

Abstract
The specific hybridization of complementary sequences is an essential property of nucleic acids, enabling diverse biological and biotechnological reactions and functions. However, the specificity of nucleic acid hybridization is compromised for long strands, except near the melting temperature. Here, we analytically derived the thermodynamic properties of a hybridization probe that would enable near-optimal single-base discrimination and perform robustly across diverse temperature, salt and concentration conditions. We rationally designed ‘toehold exchange’ probes that approximate these properties, and comprehensively tested them against five different DNA targets and 55 spurious analogues with energetically representative single-base changes (replacements, deletions and insertions). These probes produced discrimination factors between 3 and 100+ (median, 26). Without retuning, our probes function robustly from 10 °C to 37 °C, from 1 mM Mg2+ to 47 mM Mg2+, and with nucleic acid concentrations from 1 nM to 5 μM. Experiments with RNA also showed effective single-base change discrimination.
Nucleic acids are essential biomolecules, encoding and regulating the expression of hereditary information within living organisms1. The biological importance of nucleic acids has prompted the use of synthetic nucleic acid probes and primers for biology and biotechnology, for example, through the polymerase chain reaction (PCR)2, microarrays3–5 and fluorescent in situ hybridization6,7. Nucleic acids have also emerged as powerful materials in the field of nanoscale engineering8–13.
The key property of nucleic acids that renders them so useful for biology, biotechnology and bionanotechnology is the predictable and specific Watson–Crick hybridization of complementary bases. However, the thermodynamic gain of many correctly paired bases can override the thermodynamic penalty of a few mismatches, and the hybridization of long nucleic acids may be nonspecific except near the melting temperature.
High-temperature or chemical denaturation is often used to improve hybridization specificity2,6,7. At high temperatures or under chemically denaturing conditions, the standard free energy of hybridization is weaker, and marginally different sequences may be distinguished based on their binding affinities to a complement. However, operating near the melting temperature is not always feasible, such as in the case of a multiplexed system where many different hybridization reactions must proceed simultaneously. Additionally, the melting temperature depends on factors such as salinity and nucleic acid concentrations and can be difficult to predict precisely. Similarly, chemical denaturation weakens Watson–Crick base-pairing, and is subject to the same potential limitations.
Given the thermodynamic basis for discriminating closely related nucleic acid sequences near the melting temperature, it is possible to engineer frustrated complement molecules that hybridize less favourably to their intended targets than the standard complementary strands. For example, in molecular beacons14–16, the complement is flanked by extra bases and natively forms a hairpin structure. Hybridization to the beacon’s target disrupts the hairpin structure and is less thermodynamically favourable than standard hybridization; this allows for higher hybridization specificity17,18. Other probes have also aimed to reduce the thermodynamic favourability of hybridization19,20, but these are often more complex and difficult to rationally design with fine control of the thermodynamics. Although beacons and other probes can be systematically optimized for performance under any particular set of conditions (such as temperature or salinity)21, a robust hybridization probe that specifically hybridizes to its intended target across diverse conditions without retuning would be vastly more useful.
In this Article, we first present a theoretical framework for the analysis of nucleic acid hybridization specificity and derive theoretical limits, providing a benchmark for evaluating the performance of hybridization probes. Next, we analytically derive probe properties that will ensure near-optimal specificity across diverse temperatures, concentrations and salinities. Finally, we present DNA and RNA toehold exchange probes and show that they experimentally discriminate single-base changes under a wide range of conditions.
Analytic framework and probe design
Thermodynamics of hybridization specificity
Consider the hybridization of two complementary nucleic acids, target X and complement C: X + C → XC. The equilibrium constant Keq = [XC]/([X] [C]) can be calculated from the standard free energy ΔG∘, which can in turn be predicted from the sequences of X and C (ref. 22). The hybridization yield χ = [XC]/([XC] + min{[X], [C]}) denotes the fraction of the limiting reagent that exists in duplex form, and can be analytically calculated from Keq and the initial concentrations of X and C (Supplementary Text S1). More simply, χ can be written solely as a function of the concentration-adjusted equilibrium constant (Supplementary Text S1), where c denotes the initial concentration of the limiting reagent and Δn represents the change in the number of species through the course of the reaction (−1 for bimolecular hybridization). Thus, is a useful dimensionless metric for evaluating the equilibrium distribution; when , the hybridization yield χ approaches 1, and when , χ approaches 0. The concentration-adjusted standard free energy is similarly defined. Figure 1a plots χ versus ΔG′ for a standard hybridization reaction.
Figure 1. Hybridization specificity of nucleic acids.

