

Abstract
In areas without newborn screening for severe combined immunodeficiency (SCID), disease-defining infections may lead to diagnosis, and in some cases, may not be identified prior to the first year of life. We describe a female infant who presented with disseminated vaccine-acquired varicella (VZV) and vaccine-acquired rubella infections at 13 months of age. Immunological evaluations demonstrated neutropenia, isolated CD4 lymphocytopenia, the presence of CD8+ T cells, poor lymphocyte proliferation, hypergammaglobulinaemia and poor specific antibody production to VZV infection and routine immunizations. A combination of whole exome sequencing and custom-designed chromosomal microarray with exon coverage of primary immunodeficiency genes detected compound heterozygous mutations (one single nucleotide variant and one intragenic copy number variant involving one exon) within the IL7R gene. Mosaicism for wild-type allele (20–30%) was detected in pretransplant blood and buccal DNA and maternal engraftment (5–10%) demonstrated in pretransplant blood DNA. This may be responsible for the patient's unusual immunological phenotype compared to classical interleukin (IL)-7Rα deficiency. Disseminated VZV was controlled with anti-viral and immune-based therapy, and umbilical cord blood stem cell transplantation was successful. Retrospectively performed T cell receptor excision circle (TREC) analyses completed on neonatal Guthrie cards identified absent TREC. This case emphasizes the danger of live viral vaccination in severe combined immunodeficiency (SCID) patients and the importance of newborn screening to identify patients prior to high-risk exposures. It also illustrates the value of aggressive pathogen identification and treatment, the influence newborn screening can have on morbidity and mortality and the significant impact of newer genomic diagnostic tools in identifying the underlying genetic aetiology for SCID patients.
Keywords: copy number variation, IL7R, revertant mosaicism, SCID, whole exome sequencing
Introduction
Patients with severe combined immune deficiency (SCID) present commonly with opportunistic or severe recurrent infections within the first year of life, often following waning of maternal antibody at 3–8 months of age 1,2. Without haematopoietic stem cell transplantation (HSCT) or gene therapy, the condition is lethal 3–5. In a subpopulation of patients with mutations in SCID-causing genes, the combined immune deficiency may be milder due to residual T cell function 6. Despite being debilitated immunologically, these types of patients may experience their first severe or opportunistic infection beyond the first year of life and/or may present with signs of abnormal immune regulation, such as autoimmune cytopaenias, colitis, granulomata and lymphoproliferative syndromes 6. Newborn screening for SCID may detect SCID prior to manifestations of the disease, but is not universally implemented in the United States 2,7,8.
Live viral vaccine-acquired infection is a rare iatrogenic morbidity associated with delayed detection of SCID. Based on the routine childhood immunization schedule in the United States (US), this challenge may occur as early as 2 months of age with administration of the oral live rotavirus vaccine 9. Nine patients with SCID have been reported with vaccine-associated acute and subacute rotaviral diarrhoea 10. At 12 months of age, US children should receive the varicella zoster virus (VZV) and measles–mumps–rubella (MMR) live viral vaccines. Oka (vaccine) strain VZV infection has been reported in the following primary immunodeficiency diseases (PIDDs): adenosine deaminase (ADA) deficiency [Online Mendelian Inheritance in Man (OMIM) 102700], purine nucleoside phosphorylase (PNP) deficiency (OMIM 613179), natural killer T (NK T) cell deficiency and a combined PIDD of unknown aetiology 11–15. Disseminated vaccine-strain rubella (RA27/3) infection has not been reported.
We present a 13-month-old female with an atypical presentation of severe T cell deficiency combined with neutropenia as well as the unique approach taken to treat her infectious diseases and define her molecular SCID diagnosis. Both triple anti-viral therapy and VZV-specific immunoglobulins (VZIG) were utilized to control her VZV infection prior to HSCT. When the clinical phenotype does not match the known described phenotype for SCID, it may be impractical or inefficient to perform single gene sequencing, as this may delay specific diagnosis. A novel approach utilizing a combination of diagnostic genetic and genomic tools and implementing genome-wide assays, rather than single gene sequencing, was instrumental to unravelling IL7R compound heterozygous single nucleotide variant (SNV) and copy number variant (CNV) alleles as the basis of recessive gene mutations.
Materials and methods
Immunological assays
T, B and natural killer (NK) cells were measured using flow cytometric analyses, and lymphocyte proliferation assays were assessed by tritiated thymidine incorporation in a local Clinical Laboratory Improvement Amendments (CLIA)-certified laboratory 16. Paediatric and adult age-matched normal values were compared with established published values 16 and local laboratory controls. Lymphocyte proliferation assays were performed in triplicate by 20-h pulses of tritiated thymidine after 3 days stimulation with mitogens [phytohaemagglutinin (PHA) 10 μg/ml, concanvalin A (ConA) 50 μg/ml, pokeweed mitogen (PWM) 100 ng/ml] or after 5 days stimulation with antigen (tetanus 80 flocculation units/ml and Candida albicans 5 mg/ml). A normal cellular-specific immune response to antigens was defined as a stimulation index (SI) of greater than 2 and/or counts per million (cpm) values of greater than 2000. Normal control ranges were determined for mitogen-specific responses. NK cell cytotoxicity was assessed by 51Cr-release assay, as described previously 17. Total serum immunoglobulin levels were determined by nephelometry, and specific antibody titres to tetanus toxoid (Tt) and Streptococcus pneumoniae (Spn) were measured as less than two standard deviations (s.d.) below the laboratory reported mean for age-matched controls in CLIA-certified laboratories. Serum specific antibody titres were defined as protective when greater than 0·10 IU/ml to Tt and greater than 0·3 μg/ml to Spn serotypes 1, 3, 4, 5, 6B, 7F, 8, 9N, 9V, 12F, 14, 18C, 19F and 23F. Pathogen-specific serology was evaluated for the following: VZV [immunoglobulin (Ig)G and IgM] and rubella (IgG). VZV viral load was measured by quantitative PCR assay. Qualitative PCR assays were utilized to evaluate for measles, mumps and rubella infections by the National Center for Disease Control and Prevention (CDC). HIV DNA was measured by PCR assay in CLIA-certified laboratories. Enzyme-linked immunospot (ELISPOT) assay analysis was used to determine the frequency and function of T cells secreting interferon (IFN)-γ post-HSCT in response to viral antigen pepmixes™ (JPT, Berlin, Germany), as described previously 18. IFN-γ-producing cells were quantified by Zellnet Consulting (New York, NY, USA), and the frequency of spot-forming cells (SFCs) relative to the input cell number was reported.
Newborn screening
With parental informed consent, T cell receptor excision circles (TREC) assays were performed on archived newborn screening specimens, as described previously 19,20.
