

Abstract
Variation in pigmentation type and levels is a hallmark of myriad evolutionary radiations, and biologists have long been fascinated by the factors that promote and maintain variation in coloration across populations. Here, we provide insights into the genetic basis of complex and continuous patterns of colour variation in cichlid fishes, which offer a vast diversity of pigmentation patterns that have evolved in response to both natural and sexual selection. Specifically, we crossed two divergent cichlid species to generate an F2 mapping population that exhibited extensive variation in pigmentation levels and patterns. Our experimental design is robust in that it combines traditional quantitative trait locus (QTL) analysis with population genomics, which has allowed us to move efficiently from QTL interval to candidate gene. In total, we detected 41 QTL and 13 epistatic interactions that underlie melanocyte- and xanthophore-based coloration across the fins and flanks of these fishes. We also identified 2 QTL and 1 interaction for variation in the magnitude of integration among these colour traits. This finding in particular is notable as there are marked differences both within and between species with respect to the complexity of pigmentation patterns. While certain individuals are characterized by more uniform ‘integrated’ colour patterns, others exhibit many more degrees of freedom with respect to the distribution of colour ‘modules’ across the fins and flank. Our data reveal, for the first time, a genetic basis for this difference. Finally, we implicate pax3a as a mediator of continuous variation in the levels of xanthophore-based colour along the cichlid flank.
Keywords: cichlids, neural crest cells, phenotypic integration, pigmentation, sexual selection
Introduction
Variation in pigmentation is often credited as facilitating both the evolution and maintenance of biodiversity. Among animals, colour mediates myriad interactions that are critical for both survival (e.g. cryptic coloration to avoid predators, or bright coloration to deter predators) and reproductive success (e.g. male–male or male–female interaction). Because of the critical importance of coloration for the ecological and evolutionary success of animals, much recent effort has been directed towards understanding the genetic basis for variation in pigmentation types, levels and patterns.
The molecular mechanisms that underlie the development of species-specific coloration are most comprehensively understood in model systems, including Drosophila (reviewed by Kronforst et al. 2012), and instances where divergence has involved relatively simple shifts in pigmentation patterns and/or levels, including albinism in the blind cavefish (Protas et al. 2006) and melanism in various organisms through the Mc1r/Agouti pathway (reviewed by Manceau et al. 2010). The underlying theme for these and other colour polymorphisms that feature most prominently in the literature is that their genetic basis is largely Mendelian. While other studies have begun to explore the genetic basis of complex (i.e. non-Mendelian) colour variation among natural populations (Tripathi et al. 2009; Greenwood et al. 2011; Malek et al. 2012; O'Quin et al. 2013), our understanding of the specific genes that underlie this type of colour variation is far less complete, especially in vertebrate systems (as a notable exception, see Miller et al. 2007a).
Cichlid fishes offer an excellent opportunity to fill this gap in our understanding (Kocher 2004). This family is by far the most evolutionary successful vertebrate line-age, with many hundreds of species radiating within the last million years (Turner et al. 2001). A defining feature of cichlid radiations is extensive variation in coloration, with many closely related species differing only in the distribution of pigmentation across their bodies (Danley & Kocher 2001). Moreover, the complexity of colour variation can differ dramatically both among and within cichlid species. For example, certain species assemblages display relatively simple patterns of variation based on the degree of barring along the flank (e.g. Labeotropheus (Konings 2001), Fig. 1f–h), whereas other lineages exhibit many more degrees of freedom with respect to pigmentation type and distribution along the flank and fins (e.g. Tropheops (Konings 2001), Fig. 1a–e). We refer to the former as an ‘integrated’ (i.e. uniform) colour palette, and the latter as a ‘modular’ (i.e. discontinuous) colour palette (Brzozowski et al. 2012). Notably, this type of variation is also observed within species, where males and females differ with respect to the complexity of colour patterns that are expressed. We previously demonstrated that this type of variation can be explained by differences in the magnitude of integration among colour traits between the sexes, wherein males and females express the same basic pattern of pigmentation, but the colour palette is more modular in males and more integrated in females (Brzozowski et al. 2012). We suggest that a more modular colour palette in males serves the purpose of increasing contrast for courtship and/or dominance displays, whereas more integrated colour in females facilitates background matching and crypsis. Thus, differences in colour integration among sexes could facilitate the resolution of sexual antagonism within this group (Brzozowski et al. 2012). Based on these observations of differences both among species and between sexes, we expect that the genetic basis for colour trait integration will have both an autosomal and sex-linked component.
Fig. 1.

Pigmentation patterns of representative cichlid species are shown that exhibit a range of ‘complexity’ with respect to the distribution of colour along the flank. Various Tropheops species are depicted in a–e. These species express both uniform/integrated colour patterns (e.g. a–c) and more modular distributions of pigmentation (e.g. d–e). In particular, T. romandi (TR) females (a) and males (b) are either yellow or blue/black, respectively. In addition, T. mumbo zebra (TMZ) males are characterized by uniform distributions of red-orange pigment along their flank (c). Alternatively, T. gracilior (TG) and T. red cheek (TRC) males both express a more modular pigmentation pattern characterized by xanthophores distributed within relatively discrete domains along the dorsal and anterior regions of the body (d and e, respectively). Populations of Labeotropheus fuelleborni (LF) vary with respect to the degree of barring and fin pigmentation, but conspicuous bands and blotches of xanthophores along the flank figure less prominently with respect to population-level differences in this species (f–h). LF males from Chimwalani Reef (‘Cw’, f), Chinyankwazi Island (‘Ck’, g) and Makanjila Point are shown (‘Mk’, h). LF, Mk (h) were used in this experiment. Our F2 hybrids, derived from crossing TRC and LF, recovered a range of ‘modular’ colour phenotypes, including discrete bands and blotches of xanthophore-based pigmentation on the flank (i–j). Using this resource, we were able to assess the genetic basis of the modular inheritance of colour patterns. The ‘OB’ polymorphism, found in several cichlid lineages, is an extreme example of a reduction in colour trait integration (k). A rare OB male is shown here. All images of wild-caught animals provided courtesy of Ad Konings and cichlidpress.
To test this hypothesis as well as to gain more general insights into the genetic basis of cichlid coloration, we crossed two species that differed in terms of the patterns and complexity (modular/integrated) of pigmentation and performed a genetic mapping experiment using various aspects of colour as our quantitative traits. Our results offer novel insights into the genetic basis for colour trait variation and variability. Moreover, we implement a robust experimental design intended to leverage the advantages of both QTL mapping and population genomics, in order to move quickly from QTL interval to candidate gene identification. Following up on one of these candidates, we provide experimental evidence that implicates pax3a in mediating continuous variation in both the levels and patterning of cichlid coloration.
Materials and methods
Study species and experimental cross
All animals were reared and killed following protocols approved by the IACUC at Syracuse University and the University of Massachusetts. Details regarding species and husbandry are provided elsewhere (Brzozowski et al. 2012). In brief, we crossed two closely related Lake Malawi rock d welling cichlid species that differ dramatically in colour. Male Labeotropheus fuelleborni from Makanjila Point (‘Lf, Mk’ Fig. 1) are characterized by a blue body with lightly pigmented (melanocyte-based) vertical bars, a bright yellow-red dorsal fin and black leading edges on both the pelvic and anal fins. Female LF from this population are characterized by a uniform light grey-blue colour across the flank and fins, with dark leading edges on the pelvic and anal fins. Tropheops ‘red cheek’ (‘TRC’ Fig. 1) males are blue with darkly pigmented vertical bars and possess a conspicuous red/yellow blotch that covers much of their head and operculum. Their dorsal fins are yellow with a black stripe, and, similar to LF, they possess very dark leading edges on the pelvic and anal fins. Female TRC are a uniform grey-gold colour, with pale yellow fins, a dark band that runs along the dorsal fin and dark leading edges on the pelvic and anal fins. TRC animals were collected from Chizumulu Island.
A single wild-caught LF female was crossed to a single wild-caught TRC male. A full-sibling F1 family was interbred to produce 268 F2 individuals for genetic mapping. F2 individuals were reared in 10-gallon glass aquaria for 1–2 months, and then in 40-gallon glass tanks for another 6–10 months. Because of space constraints, F2 families were often combined; however, no more than 35 individuals were ever raised in one tank, and sex ratios were maintained at approximately 50:50.
Phenotypic assays
Details about the characterization of colour phenotypes are present in Brzozowski et al. (2012). Notably, not all F2 animals could be used for colour data collection (i.e. some animals were collected before suitable image data were obtained). Colour-level traits were obtained for 216 F2 animals. Briefly, fish were imaged in the left lateral view through water to eliminate glare with a Canon EOS® digital camera (Canon Inc., Lake Success, NY, USA). Two images were taken of each fish. The first picture was taken after recently euthanized fish were immersed in wet ice for 5–10 min. This procedure relaxed the chromatophores and enabled optimal visualization of overlaying black pigmentation (i.e. melanocytes). Black traits were measured from these images. Fish were then placed in an epinephrine (1 mg/mL) solution for 5 min. This solution constricted the melanocytes to better visualize the underlying red-yellow (‘R-Y’, i.e. xanthophore-based) pigmentation. A second picture was taken in the same view following epinephrine treatment and used for measuring R-Y traits.