a, Hybridization yield χ plotted against concentration-adjusted standard free energy ΔG′ = ΔG∘ + (Δn)RTln(c), where c is the concentration of the limiting species, and Δn = −1 for a standard bimolecular hybridization reaction. At room temperature, the binding of both the correct target (black dot) and the spurious target (red dot) are thermodynamically favourable and practically indistinguishable. In contrast, at the melting temperature, ΔG′ = −RTln((c/2)/(c/2)2)−RTln(c) = −0.41 kcal mol−1, the hybridization yield of the correct target is 50%, and much lower for the spurious target. b, In a hybridization-based assay or reaction, specificity is achieved when a spurious target that differs in sequence from the correct target by a single base (depicted as the red segment) does not hybridize significantly to the complement. c, The standard free energy difference (ΔΔG∘) caused by a single-base change ranges from 1.83 to 6.57 kcal mol−1, and determines the upper bound on the discrimination factor: Qmax ≡ eΔΔG∘/RT. (Graphic constructed using thermodynamic parameters by SantaLucia and Hicks22; see Supplementary Text S3 and Tables S1–S5 for detailed numerical values.) All 64 cases of single-base insertion were modelled to have identical ΔΔG∘.
A hybridization reaction or probe is specific when there is a large difference between the hybridization yield of an intended target X and that of a spurious target S (Fig. 1a). Specificity can be quantitated as discrimination factor Q = χX/χS, where χX and χS are the hybridization yields of X and S, respectively.
A fundamental upper bound on Q is prescribed by thermodynamics:
where
is the difference in standard free energies of the hybridization reaction for X and S, R is the ideal gas constant, and T is temperature (see Supplementary Text S2 for analytical proof). To investigate the limits of performance, here we only consider spurious targets S that differ from X by a single base. The ΔΔG∘ values for all single-base changes are plotted in Fig. 1c. For a typical ΔΔG∘ = 4 kcal mol−1 single-base change, Qmax = 853 at 25 °C.
Properties of a specific and robust hybridization probe
A specific and robust hybridization probe should have several functional properties: (i) high hybridization yield χX for the correct target, (ii) high discrimination factor Q against spurious targets, (iii) robustness to target and probe concentrations, (iv) robustness to temperature and (v) robustness to the chemical composition (for example, salinity) of the solvent.
There is a trade-off between high χ and high Q. For standard hybridization reactions, we prove in Supplementary Text S2 that as ΔG′ approaches +∞, Q approaches Qmax; however, χ also approaches 0. At the melting temperature of XC, ΔG′ ≈ 0, χX = 0.5 and Q >Qmax/2 (see Supplementary Text S2 for analytical proof); this is a good trade-off between specificity and yield. Hybridization probes that are designed to achieve ΔG′ ≈ 0 (for example, 1 kcal mol−1 ≥ ΔG′ ≥ −1 kcal mol−1) will similarly exhibit near-optimal specificity and reasonable hybridization yields.
Concentration robustness requires that hybridization yield χ does not have a dependence on c. Recall that χ can be expressed solely as a function of (Supplementary Text S1). In a reaction where Δn = 0, and χ will be independent of c, and therefore robust to changes in concentration.
The dependence of χ on temperature T is due to the latter’s effect on Keq:
Under standard thermodynamic models of DNA hybridization22, ΔH∘ and ΔS∘ are assumed to be temperature-invariant (dΔH∘/dT, dΔS∘/dT ≈ 0), and χ will have no dependence on T when ΔH∘ = 0. Hence, a probe that hybridizes to its target with ΔH∘ = 0 would be robust to temperature.
The effects of salts in solution on χ are also manifested through a change in Keq. There are two accepted models of the effects of cation concentrations on hybridization thermodynamics: the ‘Na+ equivalent’ model23 and the ‘tightly bound ion’ model24. In both models, the effects of cations are assumed to be purely entropic, and the entropic adjustment terms are functions of N, the total number of phosphates in double-stranded form. A reaction with no change in the total number of paired bases (ΔN = 0) is expected to feature hybridization yields that are unaffected by salt concentrations, as long as sufficient cations are present to allow DNA hybridization.
In summary, a hybridization probe is expected to robustly discriminate single-base changes across concentrations, temperatures and salinities if it reacts with its reactant with (i) little change in concentration-adjusted standard free energy (ΔG′ ≈ 0), (ii) no net change in the number of nucleic acid molecules in solution (Δn = 0), (iii) no net change in the standard enthalpy of reaction (ΔH∘ = 0) and (iv) no net change in the number of paired bases (ΔN = 0).
Toehold exchange probe design
We present toehold exchange probes that approximately satisfy all of the above criteria (Fig. 2). The hybridization of the probe (PC) to the correct target X is initiated at the green 5′ single-stranded region of C (known as a toehold25,26), proceeds through a branch migration process, and is completed via the spontaneous dissociation of the 3′ base pairs of C (blue toehold) to release single-stranded protector P (refs 26,27). The toeholds allow the forward and reverse reactions to proceed with fast kinetics. Without the toehold, the kinetics of the displacement reaction would have a half-life of months under typical experimental conditions (for example, 100 nM DNA, pH ≈ 8, T ≈ 25 °C, [Mg2+] ≈ 12 mM). In the presence of the 7 nt toeholds depicted, the rate constants of the forward and reverse reactions are up to 1 × 106 M−1 s−1, corresponding to a half-life of 10 s (ref. 26).