Genetic analyses
Genomic DNA was extracted from the patient's whole blood, prior to HSCT, and used for diagnostic whole exome sequencing (WES) and chromosomal microarray (CMA) 21,22. The variant/mutation, identified by WES, was confirmed independently by Sanger sequencing and analysed for familial segregation. CMA was performed to test for larger genomic and smaller intragenic (i.e. exonic) copy number variants (CNV) 23. The Baylor College of Medicine (BCM) CMA version 9·1.1 used is custom-designed Agilent oligo array, that contains 60 000 single nucleotide polymorphism (SNP) probes and 380 000 oligoprobes with exon coverage of approximately 4900 genes, including 200 genes reported in PIDD 24,25. The CMA genome-wide assay targets intragenic exonic CNVs, and can readily identify intragenic CNV alleles as recessive carrier states 22,23,26. Droplet PCR confirmation of the deletion was performed by designing primers and subjecting genomic DNA from each family member to analyses according to the manufacturer's recommendation (QX200; Bio-Rad Life Science Research, Hercules, CA, USA). Data were analysed using QuantaSoft version 1·4 software. For WES, 1 μg of genomic DNA in 100 μl volume was sheared into fragments of approximately 300–400 base pairs (bp) in a Covaris plate with E210 system (Covaris, Inc., Woburn, MA, USA). Genomic DNA samples were constructed into Illumina paired-end precapture libraries (Illumina Inc., San Diego, CA, USA), according to the manufacturer's protocol (Illumina Multiplexing_SamplePrep_Guide_1005361_D) with modifications as described in the BCM-HGSC Illumina Barcoded Paired-End Capture Library Preparation protocol. Libraries were prepared using Beckman robotic workstations (Biomek NXp and FXp models). The complete protocol and oligonucleotide sequences are accessible from the Human Genome Sequencing Center (HGSC) website (https://hgsc.bcm.edu/sites/default/files/documents/Illumina_Barcoded_Paired-End_Capture_Library_Preparation.pdf). Four precapture libraries were pooled together (approximately 500 ng/sample, 2 μg per pool) and hybridized in solution to the HGSC CORE design 27 (52 Mb, NimbleGen), according to the manufacturer's protocol [NimbleGen SeqCap EZ Exome Library SR User's Guide (version 2·2)], with minor revisions. Captured DNA fragments were sequenced using paired-end mode on an Illumina HiSeq 2000 platform (TruSeq SBS Kits, part no. FC-401-3001) producing 9–10 Gb per sample and achieved an average of 90% of the targeted exome bases covered to a depth of ×20 or greater. Illumina sequence analysis was performed using the HGSC Mercury analysis pipeline (http://www.tinyurl.com/HGSC-Mercury/), that addresses all aspects of data processing and analyses from the initial sequence generation on the instrument to annotated variant calls (SNPs and intra-read insertion/deletions). This pipeline uses.bcl files to then generate base-calls and base-call confidence values (qualities) using Illumina primary analysis software (casava version 1·8·0). Reads and qualities were mapped to GRCh37 human reference genome (http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/) using the Burrows–Wheeler aligner (BWA) 28 (http://bio-bwa.sourceforge.net/) producing a binary alignment/map (BAM) file 29. BAM post-processing, including insertion/deletion realignment and quality recalibration, is performed using a variety of tools (SAMtools, GATK, etc.) 30. Variants were determined using the Atlas2 suite (Atlas-SNP and Atlas-indel) to call variants and produce a variant call file (VCF) 31,32. Finally, annotation data was added to the VCF using a suite of annotation tools ‘Cassandra’ 33. Variants in known disease-related OMIM genes were selected for clinical diagnostic evaluation.
Evaluation for pre-HSCT maternal engraftment and post-HSCT donor engraftment utilized short tandem repeat (STR) analyses to differentiate patient, maternal and donor alleles. The STR analyses were performed using the Elucigene™ QST*R plus version 2 kit (Elucigene Diagnostics, Manchester, UK). The samples were prepared for STR analyses according to the manufacturer's protocol and fragment analyses performed using 3730 DNA analyser (Life Technologies, Carlsbad, CA, USA), and interpretation was completed with GeneMarker software (Softgenetics®, PA, USA). For maternal engraftment evaluation, genomic DNA was extracted from the patient's whole blood (pre-HSCT) and from buccal cells collected in repeated brush samples (post-HSCT) in comparison to whole blood from the patient's mother. Children normally inherit one set of STR markers from each parent, one paternal and one maternal STR, for each marker. In maternal engraftment, two maternal STRs for the same marker may be seen in addition to the one paternal STR, yielding three total STR genotypes for the same marker. For immune reconstitution and donor engraftment evaluation, STR analyses were performed in monocyte and granulocyte cell lines from the donor cord blood and from the patient post-HSCT.
Consent
Informed consent for research studies for the patient, parents and younger sibling were obtained through BCM Institutional Review Board approved protocols. Signed informed consent was obtained from the parents for the use of photographs and clinical data in this manuscript.
Results
Clinical presentation and initial therapy
A 13-month-old female presented with cutaneous varicella infection 3 weeks after receipt of VZV and MMR vaccines. Her past medical history included respiratory syncytial virus (RSV) bronchiolitis at 2 months of age with persistent chronic cough, hand–foot–mouth disease at 3 months of age, chronic oral mucositis since 3 months of age and recurrent episodes of acute otitis media requiring myringotomy tube placement at 9 months of age. Family history was negative for consanguinity, immune disorders, and early childhood death and disease. During a routine paediatric visit, including scheduled vaccinations at 12 months of age, a complete blood count was obtained and revealed neutropenia with absolute neutrophil count (ANC) = 100/mm3. A bone marrow aspirate was not diagnostic and showed normocellular marrow with granulocytic hyperplasia and left shift. Anti-neutrophil antibody was not detected. Despite an unclear aetiology for neutropenia, granulocyte colony-stimulating factor (G-CSF) therapy was initiated and demonstrated a modest, short-lived response to G-CSF. By 3 weeks following live viral vaccinations, vesicular lesions erupted surrounding her live viral vaccine injection site (Fig. 1a), which progressed to a diffuse vesicular rash on face, neck, trunk, extremities and eyelids. Disseminated VZV infection (Fig. 1b) without hepatitis or pneumonitis was diagnosed, and intravenous (i.v.) acyclovir was initiated.
Fig. 1.
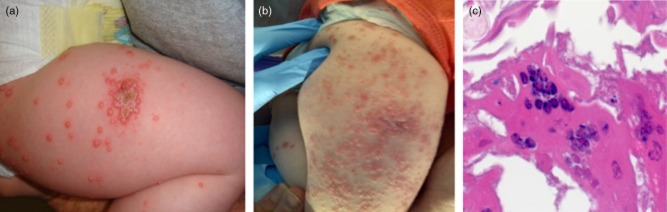
Vaccine-acquired varicella (VZV) infection: histopathology and progression of skin lesions. (a) Initial presentation: clustered vesicular lesions at VZV vaccine injection site. (b) Progression of VZV infection 2·5 weeks later: clustered lesions at VZV vaccine injection site. (c) Multi-nucleated giant cells from VZV skin lesion biopsy: demonstrated on histopathology [photomicrograph provided by National Center for Disease Control and Prevention (CDC)].
