myositis research has led to the identification of additional immune cells and pathways within inflammatory myopathy muscle tissue 77.
Such gene array studies have identified a prominent type I IFN genetic signature in DM muscle in comparison with other forms of myositis 72,78. However, expression of IFN-α and IFN-β and their inducible genes is not exclusively specific to DM and also occurs in PM 1,78, and PM patients have shown responsiveness to anti-IFN-α therapy 73,79. Furthermore pDCs, a major source of IFN-α, have been identified in the muscle tissue of patients with PM and IBM in addition to DM 77. A similar pattern of expression of IL-1α, IL-1β and TGF-β has also been demonstrated across DM, PM and IBM tissue. In particular, enhanced IL-1α expression in the endothelial cells has been noted in DM, PM and IBM tissue, highlighting these cells as a key proinflammatory cytokine source 7,70.
The key Th1 cytokine IFN-γ has been detected in the muscle tissue of all the major subtypes of inflammatory myopathy 7,28. IFN-γ and two other Th1 cytokines, IL-1β and TNF-α, were found to be markedly increased in IBM muscle compared to PM and DM tissue 39. Additionally, the IFN-γ signalling pathway has been demonstrated to be up-regulated in the invaded versus non-invaded muscle fibres in IBM tissue 56. A strong relationship between degeneration-associated markers and the expression of IFN-γ, IL-1β and TNF was observed mainly in IBM muscle tissue, compared to PM and DM 39. The Th1 response through IFN-γ secretion leads to the induction of M1 macrophages which results in further tissue damage 9. IMNM, previously described as an immune myopathy with a limited mononuclear cell infiltrate, has also been shown to display a strong M1/Th1 response within the muscle tissue 28.
Traditionally, a Th1 response was considered to be a predominant driver of disease in the inflammatory myopathies. However, the discovery of IL-17 and the Th17 lineage has drawn attention to the role of IL-17 signalling in the pathogenesis of myositis. Increased IL-17 mRNA and protein expression has been demonstrated in DM, PM and IBM muscle tissue 13. One study in PM and DM patients indicated a Th17-mediated pathway in patients who were responsive to intravenous immunoglobulin (IVIG) therapy 61. Another Th17 cytokine, IL-22, has also been detected in PM, DM and IBM tissue and found to co-express with a proportion of IL-17 cells 63,64. IL-22 was also found to correlate with disease activity in PM and DM patients 63. Furthermore IL-22 mRNA expression was down-regulated in DM patients who responded clinically to IVIG therapy, whereas IBM patients who were clinical non-responders showed no change in IL-22 expression 64.
The novel cytokine, TNF-like weak inducer of apoptosis (TWEAK), which is a member of the TNF superfamily, and its receptor FGF-inducible molecule-14 (Fn14) have been implicated in the pathogenesis of autoimmune diseases such as rheumatoid arthritis and multiple sclerosis. TWEAK has been shown to synergize with Th17 polarizing cytokines to enhance IL-17 production by Th17 cells and blockade of the TWEAK receptor Fn14 suppresses Th17 differentiation 66. More recently the TWEAK/Fn14 axis has also been implicated in myositis and muscle atrophy 67,80. Enhanced TWEAK/Fn14 mRNA and protein expression was demonstrated in IBM muscle, in contrast to PM and DM. Furthermore, siRNA inhibition of TWEAK expression restored myogenic differentiation in IBM and DM via inhibition of the NF-кB pathway 67.
Th2 cytokines such as IL-4 and IL-13 are significantly over-expressed in IBM and PM tissue in comparison to DM and healthy controls 7,59,60. Conversely, a Th2 cell infiltrate in DM muscle has been associated with a lower severity of disease 60. This is due possibly to the generation of M2 macrophages by Th2 cytokines leading to tissue repair 9. This is in contrast with the view that DM is a humorally mediated disease whereby Th2 cells would drive B cell maturation and differentiation into autoantibody-producing plasma cells, further exacerbating microvascular damage. Furthermore, there is higher expression of immunoglobulin genes in the muscle of IBM and PM patients in comparison to DM 40,64,81. By contrast, there is a more prominent IFN-inducible gene signature in DM in comparison to the other subtypes 64,74.
The cytokines TGF-β and IL-10 are considered traditionally to be anti-inflammatory; however, as discussed earlier in this review, cytokines can display both pro- and anti-inflammatory properties depending on the context. Enhanced TGF-β mRNA expression has been observed in both DM and IBM muscle, and following IVIG treatment a decrease in expression was observed in DM but not IBM 64,71. Over-expression of IL-10 mRNA in PM, IBM and DM muscle in comparison to non-myositic tissue has also been demonstrated. Again, a decrease in expression was observed in DM but not IBM tissue following IVIG therapy 46,76. The down-modulation of TGF-β and IL-10 following IVIG treatment in DM suggests a key role for these molecules in DM, but not IBM, pathogenesis.
Skin
Skin involvement is a unique characteristic of DM that distinguishes it from the other inflammatory myopathies. The pathogenesis of skin inflammation in DM is less well characterized than in muscle. Gene microarray analysis demonstrated an IFN signature in DM skin, although this study did not assign this as being ‘type I’ or ‘type II’ 82. An in-vitro study comparing muscle- and skin-derived T cells demonstrated differential cytokine production between the two tissue types. Higher levels of IL-4 and IFN-γ were secreted by muscle-derived T cells compared to skin-derived T cells and the ratio of IL-4/IFN-γ was associated with severity of muscle injury. Conversely, skin-derived T cells produced higher amounts of IL-17A than muscle-derived T cells 60. Furthermore, an additional study demonstrated an enhanced mast cell infiltrate in juvenile DM skin in comparison to paired muscle tissue 83. Over-expression of TGF-β mRNA has been described in the endomysial connective tissue of DM muscle and associated with fibrosis of the skin 64,68. Conversely, a depletion of TGF-β, IL-10 and forkhead box protein 3 (FoxP3)-positive cells has been demonstrated in DM skin, suggesting that depletion of Treg cells and associated cytokines is a key factor in the pathogenesis of the skin changes 68,69.