Figure 2. Toehold exchange probes.

a, The toehold exchange probe (PC) consists of a pre-hybridized complement strand C and a protector strand P. The probe can react with an intended target X to release P and the hybridized product XC. The probe is designed based on the sequence of the target so that the standard free energy (ΔG∘ = ΔG′) of the forward reaction is close to zero. In this example, seven new base pairs (green 5′ toehold of C) are formed, but seven existing base pairs are broken (blue 3′ toehold of C). For the sequences shown here, mathematical analysis predicts the hybridization yield χX = 0.34 at 25 °C, mimicking hybridization behaviour at close to the melting temperature of XC (Supplementary Text S4). b, Hybridization of a spurious target S with one base change is less thermodynamically favourable by +2.97 kcal mol−1, and is predicted to have χS = 0.0056. Thus, the discrimination factor Q is predicted to be 0.34/0.0056 = 61. For comparison, Qmax for ΔΔG∘ = 2.97 kcal mol−1 is 150 at 25 °C.
The blue toehold that spontaneously dissociates is designed to be similar in length (ΔN ≈ 0), base composition and thermodynamic binding strength (ΔG∘, ΔH∘) as the green toehold. Furthermore, the reaction is bimolecular with two products, so Δn = 0 and ΔG′ = ΔG∘. Consequently, this X + PC → XC + P reaction has ΔG′ ≈ 0, Δn ≈ 0, ΔH∘ ≈ 0 and ΔN ≈ 0, and is expected to be rapid and highly specific over a wide range of temperatures, salinities and nucleic acid concentrations. The standardized conceptual design and lack of complicated secondary structures (such as pseudoknots) renders the system quantitatively predictable and rationally designable. As the toehold exchange probe is a design approach that depends only on the Watson–Crick pairing property of nucleic acids, it is generalizable to RNA and other nucleic acids, such as PNA28 and LNA29.
Experimental results
The experimentally implemented toehold exchange probes (Fig. 3a) differed slightly from the theoretical construction (Fig. 2). To distinguish the hybridized products XC and SC from probe PC based on mobility in a gel, we appended 30 nt poly-T to the 5′ end of protector P. Standard DNA thermodynamics models22,30 predict that this does not significantly change the ΔG∘ of formation of the probe PC or of the released protector P.
Figure 3. Experimental demonstration of toehold exchange probes.

a, The intended DNA target sequence and the experimental probe system. Outlined in red are the positions of the one-base changes in various spurious targets. P in the experimental system has a 30 nt poly-T tail at the 5′ end (purple) to help distinguish it from other species in gel experiments. b, Native PAGE results. PC was prepared with a 2:1 ratio of P to C, and annealed with a 1 μM concentration of PC. X or S was added to achieve final concentrations of 200 nM target (X or S), 100 nM PC and 100 nM P. Reactions proceeded at 25 °C for 1 h. Four different probes were tested (for example, the 7/5 probe has a 7 nt green toehold and a 5 nt blue toehold). Middle lanes show the reaction with different spurious targets S; ‘m’, ‘d’ and ‘i’ denote mismatch, deletion and insertion, respectively (for example, in m11C, the adenine at position 11 from the 5′ end was replaced by a cytosine; see Supplementary Table S7 for sequences). The rightmost lane shows the negative control (PC only). The P band is single-stranded and stains inconsistently with SybrGold. c, Hybridization yields χ inferred from b. χX are plotted as crosses, χS as filled circles. Lines connect χ values for the same target. d, Discrimination factors Q. e, Plot of hybridization yield χ versus reaction standard free energy ΔG∘. Values of χ are plotted as crosses and filled circles against the reaction ΔG∘ calculated by NUPACK (Supplementary Text S6)31. The thick black trace shows the expected results from thermodynamic analysis (Supplementary Text S4). Adjusting all ΔG∘ by +1.5 kcal mol−1 (thin black trace) improves the agreement between model and data. f, 7/5 probe with a large excess of S (equal mixture of all 11 spurious targets). g, Fluorophore/quencher-labelled probe.
Probe performance
Our first target X was designed to have no secondary structure. Figure 3a shows its sequence and the positions of the single-base changes of spurious targets S. The probe was allowed to react with X or S at room temperature for 1 h before gel electrophoresis (1 h was verified to be sufficient to allow the reaction to proceed to equilibrium; Supplementary Fig. S3). Figure 3b shows the native polyacrylamide gel electrophoresis (PAGE) results for four different probes, labelled 7/0, 7/4, 7/5 and 7/6. Each probe has an initiation toehold of 7 nt, but differs in having a 0, 4, 5 or 6 nt blue toehold that spontaneously dissociates to release P.