Colour was quantified by first specifying 12 homologous regions (9 body and 3 fin regions) across the bodies of each fish. For each region, the total number of pixels was counted using the lasso tool in adobe photoshop cs3®. A standard set of coloured pixels from red to yellow (R-Y) and a standard set of pixels from dark grey to black were selected to count the number of R-Y or black pixels in each region. A ratio between coloured pixels and total number of pixels was then calculated. The flank was divided into nine regions (A– I), analysed individually (Fig. 2). For consistency, flank regions were designated based on specific anatomical landmarks (e.g. point of insertion for various fins). Egg spots were measured by summing the number of complete (i.e. 1.0) and incomplete (i.e. 0.5) spots for each fish. The degree of barring on the dorsal fin was assessed by scoring the amount of pigmentation between each dorsal fin spine (0–5) and summing these values (n = 15) to obtain the total amount of black in the banded region along the dorsal fin for each fish. Because colour may be influenced by size/age, we used linear regression to minimize allometric effects. Specifically, residuals for each colour trait were produced using standard length as the independent variable and used for QTL mapping.
Fig. 2.
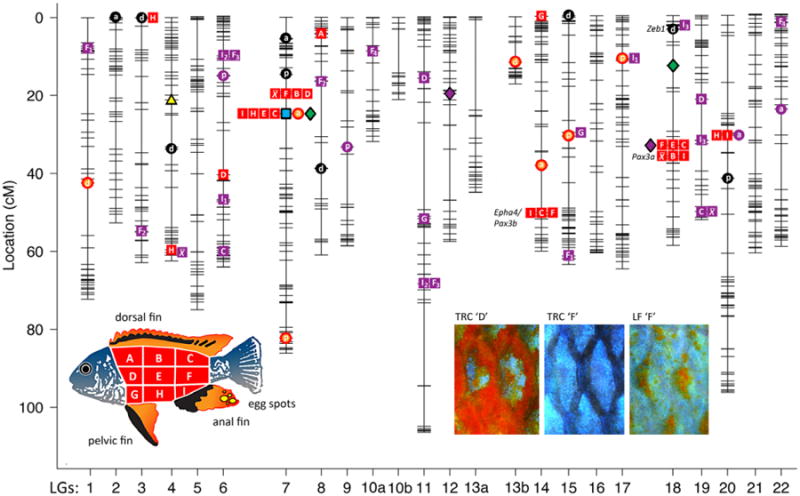
Quantitative trait locus (QTL) plot for various melanocyte- and xanthophore-based colour traits. Symbols depict traits mapped to the nearest marker. Circles represent fin traits, whereas squares represent flank traits. Orange/red circles and squares represent xanthophore-based colour. Black circles represent melanocyte-based colour. For fins, ‘a’ is the anal fin, ‘d’ is the dorsal fin, and ‘p’ is the pelvic fin. For the flank, ‘A’–‘I’ represent xanthophore-based colour variation within particular regions along the flank, as indicated in the lower left panel, while × represents average xanthophore-based colour across all flank regions. The yellow triangle indicates the QTL position for egg-spot number. The blue square indicates the QTL for sex determination. Green diamonds indicate the QTL positions for the magnitude of integration for colour traits. All purple symbols indicate epistatic interactions between loci. Subscripts are used when single traits have multiple two-way interactions. Select candidate loci are depicted to the left of their respective markers and discussed in the text. Representative scales of adult male TRC (regions ‘D’ and ‘F’) and LF (region ‘F’) are depicted to the lower right. Region ‘D’ is characterized by extensive R-Y pigmentation in TRC males (region ‘D’ in LF males looks identical to region ‘F’). Region ‘F’ is characterized by diffuse R-Y coloration in LF males and little to no R-Y pigmentation in TRC males.
Transforming integration into a quantitative trait
A covariance-based principal components analysis (PCA) was performed on colour traits. The magnitude of integration among traits can be estimated from eigenvalues, which are scalar values representing the amount of variation accounted for by each PC axis (Pavlicev et al. 2009). For PCA, investigators often present scaled eigenvalues, which are estimates of the percentage variation explained by each axis. When the covariation among traits is high (i.e. integrated), the first few PC axes will account for much of the total variance and present correspondingly high eigenvalues relative to subsequent axes. Thus, the variance among eigenvalues will be high. Alternatively, when the covariance among traits is low, eigenvalues will be distributed more evenly over many PC axes, and the variance among eigenvalues will be relatively low. To transform integration into a quantitative trait, we estimated each F2 individual's contribution to the magnitude of integration for the entire population. This was achieved by calculating the difference between the variance of scaled eigenvalues (VSE) for the whole population and that of the same population minus a particular individual. In cases where VSE goes up, it can be inferred that the removed individual detracted from the overall magnitude of integration within the population. Conversely, if VSE goes down, then the individual is assumed to have contributed to overall integration. Thus, this metric provides a relative assessment of the contribution of an individual to the population-level magnitude of integration. PCA was performed, and eigenvectors were determined from 214 F2 animals and held constant during the jack-knife procedure to prevent axes rotation. We were able to assess the magnitude of integration from the entire F2 population (i.e. both males and females), because our previous work demonstrated that male and female F2 individuals have very similar patterns of colour integration (Brzozowski et al. 2012). Holding eigenvectors constant ensured that estimates of the magnitude of integration during each jack-knife were not artificially influenced by changes in the pattern of integration. An R script for this procedure is available (Hu et al. 2014) and can be used for any suite of traits amenable to PCA.
RAD-seq genotyping
SNPs were identified and genotyped in 268 F2 as well as 20 wild-caught LF from Makanjila Point and 20 wild-caught TRC from Chizumulu Island (including the grandparents of the F2) using restriction-site-associated DNA sequencing (RAD-seq, Miller et al. 2007b). Genomic DNA was extracted from pectoral fin tissue using DN easy blood and tissue kits (Qiagen Inc. CA, USA), digested with the restriction enzyme SbfI and processed into RAD libraries following Chutimanitsakun et al. (2011). Barcoded, processed and purified DNA for individual fish was sequenced using an Illumina HiSeq 2000 (Illumina San Diego CA) and single-read (1 9 × 100 bp) sequencing chemistry. Sequencing and bioinformatics also followed Chutimanitsakun et al. (2011). Briefly, raw Illumina/Solexa data were processed using internal floragenex sequence tools and custom Perl scripts. Data from multiple Illumina/Solexa sequence channels were segregated to assign individuals using the four-nucleotide multiplex identifier (e.g. barcode). All reads were trimmed to 92 nucleotides from the 3′ end to avoid using bases with a high Illumina sequence error rate. Bowtie (Langmead et al. 2009) was used to align reads to the reference cichlid sequence (Metriaclima zebra v.0, http://cichlid.umd.edu/cichlidlabs/kocherlab/bouillabase.html), and sam-tools was used for SNP calling. Importantly, SNPs were named based on their physical position in the reference sequence, thus anchoring this resource to the Lake Malawi cichlid genome. Criteria imposed for sequence mapping included a maximum of three nucleotide mismatches, and each sequence had to anchor to a single unique position. The total number of sequence reads in the data set was 1 083 217 926. The average number of reads among individuals was 3 312 593, with an average median depth of 33×. All median base Q Scores (Phred) were well above the Q20 threshold. Likewise, a minimum Phred-scaled variant quality score of 20 was used to call SNPs. Among the 308 animals (F2 plus wild caught), 42 724 high-quality SNPs were identified. Most of these represented rare variants. Once filtered for deviations from Mendelian segregation in the F2, the final marker data set used for linkage map construction included 3087 SNPs.
Calculations of genetic divergence
The panel of 3087 SNPs was further investigated for evidence of genetic divergence between wild-caught LF and TRC. This involved calculating per-locus estimates of F statistics (FST, FSTP and FIS) following Nei (1987). A measure of population differentiation (DEST) as defined by Jost (2008) was also examined. All calculations for these metrics were performed using the r statistical programming language and using the package hierfstat (R core team). These data are presented in Table S1, Supporting information. We consider loci to exhibit a signature of divergence when FST ≥ 0.57, an empirical threshold for divergence between cichlid genera (Mims et al. 2010).
Linkage map construction and QTL analysis
To construct a genetic linkage map from our filtered data set, we used routines included within the r/qtl package as part of the r statistical language (Broman et al. 2003). Prior to map construction, individuals were checked for excessive levels of missing genotypes (40% cut-off) and for duplicate individuals using the ntyped and comparegeno functions. The intercross model was used to code for the F2 genotypes. Linkage groups were first formed with a maximum recombination frequency of 0.25 and minimum logarithm of odds (LOD) score of 8 using the formLinkageGroups function. Markers were then ordered using the order-Markers function with the Kosambi mapping function to allow for crossover. Several iterations of LOD scores and recombination frequencies (ranging from 4 to 12 for LOD, and 0.25–0.45 for recombination frequency) were used in this process and were found to produce no difference in map length. After this initial marker ordering, we used a combination of the ripple and switch.order functions to check all possible marker orders in a sliding window and changed the ordering when it resulted in a reduction in map length. Additionally, we inspected plots of the recombination frequency among markers and manually changed their order in cases where the sliding window was too small to detect ordering errors. Finally, markers that did not fall into a linkage group were dropped from the analysis. The resulting map contained 946 loci, consisted of 24 linkage groups with between 13 and 76 loci each. The average distance between markers is 1.58 cM, and the total map size is 1453.3 cM (Table S2, Supporting information). Linkage groups were numbered according to Lee et al. (2005).
Quantitative trait locus (QTL) analyses were conducted in r as described in Broman & Sen (2009). First, standard interval mapping was performed via Haley–Knott regression, and significant QTL/markers were selected as potential cofactors. These were verified by backward elimination during subsequent multiple-QTL mapping (MQM) scans using the default significance threshold of 0.02 (Arends et al. 2010). J-qtl (Smith et al. 2006; version 1.3.3) was used for two-QTL scans (epistatic interactions via Haley-Knott regressions). Logarithm of the odds (LOD) scores for interactions were calculated as the LOD for the full model minus the LOD for the additive model (Broman & Sen 2009). The variance accounted for by each QTL is calculated as 1–10−(2/n)LOD (Broman & Sen 2009) and is reported as percentage variance explained (PVE) in Table 1. Genome-wide significance thresholds (α = 0.05) for all QTL analyses were calculated by permutation tests with 1000 repeats.