Blood/serum
Similar to studies in muscle tissue, gene expression profiling of peripheral blood mononuclear cells and whole blood has identified a type 1 IFN signature in both DM and PM. This type 1 IFN profile was found to reflect disease activity in both DM and PM patients, highlighting its potential as a possible biomarker 72,84–87. The serum levels of IFN-α were found to be significantly higher in DM, PM and IMNM patients compared to IBM. Serum profiling of IBM highlighted a strong Th1 profile; flow cytometry analysis confirmed this, and showed a diminished Treg population 12.
The IL-1β and TNF-α gene pathways have been shown to be activated in a subset of PM patients 58. Both cytokines have been shown to inhibit myoblast and myotube differentiation and TNF-α has been shown to directly inhibit the expression of the myogenic microRNAs, miR-1, -133 and -206, which are heavily involved in skeletal muscle differentiation and maintenance 46.
The IL-17 gene signature was found to be enhanced in subsets of DM and PM patients over the type I IFN profile. Significantly higher levels of IL-17 and the Th17 polarizing cytokine IL-23 are produced by activated peripheral blood mononuclear cells (PBMCs) from early-stage DM and PM in comparison to samples from more established disease 13. In DM patients the percentage of circulating Th17 cells correlated positively with serum levels of creatine kinase, which is an indicator of severity of muscle injury. In comparison, Th17 cells were correlated negatively with the expression of the myogenic microRNA miR-206 65. No significant differences in IL-17 serum levels have been demonstrated between the different inflammatory myopathy subtypes 12,13. However, in DM patients IL-17 serum levels were correlated strongly with IFN gene expression and IFN-regulated chemokine levels 62. Enhanced expression of the Th17 polarizing cytokines (IL-6, IL-1β, TGF-β and IL-23) has also been demonstrated in DM patients in comparison to healthy controls 65.
Therapeutic implications
The current treatment strategy for patients with immune-mediated inflammatory myopathies involves first-line treatment with corticosteroids, alone or in combination with an immunosuppressant such as methotrexate, azathioprine or mycophenolate, and in more resistant cases intravenous immunoglobulin or biological therapy 4,88–90. B cell depletion with rituximab (anti-CD20), which interferes with autoantibody production as well as cytokine production and antigen presentation to T cells, is a very promising option, particularly for resistant cases of dermatomyositis 91–94. The TNF-α inhibitors etanercept and infliximab have shown varying responses in the treatment of DM and PM, with some cases showing no response 95–100, and lack of response in IBM 88,95. Immunomodulation with IFN-β has also been shown to be ineffective in IBM, but may be beneficial in some cases associated with chronic viral infection 101,102. Information regarding the efficacy of biological therapy in trials involving myositis patients is limited. Furthermore, interpretation of study results is restricted by the study size and duration 89,103,104. Treatment options are confounded further by reported cases of exacerbation or onset of myositis, or malignancy following treatment with TNF-α inhibitors 88. The results of these studies highlight the need for new therapeutic targets and treatment strategies.
The dissection of CD4+ T effector cell types within inflammatory and autoimmune disorders has focused drug discovery efforts on the associated cytokines of each T effector cell subset. Traditionally, CD4+ Th1 cells or CD8+ cytotoxic T cells have been considered the main players in the inflammatory myopathies. It is being recognized increasingly that a heterogeneous inflammatory infiltrate exists within the inflamed myositis muscle and specific T cell phenotypes are not exclusive to an individual disease subset 21. The three major subsets of T helper cells, Th1, Th2 and Th17, are all inhibited by abatacept [cytotoxic T lymphocyte antigen-4 immunoglobulin (CTLA-4-Ig)] which blocks the signals required for T cell activation (Fig. 2). A number of case studies describing the successful treatment of patients with DM and PM by abatacept have been reported. To date, however, a clinical trial of abatacept has not been completed 105–107.
Fig. 2.
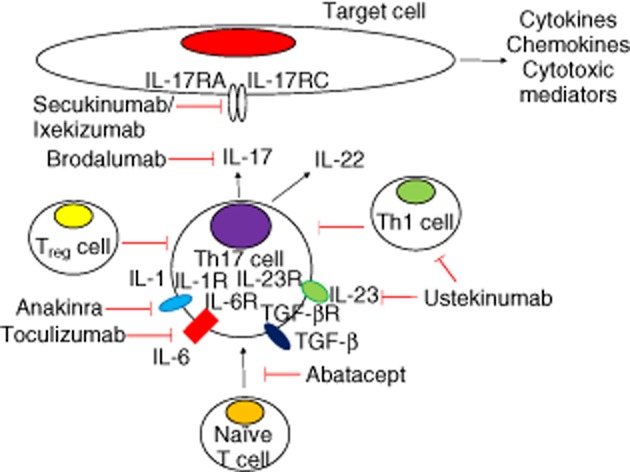
Putative cytokine targets in myositis and available blocking monoclonal antibodies. Interleukin (IL)-17RA and IL-17RC represent the A and C chains of the IL-17 receptor. IL-1R: interleukin 1 receptor; TFG-βR = transforming growth factor-β receptor; IL-23R = IL-23 receptor; IL-6R = IL-6 receptor.