The intensities of the PC, XC and SC bands appear to be directly comparable: the sum of the PC and XC or SC band intensities in each lane was roughly conserved across all lanes (6.6% standard deviation, 156 samples, Supplementary Fig. S4B). We believe this is due to both species having a similar number of double-stranded base pairs, and the fact that single-stranded nucleotides do not contribute significantly to band intensity because of their decreased efficiency of SybrGold staining. The experimentally observed hybridization yield of X was calculated as χX = {XC}/({XC} + {PC}), where {XC} and {PC} denote the band intensities of XC and PC, respectively (χS is similarly defined). Values of χ are plotted in Fig. 3c.
Discrimination factors Q were calculated for each spurious target (for example, Qm1G = χX/χm1G) and are plotted in Fig. 3d. The 7/0 probe did not achieve specificity (Q ≈ 1 for all experiments). In contrast, the 7/4, 7/5 and 7/6 probes achieved Q values between 2 and 10, 12 and 37, and 5 and 38, respectively.
The experimentally observed χ values are plotted in Fig. 3e for comparison against the χ values predicted by the reaction thermodynamics (thick black sigmoidal line), calculated using NUPACK software31. Adjusting the predicted ΔG∘ of all reactions by +1.5 kcal mol−1 significantly improves the agreement between theory and experiments. This +1.5 kcal mol−1 discrepancy may represent inaccuracy in the existing DNA thermodynamic prediction tools, specifically regarding the poly-T tail of P (ref. 30).
Theoretically, the 7/6 probes are expected to yield Q values higher than those of the 7/5 probes. In practice, however, the observed Q was lower because the χS values for the 7/6 probes were significantly higher than predicted (values not shown). Oligonucleotide synthesis errors, which remain despite purification32,33, probably contribute to spurious hybridization (Supplementary Text S5). For example, a deletion at position 12 (from the 5′ end) in the C strand would match the deletion of ‘d11’, so the corresponding PC molecule would react more favourably with the ‘d11’ spurious target than with X. Given the low values of χX observed for the 7/6 probes, a 7/7 toehold exchange probe (Fig. 3a) would be expected to produce very low hybridization yield even with the correct target X, and was therefore not tested.
Theory predicts that the hybridization yield χ will vary with the concentration of X or S, and this was experimentally verified (Supplementary Fig. S5). However, even in a large excess (200×) of spurious targets, we observed little hybridization to the probe (χS = 0.071, Fig. 3f), and spurious targets do not significantly interfere with the hybridization of correct targets (Supplementary Fig. S6).
Figure 3g presents PAGE results for a fluorescent probe, in which C is 3′-functionalized with the ROX fluorophore and P is 5′-functionalized with the Iowa Black Red Quencher (RQ). Upon hybridization to X or S, P is displaced, and the XC or SC complex fluoresces. This probe does not have a 30 nt poly-T tail on P, but its fluorophore–quencher interaction thermodynamics also make the reaction ΔG∘ difficult to predict34. Incomplete quenching of ROX by RQ caused significant background signal (as demonstrated by the visible band in the rightmost lane, ‘Control’), which decreased the quantitation sensitivity of the gel experiments. However, spectrofluorimeter experiments using these fluorophore/quencher-labelled constructs verified high discrimination factors at concentrations of 1 and 10 nM (Supplementary Fig. S7). Additionally, the kinetics of equilibration were observed to be fast (rate constant 2 × 106 M−1 s−1, 20 min to completion at 1 nM).
Target sequence generality
To show that toehold exchange probes yield high hybridization specificity for many if not most sequences, we tested four additional sets of targets and probes (Fig. 4a). The X2 and X3 targets were the DNA analogues of the let7g and mir155 microRNAs, which have important roles in cancer35. The X4 and X5 targets were designed with significant secondary structure in the branch migration region and the green toehold, respectively. All five systems tested produced hybridization yields in agreement with theory (Fig. 4b, Supplementary Figs S8–S11). Each system required a slightly different ΔG∘ adjustment (Fig. 4b, inset).
Figure 4. Results for additional DNA and RNA targets.

a, Sequences of four additional DNA targets (see Supplementary Tables S6–S8 for sequences of protectors P and spurious targets S). b, Hybridization yield versus adjusted ΔG∘. The same ΔG∘ adjustments (inset, in kcal mol−1) were applied to all reactions within a set. Systems in Fig. 3 are referred to as X1. (See Supplementary Figs S8–S11 for detailed results.) c, Distribution of observed Q for all 7/5 probes (55 data points in total). Owing to the limitations in quantifying gel band intensities, it was not possible to consistently measure Q values above 100 (see Methods). Median observed Q was 26. d, Comparison of observed Q and Q predicted based on ΔΔG∘ values (Supplementary Text S6). e, RNA target and probe. The target sequence is the RNA analogue of the X2 DNA system shown in a, and is identical to the human let7g microRNA. f, Native PAGE results. PC was prepared with a 2:1 ratio of P to C and annealed at [PC] = 3 μM. X or S was added to achieve final concentrations of 2 μM X or S, 1 μM PC and 1 μM P.