T cell studies demonstrated CD4+ T lymphocyte deficiency (Table 1) with poor T cell mitogen responses, and a SCID-like disease was suspected. Assessment of viral pathogens identified RSV-B via PCR in a nasal wash and picornavirus-like particles in stool, suggesting the persistence of RSV and hand–foot–mouth infections from earlier in life. PCR for HIV, Epstein–Barr virus (EBV) and cytomegalovirus (CMV) was negative. A skin biopsy (Fig. 1c) documented positive immunohistochemistry for VZV within lesions, with multi-nucleated keratinocytes in epidermis. On single-agent anti-viral therapy with acyclovir, her lesions progressed, and VZV viraemia was documented subsequent to initiation of acyclovir. Although multiple specimens for plasma VZV PCR were obtained while maintained on acyclovir monotherapy, the analyses were unsuccessful and not reported. Foscarnet and subsequently cidofovir were administered, as plasma VZV copies increased rapidly (Fig. 2). The patient's clinical course deteriorated, and VZV meningoencephalitis was diagnosed. During disease progression (Fig. 2), the maximum VZV measured by PCR in plasma was 751 000 copies/ml (106 copies/ml) and in cerebrospinal fluid (CSF) 977 000 copies/ml (Fig. 2). She had clinical signs of nerve entrapment and Bell's palsy associated with VZV meningoencephalitis and became symptomatic from isolated RSV. Varicella and rubella infections were determined to be vaccine-associated (Table 2a,b). Despite exposure to measles and mumps through the MMR vaccination, these viruses were never isolated from CSF, nasal wash or urine (Table 2b). Although intravenous immunoglobulin (IVIG) supplementation was given, patient VZV-specific IgG levels remained low after IVIG administration (Table 2a). Varicella zoster immunoglobulin (VZIG) was obtained as an investigational new drug, and 29 doses were delivered during her clinical course (including during myeloablation and HSCT). Due to the lack of clinical symptoms, no additional therapy was targeted to rubella. Persistent RSV infection, which was associated with chronic cough and pneumonia, was treated with palivizumab and i.v. as well as inhaled ribavirin. Ribavirin therapy was halted during myeloablation for transplant and then reinitiated during the post-transplant course. Following HSCT, acyclovir was continued as prophylaxis for 6 months and then discontinued upon documentation of donor cell engraftment and no further shedding of RSV, VZV or rubella.
Table 1.
Results of immune evaluation
| Test | Initial presentation | At time of transplant | 4 months post-transplant | 12 months post-transplant | Reference range For age | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Lymphocytes (cells/mm3, %) | ||||||||||
| Lymphocytes, total | 729 | (9·3%) | 1002 | (5·8%) | 1710 | (15·0%) | 2967 | (40·1%) | 3600–8900 | (53−75%) |
| CD3+ | 175 | (29·0%) | 90 | (9·0%) | 274 | (16·0%) | 1157 | (39·0%) | 2100–6200 | (32–51%) |
| CD4+ | 6 | (0·6%) | 1 | (0·1%) | 197 | (11·0%) | 674 | (22·7%) | 1300–3400 | (14–30%) |
| CD8+ | 153 | (21·0%) | 80 | (8·0%) | 68 | (4·0%) | 386 | (13·0%) | 620–2000 | (14–30%) |
| CD4+CD45RA+ | 7 | (1·0%) | 10 | (1·0%) | 34 | (2·0%) | 326 | (11·0%) | 134–969 | (62–90%) |
| CD19+ | 241 | (25·0%) | 721 | (72·0%) | 633 | (11·0%) | 1217 | (41·0%) | 538–1890 | (16–35%) |
| CD3−CD16+56+ | 262 | (2·0%) | 150 | (15·0%) | 718 | (42·0%) | 504 | (17·0%) | 180–1059 | (3–15%) |
| Lymphocyte proliferation (cpm) | ||||||||||
| PHA net stimulation | 60 332 | 671 | 87 242 | 177 384 | 163 500–415 807 | |||||
| Candida stimulation | 148 | n.d. | 0 | 1539 | >2000 | |||||
| Serum immunoglobulins (mg/dl) | ||||||||||
| IgG | 1370 | 811* | 1259* | 886 | 425–1054 | |||||
| IgA | 113 | n.d. | 16 | 33 | 13–116 | |||||
| IgM | 77 | n.d. | 193 | 107 | 44–155 | |||||
| Specific antibodies | ||||||||||
| Diphtheria toxin IgG (IU/ml) | 0 | 0 | n.d. | n.d. | >0·1† | |||||
| Tetanus toxoid IgG (IU/ml) | 0·1 | 0·1 | n.d. | n.d. | >0·1† | |||||
| Streptococcus pneumonia IgG serotypes (μg/ml) | 5/14 protective | 6/14 protective | n.d. | n.d. | >0·3 = protective† | |||||
| Varicella IgM (IU/ml) | 0·8 | n.d. | n.d. | n.d. | <0·9 = Neg | |||||
| Varicella IgG | Neg | n.d. | n.d. | n.d. | Pos, immune† | |||||
| Rubella IgG | Neg | n.d. | n.d. | n.d. | Pos, immune† | |||||
| HIV DNA PCR | Neg | n.d. | n.d. | n.d. | Neg | |||||
Patient on intravenous immunoglobulin (IVIG) therapy.
Post-vaccination status. NEG = negative; POS = positive; n.d. = not done; PHA = phytohaemagglutinin; Ig = immunoglobulin; PCR = polymerase chain reaction.
Fig. 2.
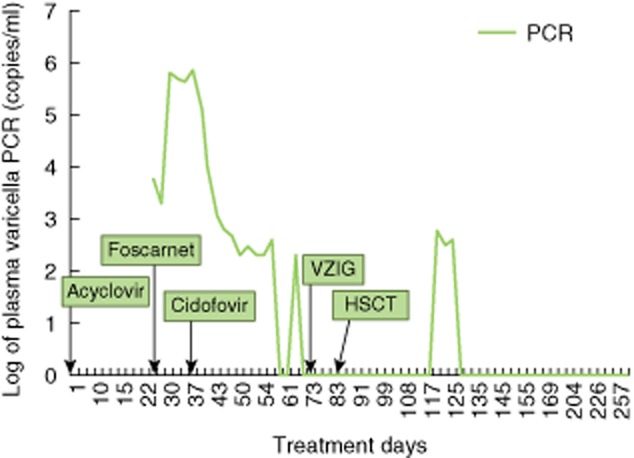
Varicella infection and response to anti-viral therapy. Disseminated varicella infection, which required multiple anti-viral therapies, was monitored by polymerase chain reaction (PCR) throughout the patient's clinical course.
Table 2a.
Serological evaluation by National Center for Disease Control and Prevention (CDC) for measles, mumps, rubella and varicella at patient age 14 months and post-IVIG therapy, but prior to VZIG
| Sample type | Measles serology | Mumps serology | Rubella serology | Varicella zoster serology | ||||
|---|---|---|---|---|---|---|---|---|
| IgM | IgG | IgM | IgG | IgM | IgG | IgM | IgG | |
| Serum | Pos | Pos | Neg | Ind | Neg | Pos | Neg | Neg |
Table 2b.
