

Supplemental digital content is available in the text.
Key Words: Alzheimer disease, Cortical neurons, EFhd2, Frontotemporal dementia, Frontotemporal lobar degeneration, Swiprosin-1, Synapses
Abstract
EFhd2 is a calcium-binding adaptor protein that has been found to be associated with pathologically aggregated tau in the brain in Alzheimer disease and in a mouse model of frontotemporal dementia. EFhd2 has cell type–specific functions, including the modulation of intracellular calcium responses, actin dynamics, and microtubule transport. Here we report that EFhd2 protein and mRNA levels are reduced in human frontal cortex tissue affected by different types of dementia with and without tau pathology. We show that EFhd2 is mainly a neuronal protein in the brain and is abundant in the forebrain. Using short hairpin RNA–mediated knockdown of EFhd2 expression in cultured cortical neurons, we demonstrate that loss of EFhd2 affects the number of synapses developed in vitro whereas it does not alter neurite outgrowth per se. Our data suggest that EFhd2 is involved in the control of synapse development and maintenance through means other than affecting neurite development. The changes in expression levels observed in human dementias might, therefore, play a significant role in disease onset and progression of dementia, which is characterized by the loss of synapses.
Dementia is a neurodegenerative disease of the aging brain. It has become widely accepted that the clinical manifestations of dementia are strongly correlated with the extent of synapse loss in affected brain regions (1–3); this pathologic hallmark is also observed in other neurodegenerative diseases (4). Alzheimer disease (AD) and several other neurodegenerative conditions, including forms of frontotemporal lobar degeneration (FTLD) such as frontotemporal dementia (FTD) with parkinsonism linked to chromosome 17 (FTDP-17) and FTLD-tau with Pick bodies (Pick disease), are classified as tauopathies. In addition to the loss of synapses, they are characterized by the presence of various types of intraneuronal aggregates of the microtubule-binding protein tau in brain regions that are involved in clinical manifestations, including dementia, aphasia, and behavioral changes (5). Fittingly, recent findings indicate that pathologic forms of tau contribute directly to synaptic degeneration even before their aggregation; mislocalization and hyperphosphorylation of tau have both been implicated in synaptic pathology in models of AD, in conjunction with amyloid-β peptides (6, 7), as well as in models of FTDs (8, 9). Other forms of FTLD such as FTLD-TDP Type B do not involve tau aggregation but are mostly characterized by intraneuronal inclusions containing the transactive response-DNA binding protein-43 (TDP-43); these disorders are clinically often associated more with semantic dementia, behavioral-variant FTD, and motor neuron disease (5, 10). Despite the existing detailed clinical descriptions of these different forms of dementia and established links to underlying genetic mutations, there is still little known about the molecular basis for their overlapping clinical manifestations and their propensity to cause neuronal and synaptic pathology in overlapping regions of the brain in the frontal and temporal lobes.
Studies in a mouse model of FTD and in the brains of humans with dementia have suggested an association of the novel calcium-binding adapter protein Swiprosin-1/EF-hand domain containing protein 2 (EFhd2) with pathologically aggregated tau (11, 12) and have revealed changes in phosphorylation of EFhd2 at Ser74 in AD frontal cortex (13). In addition, changes in EFhd2 protein levels were noted in the prefrontal cortex of patients with schizophrenia (14), the victims of suicide (15), and spinal cord lipid raft preparations from a mouse model for amyotrophic lateral sclerosis/motor neuron disease (16). Interestingly, its link to pathologic tau and phosphorylation changes in frontal cortex tissues in dementia (12, 13) and the protein expression changes in the frontal cortex reported by Martins-de-Souza et al (14) and Kekesi et al (15) hint at a possible functional role for EFhd2 cognitive processes within the frontal cortex in general.
Mechanisms for the regulation of EFhd2 expression and its cellular functions in the CNS have not yet been delineated. In immune cells, EFhd2 protein and mRNA expression were found to be under the control of signaling cascades involving Ca2+, protein kinase C, and nuclear factor-κB (17, 18). As a calcium sensor, EFhd2 has been identified as a modulator of B-cell receptor–induced Ca2+signaling (19, 20). Other studies have pointed out an involvement of EFhd2 in actin bundling and polymerization (21–23). These data support the idea that EFhd2 is involved in the spatiotemporal control of signaling events within the cell by integrating Ca2+-mediated signals and modulating the cytoskeleton. In agreement with this, it has recently been established that EFhd2 can be found in neurites as well as synapses and affect microtubule transport in neurons (24).