The discrimination factors Q achieved by the various 7/5 probes are plotted as a histogram in Fig. 4c. Except for two outliers that yielded Q ≈ 3 (X2–m17G and X5–m13G), Q ≥ 8 for all systems, with a median of Q = 26. Several spurious targets yielded no observable (χS < 0.002) hybridization to the probe; these experiments are displayed as Q = 100+ on the histogram. The experimental values of Q appear to be similar in distribution to the theoretically predicted Q (Fig. 4d). Thus, the presented toehold exchange mechanism reliably separates nucleic acid sequences differing by a single nucleotide across diverse target sequences.
RNA targets
We next tested an RNA toehold exchange probe against a synthetic RNA target with sequence corresponding to the let7g microRNA (Fig. 4e, f). In this experiment, the XC band stained less efficiently than the PC band; χ and Q were thus inferred from quantitation of the PC band alone. The concentrations used are higher than for DNA experiments due to the low staining efficiency of RNA.
Robustness
Owing to the Δn = 0 nature of the toehold exchange reaction (bimolecular with two products), the probes function robustly across oligonucleotide concentrations (Fig. 5; see Supplementary Fig. S12 for 7/4 and 7/6 probe results). We were not able to accurately achieve ΔH∘ = 0 and ΔN = 0 (no change in the number of paired bases) due to the unexpected offset of ΔG∘, but our 7/5 toehold exchange probes still managed to keep ΔH∘ and ΔN close to zero and were relatively robust to temperature and salinity (Fig. 5).
Figure 5. Performance of the 7/5 probe for the X1 target at different conditions.
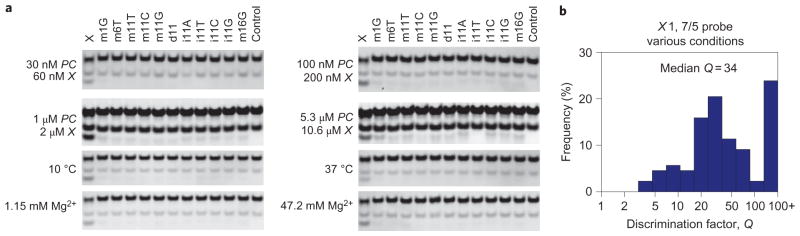
a, PAGE results. As in Fig. 3, the upper band is the unreacted PC complex, the middle band is excess P and the bottom band is the product XC or SC band. b, High discrimination factors were observed for all conditions tested (see Supplementary Figs S12–S14 for more details and results quantitation), with median Q = 34 across conditions. The histogram includes data from Fig. 3b, where the X1 7/5 probe was operated at standard conditions of 25 °C, 11.5 mM Mg2+ and 100 nM PC.
Experiments based on the X1 target and 7/5 probe showed large discrimination factors for temperatures from 10 °C to 37 °C, salinities from 1.15 mM Mg2+ to 47.2 mM Mg2+, and oligonucleotide concentrations from 30 nM to 5.3 μM (Fig. 5; Supplementary Figs S12–S14), with median Q = 34 for the X1 7/5 probe across conditions. Fluorescence measurements further verified that the toehold exchange probes effectively discriminate single-base changes at concentrations down to 1 nM (Supplementary Fig. S7).
Discussion
We have demonstrated that rationally designed toehold exchange probes achieve high hybridization specificity across a wide range of temperatures, salinities and oligonucleotide concentrations. The 7/5 probes achieve a median discrimination factor of Q = 26 against single-base-changed spurious targets, and perform robustly across all conditions tested. For comparison, analysis of our toehold exchange probes predicts a median Q of 87, and the theoretical limit of the median is Qmax = 343. The decreased performance may be attributed to (i) oligonucleotide synthesis impurities that lead to increased hybridization yield for spurious targets, (ii) inaccurate literature values of mismatch, bulge and dangle thermodynamics, and/or (iii) limitations in quantitating hybridization yields less than 0.2%. However, the discrimination factors experimentally achieved here are superior to previous hybridization probes (reported median Q ≤ 13; ref. 19).
Previously, double-stranded probes have been used for single-nucleotide polymorphism (SNP) detection in an in vitro setting36,37. However, these probes do not include the construction of the blue toehold that needs to dissociate spontaneously to release the protector, and hence spurious targets bind to the probes with strong thermodynamic favourability. Thus, these probes discriminate nucleic acids on a single-base level using initial kinetics, rather than thermodynamics. An equilibrium-based probe functions robustly regardless of secondary structure (frequent in natural nucleic acids), whereas kinetics of hybridization and strand displacement reactions depend sensitively on secondary structure32,38. To improve the specificity of the toehold-mediated strand displacement systems, previous works used competing sequences with perfect complementarity to the spurious targets to remove the spurious targets from solution36,37. In contrast, our method does not require a priori knowledge of the position or identity of the single base change.