Table 1. List of quantitative trait locus (QTL) affecting color variation in cichlids.
| Allele effects | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|||||||||||
| Traita | QTL | LG | cM | QTL interval | LOD | PVE (%) | Lf/Lf | Lf/Trc | Trc/Trc | Add | Dom |
| Sex | Sex1 | 7 | 25 | 24.7–33.7 | 26.75 | 33.79 | 0.0681 | 0.4109 | 0.9928 | −0.4624 | −0.1196 |
| Black anal fin | BLKa1 | 2 | 0 | 0.0–24.2 | 2.98* | 5.10 | −2.8066 | −1.5003 | −0.7005 | −1.0531 | 0.2532 |
| BLKa2 | 7 | 5 | 0.0–14.7 | 3.33** | 5.60 | −0.7141 | −0.8524 | −3.6182 | 1.4521 | 1.3138 | |
| Black dorsal fin | BLKd1 | 3 | 0 | 0.0–21.7 | 3.79** | 6.40 | −5.0588 | −1.8173 | −0.4383 | −2.3103 | 0.9312 |
| BLKd2 | 4 | 35 | 19.9–53.9 | 3.18* | 5.40 | −1.5495 | −4.4058 | 1.4365 | −1.4930 | −4.3493 | |
| BLKd3 | 8 | 35 | 16.4–56.9 | 3.00* | 5.10 | −6.3772 | 0.5968 | −2.0672 | −2.1550 | 4.8190 | |
| BLKd4 | 15 | 40 | 0.0–49.3 | 3.63** | 6.14 | 1.0306 | −1.6359 | −4.6706 | 2.8506 | 0.1841 | |
| BLKd5 | 18 | 5 | 0.0–12.8 | 3.84** | 6.00 | −6.0815 | 2.2889 | −9.3712 | 1.6449 | 10.0152 | |
| Black pelvic fin | BLKp1 | 7 | 15S | 10.4–33.7 | 4.99** | 8.34 | 1.1497 | −0.8653 | −3.4480 | 2.2988 | 0.2839 |
| BLKp2 | 20 | 40 | 30.7–61.0 | 3.07* | 5.21 | −0.6323 | −0.8147 | −1.8433 | 0.6055 | 0.4231 | |
| Egg spot number | ES1 | 4 | 20 | 14.0–37.7 | 3.81** | 6.45 | −0.0393 | 0.0339 | 0.3748 | −0.2071 | −0.1339 |
| RY anal fin | RYa1 | 7 | 25S | 24.7–33.7 | 15.50** | 23.70 | −19.4379 | 0.3098 | 18.9985 | −19.2182 | 0.5295 |
| RYa2 | 14 | 40 | 34.9–55.8 | 3.68** | 6.22 | 5.1754 | 5.7970 | −14.3348 | 9.7551 | 10.3767 | |
| RYae1 | 20 22 |
30 24 |
5.09** | 7.03 | Fig. S1 | ||||||
| RY dorsal fin | RYd1 | 1 | 55 | 33.0–63.3 | 3.62** | 6.12 | 1.2430 | 0.3436 | −13.5730 | 7.4080 | 6.5086 |
| RYd2 | 13b | 10 | 6.7–17.2 | 4.10** | 6.90 | −4.8916 | −7.0659 | 12.9390 | −8.9153 | −11.0896 | |
| RY pelvic fin | RYp1 | 7 | 80 | 62.3–86.2 | 3.87** | 5.26 | −10.1610 | 4.2792 | 6.4112 | −8.2861 | 6.1541 |
| RYp2 | 15 | 30 | 18.1–36.2 | 3.50** | 5.92 | −2.6955 | −2.9320 | 12.5709 | −7.6332 | −7.8697 | |
| RYp3 | 17 | 10 | 7.4–33.6 | 3.57** | 5.56 | −4.5013 | 1.6881 | 9.7224 | −7.1118 | −0.9225 | |
| RYpe1 | 6 9 |
16 34 |
3.48* | 6.80 | Fig. S1 | ||||||
| RY flank A | RYA1 | 8 | 5 | 0.0–30.6 | 3.22* | 5.46 | 6.3915 | −2.6722 | −10.6663 | 8.5289 | −0.5348 |
| RY flank B | RYB1 | 7 | 20S | 0.0–33.7 | 3.07* | 5.21 | −5.1762 | −2.2379 | 7.3529 | −6.2645 | −3.3263 |
| RYB2 | 18 | 35 | 19.5–47.7 | 3.55** | 6.01 | 9.5245 | −3.2634 | −5.7774 | 7.6509 | −5.1369 | |
| RY flank C | RYC1 | 7 | 20S | 0.0–33.7 | 4.30** | 7.23 | −7.1264 | −2.2620 | 12.2206 | −9.6735 | −4.8090 |
| RYC2 | 14 | 50 | 38.3–58.1 | 2.96* | 5.03 | −0.7288 | 6.0393 | −8.4098 | 3.8405 | 10.6086 | |
| RYC3 | 18 | 35 | 19.5–47.7 | 4.18** | 7.03 | 11.3669 | −3.4870 | −5.0070 | 8.1870 | −6.6669 | |
| RYCe1 | 6 19 |
60 50 |
3.03* | 4.98 | Fig. S1 | ||||||
| RY flank D | RYD1 | 6 | 40 | 27.3–52.3 | 3.20* | 5.43 | 4.4060 | −10.6822 | 4.3867 | 0.0097 | −15.0786 |
| RYD2 | 7 | 20S | 0.0–33.7 | 3.03* | 5.15 | -11.6265 | −6.0826 | 10.8169 | −11.2217 | −5.6778 | |
| RYDe1 | 11 19 |
16 22 |
5.58 | 8.71 | Fig. S1 | ||||||
| RY flank E | RYE1 | 7 | 20S | 19.1–33.7 | 7.89** | 12.86 | −12.0600 | −2.2977 | 10.5544 | −11.3072 | −1.5449 |
| RYE2 | 18 | 30 | 19.5–47.7 | 4.18** | 8.05 | 12.1938 | −6.3115 | −8.7216 | 10.4577 | −8.0476 | |
| RY flank F | RYF1 | 7 | 25S | 19.1–33.7 | 7.34** | 12.02 | −16.5669 | −2.1359 | 18.2965 | −17.4317 | −3.0007 |
| RYF2 | 14 | 50 | 38.3–57.7 | 3.53** | 5.97 | 1.0572 | 5.6828 | −14.5472 | 7.8022 | 12.4277 | |
| RYF3 | 18 | 30 | 19.5–47.7 | 4.11** | 6.92 | 18.8394 | −7.7350 | −10.4268 | 14.6331 | −11.9413 | |
| RYFe1 | 1 15 |
8 62 |
4.01** | 3.60 | Fig. S1 | ||||||
| RYFe2 | 3 8 |
54 20 |
4.61** | 4.20 | Fig. S1 | ||||||
| RYFe3 | 6 11 |
10 68 |
6.7** | 6.10 | Fig. S1 | ||||||
| RYFe4 | 10a 22 |
8 2 |
3.6* | 3.22 | Fig. S1 | ||||||
| RY flank G | RYG1 | 14 | 0 | 0.0–54.3 | 3.23* | 5.48 | −18.5406 | −2.0488 | −5.9430 | −6.2988 | 10.1930 |
| RYGe1 | 11 15 |
52 30 |
4.15** | 7.12 | Fig. S1 | ||||||
| RY flank H | RYG1 | 3 | 0 | 0.0–34.6 | 3.49** | 5.91 | −19.8566 | −4.0847 | −5.9276 | −6.9645 | 8.8074 |
| RYG2 | 4 | 60 | 43.6–62.0 | 3.61** | 6.10 | 4.4204 | −8.2641 | −12.8675 | 8.6439 | −4.0405 | |
| RYG3 | 7 | 25S | 19.1–36.3 | 8.92** | 14.41 | −20.1736 | −8.9966 | 13.7400 | −16.9568 | −5.7798 | |
| RYG4 | 20 | 35 | 17.2–41.8 | 3.85** | 6.50 | −9.7646 | −10.1199 | 4.0854 | −6.9250 | −7.2803 | |
| RY flank I | RYI1 | 7 | 25S | 19.1–36.3 | 9.34** | 15.04 | −16.7613 | −6.6609 | 18.4480 | −17.6047 | −7.5042 |
| RYI2 | 14 | 55 | 38.3–60.3 | 3.24* | 5.50 | −0.6242 | 0.5889 | −13.7085 | 6.5422 | 7.7552 | |
| RYI3 | 18 | 35 | 19.5–47.7 | 3.05* | 5.18 | 5.4391 | −6.2016 | −10.8185 | 8.1288 | −3.5119 | |
| RYI4 | 20 | 30 | 17.2–41.8 | 3.33** | 5.64 | −3.9901 | −7.6393 | 4.2023 | −4.0962 | −7.7454 | |
| RYIe1 | 6 17 |
46 10 |
5.29** | 5.51 | Fig. S1 | ||||||
| RYIe2 | 6 11 |
10 68 |
4.06** | 4.17 | Fig. S1 | ||||||
| RYIe3 | 18 19 |
2 28 |
3.73* | 3.81 | Fig. S1 | ||||||
| RY flank l | RYμ1 | 7 | 20S | 19.1–33.7 | 5.74** | 9.53 | −11.8115 | −5.9478 | 11.1147 | −11.4631 | −5.5993 |
| RYμ2 | 18 | 35 | 19.5–47.7 | 5.18** | 8.64 | 11.5962 | −6.5130 | −12.0087 | 11.8024 | −6.3068 | |
| RYμe1 | 4 19 |
62 50 |
3.20* | 4.41 | Fig. S1 | ||||||
| Integration All Traits | IntA1 | 7 | 30S | 24.7–42.8 | 4.61** | 7.73 | 1.28E–05 | −7.54E–07 | −1.54E–05 | 0.0000141 | 0.0000005 |
| IntA2 | 18 | 15 | 0.0–29.7 | 3.81** | 6.43 | −1.53E–05 | 1.67E–06 | 6.52E–06 | −0.0000109 | 0.0000061 | |
| IntAe1 | 12 18 |
20 36 |
4.74** | 9.69 | Fig. S1 | ||||||
| Integration Flank | IntF1 | 7 | 30S | 19.7–42.8 | 4.97** | 8.30 | 3.37E–05 | −2.90E–06 | −3.38E–05 | 0.0000337 | −0.0000029 |
| IntF2 | 18 | 15 | 1.5–29.7 | 3.52** | 5.96 | −4.07E–05 | 7.72E–06 | 1.50E–05 | −0.0000279 | 0.0000206 | |
| IntFe1 | 12 18 |
20 36 |
4.34** | 8.92 | Fig. S1 | ||||||
F2 sample size for sex and all color level QTL was 216, and 214 for both integration QTL.