Polymerase chain reaction (PCR)-based evaluation by National Center for Disease Control and Prevention (CDC) for measles, mumps, rubella and varicella at patient age 14 months, revealing vaccine-acquired viral infections
| Sample types | Real-time PCR |
Real-time PCR |
Varicella zoster PCR |
||||
|---|---|---|---|---|---|---|---|
| Measles | Mumps | Rubella | Genotype | VZV | Genotype | Copies/ml | |
| Urine | Neg | Neg | Neg | n.d. | Pos | Oka (vaccine) | 5·0 × 106 |
| Oropharyngeal swab | Neg | Neg | Pos | RA27/3 (vaccine) | Pos | Oka (vaccine) | 2·0 × 106 |
| Nasopharyngeal swab | Neg | Neg | Pos | RA27/3 (vaccine) | Pos | Oka (vaccine) | 40 000 |
| Blood | n.d. | n.d. | n.d. | n.d. | Pos | Oka (vaccine) | 5000 |
| VZV skin scrapings | n.d. | n.d. | n.d. | n.d. | Pos | Oka (vaccine) | 5·0 × 106 |
Ind = indeterminant; IVIG = intravenous immunoglobulin; n.d. = not done; Neg = negative; Oka = attenuated varicella virus strain used in vaccines; Pos = positive; RA27/3 = attenuated rubella virus strain used in vaccines; VZV = vaccine-acquired varicella; VZIG = Varicella zoster immunoglobulin; Ig = immunoglobulin.
Immunological evaluation
Results of immunological evaluations are presented in Table 1. Initial analyses of lymphocyte subsets revealed lymphopenia with virtually undetectable CD4+ and CD45RA+ (naive) T lymphocytes, age-appropriate percentage yet decreased absolute number of CD8+ T lymphocytes, age-appropriate percentage and absolute number of CD19+ lymphocytes and decreased percentage yet age-appropriate absolute number of NK cells. Proliferative T cell responses to mitogens (PHA, ConA and PWM) and antigens (Candida, tetanus and diphtheria) were very low, suggestive of very poor T cell function. NK cell function by 51Cr release assay was normal. Although serum IgG was elevated and IgM and IgA levels were normal for age, antibody titres to vaccines demonstrated five of 14 protective Spn serotypes detectable and lack of protection to tetanus toxoid and diphtheria toxin. VZV-specific IgG and IgM were negative, despite active VZV infection. Retrospective analysis of archived newborn screening specimens collected at birth and 2 weeks of age showed absence of TREC.
Immune reconstitution
A clinical diagnosis of SCID was made based on severe vaccine-acquired viral infection, CD4+ T lymphocyte deficiency and poor T cell function. Donor umbilical cord blood stem cells were identified with a 6/6 match by histocompatibility leucocyte antigen (HLA) testing. Full myeloablative conditioning for HSCT included busulfan (16 mg/kg total dose), cyclophosphamide (200 mg/kg total dose) and fludarabine (160 mg/m2 total dose). Donor engraftment was documented 1 month post-HSCT as determined by demonstration of STRs of donor origin only. Chimerism analysis by STRs was completed in monocytes and granulocytes cell lines, and our patient has demonstrated 100% engraftment in both cell lines since this initial evaluation. Anti-viral therapy was discontinued 9 months post-HSCT, and VZIG was discontinued on day +197 post-HSCT. Currently, the patient has no evidence of active disease or shedding of VZV, rubella or RSV. Table 1 demonstrates T, B and NK cell reconstitution with gradual improvement of CD4+ cells and normal lymphocyte proliferative response to mitogen stimulation with a minimally decreased proliferative response to Candida antigen. An IFN-γ ELISPOT performed on days +63 and +85 post-HSCT revealed weak but detectable T cell activity against RSV (14 and 30 SFC/1 × 106 PBMCs, respectively). VZV-directed T cell immunity by ELISPOT was assessed on days +176 and +182 post-HSCT, which revealed weak but detectable activity in peripheral blood and in once-stimulated ex-vivo expanded T cells. B cell restoration gradually demonstrated improvement with IgA class switch at 4 and 12 months post-HSCT and IgG class switch at 12 months post-HSCT. B cell challenge with killed bacterial vaccine antigens demonstrated a robust response, with protective antibody titres detected to tetanus toxoid, diphtheria toxin and 11 of 14 Spn serotypes at day +547 post-HSCT. Her resolved HSCT complications included autoimmune haemolytic anaemia and autoimmune thrombocytopenia, and she no longer requires IVIG, with the last administration at day +307 post-HSCT. Post-HSCT assessment documented normal renal and hepatic function. Formal neurodevelopmental evaluation has not been completed to date. However, upon routine assessment by the paediatrician and bone marrow transplantation team, this patient has achieved age-appropriate developmental milestones 19 months post-HSCT.
Detection of compound heterozygous mutations in the IL-7R gene
WES revealed apparent homozygosity for a single nucleotide variant (SNV) c.361dupA in exon 3 of the IL7R gene (NM_002185·3). This finding was confirmed by Sanger sequencing (Fig. 3a). The SNV was predicted to cause a frameshift, leading to a premature stop codon (p.Ile121AsnfsTer8), and would likely result in no protein product due to nonsense-mediated decay, representing a loss of function allele. Segregation analyses showed that only the father was a carrier for this SNV (Fig. 3a), leading to the conclusion that the patient was actually hemizygous for the SNV. CMA BCM version 9·1.1 revealed a deletion of exon 3 of the IL7R gene in the patient's sample. Exact positions for the intragenic deletion were as follows: arr[hg19] 5p13.2 (35862009 × 2, 35867357 − 35867581 × 1, 35867620 × 2) (Fig. 3b). The deletion was small in size, as determined by flanking non-deleted interrogating oligonucleotides, minimum 224 bp, maximum 5600 bp, and involved the entire exon 3 of the IL7R gene, and was confirmed independently by droplet PCR (Supporting information, Fig. S2). This single exon deletion was inherited from the healthy mother (Fig. 3a). Thus, the patient is compound heterozygous for a SNV and a CNV in IL7R (Supporting information, Figs S1 and S2). Among 50 000 individuals who underwent genetic evaluation with CMA testing at Medical Genetics Laboratories, Baylor College of Medicine (MGL BCM), the identical exon 3 IL7R gene deletion was detected in six others. The SNV was present on WES in 14 of 19 total reads (73%), and the wild-type allele was present on WES in five of 19 total reads (26%), a finding which was also visualized on Sanger sequencing of pre-HSCT whole blood DNA (20%) (Fig. 3a) and in buccal DNA (30%) collected post-HSCT (Supporting information, Fig. S3). In addition, using STR analyses, maternal engraftment (5–10%) was demonstrated in the child's pre-HSCT whole blood, but not in the child's buccal cells (Supporting information, Table S2, Fig. S4).