Immediate applications of the toehold exchange probes arise in biotechnological methods that rely on the specificity of nucleic acid hybridization, such as microarray analysis3,4, fluorescence in situ hybridization6,7, SNP detection39 and so on. Enzyme-based amplification methods, such as rolling circle amplification40 or PCR, can also benefit from methods to improve specificity; for example, double-stranded toehold exchange primers may improve the PCR yield in ‘high background’ systems or in highly multiplexed systems where primer mis-hybridization may be a significant concern. Finally, branch migration and strand displacement-based synthetic nucleic acid nanodevices have been demonstrated to function within living cells41,42; toehold exchange probes may also potentially find use there.
Methods
DNA oligonucleotides
The DNA and RNA oligonucleotides used in this study were purchased from Integrated DNA Technologies (IDT). DNA oligonucleotides shorter than 50 nt in length were purified by IDT using high-performance liquid chromatography (HPLC), and DNA oligonucleotides 50 nt or longer were purified by IDT using PAGE. RNA oligonucleotides underwent RNAse-free HPLC by IDT.
Standard buffer conditions
Individual DNA oligonucleotides were resuspended and stored in tris-EDTA (TE) buffer (10 mM Tris·HCl pH balanced to 8.0, with 1 mM EDTA·Na2, purchased as 20× stock from Invitrogen) at 4 °C. Directly preceding experiments, TE buffer with 62.5 mM MgCl2 was added to the sample at a ratio of 1:4, achieving a final MgCl2 concentration of 12.5 mM. Because ~1 mM of the Mg2+ is chelated by the EDTA present in solution, the free concentration of Mg2+ is estimated to be 11.5 mM.
Probe preparation
The probes always consisted of two strands, the protector and the complement. In all experiments, the probe was prepared by adding a 2× excess of protector to the complement in 1 × TE/Mg2+ buffer, and the solution was then annealed from 92 °C to room temperature over 70 min. Other experiments not displayed have shown that annealing was not necessary for probe formation.
Polyacrylamide gel electrophoresis
Native PAGE was used to evaluate hybridization yield. All experiments shown used data from 10% native PAGE. The gel solutions were prepared from 40% 19:1 acrylamide:bisacrylamide stock (J.T. Baker Analytical) in 1 × tris-acetate-EDTA buffer (TAE)/Mg2+ solution, and cast in 1.5-mm-thick plastic gel cassettes (Invitrogen).
Native loading dye containing xylene cyanol-FF (Sigma Aldrich) in 50% glycerol was added to all samples, achieving a final glycerol concentration of 10% by volume. Most gels were run at 25 °C using Novex Mini-cell chambers (Invitrogen) at 100 V for 90 min. For the temperature studies presented in Supplementary Fig. S12, the 10 °C sample was run at 100 V for 120 min in a 10 °C water bath and the 37 °C sample was run at 100 V for 70 min in a 37 °C water bath.
After running, gels containing DNA were stained with SybrGold stain (Invitrogen) for 25–45 min. The gel containing RNA was stained with SybrGreen II stain (Invitrogen) for 30 min.
Gel quantitation
Gel band quantitation, used for inferring reaction completion, made use of a Typhoon FLA 9000 gel imager. ImageQuant TL software (GE Healthcare) was subsequently used to perform band detection, background subtraction and band quantitation. Owing to the limitations of the gel imager, bands showing hybridization yields below 0.2% could not be consistently quantified (due to the SybrGold background). The hybridization yield of the correct product (for 7/5 experiments) varied between 20% and 70%; consequently, the limit for the discrimination factor Q varied between 100 and 350. To prevent reader confusion regarding different Q limits for different systems, we opted to proceed with the lowest of these numbers (100), rounding down all observed Q greater than 100 to be ‘100+’. See Supplementary Fig. S4 for further details on gel band quantitation.
Standard free energy calculations
NUPACK31 was used to calculate the standard free energies of the DNA strands and complexes. NUPACK uses a number of different parameters in its calculations; our selection of these values is detailed and justified in the following.
The temperature was set to 25 °C (the temperature at which experiments were performed). Salt concentration was set to 0.05 M Na+ and 0.0115 M Mg2+. In fact, the experimental concentration of Na+ was 0.002 M, but 0.05 M Na+ was the lowest value that NUPACK allowed. However, as Mg2+ acts as the main counterion, it is likely that this difference does not significantly change the standard free energies. The ‘dangles’ parameter was set to ‘some’, although setting it to ‘all’ would yield identical results because there were no instances of coaxial stacks in any structures.
Time-based fluorescence studies
Kinetic fluorescence measurements were performed using a PTI QuantaMaster40 spectrofluorimeter and Hellma Semi-Micro 114F spectrofluorimeter cuvettes. The excitation wavelength was set to 584 nm and the emission wavelength to 602 nm, consistent with the properties of the ROX fluorophore. Slit sizes were set at 5 nm for all monochromators. An external temperature bath maintained a reaction temperature of 25±1 °C.
