

Abstract
Mutations in the X-linked inhibitor of apoptosis (XIAP) gene have been associated with XLP-like disease, including recurrent Epstein–Barr virus (EBV)-related haemophagocytic lymphohystiocytosis (HLH), but the immunopathogenic bases of EBV-related disease in XIAP deficiency is unknown. We present the first analysis of EBV-specific T cell responses in functional XIAP deficiency. In a family of patients with a novel mutation in XIAP (G466X) leading to a late-truncated protein and varying clinical features, we identified gradual hypogammaglobulinaemia and large expansions of T cell subsets, including a prominent CD4+CD8+ population. Extensive ex-vivo analyses showed that the expanded T cell subsets were dominated by EBV-specific cells with conserved cytotoxic, proliferative and interferon (IFN)-γ secretion capacity. The EBV load in blood fluctuated and was occasionally very high, indicating that the XIAPG466X mutation could impact upon EBV latency. XIAP deficiency may unravel a new immunopathogenic mechanism in EBV-associated disease.
Keywords: cytotoxic T cells, immunodeficiency diseases, T cells, viral
Introduction
Epstein–Barr virus (EBV) is a gamma herpesvirus that co-exists with man through the establishment of a fine balance between virus replicative infection and host immune responses 1. Primary infection of a naive host leads to viral replication, increases in viral load and establishment of a lytic cycle within epithelial cells of the oropharynx 2. Coincidentally, mucosal B cells are infected, in which non-replicative infection (latency) is established 2. A vigorous CD8+ cytotoxic T lymphocyte (CTL) response is induced during the primary infection, initially to lytic and gradually also to latent viral epitopes. A less dramatic CD4+ T cell response is also detected at this stage 3,4. The immune response in healthy individuals destroys the majority of EBV-infected cells, but the virus achieves long-term persistence by down-regulating latent viral protein expression and establishing a virtually silent infection in some long-lived memory B cells 2. The establishment of long-term silent latency decreases viral antigen load, leading to waning of the T cell responses 4.
During long-term latency, sporadic reactivations of the lytic cycle occur in infected B cells, triggered by B cell activation and plasma cell differentiation signals and leading to new cycles of infection. This elicits further T cell responses, and EBV-specific memory T cells, reactive to both latent- and lytic-cycle antigens, are maintained in the periphery 4. This long-term immunosurveillance of EBV infection is critical, as clearly demonstrated by the malignant EBV-driven lymphoproliferation associated with immunosuppression such as in late HIV infection or post-transplantation 5.
EBV primary infection, occurring normally in infancy, is asymptomatic. In the benign self-limited lymphoproliferative disease, infectious mononucleosis (IM) in young persons 6, symptoms are associated with infiltrating EBV-infected B cells and EBV-specific T cells. In infants with non-functional T cells, EBV-associated haemophagocytic lymphohistiocytosis (HLH) may be manifest in different organs. The human primary immunodeficiency, the X-linked lymphoproliferative disease type 1 (XLP1), is associated with extreme sensitivity to EBV infection. The causative gene, Src-homology 2 domain containing gene 1A (SH2D1A), codes for the signalling lymphocyte activation molecule (SLAM)-associated protein (SAP) 7 (OMIM#308240). SAP is an essential adapter molecule in signalling pathways involved in cytotoxicity and approximately 50–60% of XLP patients present in childhood with severe primary EBV infection, frequently with a fatal course due to liver failure or HLH. A massive and dysregulated proliferation of CD8+ T cells incapable of exerting an effective cytotoxic response underlies the immunopathogenesis associated with EBV infection in XLP1 8–11. Twenty to 30% of XLP1 patients go on to develop malignant or non-malignant B cell lymphoproliferative disorders in relation to impaired cytotoxicity and exhaustion of EBV-specific T cells 7.
A different protein, X-linked inhibitor of apoptosis (XIAP, also named BIRC4), has been implicated in XLP2. This protein is absent or non-functional in patients with a phenotype partially resembling XLP 12 (OMIM#300365). XIAP is a member of the inhibitor of apoptosis (IAP) family that inhibits apoptosis by preventing activation of caspases 3, 7 and 9 13. XIAP deficiency may be associated with EBV-related HLH that is often recurrent and associated with hepatosplenomegaly; in contrast with SAP deficiency, so far no patients have developed lymphoma 12. The impact of XIAP deficiency on EBV cytotoxic responses and the natural course of EBV infection and the immunopathogenic mechanisms of HLH in XIAP-deficient patients are currently unknown.
In this paper we describe three kindred in a large English family in which the clinical phenotypes in several male patients are variable in type and severity 14. One non-sense mutation in XIAP, in association with a rare CD40LG polymorphismG219R, was identified in all affected males that resulted in attenuated expression of a late truncated protein (G466X) and a CD40LG protein with decreased capacity to bind to CD40. One unaffected male was found to be a carrier of the XIAPG466X alone 14. We performed an exhaustive study of the EBV-specific cellular immunity in several individuals harbouring the mutation and found a massive expansion of memory EBV-specific CD4+, CD8+ and CD4+CD8+ double-positive T cells several years after EBV primary infection. Many epitope-specific responses were identified among the responding T cells, but those to lytic cycle antigens were particularly strong. Fluctuating and occasionally very high viral loads in patients suggest that this memory ‘inflation’ results from continuous re-encounter with antigen by virus-specific T cells and that silent long-term EBV infection is disturbed in XIAPG466X individuals. This study demonstrates the fine balance struck by EBV and the immune system, and the key role of XIAP in maintaining this equilibrium.
Mutations in XIAP also result in inflammatory conditions, particularly of the gut, in children and young adults, including female carriers. Although EBV-associated haemophagocytic lymphohistiocytosis (HLH) was the original predominant clinical phenotype, other conditions rapidly became apparent 14. Recently, 17 of 25 patients were found to have presented with manifestations other than HLH. As in our original oligogenic family, these included severe infectious mononucleosis and Crohn's-like bowel disease as well as other features of a primary immunodeficiency (PID), such as antibody deficiency.
In our families the truncated version of XIAP led to early gut inflammation in one patient, late-onset Crohn's disease in one male patient and a Crohn's-like colitis in one female carrier 15. It has been reported from another centre that there are no correlations between genotype or protein expression and clinical phenotypes 16, suggesting that other genes may well be involved.
Here we update the findings on the original three families, including new findings on their clinical phenotypes, and concentrate on the anti-EBV mechanisms associated with the CD40LG polymorphism found together with truncated XIAP protein.
Methods
Case report
Initial details of five males from a large English family followed for a suspected immunodeficiency (Fig. 1) were published previously in 2011 14; more recent developments are now reported. The index patient (II-7) had non-viral acute hepatitis in childhood that evolved into idiopathic chronic liver disease with hepatosplenomegaly. He also had progressive panhypogammaglobulinaemia, though initially only a specific antibody defect against pneumococci. He then suffered from recurrent bacterial infections, developed bronchiectasis and required immunoglobulin substitution. His clinical picture was complicated by polyclonal lymphoproliferation, consisting of persistent lymphadenopathy and suspected lymphoid interstitial pneumonitis (LIP). He died at the age of 28 years due to liver failure, and later studies showed that he had two polymorphisms in HFE (homozygous for histidine at codon 63 and heterozygous for cysteine and tyrosine at codon 282). His elder brother (Fig. 1, II-5) remains clinically well, now aged 46 years. Patient III-2 (Fig. 1) suffered from a primary infection (EBV-IgM+) at the age of 7 years with hepatitis, splenomegaly and pancytopenia; he relapsed 9 months later. He recovered clinically but has persistent splenomegaly and mild hypogammaglobulinaemia with good antibody production but reduced CD4 cells. His brother (III-1) had moderate splenomegaly and mild panhypogammaglobulinaemia, but has now developed recurrent abscesses and a Crohn's like colitis at the age of 33 years and is receiving infliximab therapy. The youngest patient (III-6) presented with complicated and recurrent skin vesicles, and developed transient lymphadenopathy, splenomegaly, transient hypergammaglobulinaemia and atypical inflammatory bowel disease. He underwent haematopoietic stem cell transplantation (HSCT) successfully 12 months ago. On several occasions, all patients were found to be EBV immunoglobulin (Ig)G+ [anti-virus capsid antigen (VCA)] and cytomegalovirus (CMV) IgG–. Another related male (I-5) had a history of recurrent bacterial infections, lymphadenopathies and hepatosplenomegaly in adulthood but normal serum immunoglobulins. This patient's brother had Crohn's disease (I-3) in childhood and died aged 23 years. Five of the female carriers (I.2, I.6, II.2, II.3, II.6) are healthy, but patient III.3 had pneumonia and erythema nodosum aged 14 years with hypergammaglobulinaemia and developed recurrent skin abscesses and watery diarrhoea 14 years later. She has random inactivation of her X chromosomes, as does the other carrier with Crohn's disease due to a XIAP deficiency, in contrast to the healthy carriers 15. Informed consent was obtained from each patient and the study protocol conformed to the 1975 declaration of Helsinki and the local ethics regulations. The major histocompatibility complex (MHC) class I and class II haplotypes of the four patients included in this study are depicted in the Supporting information (Table S1).