Inhibition of IL-6, a key Th17 polarizing cytokine, by toculizumab has demonstrated efficacy in patients with refractory polymyositis 108. A clinical trial for assessing toculizumab in resistant DM and PM has been registered; however, recruitment has not been initiated (NCT02043548). Toculizumab may also be a suitable candidate for treatment of IBM, as evidenced by enhanced IL-6 expression in IBM patients, particularly those resistant to immunosuppressive therapy 76.
Blockade of IL-1 signalling using anakinra has also resulted in clinical responses in myositis patients 109–112. In a number of these patients IL-17 signalling was targeted indirectly through the reduction of IL-1-dependent Th17 differentiation 109. Ustekinumab, an approved anti-IL-23/IL-12p40 antibody, suppresses both Th1 and Th17 responses. Monoclonal antibodies targeting the IL-23p19 subunit which inhibits Th17 responses are currently in development. Over-expression of both IL-12 and IL-23 has also been observed in myositis patients, yet these therapies have not been assessed in the context of myositis 12,13.
Targeting Th17 differentiation upstream is advantageous over direct IL-17 targeting, as blocking Th17 responses abrogates not just IL-17 but also IL-22 and IFN-γ. These cytokines have been found to be co-expressed by Th17 and other IL-17-secreting cell types 57,63. Clinical trials of pharmacological inhibitors directly targeting IL-17 and its receptor are currently in progress for the treatment of a number of other autoimmune diseases 13,14. Direct IL-17 inhibition is yet to be assessed in myositis and warrants clinical trials in patients with resistant PM and DM, as well as cases of IBM which, as a group, respond poorly to conventional forms of immune therapy.
Conclusions
The introduction of biological therapy almost 20 years ago revolutionized the treatment of autoimmune disorders such as rheumatoid arthritis and multiple sclerosis by broadening the number of treatment options available. Advances in recent years leading to the identification of additional effector immune cell subsets and their associated cytokine pathways as outlined in this review have led to the development of an increasing number of monoclonal antibodies for clinical use, the most noteworthy being therapies targeting the Th17–IL-17 inflammatory axis. This holds great promise for the application of these therapies to the inflammatory myopathies, in particular for patients who respond poorly to current treatments. However, to date there have been few clinical trials and the application of biologicals in myositis has lagged behind its use in other autoimmune diseases. Controlled trials of sufficient size and duration are warranted to provide sufficient information and enable the introduction of additional therapies to the current repertoire.
Disclosure
The authors declare no competing interests.
References
- 1.Tournadre A, Lenief V, Eljaafari A, Miossec P. Immature muscle precursors are a source of interferon-beta in myositis: role of Toll-like receptor 3 activation and contribution to HLA class I up-regulation. Arthritis Rheum. 2012;64:533–541. doi: 10.1002/art.33350. [DOI] [PubMed] [Google Scholar]
- 2.Troyanov Y, Targoff IN, Tremblay JL, Goulet JR, Raymond Y, Senecal JL. Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients. Medicine (Balt) 2005;84:231–249. doi: 10.1097/01.md.0000173991.74008.b0. [DOI] [PubMed] [Google Scholar]
- 3.Dalakas MC. Pathogenesis and therapies of immune-mediated myopathies. Autoimmun Rev. 2012;11:203–206. doi: 10.1016/j.autrev.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 4.Dalakas MC. Therapeutic targets in patients with inflammatory myopathies: present approaches and a look to the future. Neuromuscul Disord. 2006;16:223–236. doi: 10.1016/j.nmd.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 5.Venalis P, Lundberg IE. Immune mechanisms in polymyositis and dermatomyositis and potential targets for therapy. Rheumatology (Oxf) 2014;53:397–405. doi: 10.1093/rheumatology/ket279. [DOI] [PubMed] [Google Scholar]
- 6.Rodríguez Cruz PM, Luo Y-B, Miller J, Junckerstorff RC, Mastaglia FL, Fabian V. An analysis of the sensitivity and specificity of MHC-I and MHC-II immunohistochemical staining in muscle biopsies for the diagnosis of inflammatory myopathies. Neuromuscul Disord. 2014 doi: 10.1016/j.nmd.2014.06.436. doi: 10.1016/j.nmd.2014.06.436. [DOI] [PubMed] [Google Scholar]
- 7.Figarella-Branger D, Civatte M, Bartoli C, Pellissier JF. Cytokines, chemokines, and cell adhesion molecules in inflammatory myopathies. Muscle Nerve. 2003;28:659–682. doi: 10.1002/mus.10462. [DOI] [PubMed] [Google Scholar]
- 8.Loell I, Lundberg IE. Can muscle regeneration fail in chronic inflammation: a weakness in inflammatory myopathies? J Intern Med. 2011;269:243–257. doi: 10.1111/j.1365-2796.2010.02334.x. [DOI] [PubMed] [Google Scholar]