Supplementary Material
Acknowledgments
The authors thank M. Dai and P-S. Loh for assistance with mathematical analysis and J. Aliperti, E. Haney, R. Jungmann and T. Schaus for helpful suggestions during manuscript preparation. This work was funded by a Wyss Institute for Biologically Inspired Engineered faculty start-up fund, an NIH Director’s New Innovator Award (1DP2OD007292), an NSF CAREER Award (CCF1054898) and an Office of Naval Research grant (N000141010827) to P.Y. D.Y.Z. is a Howard Hughes Medical Institute postdoctoral fellow, as part of the Life Sciences Research Foundation programme. There is a patent pending on the methods described in this work.
Footnotes
Author contributions
D.Y.Z. conceived the project, designed and conducted the experiments, analysed the data and wrote the manuscript. S.X.C. conducted experiments, analysed the data and edited the manuscript. P.Y. conceived, designed and supervised the study, analysed the data and wrote the manuscript.
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at www.nature.com/naturechemistry.
Supplementary information accompanies this paper at www.nature.com/naturechemistry.
Reprints and permission information is available online at http://www.nature.com/reprints.
References
- 1.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saiki RK, et al. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science. 1988;239:487–491. doi: 10.1126/science.2448875. [DOI] [PubMed] [Google Scholar]
- 3.Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 4.Gunderson KL, Steemers FJ, Lee G, Mendoza LG, Chee MS. A genome-wide scalable SNP genotyping assay using microarray technology. Nature Genet. 2005;37:549–554. doi: 10.1038/ng1547. [DOI] [PubMed] [Google Scholar]
- 5.Koltai H, Weingarten-Baror C. Specificity of DNA microarray hybridization: characterization, effectors, and approaches for data correction. Nucleic Acids Res. 2008;36:2395–2405. doi: 10.1093/nar/gkn087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeLong EF, Wickham GS, Pace NR. Phylogenetic stains: ribosomal RNA-based probes for the identification of single cells. Science. 1989;243:1360–1363. doi: 10.1126/science.2466341. [DOI] [PubMed] [Google Scholar]
- 7.Amann RI, Krumholz L, Stahl DA. Fluorescent-oligonucleotide probing of whole cells for determinative, phylogenetic, and environmental studies in microbiology. J Bacteriol. 1990;172:762–770. doi: 10.1128/jb.172.2.762-770.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seeman NC. Nanomaterials based on DNA. Annu Rev Biochem. 2010;79:65–87. doi: 10.1146/annurev-biochem-060308-102244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rothemund P. Folding DNA to create nanoscale shapes and patterns. Nature. 2006;440:297–302. doi: 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- 10.Douglas SM, et al. Self-assembly of DNA into nanoscale three-dimensional shapes. Nature. 2009;459:414–418. doi: 10.1038/nature08016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aldaye FA, Palmer AL, Sleiman HF. Assembling materials with DNA as the guide. Science. 2008;321:1795–1799. doi: 10.1126/science.1154533. [DOI] [PubMed] [Google Scholar]
- 12.Yin P, Choi HMT, Calvert CR, Pierce NA. Programming biomolecular self-assembly pathways. Nature. 2008;451:318–322. doi: 10.1038/nature06451. [DOI] [PubMed] [Google Scholar]
- 13.Zhang DY, Seelig G. Dynamic DNA nanotechnology using strand displacement reactions. Nature Chem. 2011;3:103–113. doi: 10.1038/nchem.957. [DOI] [PubMed] [Google Scholar]
- 14.Tyagi S, Kramer FR. Molecular beacons: probes that fluoresce upon hybridization. Nature Biotechnol. 1996;14:303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- 15.Tyagi S, Bratu DP, Kramer FR. Multicolor molecular beacons for allele discrimination. Nature Biotechnol. 1998;16:49–53. doi: 10.1038/nbt0198-49. [DOI] [PubMed] [Google Scholar]
- 16.Tyagi S. Imaging intracellular RNA distribution and dynamics in living cells. Nature Methods. 2009;6:331–338. doi: 10.1038/nmeth.1321. [DOI] [PubMed] [Google Scholar]
- 17.Bonnet G, Tyagi S, Libchaber A, Kramer FR. Thermodynamic basis of the enhanced specificity of structured DNA probes. Proc Natl Acad Sci USA. 1999;96:6171–6176. doi: 10.1073/pnas.96.11.6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsourkas A, Behlke MA, Rose SD, Bao G. Hybridization kinetics and thermodynamics of molecular beacons. Nucleic Acids Res. 2003;31:1319–1330. doi: 10.1093/nar/gkg212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiao Y, et al. Fluorescence detection of single-nucleotide polymorphisms with a single, self-complementary, triple-stem DNA probe. Angew Chem Int Ed. 2009;48:4354–4358. doi: 10.1002/anie.200900369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kolpashchikov DM. A binary DNA probe for highly specific nucleic acid recognition. J Am Chem Soc. 2006;128:10625–10628. doi: 10.1021/ja0628093. [DOI] [PubMed] [Google Scholar]