Fig. 1.
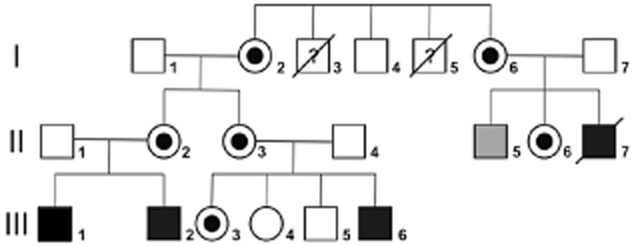
Pedigree of the family. Filled squares indicate male relatives harbouring the XIAP G466X mutation; shown in black are those with clear clinical symptoms and shown in grey those without. Circles with black centres indicate female carriers. Diagonal bars indicate deceased individuals.
Cell culture
Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood by density gradient centrifugation (Lymphoprep; Nycomed, Oslo, Norway). Naive B cells were purified from healthy donor PMBCs by staining with anti-CD19 and anti-CD27 followed by fluorescence activated cell sorter (FACS) analysis (Dako, Glostrup, Denmark). EBV-transformed lymphoblastoid cell lines (EBV-LCLs) were generated from PBMCs in the presence of cyclosporin using standard techniques.
Antibodies and reagents
Monoclonal antibodies used in flow cytometry and cell-sorting experiments were anti-CD3-peridinin chlorophyll (PerCP), CD4-allophycocyanin (APC), CD8-fluorescein isothiocyanate (FITC), CD4-phycoerythrin (PE), CD8-PerCP, CD45RA-FITC, CD45RO-PE, CD45RO-FITC, CCR7-APC, CD27-FITC, CD28-APC, Ki-67 FITC, granzyme B-APC, CD107a-FITC, interferon (IFN)γ-APC, and the corresponding isotype controls (all obtained from BD Biosciencies, Oxford, UK). The B-35 EPL loaded tetramer was obtained from the repository at the Weatherall Institute of Molecular Medicine.
Carboxyfluorescein succinimidyl ester (CFSE)-based proliferation assays
PBMCs were incubated at 1 × 107 cells/ml in phosphate-buffered saline (PBS) at 37°C for 10 min, with 1·0 M CFSE (Molecular Probes, Eugene, OR, USA). The reaction was terminated by washing the cells twice with RPMI-1640 (Gibco, Carlsbad, CA, USA) 10% human serum (R10). The cells were then resuspended at 2 × 106/ml in R10. Labelled cells (5 × 105/well; 250 μl) were cultured in a 96-well flat-bottomed plate with medium alone, phytohaemagglutinin (PHA) (5 g/ml; Sigma, St Louis, MO, USA), anti-CD3/anti-CD28/anti-CD2-coated beads (2 μl per well of 107 beads; Miltenyi Biotech, Bergisch Gladbach, Germany) or tetanus toxoid (Aventis Pasteur, Cary, NC, USA) as negative and positive controls for proliferation in a cell incubator in 37°C and 5% CO2. Viral antigens used were a cellular extract from an EBV-infected B cell line stimulated to increase lytic cycle antigens and enriched for viral proteins, and an EBV-negative control extract from the same cell line (Virusys, Sykesville, MD, USA), rubella (Advanced Biotechnologies Inc., Columbia, MD, USA), CMV (East Coast Biologies, North Berwick, ME, USA), Coxsackie (Institute Virion, Zürich, Switzerland), varicella zoster virus (VZV) (Advanced Biotechnologies), respiratory syncytial virus (RSV) (Institute Virion), B19 (VP2 recombinant protein), flu (Enzira influenza vaccine; ZLB Pharma GmbH, Marburg, Germany) and adenovirus (hexon peptide sequence). After 6 days of culture, cells were washed with PBS and stained with anti-human CD8-PE, CD4-APC and Viaprobe [7-aminoactinomycin D (7-AAD)] to exclude dead cells (BD Biosciences, San Jose, CA, USA). Flow cytometric analysis was performed on a FACSCalibur (BD Biosciences).
The number of cells that had proliferated was determined by gating on the lineage-positive CFSElo and CFSEhigh subsets. We calculated the stimulation index (SI) and considered positive responses as described previously 17. The final proliferative frequency (%) was obtained by subtracting the proliferative frequency with the antigen from the proliferative frequency without the antigen. Where the SI was <2, the proliferative frequency was set as zero.
Enzyme-linked immunospot (ELISPOT) assay
PBMCs were tested by IFN-γ ELISPOT assays (Mabtech, Stockholm, Sweden), as per the manufacturer's instructions and as described previously 17. PBMCs (2 × 105/well) were plated in anti-IFN-γ precoated 96-well plates (Millipore, Billerica, MA, USA). Medium was used as a negative control and positive controls were as for the CFSE assay, although 5 μl of anti-CD3/CD28/CD2-coated beads were used. EBV+ cell extract and controls were used as before, but at 10 g/ml final concentration. EBV-derived peptides were used at a 5 μg/ml final concentration (see Supporting information, Table S2 for complete peptide sequences) 4. The plates were developed after 14–16 h incubation using AP colour reagent (Biorad, Hercules, CA, USA) and analysed for spot-forming cells (SFCs) using an ELISPOT plate reader (ELISPOT 3·1 SR program; AID Reader System, Strasberg, Germany). Assays with high background (average 10 SFC/well in negative control wells) or insufficient PHA or T cell-stimulating beads were excluded. The frequency of IFN-γ+ T cells that were specific for each antigen was calculated by subtracting the average SFC in negative control duplicate wells from the average SFC in stimulated duplicate wells and expressed as EBV-specific IFN-γ SFCs/106 PBMC. A positive response was defined assuming a Poisson distribution, as described previously (Excel binomdist statistics program; Microsoft) 17. The mean background in the negative control wells for the control group was 1·75 spots per 200 000 cells [± standard deviation (s.d.) 1·94) and for the patients was 2·94 per 200 000 cells (± s.d. 2·4). These were not significantly different (P = 0·22).
Ex-vivo intracellular cytokine production combined with cytotoxic degranulation assay
PBMCs were resuspended at 4 × 105/ml in R10 to a final volume of 250 μl in 96-well plates. Cells were stimulated with the EBV+ cell extract and the negative control (Virusys) and anti-CD3/CD28/CD2-coated beads. Anti-CD107a FITC was added to the cells during the stimulation as described previously 18. After 2 h a Golgi blocker (brefeldin A; BD Biosciences) was added. After a total of 6 h, cells were removed, stained with CD4-PE and CD8-PerCP antibodies, fixed and permeabilized (Cytofix/cytoperm kit; BD Biosciencies). An anti-IFN-γ APC antibody (BD Biosciences) was used for intracellular staining.