- 9.Rayavarapu S, Coley W, Kinder TB, Nagaraju K. Idiopathic inflammatory myopathies: pathogenic mechanisms of muscle weakness. Skelet Muscle. 2013;3:13. doi: 10.1186/2044-5040-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yokota M, Suzuki K, Tokoyoda K, et al. Roles of mast cells in the pathogenesis of inflammatory myopathy. Arthritis Res Ther. 2014;16:R72. doi: 10.1186/ar4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bruder J, Siewert K, Obermeier B, et al. Target specificity of an autoreactive pathogenic human gammadelta-T cell receptor in myositis. J Biol Chem. 2012;287:20986–20995. doi: 10.1074/jbc.M112.356709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allenbach Y, Chaara W, Rosenzwajg M, et al. Th1 response and systemic Treg deficiency in inclusion body myositis. PLOS ONE. 2014;9:e88788. doi: 10.1371/journal.pone.0088788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moran EM, Mastaglia FL. The role of interleukin-17 in immune-mediated inflammatory myopathies and possible therapeutic implications. Neuromuscul Disord. 2014 doi: 10.1016/j.nmd.2014.06.432. doi: 10.1016/j.nmd.2014.06.432. [DOI] [PubMed] [Google Scholar]
- 14.Tournadre A, Miossec P. Interleukin-17 in inflammatory myopathies. Curr Rheumatol Rep. 2012;14:252–256. doi: 10.1007/s11926-012-0242-x. [DOI] [PubMed] [Google Scholar]
- 15.Shachar I, Karin N. The dual roles of inflammatory cytokines and chemokines in the regulation of autoimmune diseases and their clinical implications. J Leukoc Biol. 2013;93:51–61. doi: 10.1189/jlb.0612293. [DOI] [PubMed] [Google Scholar]
- 16.Tournadre A, Miossec P. A critical role for immature muscle precursors in myositis. Nat Rev Rheumatol. 2013;9:438–442. doi: 10.1038/nrrheum.2013.26. [DOI] [PubMed] [Google Scholar]
- 17.Creus KK, De Paepe B, De Bleecker JL. Idiopathic inflammatory myopathies and the classical NF-kappaB complex: current insights and implications for therapy. Autoimmun Rev. 2009;8:627–631. doi: 10.1016/j.autrev.2009.02.026. [DOI] [PubMed] [Google Scholar]
- 18.Zong M, Lundberg IE. Pathogenesis, classification and treatment of inflammatory myopathies. Nat Rev Rheumatol. 2011;7:297–306. doi: 10.1038/nrrheum.2011.39. [DOI] [PubMed] [Google Scholar]
- 19.Dalakas MC. Mechanisms of disease: signaling pathways and immunobiology of inflammatory myopathies. Nat Clin Pract Rheumatol. 2006;2:219–227. doi: 10.1038/ncprheum0140. [DOI] [PubMed] [Google Scholar]
- 20.Luo Y-B, Mastaglia FL. Dermatomyositis, polymyositis and immune-mediated necrotising myopathies. Biochim Biophys Acta. 2014 doi: 10.1016/j.bbadis.2014.05.034. doi: 10.1016/j.bbadis.2014.05.034. [DOI] [PubMed] [Google Scholar]
- 21.Fasth AE, Dastmalchi M, Rahbar A, et al. T cell infiltrates in the muscles of patients with dermatomyositis and polymyositis are dominated by CD28null T cells. J Immunol. 2009;183:4792–4799. doi: 10.4049/jimmunol.0803688. [DOI] [PubMed] [Google Scholar]
- 22.Pestronk A. Acquired immune and inflammatory myopathies: pathologic classification. Curr Opin Rheumatol. 2011;23:595–604. doi: 10.1097/BOR.0b013e32834bab42. [DOI] [PubMed] [Google Scholar]
- 23.Casciola-Rosen L, Mammen AL. Myositis autoantibodies. Curr Opin Rheumatol. 2012;24:602–608. doi: 10.1097/BOR.0b013e328358bd85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greenberg SA, Bradshaw EM, Pinkus JL, et al. Plasma cells in muscle in inclusion body myositis and polymyositis. Neurology. 2005;65:1782–1787. doi: 10.1212/01.wnl.0000187124.92826.20. [DOI] [PubMed] [Google Scholar]
- 25.Dimachkie MM. Idiopathic inflammatory myopathies. J Neuroimmunol. 2011;231:32–42. doi: 10.1016/j.jneuroim.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 26.Arahata K, Engel AG. Monoclonal antibody analysis of mononuclear cells in myopathies. V: identification and quantitation of T8+ cytotoxic and T8+ suppressor cells. Ann Neurol. 1988;23:493–499. doi: 10.1002/ana.410230511. [DOI] [PubMed] [Google Scholar]
- 27.De Paepe B, De Bleecker JL. The nonnecrotic invaded muscle fibers of polymyositis and sporadic inclusion body myositis: on the interplay of chemokines and stress proteins. Neurosci Lett. 2013;535:18–23. doi: 10.1016/j.neulet.2012.11.064. [DOI] [PubMed] [Google Scholar]
- 28.Preusse C, Goebel HH, Held J, et al. Immune-mediated necrotizing myopathy is characterized by a specific Th1-M1 polarized immune profile. Am J Pathol. 2012;181:2161–2171. doi: 10.1016/j.ajpath.2012.08.033. [DOI] [PubMed] [Google Scholar]
- 29.Needham M, Fabian V, Knezevic W, Panegyres P, Zilko P, Mastaglia FL. Progressive myopathy with up-regulation of MHC-I associated with statin therapy. Neuromuscul Disord. 2007;17:194–200. doi: 10.1016/j.nmd.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 30.Christopher-Stine L, Casciola-Rosen LA, Hong G, Chung T, Corse AM, Mammen AL. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum. 2010;62:2757–2766. doi: 10.1002/art.27572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mammen AL, Chung T, Christopher-Stine L, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum. 2011;63:713–721. doi: 10.1002/art.30156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagaraju K, Casciola-Rosen L, Lundberg I, et al. Activation of the endoplasmic reticulum stress response in autoimmune myositis: potential role in muscle fiber damage and dysfunction. Arthritis Rheum. 2005;52:1824–1835. doi: 10.1002/art.21103. [DOI] [PubMed] [Google Scholar]