Genome wide significance at P < 0.1.
Genome wide significance at P < 0.05.
Superscript ‘S’ indicates sex-linked marker; Bold-face font indicates genotype that increases the trait value.
Pax3a quantitative PCR and semiquantitative allele-specific expression
Skin tissue including overlaying scales was dissected from the flank of adult fish (regions E and F in Fig. 2). A 1 cm2 piece of tissue was used for Trizol-based RNA extraction, using bead-beating and standard manufacturer protocols (Life Technologies), including the optional addition of glycogen. Superscript III (Life Technologies) was used for reverse transcription to produce cDNA. Genomic DNA was extracted from fin tissue using the DNeasy Blood and Tissue kit (Qiagen).
Real-time quantitative PCR was used to assay pax3a expression relative to β-actin (actb) expression. Normalized cDNA was amplified by standard PCR using Phusion High Fidelity master mix (Thermo Scientific) with EvaGreen dye (Biotium), and the following primers: pax3a forward: 5′-GACGGGTTCTATCAGACCGG-3′ and reverse: 5′-CTCGTCTGAGTGACTGGCTG-3′; actb forward: 5′-CTGTCTTCCCCTCCATCGT-3′ and reverse: 5′-GATGGGGTACTTCAGGGTCA-3′. The reaction was run on a Stratagene M×3000P cycler with the following conditions: 1 cycle (98 °C, 30 s); 40 cycles (98 °C, 10 s; 65 °C, 30 s; 72 °C, 15 s; plate read); 1 cycle (95 °C, 1 min; 55 °C, 30 s; plate read every 0.5 °C; 95 °C, 30 s). Relative pax3a expression was calculated via the ΔΔCq method as described in Haimes & Kelley (2010).
Unfortunately, there was not a suitable polymorphism in pax3a to perform traditional, quantitative PCR-based, allele-specific expression. However, we adapted a previously employed strategy based on conventional Sanger sequencing (Wei et al. 2013) to allow semiquantitative analysis of allele-specific expression. For each individual, gDNA (from fin clips) and skin flank cDNA templates were amplified in parallel from nine F1 hybrid fish, and each pair of parallel reactions was performed in triplicate. A region of the 5′ UTR of the pax3a gene spanning the species-specific SNP was amplified by standard PCR using dreamtaq (Thermo Scientific) and the following primers: forward: 5′-TTTGACCAATCAGCATGCGC-3′; reverse: 5′-TCAGG TCGCTTCACACGTTT-3′. The PCR conditions were as follows: 1 cycle (95 °C, 3 min) and 32 cycles (95 °C, 20 s; 58 °C, 20 s; 72 °C, 1 min). PCR products were purified using GeneJet columns (Thermo Scientific) before Sanger sequencing with Big Dye 3.1 chemistry (Applied Biosystems) using the forward PCR primer.
Sequence chromatograms were viewed in Geneious, and the region containing the 5′ UTR SNP was converted to an image file for analysis in Adobe Photoshop. The area under the ‘C’ and ‘T’ signal peak at the SNP was selected in Photoshop and recorded in pixels, and the C/T ratio was calculated. Within each replicate, all individual C/T ratios were divided by the mean C/T ratio of the gDNA class to normalize equal amounts of the two alleles to a value of 1. Statistical tests were performed on the average of three replicates for each template within each individual.
Results and discussion
The genetic basis of sex determination
The major sex-determining locus in this cross explains nearly 34% of the variation in this trait and maps to the same locus on LG7 as other Lake Malawi cichlids (Albertson et al. 2003; Ser et al. 2010). In other cichlid pedigrees, this locus represents a male heterogametic (XY) system, although here allelic effects appear to be additive with respect to sex determination. In this cross, male F2 were assigned a score of 1 and females were designated 0. At the sex locus on LG7, TRC/TRC animals were predominantly male (mean phenotypic score of 0.99, Table 1), and LF/LF animals were predominantly female (0.07, Table 1). F2 animals that were TRC/LF at this locus had an approximately equal probability of being male or female (mean phenotypic score of 0.41, Table 1). This pattern is consistent with the observation that several interacting loci control sex determination in Lake Malawi cichlids (Ser et al. 2010). Another locus that controls sex in this group of fish is a female heterogametic (ZW) system on LG5, which maps to and segregates with a dominant ‘orange-blotch’ (OB) colour polymorphism in females. Notably, LG5 did not segregate with sex or pigmentation in this cross.
The genetic basis of colour variation: Fins
Seventeen QTL and two epistatic interactions were identified for fin traits (Fig. 2, Table 1), and these were relatively evenly distributed across the genome (epistatic effect plots for all interaction QTL are presented in Fig. S1, Supporting information). Approximately equal numbers of QTL were detected for melanocytebased (n = 9) and xanthophore-based (n = 8) colour (black and red-yellow [‘R-Y’] pigmentation, respectively), although both interactions were for xanthophore traits. Alleles inherited from Labeotropheus fuellebroni (LF, Fig. 1) were found to increase trait values for 6 QTL, while alleles from Tropheops ‘red cheek’ (TRC, Fig. 1) increased trait values for 9 QTL. This general lack of pattern with respect to allelic effects makes sense given that both species are characterized by varying amounts of black and red-yellow pigmentation across the fins. Modes of inheritance for each QTL varied from additive to dominant to overdominant (Table 1). Generally, the percent variation explained by each colour QTL was modest, between 5% and 7%.
Sex was used as a cofactor in all QTL models, but two fin QTL were nevertheless linked to the sex-determining locus on LG7 (Table 1). The xanthophore-based sex-linked QTL involved variation in anal fin pigmentation (Table 1, QTL RYa1), whereas the melanocyte-based QTL involved pelvic fin pigmentation (Table 1, QTL BLKp1). Both of these QTL explained a relatively greater percentage of the variation (23.7% and 8.3%, respectively, Table 1). For QTL RYa1, the TRC allele substantially increases the trait value, whereas the LF allele decreases the trait. This pattern makes sense given that TRC females are characterized by a yellow-pigmented anal fin (Brzozowski et al. 2012). Alternatively, the LF allele increases the trait value at QTL BLKp1, whereas the amount of black on the leading edge of the pelvic fin is decreased in individuals with the TRC allele. This pattern of inheritance does not connect as clearly to parental colour patterns, as both sexes in each species possess a black leading edge of the pelvic fin, although TRC females may have slightly less black pigmentation on the pelvic fin (Brzozowski et al. 2012).
Among R-Y fin colour traits, a single QTL of modest effect (PVE = 6.5%) was detected for egg-spot number on LG4. These bright yellow, round spots are located on the anal fin, mimic the eggs of mouth brooding females and are considered a key innovation in modern haplochromine cichlids that contribute to their fertilization (Wickler 1962) and evolutionary (Salzburger et al. 2005) success. Egg-spot number exhibits considerable variation both between and within species and is related to male body condition and dominance status (Lehtonen & Meyer 2011). Given that variation in this trait may have a strong environmental input, it is perhaps not surprising that we did not detect a more robust genetic signal for variation in egg-spot number. While the TRC allele increases egg-spot number at this locus (Table 1), we did not detect evidence for a difference in egg-spot number between our parental species (not shown). The gene csf1ra is necessary for the development of neural crest cell-derived xanthophores in zebrafish (Parichy et al. 2000) and is expressed in cichlid egg spots (Salzburger et al. 2007). However, csf1ra does not map to the QTL for egg-spot number in this cross, but rather to a scaffold anchored to LG2, which suggests that allelic variation at this locus is not associated with variation in egg-spot number.
The genetic basis of colour variation: Flank
Twenty-four R-Y QTL and 11 interactions were detected for flank colour traits (Fig. 2, Table 1). Relative to fin colour, these QTL were more clustered in the genome, with 19 of 24 (80%) flank colour QTL mapping to four discrete intervals on LGs 7, 14, 18 and 20 (Fig. 2, Table 1). Alternatively, only 5 of 17 (29%) fin colour QTL overlapped. One large cluster (i.e. 8 QTL) mapped to the sex-determining locus on LG7. Variation in all but two anterior flank regions (A and G) mapped to this interval, and in each case, the TRC allele (which was the male in the F0 generation) increased the level of R-Y pigmentation. A second large cluster of QTL for more posterior R-Y coloration (i.e. 6 QTL) mapped to LG18. Notably, for these QTL, the LF allele contributed to an increase in R-Y pigmentation. A small cluster of QTL (i.e. 3 QTL) was detected for the posterior-most regions of the flank (C, F, I) on LG14. Here, heterozygous animals had the highest trait values, followed closely by animals with two LF alleles, whereas animals homozygous for the TRC allele at this locus had the lowest trait values. Finally, two QTL for the ventral ‘belly’ region of the flank (H, I) mapped to LG20. Animals with two TRC alleles at this locus had the highest trait value. Excluding the sex-linked QTL, LF alleles contributed to an overall higher level of R-Y pigmentation for 10 of 16 (63%) QTL, whereas TRC alleles contributed to increased R-Y levels for 4 of 16 (25%) QTL. At a superficial level, this may seem counterintuitive as adult TRC males are characterized by a conspicuous R-Y blotch covering most of their head and anterior flank (Fig. 2, region ‘D’). However, the posterior flank of TRC males is largely devoid of xanthophores (Fig. 2, region ‘F’), suggesting that this cell lineage is restricted to the anterior region of the body in TRC. LF males, on the other hand, possess low levels of R-Y pigmentation across much of their flank (Fig. 2, ‘F’). It is therefore not surprising that LF alleles contribute to an overall increase in R-Y pigmentation in the F2, especially for more posterior regions of the flank.