Flow cytometry-based cytotoxic assay
PBMCs from patient III-6 were stained with the B35-EPL loaded tetramer (Dako) and tetramer-positive (tetramer+) cells were isolated by cell sorting. Cells were expanded in vitro by co-culture with an irradiated control B35+ EBV-LCL obtained from a healthy donor and pulsed with the EPL peptide, in the presence of irradiated allogenic feeder cells, PHA and interleukin (IL)-2. After 12 days, the cell line was confirmed to contain exclusively (>99%) CD8+ tetramer+ cells. Specific cytotoxicity towards EBV+ B cells presenting the BZLF1-derived EPL peptide was analysed, using the same B35+ EBV-LCL and a modified method described previously 18. Fewer than 2% of cells were positive for the lytic cycle protein BZLF1 in the target cell line. Cells were labelled with either CFSE (green) or tetramethylrhodamine, methyl ester (TMRM) (red) (both from Molecular Probes). CFSE staining was performed as described above. For TMRM staining 2 × 106 cells/ml were incubated in prewarmed RPMI-10% fetal calf serum (FCS) supplemented with 5 μM TMRM at 37°C for 30 min, washed twice in prewarmed R10 and then incubated in R10 alone for 1 h prior to a final wash. Cells labelled with TMRM were pulsed with the B35-restricted peptide (EPLPQGQLTAY) and washed three times with R10, or mock-pulsed in parallel. For cytotoxic assays 80 000 cells with a 2:3 TMRM-labelled versus CFSE-labelled ratio of B35+ EBV+ cells were distributed in flat 96-well plates. CD8+ B35-EPL tetramer+ cells were added to the given effector : target (E : T) ratios and incubated for 6 h at 37°C 5% CO2. The decrease in the percentage of live peptide-pulsed TMRM-labelled cells (red) relative to the unpulsed CFSE-labelled cells (green) was measured as an indication of specific CD8+-mediated cytotoxicity. Experiments were performed in triplicate for each given E : T ratio, including parallel assays with mock-pulsed TMRM-labelled cells to control for non-specific toxicity of TMRM on the EBV-LCL. Specific lysis on the TMRM-labelled cells was calculated as follows:
EBV viral load determination by real-time–quantitativepolymerase chain reaction (RT–qPCR) assay
DNA was extracted from PBMCs using DNeasy Tissue kits (Qiagen Ltd, Crawley, UK). EBV load was determined using a multiplex real-time PCR assay as described previously to amplify the EBV DNA polymerase gene and the β2-microglobulin gene as an endogenous control to allow determination of input cellular DNA 19. A standard curve for EBV genome copy number was determined by analysis of serial dilutions of the diploid Namalwa cell line, known to contain two copies of EBV per cell. All standards and test samples were analysed in duplicate. The threshold of detection for the viral genome using this assay is two viral copies per PCR well. TaqMan primers and probes used were as follows: EBV POL (BALF5); forward: CTT TGG CGC GGA TCC TC, reverse: AGT CCT TCT TGG CTA GTC TGT TGA C, POL probe (FAM): CAT CAA GAA GCT GCT GGC GGC C; 2 m; forward: GGA ATT GAT TTG GGA GAG CAT C, reverse: CAG GTC CTG GCT CTA CAA TTT ACT AA, POL probe (FAM): AGT GTG ACT GGG CAG ATC ATC CAC CTT C.
Statistical analysis
Pooled data are presented as means and error bars represent standard deviation. The unpaired t-test was performed using Prism version 3 software (GraphPad, San Diego, CA, USA). Differences were considered statistically significant when P < 0·05 20.
Results
Molecular studies of SAP and XIAP genes and haplotype determination
Null mutations in the XIAP gene may lead to HLH, colitis or hypogammaglobulinaemia 12,21. Five males, including the one deceased (II-7) and his unaffected brother, were found to harbour a non-sense hemizygous guanosine to thymidine transversion at position 1396 of the cDNA 14, resulting in a premature stop codon at amino acid position 466 (G466X) of the XIAP sequence leading to the expression of a truncated XIAP. X-linked recessive inheritance (Fig. 1) was confirmed by finding the same mutations in their mothers and in a sixth carrier (sister – III.3), who has since developed a Crohn's like disease similar (although of late onset) to her brother (III.6). This mutation was found in association with a rare polymorphism in CD40LG 14 in all patients but not in healthy subject II.5, who carried XIAPG466X alone. The absence of the CD40LG polymorphism in this individual enabled the role of the truncated XIAP protein alone to be tested in the unaffected male.
Effect on T cell distributions
Despite the variable clinical symptoms in the patients with the XIAPG466X mutation, the immunological abnormalities were similar in the patients who were more than 10 years old. Persistently reduced or inverted CD4/CD8 ratios were observed due to increased absolute numbers of CD8+ T cells; this was a markedly persistent feature in patients III-1 and III-2 (Table 1) and in the healthy brother II.5, who lacked the CD40LG polymorphism. B and natural killer (NK) cells were within normal limits 14. NK T cell numbers were marginally reduced (data not shown). Further analysis of the CD4+ subpopulations showed reduced percentages of naive CD4+ T cells (CD45RA+, CCR7+) and increased effector memory cells (CD45RO+, CCR7−) in patients III-1 and III-2. In addition, patient III-2 maintained an expansion of CD3+CD4+CD8+ double-positive T cells over 10 years (up to 14% of the CD3+ T cells); the majority of these cells have a phenotype consistent with antigen-experienced effector memory cells and contain cytotoxic granules (granzyme B+) (Fig. 2).
Table 1.
Immunological evaluation in X-linked inhibitor of apoptosis (XIAP)G466X patients and female carriers
| Patients |
Carriers |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| III-1*** | III-2 | III-6 | II-5* | II-2 | III-3** | II-3 | II-6 | I-2 | Normal range | |
| T cells CD4+ % | 17 | 30 | 43 | 40 | 39 | 48 | 52 | 51 | 52 | 31–60% |
| T cells CD4+ abs | 442 | 691 | 857 | 801 | 866 | 1025 | 1071 | 1051 | 576 | 410–1590 |
| CD4+ (RA+/CCR7+) | 9·98 | 5·97 | 56·23 | 13·54 | 29·6 | 36·09 | 11·59 | 26·61 | 35·88 | 12·2–61·5 |
| CD4+ (RA−/CCR7+) | 42·76 | 22·91 | 21·05 | 62·63 | 57·43 | 36·25 | 69·41 | 46·83 | 39·54 | 27·2–51·4 |
| CD4+ (RA−/CCR7−) | 46·39 | 70·58 | 20·3 | 23·63 | 12·74 | 27·4 | 13·99 | 25·4 | 22·09 | 7·4–35·6 |
| T cells CD8+ % | 70 | 69 | 34 | 48 | 26 | 30 | 20 | 27 | 19 | 13–41% |
| T cells CD8+ abs | 1828 | 1592 | 677 | 961 | 583 | 640 | 409 | 547 | 204 | 190–1140 |
| CD8+ (CD28+/CD27+) | 58·6 | 69·1 | 69·7 | 62·6 | 82·58 | 70·05 | 87·66 | 75·69 | 70·13 | 34–90·2 |
| CD8+ (CD28−/CD27+) | 21·42 | 5·05 | 19·53 | 17·26 | 6·27 | 23·4 | 7·34 | 16·71 | 13·55 | 5·2–23·2 |
| CD8+ (CD28−/CD27−) | 14·33 | 8·11 | 9·64 | 16·06 | 4·92 | 4·9 | 2·48 | 5·92 | 9·7 | 1·3–56·1 |
| CD3+ (CD4+/CD8+) | 0 | 15 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0·2–8·2% |
| CD4/CD8 ratio | 0·24 | 0·43 | 1·3 | 0·83 | 1·5 | 1·6 | 2·6 | 1·9 | 2·7 | 0·9–4·5 |
| CD19+ % | 9 | 9 | 15 | 5 | 19 | 11 | 9 | 11 | 7 | 6–25% |
| CD19+ abs | 236 | 213 | 304 | 99 | 397 | 227 | 216 | 213 | 74 | 90–660 |
| NK cells % | 3 | 5 | 6 | 5 | 12 | 10 | 16 | 8 | 22 | 5–27% |
| NK cells abs | 80 | 108 | 128 | 91 | 244 | 215 | 380 | 147 | 233 | 90–590 |
II.5 is healthy, aged 48 years and lacks the CD40LG polymorphism.