- 33.Grundtman C, Lundberg IE. Pathogenesis of idiopathic inflammatory myopathies. Curr Rheumatol Rep. 2006;8:188–195. doi: 10.1007/s11926-996-0024-4. [DOI] [PubMed] [Google Scholar]
- 34.Arahata K, Engel AG. Monoclonal antibody analysis of mononuclear cells in myopathies. IV: cell-mediated cytotoxicity and muscle fiber necrosis. Ann Neurol. 1988;23:168–173. doi: 10.1002/ana.410230210. [DOI] [PubMed] [Google Scholar]
- 35.Salajegheh M, Rakocevic G, Raju R, Shatunov A, Goldfarb LG, Dalakas MC. T cell receptor profiling in muscle and blood lymphocytes in sporadic inclusion body myositis. Neurology. 2007;69:1672–1679. doi: 10.1212/01.wnl.0000265398.77681.09. [DOI] [PubMed] [Google Scholar]
- 36.Benveniste O, Herson S, Salomon B, et al. Long-term persistence of clonally expanded T cells in patients with polymyositis. Ann Neurol. 2004;56:867–872. doi: 10.1002/ana.20293. [DOI] [PubMed] [Google Scholar]
- 37.Amemiya K, Granger RP, Dalakas MC. Clonal restriction of T-cell receptor expression by infiltrating lymphocytes in inclusion body myositis persists over time. Studies in repeated muscle biopsies. Brain. 2000;123(Pt 10):2030–2039. doi: 10.1093/brain/123.10.2030. [DOI] [PubMed] [Google Scholar]
- 38.Askanas V, Engel WK, Nogalska A. Pathogenic considerations in sporadic inclusion-body myositis, a degenerative muscle disease associated with aging and abnormalities of myoproteostasis. J Neuropathol Exp Neurol. 2012;71:680–693. doi: 10.1097/NEN.0b013e31826183c8. [DOI] [PubMed] [Google Scholar]
- 39.Schmidt J, Barthel K, Wrede A, Salajegheh M, Bahr M, Dalakas MC. Interrelation of inflammation and APP in sIBM: IL-1 beta induces accumulation of beta-amyloid in skeletal muscle. Brain. 2008;131:1228–1240. doi: 10.1093/brain/awn053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greenberg SA, Pinkus JL, Pinkus GS, et al. Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol. 2005;57:664–678. doi: 10.1002/ana.20464. [DOI] [PubMed] [Google Scholar]
- 41.Stenzel W, Goebel HH, Aronica E. Review: immune-mediated necrotizing myopathies – a heterogeneous group of diseases with specific myopathological features. Neuropathol Appl Neurobiol. 2012;38:632–646. doi: 10.1111/j.1365-2990.2012.01302.x. [DOI] [PubMed] [Google Scholar]
- 42.Rojana-udomsart A, Mitrpant C, Bundell C, et al. Complement-mediated muscle cell lysis: a possible mechanism of myonecrosis in anti-SRP associated necrotizing myopathy (ASANM) J Neuroimmunol. 2013;264:65–70. doi: 10.1016/j.jneuroim.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 43.Mammen AL, Casciola-Rosen LA, Hall JC, Christopher-Stine L, Corse AM, Rosen A. Expression of the dermatomyositis autoantigen Mi-2 in regenerating muscle. Arthritis Rheum. 2009;60:3784–3793. doi: 10.1002/art.24977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Casciola-Rosen L, Nagaraju K, Plotz P, et al. Enhanced autoantigen expression in regenerating muscle cells in idiopathic inflammatory myopathy. J Exp Med. 2005;201:591–601. doi: 10.1084/jem.20041367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O'Neill LA, Sheedy FJ, McCoy CE. MicroRNAs: the fine-tuners of Toll-like receptor signalling. Nat Rev Immunol. 2011;11:163–175. doi: 10.1038/nri2957. [DOI] [PubMed] [Google Scholar]
- 46.Georgantas RW, Streicher K, Greenberg SA, et al. Inhibition of myogenic microRNAs 1, 133, and 206 by inflammatory cytokines links inflammation and muscle degeneration in adult inflammatory myopathies. Arthritis Rheumatol. 2014;66:1022–1033. doi: 10.1002/art.38292. [DOI] [PubMed] [Google Scholar]
- 47.Fearon U. Interleukin-27: a master regulator in inflammation. Arthritis Rheum. 2011;63:2157–2160. doi: 10.1002/art.30322. [DOI] [PubMed] [Google Scholar]
- 48.Miossec P, Kolls JK. Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discov. 2012;11:763–776. doi: 10.1038/nrd3794. [DOI] [PubMed] [Google Scholar]
- 49.Wilson CB, Rowell E, Sekimata M. Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol. 2009;9:91–105. doi: 10.1038/nri2487. [DOI] [PubMed] [Google Scholar]
- 50.Elliott MJ, Maini RN, Feldmann M, et al. Randomised double-blind comparison of chimeric monoclonal antibody to tumour necrosis factor alpha (cA2) versus placebo in rheumatoid arthritis. Lancet. 1994;344:1105–1110. doi: 10.1016/s0140-6736(94)90628-9. [DOI] [PubMed] [Google Scholar]