Fig. 3.

(a) Family pedigree showing the variants in the IL7R gene for each of the family members. Sanger sequencing results (top portion of inset boxes) and chromosomal microarray results (bottom portion of inset boxes, mother and patient). Whole exome sequencing (WES) of whole blood DNA identified a single nucleotide variant (SNV), the frameshift mutation c.361dupA (arrow) in exon 3 of the IL7R gene in the patient, which was inherited from the healthy father. The patient was hemizygous for the SNV, but with minor peaks indicating the presence of wild-type (WT) allele as well. While the mother's sequence was WT on Sanger sequencing, chromosomal microarray (CMA) of the patient's sample identified an intragenic copy number variant (CNV) deletion involving entire exon 3 of the IL7R gene, which was inherited from the unaffected mother. The patient has a younger, healthy sibling. (b) Index patient's chromosomal microarray results, in detail. Chromosomal microarray results showing deletion signal for the three probes in exon 3 in the IL7R gene detected in the patient's DNA sample. Exons are marked by their numbers and positioned corresponding to the CMA probes and their genomic position.
Discussion
IL-7Rα deficiency (OMIM 608971) is an autosomal recessive disease described as the third most common type of SCID in the United States 4. It typically presents as T–B+ NK+ immunophenotype and with opportunistic infections within the first year of life 2,34. IL-7 is essential in lymphopoiesis and regulation of peripheral T cells. The interleukin-7 receptor is expressed on the lymphocyte surface as a heterodimer, consisting of two subunits, IL-7Rα chain (CD127) and common-γ chain (CD132). Binding of IL-7 to IL-7Rα chain leads to dimerization with the common-γ chain and subsequent activation of the Janus kinase–signal transducer and activator of transcription (JAK–STAT) pathway 35.
This patient's immunophenotype was not classic for IL-7Rα deficiency, including significant neutropenia (ANC < 100/mm3) and absence of CD4+ T cells, but the presence of CD8+ T cells, and hypergammaglobulinaemia. Hypergammaglobulinaemia was also reported in another child with IL-7Rα-deficient SCID and maternal engraftment 36. Neutropenia, not commonly associated with IL-7Rα-deficient SCID, was the most prominent abnormality identified in this patient prior to presentation with severe infection 36,37. It is plausible that IL-7 may play a role in myelopoiesis, as IL-7 stimulation of T cells induces production of granulocyte–macrophage colony-stimulating factor (GM-CSF) 38. IL-7Rα deficiency has been reported previously in a child with persistent oral ulcers from a young age, similar to our patient, with oral mucositis from 3 months of age 39. Although there was no documented neutropenia in this patient, they may have had mucositis concurrent with unidentified neutropenia that may not have been as profound as in our patient. The causative aetiology of our patient's neutropenia remains unclear, and bone marrow findings were non-specific. Neutropenia, CD4+ lymphocytopenia and hypergammaglobulinaemia can be observed in HIV infection, which was promptly excluded in this patient by HIV-1/2-specific serology and DNA PCR assays.
Importantly, our patient presented with disseminated vaccine-strain varicella. The estimated rate of Oka (vaccine) strain VZV infection in the United States occurs in one case per 10 000 vaccines administered 40. Although there are limited data for VZV-associated mortality in immunocompromised patients, the mortality rate in paediatric oncology patients prior to the advent of anti-viral therapy was estimated to be 7–55% 40,41. Varicella resistance to acyclovir has been reported in immunocompromised patients. Foscarnet, a direct viral DNA polymerase inhibitor, is the drug of choice for patients with disseminated acyclovir-resistant varicella 42. Cidofovir is recommended for acyclovir- and foscarnet-resistant strains 43. VZIG is a purified pooled anti-IgG to VZV, and prophylaxis is recommended up to 10 days following VZV exposure in immunocompromised patients 44. Aggressive triple anti-viral therapy and targeted VZV-specific immune-based therapy likely resulted in VZV control and suppression, even in the face of full myeloablation and transplant in this patient. Vigilance for known adverse events resulted in minimal organ damage from this therapy.
Our patient also had vaccine-strain rubella (RA27/3) infection, which has not been reported previously. There is no known effective treatment for rubella infection, yet our patient did not have significant signs and symptoms consistent with this infection. Given that the isolated rubella was an attenuated vaccine-strain virus, this may account for the lack of overt signs of rubella infection in our patient, yet treatment with multiple anti-viral medications and IVIG may have played a role in reducing viral load.
In recent years, an increasing number of states have implemented newborn screening (NBS) for SCID by measuring TREC, a marker for naive T cell production in the thymus during T cell receptor rearrangement, from dried blood spots on NBS Guthrie cards 8. All known SCID-causing genes share a common defect of partial or total absence of T cells. With early diagnosis of SCID and subsequent HSCT at an early age, infants may have decreased morbidity and mortality. Upon retrospective analyses of our patient's NBS samples (at birth and 2 weeks of age), TREC were confirmed to be absent – despite the presence of CD8+ cells. Had NBS been implemented in Texas prior to our patient's birth, her T cell lymphocytopenia would have been detected, thus allowing for early diagnosis, avoidance of live viral vaccine delivery and timely immune reconstitution options. This would likely have prevented our patient's complicated clinical course.
With the application of tandem gene sequencing and copy number variability assessment, our patient received a definitive genetic diagnosis. To our knowledge, disease-causing copy number variants of the IL7R gene have not been described previously. Copy number variant testing is currently not part of standard genetic testing for suspected congenital immunodeficiency; hence, causative genetic alterations may not be identified. Autosomal CNVs are undetectable by PCR–single-strand conformational polymorphism (SSCP) testing or Sanger sequencing, but can be detected by quantitative analyses, such as multiplex ligation-dependent probe amplification (MLPA) or CMA with exon coverage of the genes of interest (PIDD genes) 45.
Whole exome sequencing, as well as targeted exome sequencing, with more complete gene coverage and increased number of reads, is currently not sufficient to detect small, intragenic CNVs 46. With sufficient coverage and number of reads of the region of interest, bioinformatics tools may allow for detection of small (fewer than 4 exons) intragenic CNVs 47. Currently, in patients with unidentified genetic diagnosis for SCID, the combination of whole exome sequencing and CMA utilizing probes covering all exons of PIDD genes, improves genetic diagnostics.
This is the first report of an intragenic CNV in an IL-7Rα-deficient SCID. Larger CNVs spanning several genes have been reported previously in other PIDDs, such as deletion of 16p11.2 in autosomal recessive Coronin-1a deficiency (OMIM 615401) and of 22q11.2 in DiGeorge syndrome (OMIM 188400) 48. Intragenic CNVs have been reported in autosomal recessive DOCK8 deficiency (OMIM 243700) and autosomal dominant PLCG2-related CVID (OMIM 614878) 49,50. The exon 3 deletion in IL7R may have occurred de novo in our patient's healthy mother or may have been inherited. Interestingly, the same exon 3 deletion has also been detected in samples from several individuals in our patient population, and deletions in IL7R have been reported in the Database of Genomic Variants (http://dgv.tcag.ca) 51. Thus, a recessive CNV carrier state for this gene may be present in subsets of the general population 52.