III-3 has developed atypical severe inflammatory bowel disease.
III.1 has developed a Crohn's like disease and recurrent abscesses. NK = natural killer; % = percentage within the lymphocyte gate; abs = absolute number in cells mm3.
Fig. 2.
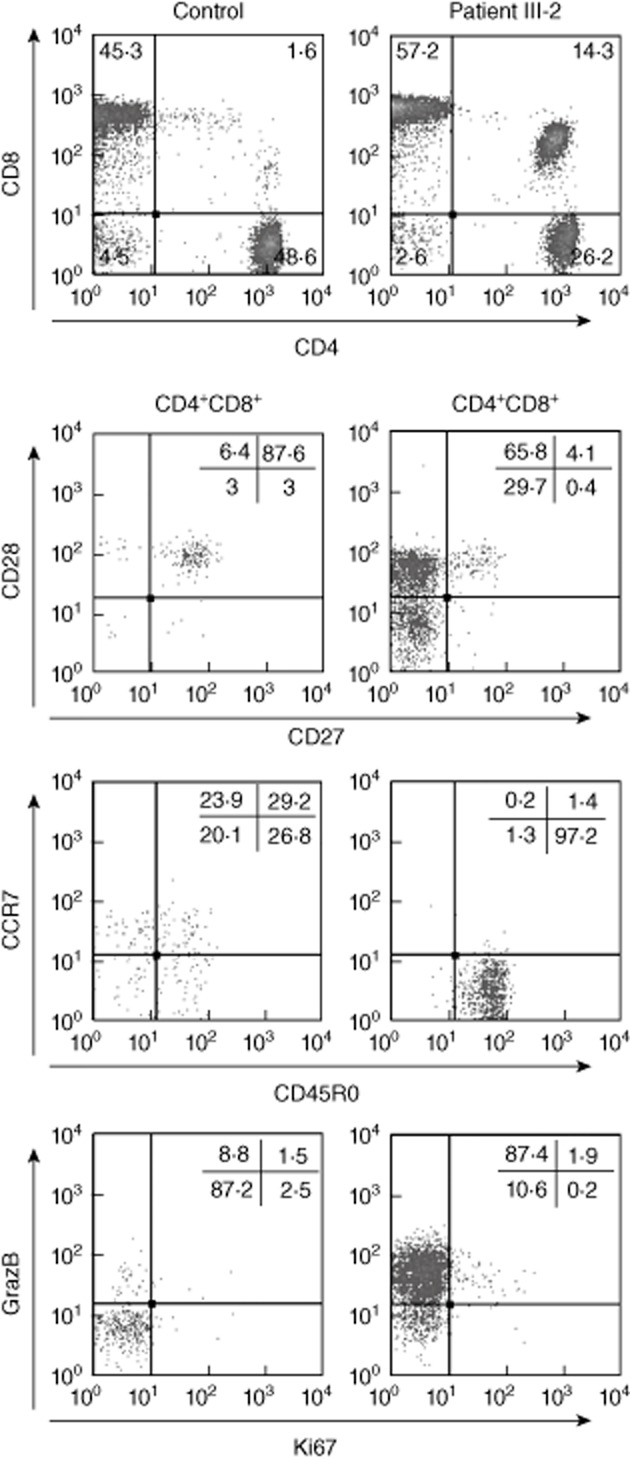
Frequency and phenotype of persistently expanded double-positive T cells in patient III-2. Phenotypical analysis of the CD4+CD8+ double-positive T cell subpopulation in patient III-2 (right column) in comparison to one representative from a cohort of Epstein–Barr virus (EBV)+ healthy adult carriers (left column). Percentages in the upper panel are within the CD3+ lymphocyte gate; in the lower three, double panels are within the CD4+CD8+ double-positive cells.
EBV-specific responses by proliferation
Patients with null XIAP mutations present with variable disease phenotypes after EBV infection, ranging from hepatosplenomegaly to fatal haemophagocytic lymphohistiocytosis (HLH) 22. The role of EBV in these families with truncated XIAP was examined to try to define the role of XIAP in the immune response to EBV. The expanded CD8+ and CD4+CD8+ T cells (Fig. 2) suggested an exaggerated immune response to a virus (or viruses), most likely to be EBV. To confirm this, we used an assay that allows identification and measurement of T cells that proliferate in vitro in response to a variety of recall antigens. PBMCs from patient III-2 were activated with a panel of common viral antigens or protein lysates from virus-infected cell lines, to study both CD4+ and CD8+ responses simultaneously. As seen in Fig. 3a, CD4+ cells from patient III-2 responded by proliferation to five viruses but an EBV+ cell lysate was the only one that induced a marked proliferation in all three CD4+, CD8+ and CD4+CD8+ subpopulations. This suggested that EBV-specific cells were greatly over-represented within these individual different T cell subpopulations compared to cells specific for other viruses. This assay was repeated with a different blood sample 4 months later, as well as twice with independent samples from patient III-1: similar results were obtained (data not shown). The CD4+ proliferative responses induced by the EBV+ cell lysate in patients III-1 and III-2 were compared to a control group of EBV healthy carriers and found to be significantly higher (P = 0·00003) (Fig. 3b).
Fig. 3.

Epstein–Barr virus (EBV)-specific T cell responses in X-linked inhibitor of apoptosis (XIAP)G466X patients. (a) Proliferation of lymphocytes from patient III-2 in resting (negative) or stimulated conditions with different viral antigens. Numbers indicate percentages of cells with decreased carboxyfluorescein succinimidyl ester (CFSE) staining. A representative example of one of two independent blood samples is shown. (b) Comparison of EBV-induced proliferative responses between patients III-1 and III-2 and a group of 23 EBV+ age-matched healthy carriers. The percentage of cells with decreased CFSE staining in response to the EBV+ lysate minus the percentage in the negative conditions is indicated. (c) Ex-vivo interferon (IFN)-γ release in response to 6 h stimulation with an EBV+ cell lysate in patients in comparison to a normal control group. Results represent the number of spots per 106 cells after subtracting the spots identified with an EBV− cell lysate. The means of two independent blood samples from each of the patients are included in the analysis and a cohort of 41 age-matched controls used for comparison. (d) Ex-vivo IFN production and cytotoxic degranulation (CD107a membrane expression) in peripheral blood mononuclear cells (PBMCs) in response to 6 h stimulation with an EBV− cell lysate (negative control), EBV+ cell lysate or anti-CD3–CD28–CD2 coated beads (positive control) in patients III-1 and III-2. Numbers in each quadrant indicate percentage of cells calculated within each T cell subpopulation. A representative example from one of three independent blood samples obtained within a year is shown for each patient.
EBV-specific responses by IFN-γ production
We also examined IFN-γ secretion in response to EBV, to determine if this was increased in parallel to the increased in-vitro proliferative responses to EBV. PBMCs from the four available XIAPG466X patients were stimulated with the EBV+ cell lysate and an EBV– control lysate, and the ex-vivo ability to release IFN-γ was analysed by ELISPOT assay. A positive response was detected in the four XIAPG466X patients in two separate samples; those using PBMCs from patients III-1 and III-2 were much higher than with cells from individuals from the other two kindred (Fig. 3c). In order to identify and enumerate the EBV-specific T cells producing IFN-γ in the ex-vivo ELISPOT assay, we performed an ex-vivo intracellular cytokine staining assay (ICS) in patients III-1 and III-2. Detection of surface expression of CD107a was included to study the cytotoxic capacity of the IFN-γ producer cells 18, as this protein is present in the membrane of cytotoxic granules and is detectable on the cytoplasmic membrane upon degranulation. As shown in Fig. 3d, a considerable proportion of the CD4+CD8+ double-positive T cells in patients III-2 expressed CD107a and/or produced IFN-γ upon stimulation with the EBV+ cell lysate. This indicates that these EBV-specific cells are not only able to proliferate (Fig. 3a) but potentially exert cytotoxic function. In comparison with healthy EBV carriers and patients with IM in similar experiments 23, CD4+ EBV-specific responses were strikingly strong in both patients III-1 and III-2. The frequency of cells responding by IFN-γ production from patients III-1 and III-2 were 10·7 and 17·2% of CD4+ T cells, respectively, compared with 0·04–5·2% (mean 1·4%) of CD4+ T cells in 36 acute cases of IM, and only 0·05–1·26% (mean 0·34%) of 23 responders of 28 healthy EBV carriers. Additionally, a significant proportion of CD4+ T cells also acquired membrane staining of CD107a, suggesting cytotoxic capacity (Fig. 3d).