- 51.Bartee E, McFadden G. Cytokine synergy: an underappreciated contributor to innate anti-viral immunity. Cytokine. 2013;63:237–240. doi: 10.1016/j.cyto.2013.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moran EM, Mullan R, McCormick J, et al. Human rheumatoid arthritis tissue production of IL-17A drives matrix and cartilage degradation: synergy with tumour necrosis factor-alpha, Oncostatin M and response to biologic therapies. Arthritis Res Ther. 2009;11:R113. doi: 10.1186/ar2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Granet C, Miossec P. Combination of the pro-inflammatory cytokines IL-1, TNF-alpha and IL-17 leads to enhanced expression and additional recruitment of AP-1 family members, Egr-1 and NF-kappaB in osteoblast-like cells. Cytokine. 2004;26:169–177. doi: 10.1016/j.cyto.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 54.Smilek DE, Ehlers MR, Nepom GT. Restoring the balance: immunotherapeutic combinations for autoimmune disease. Dis Model Mech. 2014;7:503–513. doi: 10.1242/dmm.015099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Greenberg SA. Type 1 interferons and myositis. Arthritis Res Ther. 2010;12(Suppl 1):S4. doi: 10.1186/ar2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ivanidze J, Hoffmann R, Lochmuller H, Engel AG, Hohlfeld R, Dornmair K. Inclusion body myositis: laser microdissection reveals differential up-regulation of IFN-gamma signaling cascade in attacked versus nonattacked myofibers. Am J Pathol. 2011;179:1347–1359. doi: 10.1016/j.ajpath.2011.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zong M, Dorph C, Dastmalchi M, et al. Anakinra treatment in patients with refractory inflammatory myopathies and possible predictive response biomarkers: a mechanistic study with 12 months follow-up. Ann Rheum Dis. 2014;73:913–920. doi: 10.1136/annrheumdis-2012-202857. [DOI] [PubMed] [Google Scholar]
- 58.Higgs BW, Zhu W, Richman L, et al. Identification of activated cytokine pathways in the blood of systemic lupus erythematosus, myositis, rheumatoid arthritis, and scleroderma patients. Int J Rheum Dis. 2012;15:25–35. doi: 10.1111/j.1756-185X.2011.01654.x. [DOI] [PubMed] [Google Scholar]
- 59.Caproni M, Torchia D, Cardinali C, et al. Infiltrating cells, related cytokines and chemokine receptors in lesional skin of patients with dermatomyositis. Br J Dermatol. 2004;151:784–791. doi: 10.1111/j.1365-2133.2004.06144.x. [DOI] [PubMed] [Google Scholar]
- 60.Fujiyama T, Ito T, Ogawa N, Suda T, Tokura Y, Hashizume H. Preferential infiltration of IL-4-producing CXCR4 T cells in the lesional muscle but not skin of patients with dermatomyositis. Clin Exp Immunol. 2014;177:110–120. doi: 10.1111/cei.12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tournadre A, Porcherot M, Cherin P, Marie I, Hachulla E, Miossec P. Th1 and Th17 balance in inflammatory myopathies: interaction with dendritic cells and possible link with response to high-dose immunoglobulins. Cytokine. 2009;46:297–301. doi: 10.1016/j.cyto.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 62.Allenbach Y, Rosenzwajg M, Prevel N, et al. P5.27 evidence for the implication of Th-1 and Treg cells but not Th-17 in sporadic inclusion body myositis. Neuromuscul Disord. 2011;21:732. [Google Scholar]
- 63.Ciccia F, Rizzo A, Alessandro R, et al. Activated IL-22 pathway occurs in the muscle tissues of patients with polymyositis or dermatomyositis and is correlated with disease activity. Rheumatology (Oxf) 2014;53:1307–1312. doi: 10.1093/rheumatology/keu005. [DOI] [PubMed] [Google Scholar]
- 64.Raju R, Dalakas MC. Gene expression profile in the muscles of patients with inflammatory myopathies: effect of therapy with IVIg and biological validation of clinically relevant genes. Brain. 2005;128:1887–1896. doi: 10.1093/brain/awh518. [DOI] [PubMed] [Google Scholar]
- 65.Tang X, Tian X, Zhang Y, et al. Correlation between the frequency of Th17 cell and the expression of microRNA-206 in patients with dermatomyositis. Clin Dev Immunol. 2013;2013:345347–345354. doi: 10.1155/2013/345347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Park J-S, Park M-K, Lee S-Y, et al. TWEAK promotes the production of interleukin-17 in rheumatoid arthritis. Cytokine. 2012;60:143–149. doi: 10.1016/j.cyto.2012.06.285. [DOI] [PubMed] [Google Scholar]
- 67.Morosetti R, Gliubizzi C, Sancricca C, et al. TWEAK in inclusion-body myositis muscle: possible pathogenic role of a cytokine inhibiting myogenesis. Am J Pathol. 2012;180:1603–1613. doi: 10.1016/j.ajpath.2011.12.027. [DOI] [PubMed] [Google Scholar]
- 68.Antiga E, Kretz CC, Klembt R, et al. Characterization of regulatory T cells in patients with dermatomyositis. J Autoimmun. 2010;35:342–350. doi: 10.1016/j.jaut.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 69.Solomon GJ, Magro CM. Foxp3 expression in cutaneous T-cell lymphocytic infiltrates. J Cutan Pathol. 2008;35:1032–1039. doi: 10.1111/j.1600-0560.2007.00969.x. [DOI] [PubMed] [Google Scholar]
- 70.Lundberg I, Ulfgren AK, Nyberg P, Andersson U, Klareskog L. Cytokine production in muscle tissue of patients with idiopathic inflammatory myopathies. Arthritis Rheum. 1997;40:865–874. doi: 10.1002/art.1780400514. [DOI] [PubMed] [Google Scholar]
- 71.Amemiya K, Semino-Mora C, Granger RP, Dalakas MC. Downregulation of TGF-beta1 mRNA and protein in the muscles of patients with inflammatory myopathies after treatment with high-dose intravenous immunoglobulin. Clin Immunol. 2000;94:99–104. doi: 10.1006/clim.1999.4823. [DOI] [PubMed] [Google Scholar]