Although this patient inherited one mutant IL7R exon 3 allele from each parent, a wild-type allele was identified unexpectedly on WES using DNA from whole blood obtained pre-HSCT and also identified in buccal samples. This wild-type allele in the c.361dupA SNV position was present in five of 19 (26%) WES reads, and was also evident on Sanger sequencing (Fig. 3a). Sequencing of buccal samples showed a similar percentage of wild-type allele present (Supporting information, Fig. S3, Table S1). The unexpected, non-inherited wild-type allele may be the result of a spontaneous somatic revertant correcting the inherited SNV in one cell lineage during early embryonic development and may subsequently cause the mosaicism to be present in several cell lines and tissues. Revertant somatic mosaicism, previously thought to be rare, is a phenomenon of ‘natural gene therapy’ recognized in PIDDs and in heritable skin diseases 53. Revertant mosaicism confined to lymphohaematopoietic stem cells has been observed in ADA deficiency (OMIM 102700), X-linked SCID (OMIM 300400), leucocyte adhesion deficiency type I (OMIM 116920), Wiskott–Aldrich syndrome (OMIM 301000) and in Fanconi anaemia patients with FANCA mutations (OMIM 227650) 54–59. Although both CD4+ and CD8+ T cells are typically absent in IL-7Rα deficiency, CD8+ T cells were present in our patient, leading to the hypothesis that the revertant mutation was present in select subclones of CD8+ cells. However, two maternal STR peaks were observed for several markers in the patient's pre-HSCT blood, consistent with maternal engraftment of a leucocyte subset. Only DNA extracted from whole blood was available, preventing definitive derivation of the patient's CD8+ T cells. The amount of maternal engraftment in whole blood was 5–10%, which is nearly below the detection limit of STR analyses and is considered a small percentage compared to the larger percentage of maternal engraftment observed in other SCID patients (Supporting information, Fig. S4). The mosaicism for the wild-type allele and the maternal engraftment may have contributed to the immunological phenotype observed and the delayed onset of infectious complications.
Conclusion
This child with disseminated vaccine-strain VZV and rubella infections is the first SCID case reported with an intragenic CNV and reversion mutation in the IL7R gene. Due to delayed SCID diagnosis, this patient received live viral vaccines, which would have otherwise been avoided. In this case, the retrospective analysis of TREC demonstrates how early detection of T cell abnormalities by NBS is an essential tool in preventing morbidity. Through the combination of CMA and WES, our patient was found to have IL-7Rα deficiency and was compound heterozygous for the following two deleterious IL7R gene variants: a single nucleotide variant (SNV) located within exon 3 and a CNV deletion involving entire exon 3 of the IL7R gene. Maternal engraftment, somatic revertant mosaicism or both phenomena may explain the immunophenotype and delayed disease onset in this patient. Our case demonstrates that the combination of newer genetic diagnostic tools, such as WES combined with CMA, can identify novel genetic aetiologies of SCID and broaden our knowledge of PIDD immunophenotypes. It also re-emphasizes the importance of avoiding live viral vaccination in SCID patients and the value of NBS for SCID.
Acknowledgments
The authors are grateful to the family for their participation in this study. We thank Texas Department of Health Services Newborn Screening Program for retrieval of newborn screening specimens. We gratefully acknowledge the following individuals at the CDC for their respective roles in evaluation of vaccine-acquired viral infections: Dr. C. Paddock of the NCEZID, Division of High-Consequence Pathogens and Pathology, Infectious Diseases Pathology Branch, and the following individuals and their respective teams in the NCIRD, Division of Viral Diseases, Measles, Mumps, Rubella, and Herpesvirus Laboratory Branch: Dr. D.S. Schmid and K.W. Radford, Herpesvirus Team, Dr. J. Icenogle and E. Abernaty, MS, Rubella Team, and Dr. P. Rota and Dr. R. McNall, Measles Team. Sincere thanks to Dr C. Bollard, Dr A. Leen, Dr A. Papadopoulou and Dr H. Tashiro for evaluation of immune reconstitution by ELISPOT assay and editing contributions. Special thanks are due to Lillian Gundersen for technical assistance and to Dr S. Nicholas for editing contributions. The Baylor-Hopkins Center for Mendelian Genomics is funded by the National Human Genome Research Institute (U54HG006542 and U54HG003273), National Institutes of Health, USA.
Disclosure
The Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from molecular genetic testing offered in the Medical Genetics Laboratories. J. R. L. holds stock ownership in 23andMe, Inc., and is a co-inventor on multiple United States and European patents related to molecular diagnostics. The other authors have no personal conflicts of interest to declare.
Supporting Information
Additional Supporting information may be found in the online version of this article at the publisher's web-site:
Fig. S1. Chromosomal localization and genomic structure of the IL7R gene with illustration of the genetic alterations in IL7R causing the patient's disease.
Fig. S2. Droplet polymerase chain reaction (PCR) quantitation of IL7R exon 3.
Fig. S3. Sanger sequencing results from patient's blood and buccal tissue.
Fig. S4. Results of short tandem repeat (STR) testing for maternal engraftment.
Table S1. Mosaicism in blood and buccal DNA.
Table S2. Short tandem repeats (STRs) of the patient's pre-haematopoietic stem cell transplantation (HSCT) blood DNA.