Definition of the EBV epitope-specific T cell responses
The results presented above indicate an ‘inflated’ EBV specific T cell response in patients III-1 and III-2 analogous to T cell responses observed in CMV chronic infections 24. The type of EBV antigen preparation used above induces CD4+ T cell responses preferentially 23. To examine the CD8+ and CD4+ T cell responses more precisely, PBMCs from all the XIAPG466X patients were stimulated with a panel of class I and II optimal viral epitopes from lytic and latent proteins known to induce T cell responses in human leucocyte antigen (HLA)-matched IM patients and/or healthy EBV carriers 4. The complete sequence of these peptides is given in Supporting information, Table S2. The ability of a given peptide to induce ex-vivo release of IFN-γ was analysed using an ELISPOT assay in parallel with a number of controls, including the whole EBV+ cell lysate. A number of epitopes from lytic and latent cycle proteins induced significant reactivity in all four XIAPG466X patients tested (Table 2). The responses were particularly intense in comparison to those in otherwise healthy EBV+ individuals, in whom the range of mean positive results for each individual peptide was 50–693 spots per 106 cells in a similar study 25.
Table 2.
Epstein–Barr virus (EBV) epitope-specific interferon (IFN)-γ production measured by enzyme-linked immunospot (ELISPOT) assay in four X-linked inhibitor of apoptosis (XIAP)G466X patients
| MHC class I | ||||||||
|---|---|---|---|---|---|---|---|---|
| II-5 | A 0201 | A 0301 | B 3501 | |||||
| Peptide | GLC | CLG | FLY | YVL | RLR | EPL | YPL | HPV |
| Protein | BMLF1 | LMP2 | LMP2 | BRLF1 | EBNA3 | BZLF1 | EBNA3 | EBNA1 |
| Cycle | Lytic | Latent | Latent | Lytic | Latent | Lytic | Latent | Latent |
| ELISPOT | 25 | 88 | 88 | 15 | 63 | 1500 | 60 | 183 |
| III-1 | B 4501 | B 4001 | ||||||
| Peptide | AEN | IED | SEN | |||||
| Protein | BRLF1 | LMP2 | BZLF1 | |||||
| Cycle | Lytic | Latent | Lytic | |||||
| ELISPOT | 1478 | 1478 | 133 | |||||
| III-2 | B 4501 | B 4001 | ||||||
| Peptide | AEN | IED | SEN | |||||
| Protein | BRLF1 | LMP2 | BZLF1 | |||||
| Cycle | Lytic | Latent | Lytic | |||||
| ELISPOT | 1498 | 1498 | 73 | |||||
| III-6 | A 1101 | B 3501 | ||||||
| Peptide | IVT | AVF | EPL | YPL | HPV | |||
| Protein | EBNA3B | EBNA 3B | BZLF1 | EBNA3 | EBNA1 | |||
| Cycle | Latent | Latent | Lytic | Latent | Latent | |||
| ELISPOT | 228 | 268 | 1493 | 28 | 60 | |||
| MHC class II | ||||||||
|---|---|---|---|---|---|---|---|---|
| II-5 | DRB1 0101 | DQB1 0501 | ||||||
| Peptide | TSL | GPW | SDD | |||||
| Protein | EBNA1 | EBNA3A | EBNA3C | |||||
| Cycle | Latent | Latent | Latent | |||||
| ELISPOT | 0 | 0 | 0 | |||||
| III-1 | DRB1 0401 | DRB1 0701 | DQB1 0201 | |||||
| Peptide | GQT | TVV | PYY | PRS | PPW | |||
| Protein | EBNA2 | BHRF1 | BHRF1 | EBNA2 | EBNA1 | |||
| Cycle | Latent | Lytic | Lytic | Latent | Latent | |||
| ELISPOT | 220 | 193 | 225 | 273 | 203 | |||
| III-2 | DRB1 0401 | DRB1 0701 | DQB1 0201 | |||||
| Peptide | GQT | TVV | PYY | PRS | PPW | |||
| Protein | EBNA2 | BHRF1 | BHRF1 | EBNA2 | EBNA1 | |||
| Cycle | Latent | Lytic | Lytic | Latent | Latent | |||
| ELISPOT | 150 | 120 | 148 | 250 | 170 | |||
| III-6 | DRB1 0701 | DQB1 0201 | ||||||
| Peptide | PRS | PPW | ||||||
| Protein | EBNA2 | EBNA1 | ||||||
| Cycle | Latent | Latent | ||||||
| ELISPOT | 80 | 55 | ||||||
ELISPOT = number of spots per 106 peripheral blood mononuclear cells (PBMCs); MHC = major histocompatibility complex.
In patients III-6 and II-5, who share a B35 MHC class I haplotype, particularly strong ELISPOT responses (>1400 spots per 106 cells) were elicited against the BZLF1-derived epitope (EPL), known to be immunodominant in B35+ individuals 4. To determine the exact percentage of CD8+ circulating T cells specific for this peptide in these patients, B35-PE labelled class I tetramers loaded with the EPL peptide were used in a flow cytometric analysis with fresh PBMCs. Of the CD8+ T cells, 5·9% (2·6% of the total lymphocyte population) from the healthy individual II-5 were stained by the tetramer, whereas in patient III-6 (with recurrent viral infections and early-onset inflammatory bowel disease), the percentages were even higher, 14·5% (3·75% of the total lymphocyte population) (Fig. 4a). Individual lytic-cycle epitope-specific subpopulations can account for up to 40% of the CD8+ cells in IM patients and decrease to 0·2–2% CD8+ cells in healthy long-term carriers 4. The immunophenotypical characteristics of tetramer-positive cells from patient III-6 were analysed and shown to be CD27+CD28−, CD27−CD28−, typical of intermediate and late differentiated CD8 cells and CD45RO+CCR7−, indicating effector-memory cells. Increased numbers of activated (CD38+ or DR+) and cytotoxic (granzyme B+) cells were also observed within the tetramer-positive cells (Fig. 4b), confirming that tetramer-positive cells were activated and antigen-experienced cells. A substantial proportion of CD8+ cells proliferated in response to the BZLF1-derived EPL peptide, as demonstrated by a CFSE-based assay (Fig. 4c). More importantly, there was a higher proportion of EPL-specific cells that were potentially cytotoxic, as indicated by the percentage of positive cells in an ex-vivo IFN-γ ICS and CD107a expression assay in which PBMCs were stimulated with the EPL peptide (Fig. 4d).
Fig. 4.
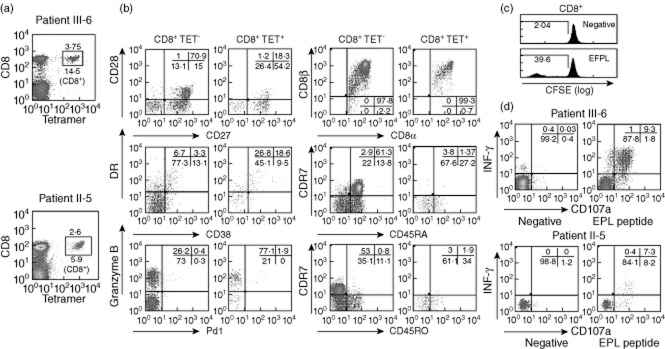
Phenotype and function of CD8+ T cells specific for lytic-cycle antigens. (a) Percentage of cells stained with a B35 class-I tetramer loaded with the BZLF1-derived expressed protein ligation (EPL) peptide and conjugated with phycoerythrin (PE) in patients III-6 and II-6. Numbers indicate percentage of positive cells within the lymphocyte gate and within the CD8+ T cell subpopulation. (b) Comparison of the phenotypical characteristics of CD8+ tetramer-positive and CD8+ tetramer-negative cells from Fig. 4a in patient III-6. (c) In-vitro carboxyfluorescein succinimidyl ester (CFSE)-based proliferation assay of peripheral blood mononuclear cells (PBMCs) from patient III-6 in response to the BZLF1-derived peptide EPL gated to show the CD8+ population. (d) Ex-vivo intracellular staining for interferon (IFN)-γ production and cytotoxic degranulation (CD107a surface expression) after 6 h of activation of PBMCs with the BZLF1-derived EPL peptide in patient III-6 and II-5. Numbers indicate percentage of cells within the CD8+ subpopulation.