- 72.Baechler E, Bilgic H, Reed A. Type I interferon pathway in adult and juvenile dermatomyositis. Arthritis Res Ther. 2011;13:249. doi: 10.1186/ar3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Higgs BW, Zhu W, Morehouse C, et al. A phase 1b clinical trial evaluating sifalimumab, an anti-IFN-alpha monoclonal antibody, shows target neutralisation of a type I IFN signature in blood of dermatomyositis and polymyositis patients. Ann Rheum Dis. 2014;73:256–262. doi: 10.1136/annrheumdis-2012-202794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tezak Z, Hoffman EP, Lutz JL, et al. Gene expression profiling in DQA1*0501+ children with untreated dermatomyositis: a novel model of pathogenesis. J Immunol. 2002;168:4154–4163. doi: 10.4049/jimmunol.168.8.4154. [DOI] [PubMed] [Google Scholar]
- 75.Guo X, Higgs BW, Rebelatto M, et al. Suppression of soluble T cell-associated proteins by an anti-interferon-α monoclonal antibody in adult patients with dermatomyositis or polymyositis. Rheumatology (Oxf) 2014;53:686–695. doi: 10.1093/rheumatology/ket413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zschuntzsch J, Voss J, Creus K, et al. Provision of an explanation for the inefficacy of immunotherapy in sporadic inclusion body myositis: quantitative assessment of inflammation and beta-amyloid in the muscle. Arthritis Rheum. 2012;64:4094–4103. doi: 10.1002/art.37692. [DOI] [PubMed] [Google Scholar]
- 77.Greenberg SA. A gene expression approach to study perturbed pathways in myositis. Curr Opin Rheumatol. 2007;19:536–541. doi: 10.1097/BOR.0b013e3282efe261. [DOI] [PubMed] [Google Scholar]
- 78.Greenberg SA, Higgs BW, Morehouse C, et al. Relationship between disease activity and type 1 interferon- and other cytokine-inducible gene expression in blood in dermatomyositis and polymyositis. Genes Immun. 2012;13:207–213. doi: 10.1038/gene.2011.61. [DOI] [PubMed] [Google Scholar]
- 79.Guo X, Higgs BW, Rebelatto M, et al. Suppression of soluble T cell-associated proteins by an anti-interferon-alpha monoclonal antibody in adult patients with dermatomyositis or polymyositis. Rheumatology (Oxf) 2014;53:686–695. doi: 10.1093/rheumatology/ket413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sato S, Ogura Y, Kumar A. TWEAK/Fn14 signaling axis mediates skeletal muscle atrophy and metabolic dysfunction. Front Immunol. 2014;5:18. doi: 10.3389/fimmu.2014.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Greenberg SA. DNA microarray gene expression analysis technology and its application to neurological disorders. Neurology. 2001;57:755–761. doi: 10.1212/wnl.57.5.755. [DOI] [PubMed] [Google Scholar]
- 82.Wong D, Kea B, Pesich R, et al. Interferon and biologic signatures in dermatomyositis skin: specificity and heterogeneity across diseases. PLOS ONE. 2012;7:e29161. doi: 10.1371/journal.pone.0029161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shrestha S, Wershil B, Sarwark JF, Niewold TB, Philipp T, Pachman LM. Lesional and nonlesional skin from patients with untreated juvenile dermatomyositis displays increased numbers of mast cells and mature plasmacytoid dendritic cells. Arthritis Rheum. 2010;62:2813–2822. doi: 10.1002/art.27529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.O'Connor KA, Abbott KA, Sabin B, Kuroda M, Pachman LM. MxA gene expression in juvenile dermatomyositis peripheral blood mononuclear cells: association with muscle involvement. Clin Immunol. 2006;120:319–325. doi: 10.1016/j.clim.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Walsh RJ, Kong SW, Yao Y, et al. Type I interferon-inducible gene expression in blood is present and reflects disease activity in dermatomyositis and polymyositis. Arthritis Rheum. 2007;56:3784–3792. doi: 10.1002/art.22928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bilgic H, Ytterberg SR, Amin S, et al. Interleukin-6 and type I interferon-regulated genes and chemokines mark disease activity in dermatomyositis. Arthritis Rheum. 2009;60:3436–3446. doi: 10.1002/art.24936. [DOI] [PubMed] [Google Scholar]
- 87.Liao AP, Salajegheh M, Nazareno R, Kagan JC, Jubin RG, Greenberg SA. Interferon beta is associated with type 1 interferon-inducible gene expression in dermatomyositis. Ann Rheum Dis. 2011;70:831–836. doi: 10.1136/ard.2010.139949. [DOI] [PubMed] [Google Scholar]
- 88.Albayda J, Christopher-Stine L. Novel approaches in the treatment of myositis and myopathies. Ther Adv Musculoskelet Dis. 2012;4:369–377. doi: 10.1177/1759720X12447705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Carstens PO, Schmidt J. Diagnosis, pathogenesis and treatment of myositis: recent advances. Clin Exp Immunol. 2014;175:349–358. doi: 10.1111/cei.12194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dalakas MC. Therapeutic advances and future prospects in immune-mediated inflammatory myopathies. Ther Adv Neurol Disord. 2008;1:157–166. doi: 10.1177/1756285608097463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Couderc M, Gottenberg JE, Mariette X, et al. Efficacy and safety of rituximab in the treatment of refractory inflammatory myopathies in adults: results from the AIR registry. Rheumatology (Oxf) 2011;50:2283–2289. doi: 10.1093/rheumatology/ker305. [DOI] [PubMed] [Google Scholar]