The genetic basis of variation in the magnitude of integration of colour traits
Phenotypic integration is an important trait with respect to organismal evolution, as species and populations can vary considerably with respect to the patterns of covariation among traits. Unfortunately, progress into the genetic basis for phenotypic integration has been hindered by the lack of an individual metric for this inherently population-level phenomenon. We have recently developed a solution to this problem through the implementation of a method based on PCA that measures an individual's contribution to population-level integration levels (e.g. Parsons et al. 2012; Hu et al. 2014). Specifically, we performed a controlled jack-knife approach where the magnitude of integration is first estimated for the population, and then an individual is removed from the population and levels are re-estimated. The difference between estimates reflects an individual's contribution to trait integration. By repeating this process over the entire population, integration, which is by definition a population-level metric, can be turned into a quantitative trait suitable for genetic mapping. Utilizing this resource, we are able to test predictions about the genetic basis for this important trait. Specifically, based on previous analyses and observations showing different patterns of colour integration both between sexes and among species (Brzozowski et al. 2012), we predicted that variation in the magnitude of integration will map to both sex-linked and non-sex-linked loci.
We identified two QTL and one significant interaction for colour trait integration (green and purple diamonds, Fig. 2, Table 1), and these were the same regardless of whether integration was estimated using all colour traits (fins and flank regions A–I) or just flank traits (Table 1). One QTL was linked to the sex-determining locus on LG7, whereas the other was on LG18. At the sex-determining locus, the LF allele (female in the F0) increased integration, whereas the TRC/male allele decreased integration. These data are consistent with our previous observation that females express an over-all more integrated colour palette while pigmentation in males is more modular (Brzozowski et al. 2012). They also suggest a linkage between levels and patterns of pigmentation in this cross, given that multiple flank coloration QTL also map to this same locus (Fig. 2). The LF/female allele decreases the level of R-Y pigmentation across the flank, which should lead to a concomitant increase in overall R-Y colour integration (i.e. a uniform decrease in xanthophore expression over the flank). The TRC/male allele, on the other hand, both increases R-Y levels and decreases R-Y integration. This is because not all F2 males express uniformly high levels of R-Y colour, but rather, males may possess relatively higher levels of R-Y pigmentation in various, but discrete, regions along the flank. Thus, while average R-Y colour is higher in males, they also exhibit a more ‘modular’ inheritance of R-Y colour patterns (Brzozowski et al. 2012).
We have previously suggested that this trade-off with respect to colour trait integration among males (modular) and females (integrated) represents a means through which species mitigate sexual antagonism (Brzozowski et al. 2012). Specifically, we posited that relatively high levels of integration should be optimal for background matching in females, who are under strong natural selection for crypsis, whereas low levels of integration (i.e. increased modularity) should be optimal for the expression of a more conspicuous colour palette during competitive interactions between males and/or signalling to females during courtship (i.e. intra-and intersexual selection). This hypothesis predicts that variation in the magnitude of integration should be sex linked (Brzozowski et al. 2012), which is consistent with the genotype–phenotype map presented here.
We also detected a second QTL for the magnitude of integration on LG18 (Fig. 2). This locus is linked to neither sex nor any other colour QTL (i.e. it maps to a distinct region on LG18 from the large QTL cluster discussed earlier), which is consistent with the idea that variation in the magnitude of phenotypic integration can be regulated independently from sex determination or levels of pigmentation. This finding is also consistent with observations in the wild that certain species and lineages differ with respect to the complexity/modularity of expressed colour patterns (Fig. 1).
We identified a single interaction between loci on LGs 12 and 18 that affects the magnitude of colour trait integration (purple diamond, Fig. 2). This interaction further illustrates the complex relationship between pigmentation levels and patterns. Specifically, the interaction locus on LG18 colocalizes with the large cluster of R-Y QTL previously discussed. Thus, a QTL that determines the levels of pigmentation across multiple regions of the flank interacts with a second locus to affect whether pigmentation levels are expressed in a uniform or discontinuous manner across those regions. The locus on LG12 does not segregate with any colour trait, even with a very low (i.e. 0.1 chromosome-wide) statistical threshold, which suggests that it affects integration without simultaneously altering pigmentation levels.
Recent work in cichlids has identified the causative locus for an extreme loss of colour trait integration. The orange-blotch (OB) colour polymorphism is found in several rock-dwelling Lake Malawi cichlid species, maps to LG5 and is almost exclusive to females (although in rare cases male OBs can be found, Fig. 1). It is characterized by the development of variably sized and randomly positioned black blotches across the flank and fins and is thought to enhance background matching against the pock-marked rocks that are common along the near-shore rocky habitat (Roberts et al. 2009). The OB phenotype essentially breaks apart the ‘normal’ integrated colour pattern in these fishes, which is characterized by the development of a highly organized series of dark vertical bars on top of an overall brown to grey body. High-resolution association mapping coupled with expression analyses implicated pax7 as the causative gene underlying this polymorphism (Roberts et al. 2009). Notably, while pax7 is not implicated in regulating pigment variation in this cross, another member of the same gene subfamily, pax3a, is.
Pax3a mediates levels and integration of cichlid colour along the flank
The large QTL cluster for R-Y colour levels along the flank consistently maps to a ∼28-cM interval on LG18 (i.e. 19.5–47.7 cM, Table 1). In terms of genetic distance, this locus is fairly broad; however, when anchored to the Lake Malawi cichlid genome assembly (Metriaclima v.0), the corresponding physical distance becomes more tractable. Specifically, ∼25 cM of this interval corresponds to just over 9 Mb on scaffold 6. The remaining ∼3 cM corresponds to just under 1 Mb on scaffold 173. From this ∼10 Mb of sequence, our genome scan identified 366 polymorphisms (289 from scaffold 6, and 77 from scaffold 173), or one SNP every ∼27 kb. It is important to note that RAD-tag markers are based on sequences that align to a reference genome, and so any gaps in the genome will also be reflected in our data. Thus, we cannot rule out the possibility that there is additional sequence in this region that has been missed by our analyses. Nevertheless, of the 366 detected SNPs, 17 exhibited FST values greater than 0.57, an empirical threshold for divergence between cichlid genera estimated from multilocus SNP data (Mims et al. 2010). Most of these SNPs map to loci with no known role in neural crest or pigment cell development (Table S3, Supporting information). However, an SNP at the centre of the QTL peak localizes to the 5′ UTR of pax3a and is alternatively fixed between LF (n = 20) and TRC (n = 20) (FST = 1.00). We resequenced this genetic locus (including introns and exons) in a panel of five wild-caught LF and five wild-caught TRC animals. While a handful of other rare polymorphisms were found, none were nonsynonymous, which suggests that any pax3a-mediated variation in colour is due to changes in the regulatory properties of this gene. Moreover, only the polymorphism within the 5′ UTR was found to segregate in this cross. We resequenced this SNP in a panel of 25 wild-caught LF from five populations, which exhibited variation in the R-Y colour on the fins, but not the flank. All LF sequenced were invariant at this locus, and thus, this polymorphism does not segregate with xanthophore-based fin colour in this species. Alternatively, when resequenced in a panel of seven Tropheops species (n = 47 individuals) that varied in the extent of R-Y coloration along the flank, this SNP did segregate. Specifically, species in which males are characterized by extensive R-Y colour on their flank, T. gracilior (n = 10) and T. mumbo zebra (n = 10) (Figs 1 and 3), were nearly fixed for the LF allele (FST = 0.92 when compared to TRC). These data are consistent with the observation that the LF allele increases R-Y levels in the mapping cross.
Fig. 3.
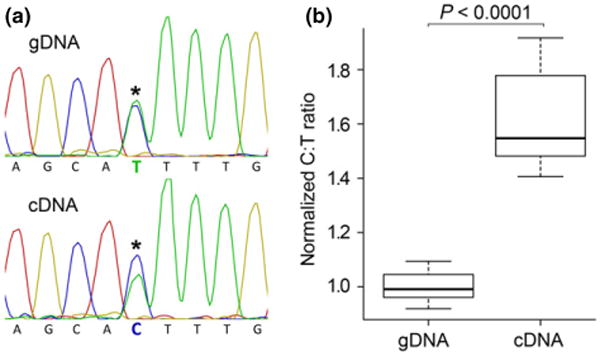
Allele-specific expression of pax3a. (a) Conventional Sanger sequencing chromatograms spanning an SNP in the 5′ UTR of the pax3a gene, from genomic DNA and flank skin cDNA (from region ‘E–F’; see Fig. 2) from a single hybrid individual. In the cDNA sequence (bottom), the C signal is greater than that of the T. A genomic DNA sequence trace (top) serves as a control for equal loading of the two alleles. (b) Semiquantitative analysis of chromatogram signal ratios at the 5′ UTR SNP (see Materials and methods), comparing gDNA and cDNA in interspecific hybrids (n = 9, P > 0.0001, paired t-test). In all heterozygotes tested, the C allele (from Labeotropheus) is found at higher levels in skin flank cDNA.