References
- 1.Stiehm ER, Fudenberg HH. Serum levels of immune globulins in health and disease: a survey. Pediatrics. 1966;37:715–727. [PubMed] [Google Scholar]
- 2.Puck JM. The case for newborn screening for severe combined immunodeficiency and related disorders. Ann NY Acad Sci. 2011;1246:108–117. doi: 10.1111/j.1749-6632.2011.06346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buckley RH. The long quest for neonatal screening for severe combined immunodeficiency. J Allergy Clin Immunol. 2012;129:597–604. doi: 10.1016/j.jaci.2011.12.964. quiz 5–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Puck JM. Laboratory technology for population-based screening for severe combined immunodeficiency in neonates: the winner is T-cell receptor excision circles. J Allergy Clin Immunol. 2012;129:607–616. doi: 10.1016/j.jaci.2012.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Notarangelo LD. Primary immunodeficiencies. J Allergy Clin Immunol. 2010;125(2 Suppl 2):S182–194. doi: 10.1016/j.jaci.2009.07.053. [DOI] [PubMed] [Google Scholar]
- 6.Felgentreff K, Perez-Becker R, Speckmann C, et al. Clinical and immunological manifestations of patients with atypical severe combined immunodeficiency. Clinical Immunology. 2011;141:73–82. doi: 10.1016/j.clim.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 7.Dvorak CC, Cowan MJ, Logan BR, et al. The natural history of children with severe combined immunodeficiency: baseline features of the first fifty patients of the primary immune deficiency treatment consortium prospective study 6901. J Clin Immunol. 2013;33:1156–1164. doi: 10.1007/s10875-013-9917-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Verbsky JW, Baker MW, Grossman WJ, et al. Newborn screening for severe combined immunodeficiency; the Wisconsin experience (2008–2011) J Clin Immunol. 2012;32:82–88. doi: 10.1007/s10875-011-9609-4. [DOI] [PubMed] [Google Scholar]
- 9.Centers for Disease Control and Prevention. Advisory Committee on Immunization Practices (ACIP) recommended immunization schedules for persons aged 0 through 18 years and adults aged 19 years and older – United States, 2013. MMWR Surveill Summ. 2013;62(Suppl. 1):2–8. [PubMed] [Google Scholar]
- 10.Patel NC, Hertel PM, Estes MK, et al. Vaccine-acquired rotavirus in infants with severe combined immunodeficiency. N Engl J Med. 2010;362:314–319. doi: 10.1056/NEJMoa0904485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghaffar F, Carrick K, Rogers BB, Margraf LR, Krisher K, Ramilo O. Disseminated infection with varicella-zoster virus vaccine strain presenting as hepatitis in a child with adenosine deaminase deficiency. Pediatr Infect Dis J. 2000;19:764–766. doi: 10.1097/00006454-200008000-00022. [DOI] [PubMed] [Google Scholar]
- 12.Banzhoff A, Schauer U, Riedel F, Gahr M, Rieger CH. Fatal varicella in a 5-year-old boy. Eur J Pediatr. 1997;156:333–334. doi: 10.1007/s004310050612. [DOI] [PubMed] [Google Scholar]
- 13.Levy O, Orange JS, Hibberd P, et al. Disseminated varicella infection due to the vaccine strain of varicella-zoster virus, in a patient with a novel deficiency in natural killer T cells. J Infect Dis. 2003;188:948–953. doi: 10.1086/378503. [DOI] [PubMed] [Google Scholar]
- 14.Banovic T, Yanilla M, Simmons R, et al. Disseminated varicella infection caused by varicella vaccine strain in a child with low invariant natural killer T cells and diminished CD1d expression. J Infect Dis. 2011;204:1893–1901. doi: 10.1093/infdis/jir660. [DOI] [PubMed] [Google Scholar]
- 15.Jean-Philippe P, Freedman A, Chang MW, et al. Severe varicella caused by varicella-vaccine strain in a child with significant T-cell dysfunction. Pediatrics. 2007;120:e1345–1349. doi: 10.1542/peds.2004-1681. [DOI] [PubMed] [Google Scholar]
- 16.Shearer WT, Rosenblatt HM, Gelman RS, et al. Lymphocyte subsets in healthy children from birth through 18 years of age: the Pediatric AIDS Clinical Trials Group P1009 study. J Allergy Clin Immunol. 2003;112:973–980. doi: 10.1016/j.jaci.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 17.Grier JT, Forbes LR, Monaco-Shawver L, et al. Human immunodeficiency-causing mutation defines CD16 in spontaneous NK cell cytotoxicity. J Clin Invest. 2012;122:3769–3780. doi: 10.1172/JCI64837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leen AM, Myers GD, Sili U, et al. Monoculture-derived T lymphocytes specific for multiple viruses expand and produce clinically relevant effects in immunocompromised individuals. Nat Med. 2006;12:1160–1166. doi: 10.1038/nm1475. [DOI] [PubMed] [Google Scholar]
- 19.Routes JM, Grossman WJ, Verbsky J, et al. Statewide newborn screening for severe T-cell lymphopenia. JAMA. 2009;302:2465–2470. doi: 10.1001/jama.2009.1806. [DOI] [PubMed] [Google Scholar]
- 20.Baker MW, Grossman WJ, Laessig RH, et al. Development of a routine newborn screening protocol for severe combined immunodeficiency. J Allergy Clin Immunol. 2009;124:522–527. doi: 10.1016/j.jaci.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 21.Lupski JR, Gonzaga-Jauregui C, Yang Y, et al. Exome sequencing resolves apparent incidental findings and reveals further complexity of SH3TC2 variant alleles causing Charcot–Marie–Tooth neuropathy. Genome Med. 2013;5:57. doi: 10.1186/gm461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheung SW, Shaw CA, Yu W, et al. Development and validation of a CGH microarray for clinical cytogenetic diagnosis. Genet Med. 2005;7:422–432. doi: 10.1097/01.gim.0000170992.63691.32. [DOI] [PubMed] [Google Scholar]
- 23.Boone PM, Bacino CA, Shaw CA, et al. Detection of clinically relevant exonic copy-number changes by array CGH. Hum Mutat. 2010;31:1326–1442. doi: 10.1002/humu.21360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Al-Herz W, Bousfiha A, Casanova JL, et al. Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency. Front Immunol. 2011;2:54. doi: 10.3389/fimmu.2011.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keerthikumar S, Raju R, Kandasamy K, et al. RAPID: Resource of Asian Primary Immunodeficiency Diseases. Nucleic Acids Res. 2009;37(database issue):D863–D867. doi: 10.1093/nar/gkn682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boone PM, Campbell IM, Baggett BC, et al. Deletions of recessive disease genes: CNV contribution to carrier states and disease-causing alleles. Genome Res. 2013;23:1383–1394. doi: 10.1101/gr.156075.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bainbridge MN, Wang M, Wu Y, et al. Targeted enrichment beyond the consensus coding DNA sequence exome reveals exons with higher variant densities. Genome Biol. 2011;12:R68. doi: 10.1186/gb-2011-12-7-r68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Challis D, Yu J, Evani US, et al. An integrative variant analysis suite for whole exome next-generation sequencing data. BMC Bioinformatics. 2012;13:8. doi: 10.1186/1471-2105-13-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Danecek P, Auton A, Abecasis G, et al. The variant call format and VCF tools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bainbridge MN, Wiszniewski W, Murdock DR, et al. Whole-genome sequencing for optimized patient management. Sci Transl Med. 2011;3:87re3. doi: 10.1126/scitranslmed.3002243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buckley RH. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu Rev Immunol. 2004;22:625–655. doi: 10.1146/annurev.immunol.22.012703.104614. [DOI] [PubMed] [Google Scholar]