- 92.de Visser M. The efficacy of rituximab in refractory myositis: the jury is still out. Arthritis Rheum. 2013;65:303–306. doi: 10.1002/art.37758. [DOI] [PubMed] [Google Scholar]
- 93.Oddis CV, Reed AM, Aggarwal R, et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: a randomized, placebo-phase trial. Arthritis Rheum. 2013;65:314–324. doi: 10.1002/art.37754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Levine TD. Rituximab in the treatment of dermatomyositis: an open-label pilot study. Arthritis Rheum. 2005;52:601–607. doi: 10.1002/art.20849. [DOI] [PubMed] [Google Scholar]
- 95.Barohn RJ, Herbelin L, Kissel JT, et al. Pilot trial of etanercept in the treatment of inclusion-body myositis. Neurology. 2006;66:S123–124. doi: 10.1212/01.wnl.0000192258.32408.54. [DOI] [PubMed] [Google Scholar]
- 96.Selva-O'Callaghan A, Martinez-Costa X, Solans-Laque R, Mauri M, Capdevila JA, Vilardell-Tarres M. Refractory adult dermatomyositis with pneumatosis cystoides intestinalis treated with infliximab. Rheumatology (Oxf) 2004;43:1196–1197. doi: 10.1093/rheumatology/keh285. [DOI] [PubMed] [Google Scholar]
- 97.Labioche I, Liozon E, Weschler B, Loustaud-Ratti V, Soria P, Vidal E. Refractory polymyositis responding to infliximab: extended follow-up. Rheumatology (Oxf) 2004;43:531–532. doi: 10.1093/rheumatology/keh079. [DOI] [PubMed] [Google Scholar]
- 98.Korkmaz C, Temiz G, Cetinbas F, Buyukkidan B. Successful treatment of alveolar hypoventilation due to dermatomyositis with anti-tumour necrosis factor-alpha. Rheumatology (Oxf) 2004;43:937–938. doi: 10.1093/rheumatology/keh226. [DOI] [PubMed] [Google Scholar]
- 99.Hengstman GJ, van den Hoogen FH, Barrera P, et al. Successful treatment of dermatomyositis and polymyositis with anti-tumor-necrosis-factor-alpha: preliminary observations. Eur Neurol. 2003;50:10–15. doi: 10.1159/000070852. [DOI] [PubMed] [Google Scholar]
- 100.Muscle Study Group. A randomized, pilot trial of etanercept in dermatomyositis. Ann Neurol. 2011;70:427–436. doi: 10.1002/ana.22477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Muscle Study Group. Randomized pilot trial of betaINF1a (Avonex) in patients with inclusion body myositis. Neurology. 2001;57:1566–1570. doi: 10.1212/wnl.57.9.1566. [DOI] [PubMed] [Google Scholar]
- 102.Yakushiji Y, Satoh J, Yukitake M, et al. Interferon beta-responsive inclusion body myositis in a hepatitis C virus carrier. Neurology. 2004;63:587–588. doi: 10.1212/01.wnl.0000133411.10574.ba. [DOI] [PubMed] [Google Scholar]
- 103.Aggarwal R, Oddis CV. Therapeutic approaches in myositis. Curr Rheumatol Rep. 2011;13:182–191. doi: 10.1007/s11926-011-0172-z. [DOI] [PubMed] [Google Scholar]
- 104.Greenberg SA. Pathogenesis and therapy of inclusion body myositis. Curr Opin Neurol. 2012;25:630–639. doi: 10.1097/WCO.0b013e328357f211. [DOI] [PubMed] [Google Scholar]
- 105.Musuruana JL, Cavallasca JA. Abatacept for treatment of refractory polymyositis. Joint Bone Spine. 2011;78:431–432. doi: 10.1016/j.jbspin.2011.03.022. [DOI] [PubMed] [Google Scholar]
- 106.Arabshahi B, Silverman RA, Jones OY, Rider LG. Abatacept and sodium thiosulfate for treatment of recalcitrant juvenile dermatomyositis complicated by ulceration and calcinosis. J Pediatr. 2012;160:520–522. doi: 10.1016/j.jpeds.2011.11.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kerola AM, Kauppi MJ. Abatacept as a successful therapy for myositis – a case-based review. Clin Rheumatol. 2014 doi: 10.1007/s10067-014-2507-4. doi: 10.1007/s10067-014-2507-4. [DOI] [PubMed] [Google Scholar]
- 108.Narazaki M, Hagihara K, Shima Y, Ogata A, Kishimoto T, Tanaka T. Therapeutic effect of tocilizumab on two patients with polymyositis. Rheumatology (Oxf) 2011;50:1344–1346. doi: 10.1093/rheumatology/ker152. [DOI] [PubMed] [Google Scholar]
- 109.Zong M, Dorph C, Dastmalchi M, et al. Anakinra treatment in patients with refractory inflammatory myopathies and possible predictive response biomarkers: a mechanistic study with 12 months follow-up. Ann Rheum Dis. 2013;73:913–920. doi: 10.1136/annrheumdis-2012-202857. [DOI] [PubMed] [Google Scholar]
- 110.Kosmidis ML, Alexopoulos H, Tzioufas AG, Dalakas MC. The effect of anakinra, an IL1 receptor antagonist, in patients with sporadic inclusion body myositis (sIBM): a small pilot study. J Neurol Sci. 2013;334:123–125. doi: 10.1016/j.jns.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 111.Furlan A, Botsios C, Ruffatti A, Todesco S, Punzi L. Antisynthetase syndrome with refractory polyarthritis and fever successfully treated with the IL-1 receptor antagonist, anakinra: a case report. Joint Bone Spine. 2008;75:366–367. doi: 10.1016/j.jbspin.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 112.Estublier C, Stankovic Stojanovic K, Bergerot JF, Broussolle C, Seve P. Myositis in a patient with familial Mediterranean fever and spondyloarthritis successfully treated with anakinra. Joint Bone Spine. 2013;80:645–649. doi: 10.1016/j.jbspin.2013.03.004. [DOI] [PubMed] [Google Scholar]