- 21.Dave N, Liu J. Fast molecular beacon hybridization in organic solvents with improved target specificity. J Phys Chem B. 2010;114:15694–15699. doi: 10.1021/jp106754k. [DOI] [PubMed] [Google Scholar]
- 22.SantaLucia J, Hicks D. The thermodynamics of DNA structural motifs. Ann Rev Biophys Biomol Struct. 2004;33:415–440. doi: 10.1146/annurev.biophys.32.110601.141800. [DOI] [PubMed] [Google Scholar]
- 23.Peyret N. Doctoral thesis. Wayne State University; 2000. Prediction of Nucleic Acid Hybridization: Parameters and Algorithms. [Google Scholar]
- 24.Tan ZJ, Chen SJ. Nucleic acid helix stability: effects of salt concentration, cation valence and size, and chain length. Biophys J. 2006;90:1175–1190. doi: 10.1529/biophysj.105.070904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yurke B, Turberfield AJ, Mills AP, Simmel FC, Neumann JL. A DNA-fuelled molecular machine made of DNA. Nature. 2000;406:605–608. doi: 10.1038/35020524. [DOI] [PubMed] [Google Scholar]
- 26.Zhang DY, Winfree E. Control of DNA strand displacement kinetics using toehold exchange. J Am Chem Soc. 2009;131:17303–17314. doi: 10.1021/ja906987s. [DOI] [PubMed] [Google Scholar]
- 27.Zhang DY, Turberfield AJ, Yurke B, Winfree E. Engineering entropy-driven reactions and networks catalyzed by DNA. Science. 2007;318:1121–1125. doi: 10.1126/science.1148532. [DOI] [PubMed] [Google Scholar]
- 28.He G, Rapireddy S, Bahal R, Sahu B, Ly DH. Strand invasion of extended, mixed-sequence B-DNA by γPNAs. J Am Chem Soc. 2009;131:12088–12090. doi: 10.1021/ja900228j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Petersen M, Wengel J. LNA: a versatile tool for therapeutics and genomics. Trends Biotechnol. 2003;21:74–81. doi: 10.1016/S0167-7799(02)00038-0. [DOI] [PubMed] [Google Scholar]
- 30.Bommarito S, Peyret N, SantaLucia J. Thermodynamic parameters for DNA sequences with dangling ends. Nucleic Acids Res. 2000;28:1929–1934. doi: 10.1093/nar/28.9.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dirks RM, Bois JS, Schaeffer JM, Winfree E, Pierce NA. Thermodynamic analysis of interacting nucleic acid strands. SIAM Rev. 2007;49:65–88. [Google Scholar]
- 32.Zhang DY, Winfree E. Robustness and modularity properties of a non-covalent DNA catalytic reaction. Nucleic Acids Res. 2010;38:4182–4197. doi: 10.1093/nar/gkq088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Temsamani J, Kubert M, Agrawal S. Sequence identity of the n–1 product of a synthetic oligonucleotide. Nucleic Acids Res. 1995;23:1841–1844. doi: 10.1093/nar/23.11.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marras SA, Kramer FR, Tyagi S. Efficiencies of fluorescence resonance energy transfer and contact-mediated quenching in oligonucleotide probes. Nucleic Acids Res. 2002;30:e122. doi: 10.1093/nar/gnf121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu J, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 36.Li Q, Luan G, Guo Q, Liang J. A new class of homogeneous nucleic acid probe based on specific displacement hybridization. Nucleic Acids Res. 2002;30:e5. doi: 10.1093/nar/30.2.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Subramanian HKK, Chakraborty B, Sha R, Seeman NC. The label-free unambiguous detection and symbolic display of single nucleotide polymorphisms on DNA origami. Nano Lett. 2010;11:910–913. doi: 10.1021/nl104555t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao Y, Wolf LK, Georgiadis RM. Secondary structure effects on DNA hybridization kinetics: a solution versus surface comparison. Nucleic Acids Res. 2006;34:3370–3377. doi: 10.1093/nar/gkl422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim S, Misra A. SNP genotyping: technologies and biomedical applications. Annu Rev Biomed Eng. 2007;9:289–320. doi: 10.1146/annurev.bioeng.9.060906.152037. [DOI] [PubMed] [Google Scholar]
- 40.Lizardi PM, et al. Mutation detection and single-molecule counting using isothermal rolling-circle amplification. Nature Genet. 1998;19:225–232. doi: 10.1038/898. [DOI] [PubMed] [Google Scholar]
- 41.Isaacs FJ, et al. Engineered riboregulators enable post-transcriptional control of gene expression. Nature Biotechnol. 2004;22:841–847. doi: 10.1038/nbt986. [DOI] [PubMed] [Google Scholar]
- 42.Venkataraman S, Dirks RM, Ueda CT, Pierce N. Selective cell death mediated by small conditional RNAs. Proc Natl Acad Sci USA. 2010;107:16777–16782. doi: 10.1073/pnas.1006377107. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.