- 35.Mazzucchelli R, Durum SK. Interleukin-7 receptor expression: intelligent design. Nat Rev Immunol. 2007;7:144–154. doi: 10.1038/nri2023. [DOI] [PubMed] [Google Scholar]
- 36.Giliani S, Mori L, de Saint Basile G, et al. Interleukin-7 receptor alpha (IL-7Ralpha) deficiency: cellular and molecular bases. Analysis of clinical, immunological, and molecular features in 16 novel patients. Immunol Rev. 2005;203:110–126. doi: 10.1111/j.0105-2896.2005.00234.x. [DOI] [PubMed] [Google Scholar]
- 37.Sokolic R. Neutropenia in primary immunodeficiency. Curr Opin Hematol. 2013;20:55–65. doi: 10.1097/MOH.0b013e32835aef1c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aiello FB, Keller JR, Klarmann KD, Dranoff G, Mazzucchelli R, Durum SK. IL-7 induces myelopoiesis and erythropoiesis. J Immunol. 2007;178:1553–1563. doi: 10.4049/jimmunol.178.3.1553. [DOI] [PubMed] [Google Scholar]
- 39.Roifman CM, Zhang J, Chitayat D, Sharfe N. A partial deficiency of interleukin-7R alpha is sufficient to abrogate T-cell development and cause severe combined immunodeficiency. Blood. 2000;96:2803–2807. [PubMed] [Google Scholar]
- 40.Sharrar RG, LaRussa P, Galea SA, et al. The postmarketing safety profile of varicella vaccine. Vaccine. 2000;19:916–923. doi: 10.1016/s0264-410x(00)00297-8. [DOI] [PubMed] [Google Scholar]
- 41.Wiegering V, Schick J, Beer M, et al. Varicella-zoster virus infections in immunocompromised patients – a single centre 6-years analysis. BMC Pediatr. 2011;11:31. doi: 10.1186/1471-2431-11-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hatchette T, Tipples GA, Peters G, Alsuwaidi A, Zhou J, Mailman TL. Foscarnet salvage therapy for acyclovir-resistant varicella zoster: report of a novel thymidine kinase mutation and review of the literature. Pediatr Infect Dis J. 2008;27:75–77. doi: 10.1097/INF.0b013e3181598315. [DOI] [PubMed] [Google Scholar]
- 43.Andrei G, Topalis D, Fiten P, et al. In vitro-selected drug-resistant varicella-zoster virus mutants in the thymidine kinase and DNA polymerase genes yield novel phenotype–genotype associations and highlight differences between antiherpesvirus drugs. J Virol. 2012;86:2641–2652. doi: 10.1128/JVI.06620-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Food and Drug Administration. 2006. Varicella zoster immune globulin (VZIG) anticipated short supply and alternate product availability under an investigational new drug application expanded access protocol. Silver Spring, MD: US Department of Health and Human Services, Food and Drug Administration. Available at: http://www.fda.gov/biologicsbloodvaccines/safetyavailability/ucm176029.htm (accessed 11 July 2014)
- 45.Eijk-Van Os PG, Schouten JP. Multiplex ligation-dependent probe amplification [MLPA(R)] for the detection of copy number variation in genomic sequences. Methods Mol Biol. 2011;688:97–126. doi: 10.1007/978-1-60761-947-5_8. [DOI] [PubMed] [Google Scholar]
- 46.de Ligt J, Boone PM, Pfundt R, et al. Detection of clinically relevant copy number variants with whole-exome sequencing. Hum Mutat. 2013;34:1439–1448. doi: 10.1002/humu.22387. [DOI] [PubMed] [Google Scholar]
- 47.Wu J, Grzeda KR, Stewart C, et al. Copy number variation detection from 1000 Genomes Project exon capture sequencing data. BMC Bioinformatics. 2012;13:305. doi: 10.1186/1471-2105-13-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shiow LR, Paris K, Akana MC, Cyster JG, Sorensen RU, Puck JM. Severe combined immunodeficiency (SCID) and attention deficit hyperactivity disorder (ADHD) associated with a Coronin-1A mutation and a chromosome 16p11.2 deletion. Clin Immunol. 2009;131:24–30. doi: 10.1016/j.clim.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Q, Davis JC, Lamborn IT, et al. Combined immunodeficiency associated with DOCK8 mutations. N Engl J Med. 2009;361:2046–2055. doi: 10.1056/NEJMoa0905506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ombrello MJ, Remmers EF, Sun G, et al. Cold urticaria, immunodeficiency, and autoimmunity related to PLCG2 deletions. N Engl J Med. 2012;366:330–338. doi: 10.1056/NEJMoa1102140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.MacDonald JR, Ziman R, Yuen RK, Feuk L, Scherer SW. The database of genomic variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 2013;42:986–992. doi: 10.1093/nar/gkt958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wiszniewski W, Hunter JV, Hanchard NA, et al. TM4SF20 ancestral deletion and susceptibility to a pediatric disorder of early language delay and cerebral white matter hyperintensities. Am J Hum Genet. 2013;93:197–210. doi: 10.1016/j.ajhg.2013.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jonkman MF, Sceffer H, Stulp R, et al. Revertant mosaicism in epidermolysis bullosa caused by mitotic gene conversion. Cell. 1997;88:543–551. doi: 10.1016/s0092-8674(00)81894-2. [DOI] [PubMed] [Google Scholar]
- 54.Hirschhorn R, Yang DR, Puck JM, Huie ML, Jiang CK, Kurlandsky LE. Spontaneous in vivo reversion to normal of an inherited mutation in a patient with adenosine deaminase deficiency. Nat Genet. 1996;13:290–295. doi: 10.1038/ng0796-290. [DOI] [PubMed] [Google Scholar]
- 55.Stephan V, Wahn V, Le Deist F, et al. Atypical X-linked severe combined immunodeficiency due to possible spontaneous reversion of the genetic defect in T cells. N Engl J Med. 1996;335:1563–1567. doi: 10.1056/NEJM199611213352104. [DOI] [PubMed] [Google Scholar]
- 56.Tone Y, Wada T, Shibata F, et al. Somatic revertant mosaicism in a patient with leukocyte adhesion deficiency type 1. Blood. 2007;109:1182–1184. doi: 10.1182/blood-2007-08-039057. [DOI] [PubMed] [Google Scholar]
- 57.Wada T, Konno A, Schurman SH, et al. Second-site mutation in the Wiskott–Aldrich syndrome (WAS) protein gene causes somatic mosaicism in two WAS siblings. J Clin Invest. 2003;111:1389–1397. doi: 10.1172/JCI15485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gregory JJ, Jr, Wagner JE, Verlander PC, et al. Somatic mosaicism in Fanconi anemia: evidence of genotypic reversion in lymphohematopoietic stem cells. Proc Natl Acad Sci USA. 2001;98:2532–2537. doi: 10.1073/pnas.051609898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gross M, Hanenberg H, Lobitz S, et al. Reverse mosaicism in Fanconi anemia: natural gene therapy via molecular self-correction. Cytogenet Genome Res. 2002;98:126–135. doi: 10.1159/000069805. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Chromosomal localization and genomic structure of the IL7R gene with illustration of the genetic alterations in IL7R causing the patient's disease.
Fig. S2. Droplet polymerase chain reaction (PCR) quantitation of IL7R exon 3.
Fig. S3. Sanger sequencing results from patient's blood and buccal tissue.
Fig. S4. Results of short tandem repeat (STR) testing for maternal engraftment.
Table S1. Mosaicism in blood and buccal DNA.
Table S2. Short tandem repeats (STRs) of the patient's pre-haematopoietic stem cell transplantation (HSCT) blood DNA.