We next examined the hypothesis that differences in pax3a expression modulate flank colour. Both LF and TRC show variation in the amount of xanthophore-based pigment across the flank, and relative levels of pax3a transcript from regions E-F correlate to xanthophore counts in individuals of both species (Fig. S2, Supporting information). As such differences may not be causative, we developed an allele-specific assay in a set of F1 hybrids that were heterozygous for the 5′UTR SNP to examine expression differences between LF and TRC alleles of pax3a in xanthophore-containing tissue from the flanks of interspecific hybrids. While allele-specific assays based on pyrosequencing data are the standard in the field (Wittkopp 2011), not all systems are amenable to such an approach. In particular, when comparing recently diverged species/populations, nucleotide changes within coding sequence are less likely to occur, which is the case for pax3a in this cross. While the C/T SNP discussed earlier does occur within the 5′UTR, it is associated with a 4-mer poly-T repeat and thus is recalcitrant to allele-specific expression analysis based on pyrosequencing. As an alternative, we adapted a previously employed strategy based on conventional Sanger sequencing (Wei et al. 2013) to allow for the semiquantitative analysis of allele-specific expression, using genomic DNA as a balanced allele control. For each of nine heterozygous individuals, the LF allele was consistently found at relatively higher levels than the TRC allele in F1 flank skin cDNA from region E-F (Fig. 3). These data are consistent with the hypothesis that species-specific cis-regulatory differences at pax3a underlie the genetic signal for flank colour in our hybrid mapping population. Moreover, we suggest that while not as rigorous as more quantitative assays of allele-specific expression, this method provides a viable (albeit semiquantitative) alternative for systems in which suitable polymorphisms are lacking.
Taken together with the QTL results, these expression data suggest a model wherein the LF allele produces higher levels of pax3a transcript in the flank, thus increasing R-Y levels. This may be due to an expansion of xanthophores at the expense of melanocytes. It has recently been demonstrated in zebrafish that pax3a is necessary for xanthophore fate specification from a neural crest cell progenitor population (Minchin & Hughes 2008). In particular, pax3a was shown to mediate a fate switch between melanocytes and xanthophores. Under this model, pax3-mediated colour variation in nature should involve a trade-off between R-Y and black coloration in the affected region, and this appears to be the case in the fish examined here. Specifically, TRC males possess many melanocytes and very few xanthophores in posterior regions of the flank, whereas LF possess fewer melanocytes and many more xanthophores (Fig. 4). There is also a very clear negative relationship between R-Y and black pigmentation along the flank in our F2 mapping population (r = −0.318, P < 0.001). That is, males with high levels of R-Y pigmentation have relatively little black on their flanks, and vice versa (Fig. 4). These data implicate pax3a in mediating the trade-off between R-Y and black coloration in this cross. Notably, the pax3a locus is also implicated in the epistatic relationship that underlies variation in integration levels, which suggests that this factor may be involved in mediating both levels (i.e. high/low) and patterns (i.e. integrated/modular) of R-Y pigmentation.
Fig. 4.
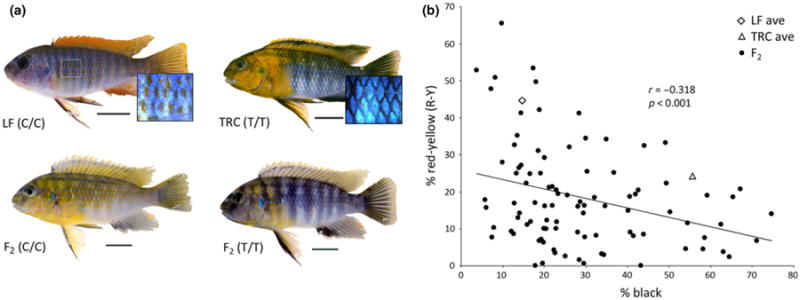
An apparent trade-off between melanocyte- and xanthophore-based coloration in cichlids. (a) Conspicuous xanthophore-based pigmentation is largely restricted to the head and anterior body of TRC males. Alternatively, male LF express xanthophores along their entire flank. High-resolution scale images are taken from region ‘F’ of LF and TRC males. LF animals are fixed for the ‘C’ allele at pax3a, whereas TRC are fixed for the ‘T’ allele. F2 males derived from crossing LF and TRC exhibit a range of colour phenotypes, but notably those that are homozygous for the ‘C’ allele possess large amounts of xanthophore-based pigmentation along the flank, while F2 that are homozygous for the ‘T’ allele are largely devoid of xanthophores on the posterior flank. LF, TRC and F2 animals shown here were laboratory reared. Scale bars indicate 1 cm. (b) A statistically significant negative relationship between the amount of R-Y and black pigment along the flank is noted for the F2. Animals with a relatively high amount of R-Y on their flanks have little melanocyte-based pigmentation and possess the LF/‘C’ allele. Alternatively, F2 animals with few xanthophores on their flank have correspondingly more melanocytes and possess the TRC/‘T’ allele. This negative relationship is consistent with the known role for pax3a in mediating a switch between xanthophore and melanocyte fate in the progenitor chromatophore stem cell population.
Integrating population genomics and QTL analyses to identify candidate genes
Quantitative trait locus (QTL) analyses have traditionally been limited in their ability to identify causative genes involved in adaptive divergence, especially for complex traits in vertebrate systems. Population genomics is a fast and efficient way to identify loci under selection, but linking these loci to complex phenotypes that show continuous variation is not straightforward. Here, we apply an experimental design that capitalizes on the advantages while mitigating the disadvantages of each approach. The power and utility of such a combined approach has been demonstrated in several recent studies (e.g. Rogers & Bernatchez 2007; Cadic et al. 2013; Berner et al. 2014), and we have previously referred to this methodology as ‘selection signature QTL’ (ssQTL), as each QTL is explicitly linked to a set of divergent loci between species/populations (Parsons & Albertson 2013).
The method begins with the genotyping of SNPs across the genomes of divergent populations to identify loci that exhibit the signature of divergence (in this case FST ≥ 0.57 when comparing LF and TRC). Divergent loci are then used in the construction of a high-density genetic linkage map from a laboratory cross between individuals from the same two populations. In this way, traditional QTL mapping has the potential to link divergent loci to specific phenotypes segregating in the mapping population. The potential of this method in terms of the efficient identification of candidate genes is illustrated for the pax3a locus above.
In addition, a QTL for the degree of black barring along the dorsal fin mapped to a 13-cM interval on LG18 (Fig. 5). This interval corresponds to several smaller scaffolds, and approximately 9 Mb of sequence. We identified 661 polymorphisms within this sequence, or 1 SNP every ∼13.6 kb (Fig. 5; Table S4, Supporting information). Of these, 28 exhibited FST values over 0.57, including an intronic SNP within the gene zeb1a. This SNP exhibited an FST of 0.95 between natural populations (n = 20 LF, n = 20 TRC). Zeb1 has been shown to directly bind to the promoter region and repress the expression of mitf (Liu et al. 2009), which is an evolutionarily conserved transcription factor and key regulator of pigment synthesis (reviewed by Sommer 2011). Zebrafish that harbour a loss-of-function mutation within mitf exhibit a complete lack of neural crest cell-derived pigmentation (reviewed by Rawls et al. 2001). While levels of pigmentation segregated for all of the fins in this cross, variation in the amount of black was only linked to zeb1a for the dorsal fin. We therefore consider this locus to be an ssQTL that implicates zeb1a as a mediator of black barring on the dorsal fin.
Fig. 5.
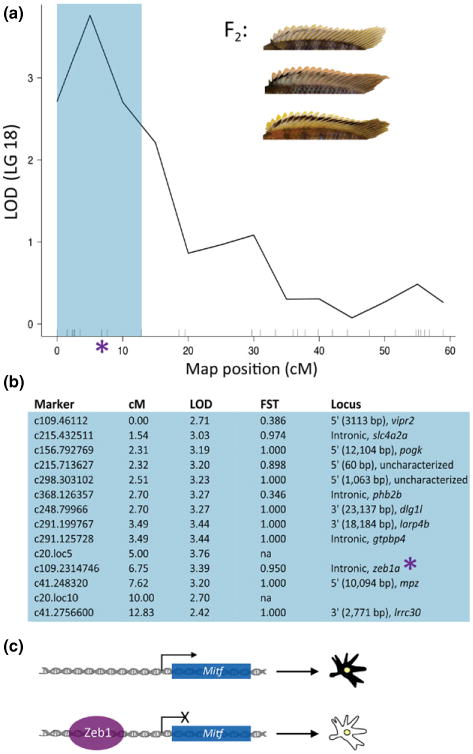
Example of a combined mapping (i.e. ‘ssQTL’) approach to candidate gene identification. (a) QTL plot (LG18) for the extent of barring along the dorsal fin. Blue-shaded region represents the 95% confidence interval for the QTL. The inset shows variation in this trait in the F2 population. (b) Markers within this interval are listed. Each marker is named according to its physical position within the genome. For instance, marker c109.2314746 corresponds to an SNP at the 2 314 746 base pair on scaffold 109 of the cichlid genome (Metriaclima v0). For each marker, the map position (cM) and LOD score are given. In addition, FST values, calculated from genotyping 20 LF and 20 TRC from natural populations, are also listed for each marker. Finally, the name of the nearest gene and relative position of the SNP to that gene are provided. A list of all of the SNPs with high FST values detected in our genome scan that correspond to this interval is provided in Table S4 (Supporting information). Among the markers in this interval is an SNP that is nearly alternatively fixed between LF and TRC and is found within the intron of the gene zeb1a (asterisk, a–b). (c) The protein encoded by zeb1 has been shown to regulate pigment synthesis by directly binding to and repressing the expression of mitf, a transcription factor whose expression is necessary and sufficient for melanocyte specification from neural crest cell progenitors. We suggest that zeb1a is a good candidate for regulating pigmentation variation among cichlids.