Demonstration of killing capacity by XIAPG466X CD8+ T cells
We analysed the capacity of CD8 cells to kill using a flow cytometry-based killing assay described previously 18. XIAPG466X CD8+ T cells specific for the B35- restricted lytic peptide EPL from patient III-6 were selected with tetramers and expanded in vitro. B35+ EBV-LCL cells were labelled with either a green or red fluorochrome, and only red cells were loaded with the EPL peptide. As shown in Fig. 5, upon incubation with cells from a B35+ EBV-LCL, XIAPG466X CD8+ T cells are able to kill the target B cells in a peptide-specific manner.
Fig. 5.
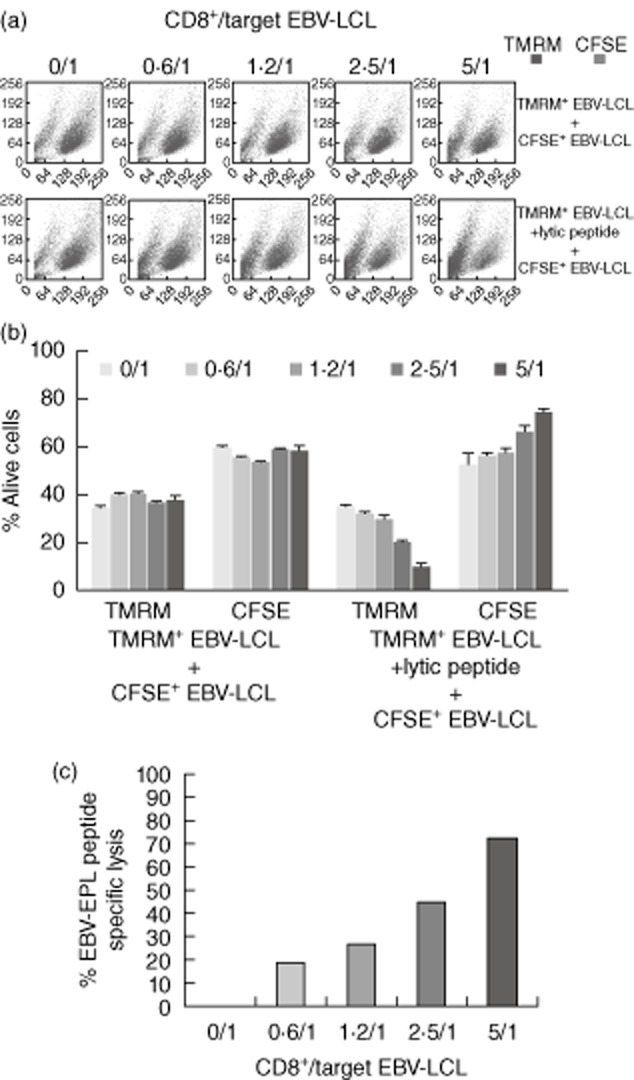
Epstein–Barr virus (EBV)-specific cytotoxic activity of X-linked inhibitor of apoptosis (XIAP)G466X CD8+ T cells. (a) tetramethylrhodamine methyl ester (TMRM) and carboxyfluorescein succinimidyl ester (CFSE)-labelled cells within the forward-/side-scatter profile when increased numbers of B35-expressed protein ligation (EPL) tetramer+ CD8+ T cells are added to the co-culture. In the upper panel neither population was pulsed with the EPL peptide, whereas in the lower panel TMRM+ cells were pulsed with the EPL peptide. A representative example of the results obtained in one of the triplicates from one experiment is shown. (b) Percentages of live cells for each experimental condition. Decreased percentages of live EPL-loaded TMRM+ cells are observed when increase ratios of EPL-specific CD8+ T cells are added to the cell culture. The experiments were performed in triplicate. (c) Specific lysis of B cells pulsed with B35-EPL peptide, at the effector : target (E : T) ratios indicated on the graph, calculated as described in Methods.
EBV viral load
The observed expansion of EBV-specific T cells and the strong ex-vivo responses to lytic epitopes in the ELISPOT assay suggested that the establishment of EBV latent carrier status is disturbed in these patients. Serial analyses of EBV viral load were performed in cells from stored peripheral blood samples from all patients, including the index case, at different times spanning 10 years of follow-up. The EBV-polymerase gene was amplified by real-time quantitative PCR in DNA isolated from 21 different samples of patients' PBMCs. The EBV genome could be amplified in only four samples (Table 3), although the level was within the normal range observed in healthy carriers in three of these positive results 26. In patient III-2 a sample from 2006 showed no positive amplification, but a subsequent sample taken in 2007 demonstrated a high viral load of 2·75 log copies/μg DNA, well above the range in healthy EBV carriers when detectable (0·9–1·8 log copies/μg DNA) and within the range observed in IM patients (1·6–3·8 log copies/μg DNA) 26, without changes in his clinical condition.
Table 3.
Epstein–Barr virus (EBV) viral load* in X-linked inhibitor of apoptosis (XIAP)G466X patients (1998–2008)
| 1998 | 1999 | 2003 | 2004 | 2005 | 2006 | 2007 | 2008 | |
|---|---|---|---|---|---|---|---|---|
| Pat II-7 | 0 | 0 | ||||||
| Pat II-5 | 0 | 0 | 0 | |||||
| Pat III-1 | 0 | 0·15 | 0 | 1·3 | ||||
| Pat III-2 | 0 | 0 | 0 | 2·75 | 0·82 | |||
| Pat III-6 | 0 | 0 | 0 | 0 |
Log copies/μg DNA [peripheral blood mononuclear cells (PBMCs)].
Discussion
The impact of XIAP deficiency on the immune response to EBV is unknown 12. We have characterized extensively the EBV-specific T cell responses in a family presenting a XIAPG466X mutation and have found evidence of an abnormal course of latent infection with greatly increased EBV-specific T cells with conserved cytotoxic capacity. A major finding in the immune evaluation of this family was a constant CD8+ lymphocytosis in patients III-1 and III-2 over the years (Table 1) and even a high proportion of CD8+ cells in the otherwise healthy individual, highly suggestive of abnormal control of viral infection. The differentially strong proliferative response induced by EBV in different lymphocyte subpopulations (Fig. 3a,b) and the massive IFN-γ response in the ELISPOT assay (Fig. 3c) indicated that EBV was responsible for this expansion.