Finally, the R-Y QTL cluster on LG 14 consistently maps to a ∼20-cM interval that is anchored to several physical scaffolds and approximately 11.8 Mb of sequence (Table S5, Supporting information). The effect of this QTL on colour appears to be limited to more caudal regions of the flank (i.e. regions C, F, I), whereas the effects of R-Y QTL on LG18 extend more anteriorly. We identified 186 SNPs within the 11.8 Mb stretch of sequence that corresponds to the QTL on LG14, with 26 exhibiting high FST values. Among these is an SNP adjacent to the pax3a paralog, pax3b. Less than 150 kb away is another SNP adjacent to ephrin a4. Both SNPs are alternatively fixed between LF and TRC (n = 20 LF, n = 20 TRC), and both genes are viable candidates for trunk pigmentation. Ephrins are well-characterized mediators of neural crest cell migration in both the head and trunk (reviewed by Kuriyama & Mayor 2008), and pax3b has been shown to play a complementary role to pax3a in xanthophore development in zebrafish (Minchin & Hughes 2008). Disentangling the specific molecular basis for this QTL's affect on R-Y colour variation along the cichlid flank will require further experimentation; however, it is notable that paralogous regions of the cichlid genome are both linked to QTL for R-Y phenotypes.
As Tables S4 and S5 (Supporting information) illustrate, there are a few instances where markers from the same physical scaffold are not adjacent on the linkage map. This tended to occur when dealing with short scaffolds (e.g. <1 Mb) and between closely spaced markers along the genetic map (e.g. <1 cM). Markers from larger scaffolds were nearly always contiguous. Thus, the discrepancy between physical and genetic maps may be due to either genotyping error or errors/gaps in the genome assembly.
While cross-referencing population genomic and mapping data makes a strong case to follow up on these and other candidates, demonstrating causation will require more in the way of future research. Moreover, there is room for improvement in this experimental design, in terms of both increasing the number of recombination events (e.g. F3, F4 or F5) and the identification of additional divergent markers among wild-caught animals. Nevertheless, even with this modest design, this approach has yielded a number of legitimate candidate genes to be followed up in future studies.
Conclusion
How does biodiversity arise, and how is it maintained over time? Over 150 years after the publication of Darwin's The Origin of Species, we are still only beginning to understand the genetic basis for evolutionary change. With respect to colour, considerable inroads have been made to understand the molecular basis for variation in relatively simple colour polymorphisms (Manceau et al. 2010; Kronforst et al. 2012); however, the vast majority of colour variation among species is complex. An open question in evolutionary biology is whether the mechanisms that underlie discrete polymorphisms [e.g. oca2 (Protas et al. 2006), mc1r (Uy et al. 2009), pax7 (Roberts et al. 2009)] also regulate continuous variation among traits. There is some evidence to suggest that, in certain cases involving coloration, it may. For example, allelic variation in mc1r is associated with a quantitative reduction in pigmentation in blind cavefish (Gross et al. 2009). In addition, while pax7 is not implicated in mediating colour variation in this cross, a member of the same gene subfamily, pax3a (and possibly pax3b), is. Both pax3 and pax7 are involved in xanthophore development in zebrafish (Minchin & Hughes 2008), and data presented here implicate this gene subfamily in mediating both saltatory and continuous variation in colour patterns. Additional work in this and other systems will continue to shed light on this important question.
We also suggest that genetic analyses of more complex colour patterns are needed in general. Integration is an especially important trait to consider along these lines, as it relates to the evolutionary potential (i.e. evolvability) of a trait (Klingenberg 2008). Historically, gaining genetic inroads into this trait has been difficult (but see relationship QTL in Cheverud et al. 2004; Pavlicev et al. 2008). Here, we offer a new, relatively straightforward method in which to assess the genetic basis of this important trait. This individual metric of integration combined with the ssQTL approach illustrated earlier holds great promise to advance a better understanding of the specific genetic mechanisms that promote diversity and effect trait evolvability.
Supplementary Material
Fig. S1 Effect plots from all significant (i.e., 0.05 genome-wide level) two-way interactions are shown.
Fig. S2 A positive association is noted between relative pax3a transcript levels and xanthophore count in both LF and TRC.
Table S1 Various measures of genetic differentiation across SNPs derived from genotyping twenty wild caught LF and twenty wild caught TRC.
Table S2 Genetic linkage map for the LFxTRC F2 cross.
Table S3 Xanthophore-based color QTL interval for the pax3a locus on LG18.
Table S4 Dorsal fin melanocyte-based color QTL interval on LG18.
Table S5 Xanthophore-based flank color QTL interval on LG14.
Acknowledgments
We thank Brooke Stebbins for assistance with genotyping, as well as past and present members of the Albertson lab for helpful comments on various aspects of this project. This work was supported by a grant to R. C. A. from the NSF (IOS-1054909).
R.C.A. obtained funding and conceived project. R.C.A., R.B.R. and K.J.P. designed all experiments. R.C.A. performed genetic mapping and analyzed genetic/genomic data. K.E.P. performed all genotyping around pax3a. Y.H. obtained integration metric for mapping. K.P.C. and R.B.R. performed and analyzed qPCR and allele-specific expression experiments. K.J.P. constructed the genetic linkage map and performed FST scans. R.C.A. wrote the manuscript with important contributions from R.B.R. and K.J.P. All authors read and provided input on the manuscript.
Footnotes
Supporting information: Additional supporting information may be found in the online version of this article.
Data accessibility: Tropheops sp. ‘red cheek’ pax3a cDNA: GenBank accession no. KM272327 Labeotropheus fuelleborni pax3a cDNA: GenBank accession no. KM272328 Genotypic and phenotypic data for QTL analyses (formatted for R-qtl); pax3a qPCR data; pax3a allele-specific expression data: Dryad provisional DOI: 10.5061/dryad.7sr73.
References
- Albertson RC, Streelman JT, Kocher TD. Directional selection has shaped the oral jaws of Lake Malawi cichlid fishes. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:5252–5257. doi: 10.1073/pnas.0930235100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arends D, Prins P, Jansen RC, Broman KW. R/qtl: High-throughput multiple QTL mapping. Bioinformatics. 2010;26:2990–2992. doi: 10.1093/bioinformatics/btq565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berner D, Moser D, Roesti M, Buescher H, Salzburger W. Genetic architecture of skeletal evolution in European lake and stream stickleback. Evolution. 2014;68:1792–1805. doi: 10.1111/evo.12390. [DOI] [PubMed] [Google Scholar]
- Broman KW, Sen S. A guide to QTL mapping with R/qtl. Springer; Dordrecht, Heidelberg, London, New York: 2009. [Google Scholar]
- Broman KW, Wu H, Sen S, Churchill GA. R/qtl: QTL mapping in experimental crosses. Bioinformatics. 2003;19:889–890. doi: 10.1093/bioinformatics/btg112. [DOI] [PubMed] [Google Scholar]
- Brzozowski F, Roscoe J, Parsons K, Albertson RC. Sexually dimorphic levels of color trait integration and the resolution of sexual conflict in Lake Malawi cichlids. Journal of Experimental Zoology Part B, Molecular and Developmental Evolution. 2012;318:268–278. doi: 10.1002/jez.b.22443. [DOI] [PubMed] [Google Scholar]
- Cadic E, Coque M, Vear F, et al. Combined linkage and association mapping of flowering time in Sunflower (Helianthus annuus L.) Theoretical and Applied Genetics. 2013;126:1337–1356. doi: 10.1007/s00122-013-2056-2. [DOI] [PubMed] [Google Scholar]
- Cheverud JM, Ehrich TH, Vaughn TT, Koreishi SF, Linsey RB, Pletscher LS. Pleiotropic effects on mandibular morphology II: differential epistasis and genetic variation in morphological integration. Journal of Experimental Zoology Part B, Molecular and Developmental Evolution. 2004;302:424–435. doi: 10.1002/jez.b.21008. [DOI] [PubMed] [Google Scholar]
- Chutimanitsakun Y, Nipper RW, Cuesta-Marcos A, et al. Construction and application for QTL analysis of a Restriction Site Associated DNA (RAD) linkage map in barley. BMC Genomics. 2011;12:4. doi: 10.1186/1471-2164-12-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danley PD, Kocher TD. Speciation in rapidly diverging systems: lessons from Lake Malawi. Molecular Ecology. 2001;10:1075–1086. doi: 10.1046/j.1365-294x.2001.01283.x. [DOI] [PubMed] [Google Scholar]
- Greenwood AK, Jones FC, Chan YF, et al. The genetic basis of divergent pigment patterns in juvenile threespine sticklebacks. Heredity. 2011;107:155–166. doi: 10.1038/hdy.2011.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross JB, Borowsky R, Tabin CJ. A novel role for Mc1r in the parallel evolution of depigmentation in independent populations of the cavefish Astyanax mexicanus. PLoS Genetics. 2009;5:e1000326. doi: 10.1371/journal.pgen.1000326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haimes J, Kelley M. Demonstration of a ΔΔCq calculation method to compute relative gene expression from qPCR data. Thermo Fisher Scientific Tech Note. 2010 http://dharmacon.gelifesciences.com/uploadedFiles/Resources/tech-note-demonstration-of-a-cq-calculation-method-using-solaris-qpcr-as-says.pdf.