These results reinforced the previous observation of particular susceptibility in XIAP- deficient patients to EBV 12 and prompted us to study in detail the intensity and functionality of EBV-specific T cell responses in XIAPG466X patients. Ex-vivo IFN-γ−ICS and cytotoxic degranulation (CD107a expression) assays showed how the persistent expansion of antigen-experienced T cells, not only CD4+ and CD8+ but also CD4+CD8+ double-positive T cells observed in patient III-2, were composed mainly of EBV-responding cells (Fig. 3d). Double-positive T cells are present in peripheral blood in healthy controls at very low percentages, but this subpopulation is increased in persistent viral infections 27, including acute EBV-IM 28. Secondly, a strikingly high proportion of CD4+ T cells responded to the whole EBV+ cell lysate with IFN-γ production and evidence of cytotoxic degranulation (Fig. 3d). CD4+ T cell responses are detectable in both the primary and the latent EBV infection, albeit to a much lesser extent than CD8+ CTLs, and have been shown to exert cytolytic action and IFN-γ production against EBV-LCLs, suggesting that CD4+ T cells also contribute to the destruction of EBV-infected cells in vivo 29–31. Abnormally large subpopulations of EBV-specific CD4+ and CD4+CD8+ T cells were present in patients III-1 and III-2, in contrast to surviving SAP-deficient patients. The ELISPOT assays revealed responses to several MHC class I and II EBV-epitopes, but a consistent feature in all the XIAPG466X individuals was remarkably strong CD8+ responses to BZLF1 or BRLF1 early lytic antigens (Table 2). Although the IFN-γ response to the whole EBV+ cell lysate (an antigen preparation which is relatively inefficient at stimulation of CD8+ cells) had not been significantly high in patients II-5 and III-6, the B35-restricted BZLF1-derived EPL peptide induced a very strong response in the ELISPOT, later confirmed by CFSE-proliferation and ICS-degranulation assays (Table 2, Fig. 4c,d). Although total numbers of CD8+ T cells are not as high as in their affected cousins III-1 and III-2, patients II-5 and III-6 show a significant disturbance in the CD8+ T cell repertoire, in which EBV-specific cells are over-represented in comparison to healthy EBV carriers. A substantial CD4+ T cell response to lytic cycle epitopes of BHRF1 in patients III-1 and III-2 (Table 2) suggests ongoing exposure to these antigens. EBV-specific CD4 and CD8 responses are not identical in all individuals harbouring the XIAPG466X mutation: not surprisingly, as the subjects are from three different but related kindred, and therefore, at the very least, MHC types differ.
We found no evidence of impaired function in EBV-specific XIAPG466X T cells, which are able to proliferate, produce IFN-γ and activate cytotoxic degranulation when stimulated with EBV+ cell lysates and EBV-derived peptides. In contrast to SAP-deficient patients 10,11, XIAPG466X CD8+ peptide-specific T cells are able to destroy peptide-loaded EBV-LCL targets (Fig. 5). Normal percentages and absolute numbers of NK cells are found in XIAP-deficient patients. Furthermore, NK cytotoxicity mediated by 2B4 stimulation is conserved in XIAP-deficient patients 12. More recently, cytotoxic degranulation in response to typical NK target cells has been demonstrated to be normal in XIAP-deficient patients 32 which, taken together with this study, may help to explain the absence of lymphomas in XIAP-deficient patients. However, longer follow-up of XIAP-deficient patients is needed to exclude EBV-related malignant complications.
EBV-specific T cell responses in XIAPG466X patients revealed abnormally expanded T cell specific subpopulations with remarkably strong responses to peptides derived from proteins of the EBV lytic cycle. In the absence of an obvious functional defect in T cells, frequent chronic antigen re-encounter leading to ‘memory inflation’ of the normal EBV– responses seems a plausible mechanism. Patient III-2 is a typical example in whom massive and persistent expansion of EBV-T cells is found 18 years after well-documented acute primary EBV infection with a fluctuant EBV viral load.
The phenotypes of SAP- and XIAP-deficient mice are different 33, as in humans. XIAP-deficient cells were more susceptible to death upon infection with the murine MHV-68, a gammaherpes virus, and produced more infectious virus than control cells in a XIAP-dependent manner. The EBV latency mechanism induces resistance to apoptosis in infected B cells by inducing increased expression of anti-apoptotic proteins such as XIAP via up-regulation of basal NF-kB activity 13,34. Failure to induce EBV latency could underlie the increased availability of EBV antigens leading to expanded T cell responses in XIAP-deficient patients. Because concomitant infections can induce reactivation of the EBV lytic cycle 35, it is tempting to speculate that episodes of increased EBV load could coincide with common viral or bacterial infections inducing B cell activation in XIAPG466X patients. In a recent study, co-stimulation of cells PBMCs with Toll-like receptor (TLR)-7/8 and TLR-9 agonists increased significantly the frequency of EBV transformed B cells (P < 0·001) 36.
Not all individuals with the XIAPG466X mutations present clinical symptoms, although we found abnormal EBV-specific immunological findings in all. Of note is the fact that a rare polymorphism in the CD40 ligand (CD154) gene (CD40LGG219R), described previously in healthy individuals 37, is present in the symptomatic patients but not in the asymptomatic individual II-5. Although the healthy individual II-5, who had only the XIAPG466X mutation, exhibited some abnormalities in the EBV-specifc CD8+ responses, those are significantly weaker than in patients III-1, III-2 and III-6. This could suggest that the CD40LG219R polymorphism participates in the inflated T cell responses reported in the patients. Our previous results suggested that both the XIAP and CD40LG variants were needed for the expression of the variable clinical phenotypes in this family, although interplay with other genes or environmental factors will also be a factor. Further analysis of these and other families using next-generation sequencing will contribute further to dissection of the clinical phenotype.
Acknowledgments
We thank all the patients and their families for their invaluable contribution to this study, also our colleagues in the Oxford University Hospitals, namely Dr S. Misbah, Professor A. Pollard, N. Bentley, J. Burton and C. Ross for assistance with samples and patients. We thank Professor B. Ferry and her staff in Laboratory Immunology for diagnostic immunoglobulin and antibody levels and for providing several laboratory reagents, and Professor Martin Rowe and Dr Holm Uhlig for their thoughtful comments on the manuscript. This study was funded by the NIHR Oxford Biomedical Research Centre Programme and by the UK Primary Immunodeficiency Association (PIA) Center of Excellence award, Jeffrey Model Foundation NYC, Baxter Healthcare LA and the Oxfordshire Health Service Research Committee.
Author contributions
H. C. and S. P. managed the patients, E. L.-G., M. S., A. K. K., C. W. and S. S. performed the experiments, H. L. and S. R. assisted, E. L.-G., A. H., C. F., A. R., S. L., P. K. and H. C. designed the study, E. L.-G., H. C. and A. H. wrote the paper.
Disclosure
The authors have no conflicts of interest.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web-site:
Table S1. Class I and II haplotypes in X-linked inhibitor of apoptosis (XIAP)-G466X patients.
Table S2. Peptide sequences.