- Hu Y, Parsons KJ, Albertson RC. Evolvability of the cichlid jaw: new tools provide insights into the genetic basis of phenotypic integration. Evolutionary Biology. 2014;41:145–153. [Google Scholar]
- Jost L. GST and its relatives do not measure differentiation. Molecular Ecology. 2008;17:4015–4026. doi: 10.1111/j.1365-294x.2008.03887.x. [DOI] [PubMed] [Google Scholar]
- Klingenberg C. Morphological integration and developmental modularity. Annual Review of Ecology, Evolution, and Systematics. 2008;39:115–132. [Google Scholar]
- Kocher TD. Adaptive evolution and explosive speciation: the cichlid fish model. Nature Reviews Genetics. 2004;5:288–298. doi: 10.1038/nrg1316. [DOI] [PubMed] [Google Scholar]
- Konings A. Malawi Cichlids in Their Natural Habitat. 3rd. Cichlid Press; El Paso, Texes: 2001. [Google Scholar]
- Kronforst MR, Barsh GS, Kopp A, et al. Unraveling the thread of nature's tapestry: the genetics of diversity and convergence in animal pigmentation. Pigment Cell Melanoma Research. 2012;25:411–433. doi: 10.1111/j.1755-148X.2012.01014.x. [DOI] [PubMed] [Google Scholar]
- Kuriyama S, Mayor R. Molecular analysis of neural crest migration. Philosophical Transactions of the Royal Society of London Series B, Biological sciences. 2008;363:1349–1362. doi: 10.1098/rstb.2007.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BY, Lee WJ, Streelman JT, et al. A second-generation genetic linkage map of tilapia (Oreochromis spp.) Genetics. 2005;170:237–244. doi: 10.1534/genetics.104.035022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtonen TK, Meyer A. Heritability and adaptive significance of the number of egg-dummies in the cichlid fish Astatotilapia burtoni. Proceedings Biological Sciences. 2011;278:2318–2324. doi: 10.1098/rspb.2010.2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Ye F, Li Q, et al. Zeb1 represses Mitf and regulates pigment synthesis, cell proliferation, and epithelial morphology. Investigative Ophthalmology & Visual Science. 2009;50:5080–5088. doi: 10.1167/iovs.08-2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malek TB, Boughman JW, Dworkin I, Peichel CL. Admixture mapping of male nuptial colour and body shape in a recently formed hybrid population of threespine stickleback. Molecular Ecology. 2012;21:5265–5279. doi: 10.1111/j.1365-294X.2012.05660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manceau M, Domingues VS, Linnen CR, Rosenblum EB, Hoekstra HE. Convergence in pigmentation at multiple levels: mutations, genes and function. Philosophical Transactions of the Royal Society of London Series B, Biological sciences. 2010;365:2439–2450. doi: 10.1098/rstb.2010.0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CT, Beleza S, Pollen AA, et al. cis-regulatory changes in Kit ligand expression and parallel evolution of pigmentation in sticklebacks and humans. Cell. 2007a;131:1179–1189. doi: 10.1016/j.cell.2007.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MR, Dunham JP, Amores A, Cresko WA, Johnson EA. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Research. 2007b;17:240–248. doi: 10.1101/gr.5681207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mims MC, Hulsey DC, Fitzpatrick BM, Streelman JT. Geography disentangles introgression from ancestral polymorphism in Lake Malawi cichlids. Molecular Ecology. 2010;19:940–951. doi: 10.1111/j.1365-294X.2010.04529.x. [DOI] [PubMed] [Google Scholar]
- Minchin JE, Hughes SM. Sequential actions of Pax3 and Pax7 drive xanthophore development in zebrafish neural crest. Journal of Developmental Biology. 2008;317:508–522. doi: 10.1016/j.ydbio.2008.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M. Molecular Evolutionary Genetics. Columbia University Press; New York, NY: 1987. [Google Scholar]
- O'Quin CT, Drilea AC, Conte MA, Kocher TD. Mapping of pigmentation QTL on an anchored genome assembly of the cichlid fish, Metriaclima zebra. BMC Genomics. 2013;14:287. doi: 10.1186/1471-2164-14-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parichy DM, Ransom DG, Paw B, Zon LI, Johnson SL. An orthologue of the kit-related gene fms is required for development of neural crest-derived xanthophores and a subpopulation of adult melanocytes in the zebrafish, Danio rerio. Development. 2000;127:3031–3044. doi: 10.1242/dev.127.14.3031. [DOI] [PubMed] [Google Scholar]
- Parsons KJ, Albertson RC. Unifying and generalizing the two strands of evo-devo. Trends in Ecology & Evolution. 2013;28:584–591. doi: 10.1016/j.tree.2013.06.009. [DOI] [PubMed] [Google Scholar]
- Parsons KJ, Marquez E, Albertson RC. Constraint and opportunity: the genetic basis and evolution of modularity in the cichlid mandible. The American Naturalist. 2012;179:64–78. doi: 10.1086/663200. [DOI] [PubMed] [Google Scholar]
- Pavlicev M, Kenney-Hunt JP, Norgard EA, Roseman CC, Wolf JB, Cheverud JM. Genetic variation in pleiotropy: differential epistasis as a source of variation in the allometric relationship between long bone lengths and body weight. Evolution. 2008;62:199–213. doi: 10.1111/j.1558-5646.2007.00255.x. [DOI] [PubMed] [Google Scholar]
- Pavlicev M, Cheverud JM, Wagner GP. Measuring morphological integration using eigenvalue variance. Evolutionary Biology. 2009;36:157–170. [Google Scholar]
- Protas ME, Hersey C, Kochanek D, et al. Genetic analysis of cavefish reveals molecular convergence in the evolution of albinism. Nature Genetics. 2006;38:107–111. doi: 10.1038/ng1700. [DOI] [PubMed] [Google Scholar]
- Rawls JF, Mellgren EM, Johnson SL. How the zebrafish gets its stripes. Journal of Developmental Biology. 2001;240:301–314. doi: 10.1006/dbio.2001.0418. [DOI] [PubMed] [Google Scholar]
- Roberts RB, Ser JR, Kocher TD. Sexual conflict resolved by invasion of a novel sex determiner in Lake Malawi cichlid fishes. Science. 2009;326:998–1001. doi: 10.1126/science.1174705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers SM, Bernatchez L. The genetic architecture of ecological speciation and the association with signatures of selection in natural lake whitefish (Coregonus sp. Salmonidae) species pairs. Molecular Biology and Evolution. 2007;24:1423–1438. doi: 10.1093/molbev/msm066. [DOI] [PubMed] [Google Scholar]
- Salzburger W, Mack T, Verheyen E, Meyer A. Out of Tanganyika: genesis, explosive speciation, key-innovations and phylogeography of the haplochromine cichlid fishes. BMC Evolutionary Biology. 2005;5:17. doi: 10.1186/1471-2148-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzburger W, Braasch I, Meyer A. Adaptive sequence evolution in a color gene involved in the formation of the characteristic egg-dummies of male haplochromine cichlid fishes. BMC Biology. 2007;5:51. doi: 10.1186/1741-7007-5-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ser JR, Roberts RB, Kocher TD. Multiple interacting loci control sex determination in lake Malawi cichlid fish. Evolution. 2010;64:486–501. doi: 10.1111/j.1558-5646.2009.00871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R, Sheppard K, DiPetrillo K, Churchill G. Quantitative trait locus analysis using J/qtl. Methods in Molecular Biology. 2006;573:175–188. doi: 10.1007/978-1-60761-247-6_10. [DOI] [PubMed] [Google Scholar]
- Sommer L. Generation of melanocytes from neural crest cells. Pigment Cell Melanoma Research. 2011;24:411–421. doi: 10.1111/j.1755-148X.2011.00834.x. [DOI] [PubMed] [Google Scholar]
- Tripathi N, Hoffmann M, Willing EM, Lanz C, Weigel D, Dreyer C. Genetic linkage map of the guppy, Poecilia reticulata, and quantitative trait loci analysis of male size and colour variation. Proceedings Biological Sciences. 2009;276:2195–2208. doi: 10.1098/rspb.2008.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner GF, Seehausen O, Knight ME, Allender CJ, Robinson RL. How many species of cichlid fishes are there in African lakes? Molecular Ecology. 2001;10:793–806. doi: 10.1046/j.1365-294x.2001.01200.x. [DOI] [PubMed] [Google Scholar]
- Uy JA, Moyle RG, Filardi CE, Cheviron ZA. Difference in plumage color used in species recognition between incipient species is linked to a single amino acid substitution in the melanocortin-1 receptor. The American Naturalist. 2009;174:244–254. doi: 10.1086/600084. [DOI] [PubMed] [Google Scholar]
- Wei QX, Claus R, Hielscher T, et al. Germline allele-specific expression of DAPK1 in chronic lymphocytic leukemia. PLoS ONE. 2013;8:e55261. doi: 10.1371/journal.pone.0055261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickler W. ‘Egg-dummies’ as natural releasers in mouth-breeding cichlids. Nature. 1962;194:1092–1093. [Google Scholar]
- Wittkopp PJ. Using pyrosequencing to measure allele-specific mRNA abundance and infer the effects of cis- and trans-regulatory differences. Methods in Molecular Biology. 2011;772:297–317. doi: 10.1007/978-1-61779-228-1_18. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Effect plots from all significant (i.e., 0.05 genome-wide level) two-way interactions are shown.
Fig. S2 A positive association is noted between relative pax3a transcript levels and xanthophore count in both LF and TRC.
Table S1 Various measures of genetic differentiation across SNPs derived from genotyping twenty wild caught LF and twenty wild caught TRC.
Table S2 Genetic linkage map for the LFxTRC F2 cross.
Table S3 Xanthophore-based color QTL interval for the pax3a locus on LG18.
Table S4 Dorsal fin melanocyte-based color QTL interval on LG18.
Table S5 Xanthophore-based flank color QTL interval on LG14.