References
- 1.Young LS, Rickinson AB. Epstein–Barr virus: 40 years on. Nat Rev Cancer. 2004;4:757–768. doi: 10.1038/nrc1452. [DOI] [PubMed] [Google Scholar]
- 2.Kuppers R. B cells under influence: transformation of B cells by Epstein–Barr virus. Nat Rev Immunol. 2003;3:801–812. doi: 10.1038/nri1201. [DOI] [PubMed] [Google Scholar]
- 3.Callan MF. The immune response to Epstein–Barr virus. Microbes Infect. 2004;6:937–945. doi: 10.1016/j.micinf.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 4.Hislop AD, Taylor GS, Sauce D, Rickinson AB. Cellular responses to viral infection in humans: lessons from Epstein–Barr virus. Annu Rev Immunol. 2007;25:587–617. doi: 10.1146/annurev.immunol.25.022106.141553. [DOI] [PubMed] [Google Scholar]
- 5.Rickinson AB, Kieff E. Epstein–Barr virus. In: Knipe DM, Howley PM, Griffin DE, et al., editors. Fields virology. 4th edn. Philadelphia, PA: Lippincott Williams & Wilkins; 2001. pp. 2575–2627. [Google Scholar]
- 6.Ebell MH. Epstein–Barr virus infectious mononucleosis. Am Fam Physician. 2004;70:1279–1287. [PubMed] [Google Scholar]
- 7.Nichols KE, Ma CS, Cannons JL, Schwartzberg PL, Tangye SG. Molecular and cellular pathogenesis of X-linked lymphoproliferative disease. Immunol Rev. 2005;203:180–199. doi: 10.1111/j.0105-2896.2005.00230.x. [DOI] [PubMed] [Google Scholar]
- 8.Parolini S, Bottino C, Falco M, et al. X-linked lymphoproliferative disease. 2B4 molecules displaying inhibitory rather than activating function are responsible for the inability of natural killer cells to kill Epstein–Barr virus-infected cells. J Exp Med. 2000;192:337–346. doi: 10.1084/jem.192.3.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bottino C, Falco M, Parolini S, et al. NTB-A [correction of GNTB-A], a novel SH2D1A-associated surface molecule contributing to the inability of natural killer cells to kill Epstein–Barr virus-infected B cells in X-linked lymphoproliferative disease. J Exp Med. 2001;194:235–246. doi: 10.1084/jem.194.3.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharifi R, Sinclair JC, Gilmour KC, et al. SAP mediates specific cytotoxic T-cell functions in X-linked lymphoproliferative disease. Blood. 2004;103:3821–3827. doi: 10.1182/blood-2003-09-3359. [DOI] [PubMed] [Google Scholar]
- 11.Dupre L, Andolfi G, Tangye SG, et al. SAP controls the cytolytic activity of CD8+ T cells against EBV-infected cells. Blood. 2005;105:4383–4389. doi: 10.1182/blood-2004-08-3269. [DOI] [PubMed] [Google Scholar]
- 12.Rigaud S, Fondaneche MC, Lambert N, et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature. 2006;444:110–114. doi: 10.1038/nature05257. [DOI] [PubMed] [Google Scholar]
- 13.Salvesen GS, Duckett CS. IAP proteins: blocking the road to death's door. Nat Rev Mol Cell Biol. 2002;3:401–410. doi: 10.1038/nrm830. [DOI] [PubMed] [Google Scholar]
- 14.Rigaud S, Lopez-Granados E, Sibéril S, et al. Human X-linked variable immunodeficiency caused by a hypomorphic mutation in XIAP in association with a rare polymorphism in CD40LG. Blood. 2011;118:252–261. doi: 10.1182/blood-2011-01-328849. [DOI] [PubMed] [Google Scholar]
- 15.Aguilar C, Lenoir C, Lambert N, et al. XIAP deficiency is associated with severe Crohn's disease and defective NOD2 function. J Allergy Clin Immunol. 2014 in press. [Google Scholar]
- 16.Speckmann C, Lehmberg K, Albert MH, et al. X-linked inhibitor of apoptosis (XIAP) deficiency: the spectrum of presenting manifestations beyond hemophagocytic lymphohistiocytosis. Clin Immunol. 2013;149:133–141. doi: 10.1016/j.clim.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 17.Semmo N, Krashias G, Willberg C, Klenerman P. Analysis of the relationship between cytokine secretion and proliferative capacity in hepatitis C virus infection. J Viral Hepat. 2007;14:492–502. doi: 10.1111/j.1365-2893.2007.00842.x. [DOI] [PubMed] [Google Scholar]
- 18.Betts MR, Brenchley JM, Price DA, et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003;281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 19.Junying J, Herrmann K, Davies G, et al. Absence of Epstein–Barr virus DNA in the tumor cells of European hepatocellular carcinoma. Virology. 2003;306:236–243. doi: 10.1016/s0042-6822(02)00027-2. [DOI] [PubMed] [Google Scholar]
- 20.Ressing ME, Keating SE, van Leeuwen D, et al. Impaired transporter associated with antigen processing-dependent peptide transport during productive EBV infection. J Immunol. 2005;174:6829–6838. doi: 10.4049/jimmunol.174.11.6829. [DOI] [PubMed] [Google Scholar]
- 21.Yang X, Kanegane H, Nishidam N, et al. Clinical and genetic characteristics of XIAP deficiency in Japan. J Clin Immunol. 2012;32:411–420. doi: 10.1007/s10875-011-9638-z. [DOI] [PubMed] [Google Scholar]
- 22.Pachlopnik Schmid J, Canioni D, Moshous D, et al. Clinical similarities and differences of patients with X-linked lymphoproliferative syndrome type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAP deficiency) Blood. 2011;117:1522–1529. doi: 10.1182/blood-2010-07-298372. [DOI] [PubMed] [Google Scholar]
- 23.Amyes E, Hatton C, Montamat-Sicotte D, et al. Characterization of the CD4+ T cell response to Epstein–Barr virus during primary and persistent infection. J Exp Med. 2003;198:903–911. doi: 10.1084/jem.20022058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karrer U, Sierro S, Wagner M, et al. Memory inflation: continuous accumulation of antiviral CD8+ T cells over time. J Immunol. 2003;170:2022–2029. doi: 10.4049/jimmunol.170.4.2022. [DOI] [PubMed] [Google Scholar]
- 25.Bihl F, Frahm N, Di Giammarino L, et al. Impact of HLA-B alleles, epitope binding affinity, functional avidity, and viral coinfection on the immunodominance of virus-specific CTL responses. J Immunol. 2006;176:4094–4101. doi: 10.4049/jimmunol.176.7.4094. [DOI] [PubMed] [Google Scholar]
- 26.Fafi-Kremer S, Morand P, Brion JP, et al. Long-term shedding of infectious Epstein–Barr virus after infectious mononucleosis. J Infect Dis. 2005;191:985–989. doi: 10.1086/428097. [DOI] [PubMed] [Google Scholar]
- 27.Nascimbeni M, Shin EC, Chiriboga L, Kleiner DE, Rehermann B. Peripheral CD4(+)CD8(+) T cells are differentiated effector memory cells with antiviral functions. Blood. 2004;104:478–486. doi: 10.1182/blood-2003-12-4395. [DOI] [PubMed] [Google Scholar]
- 28.Ortolani C, Forti E, Radin E, Cibin R, Cossarizza A. Cytofluorimetric identification of two populations of double positive (CD4+,CD8+) T lymphocytes in human peripheral blood. Biochem Biophys Res Commun. 1993;191:601–609. doi: 10.1006/bbrc.1993.1260. [DOI] [PubMed] [Google Scholar]
- 29.Landais E, Saulquin X, Scotet E, et al. Direct killing of Epstein–Barr virus (EBV)-infected B cells by CD4 T cells directed against the EBV lytic protein BHRF1. Blood. 2004;103:1408–1416. doi: 10.1182/blood-2003-03-0930. [DOI] [PubMed] [Google Scholar]
- 30.Adhikary D, Behrends U, Moosmann A, Witter K, Bornkamm GW, Mautner J. Control of Epstein–Barr virus infection in vitro by T helper cells specific for virion glycoproteins. J Exp Med. 2006;203:995–1006. doi: 10.1084/jem.20051287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heller KN, Gurer C, Munz C. Virus-specific CD4+ T cells: ready for direct attack. J Exp Med. 2006;203:805–808. doi: 10.1084/jem.20060215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marsh R, Madden L, Kitchen BJ, et al. XIAP deficiency: a unique primary immunodeficiency best classified as X-linked familial hemophagocytic lymphohistiocytosis and not as X-linked lymphoproliferative disease. Blood. 2010;116:1079–1082. doi: 10.1182/blood-2010-01-256099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rumble J, Oetjen KA, Stein PL, Schwartzberg PL, Moore BB, Duckett CS. Phenotypic differences between mice deficient in XIAP and SAP, two factors targeted in X-linked lymphoproliferative syndrome (XLP) Cell Immunol. 2009;259:82–89. doi: 10.1016/j.cellimm.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang S, Rowe M, Lundgren E. Expression of the Epstein–Barr virus transforming protein LMP1 causes a rapid and transient stimulation of the Bcl-2 homologue Mcl-1 levels in B-cell lines. Cancer Res. 1996;56:4610–4613. [PubMed] [Google Scholar]
- 35.Chene A, Donati D, Guerreiro-Cacais AO, et al. A molecular link between malaria and Epstein–Barr virus reactivation. PLOS Pathog. 2007;3:e80. doi: 10.1371/journal.ppat.0030080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Younesi V, Shirazi FG, Memarian A, Amanzadeh A, Jeddi-Tehrani M, Shokri F. Assessment of the effect of TLR7/8, TLR9 agonists and CD40 ligand on the transformation efficiency of Epstein–Barr virus in human B lymphocytes by limiting dilution assay. Cytotechnology. 2014;66:95–105. doi: 10.1007/s10616-013-9542-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin Q, Rohrer J, Allen RC, et al. A single strand conformation polymorphism study of CD40 ligand. Efficient mutation analysis and carrier detection for X-linked hyper IgM syndrome. J Clin Invest. 1996;97:196–201. doi: 10.1172/JCI118389. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Class I and II haplotypes in X-linked inhibitor of apoptosis (XIAP)-G466X patients.
Table S2. Peptide sequences.