Here, we report that EFhd2 expression levels are altered in the human frontal cortex affected by different types of dementia with and without tau pathology. Furthermore, we present evidence that EFhd2 is directly or indirectly involved in the formation of synapses in cortical neurons in vitro.
MATERIALS AND METHODS
Reagents and Antibodies
All chemicals were purchased from Sigma (Gillingham, Dorset, UK), and all cell culture reagents were purchased from Invitrogen (Paisley, UK), unless stated otherwise.
Ethics Statement (Animals and Human Tissue)
Human brain tissue and paraffin sections were obtained from the Manchester Brain Bank (approved by Newcastle and North Tyneside 1 Research Ethics Committee [09/H0906/52]) and the Queen Square Brain Bank for Neurological Disorders (approved by the London Research Ethics Committee [08/H0718/54]), and human tissue–related work was approved by the University of St. Andrews Teaching and Research Ethics Committee. Detailed data on the cases used in this study are shown in the Table.
TABLE.
Patient Data for Brain Tissues Used in This Study
Wild-type CD1 mice (Charles River Laboratories) were housed in accordance with Schedule 1 of the UK Animals (Scientific Procedures) Act 1986, and procedures were reviewed and approved by the University of St. Andrews Animal Welfare and Ethics Committee.
Plasmids and Cloning
A short hairpin RNA (shRNA) sequence targeting EFhd2 (CGTTTGCCTCAGCGGATA) or a nonrelevant shRNA sequence targeting green fluorescent protein (GFP) were cloned as inverted repeats into the lentiviral vector pLKO.1 after Age1/EcoR1 digest and calf intestinal phosphatase treatment. Lentiviral envelope vector pSD11 (VSV-G) and packaging vector pSD16 (Gag-Pro-Pol, RRE-TAT-Rev) were a kind gift from Dr Paul Reynolds (University of St. Andrews, St. Andrews, UK). Lentiviral vectors for the expression of enhanced GFP (EGFP)-EFhd2 or EGFP alone were a kind gift from Dr Chan-Duk Jun (Gwangju Institute of Science and Technology, Gwangju, Republic of Korea).
Cell Culture and Lentivirus Production
Lentigrade 293T cells were cultured in Dulbecco modified Eagle medium supplemented with 10% fetal calf serum, 2 mmol/L glutamine, 1 mmol/L sodium pyruvate, 100 units/mL penicillin, and 100 μg/mL streptomycin. A total of 4 × 106 cells were plated in 10-cm culture dishes and cotransfected with 2.1 μg pSD11, 6.2 μg pSD16, and 5.7 μg lentiviral transfer vectors. At 24 hours posttransfection, cells were rinsed once with PBS, and 4.5 mL neuronal culture medium was added to produce lentiviral supernatants. The supernatants were removed after 24 hours, and 4.5 mL neuronal culture medium was added again for 24 hours. Lentiviral supernatants were cleared by filtration through 0.45-μm syringe filters, snap frozen in liquid N2, and stored at −80°C. Lentiviral titering was performed on 293T cells according to standard protocols. Primary cortical neurons were infected with approximately 105 TU/mL of lentiviruses diluted in fresh neuronal culture medium (theoretical multiplicity of infection of 1–3) for 4 to 6 hours at 37°C at least 48 hours before analysis.
Primary Neuron Culture
Primary neuronal cultures were prepared from Embryonic Day 15 mouse embryos. Brains were dissected and placed into ice-cold dissection medium (Hanks balanced salt solution [HBSS] w/o Ca2+/Mg2+, 7 mmol/L HEPES, 10 mmol/L MgCl2, 2 mmol/L GlutaMax, 50 units/mL penicillin, 50 μg/mL streptomycin). Meninges were removed and cortices were separated from the cerebellum and diencephalon and transferred to ice-cold dissection medium in a 15-mL centrifuge tube. Dissection medium was replaced 0.05% trypsin/0.02% EDTA in HBSS w/o Ca2+/Mg2+and incubated for 25 minutes at 37°C. One volume of 0.001% DNAse1 in HBSS was added, the contents were mixed by inversion, and the tissue was left to settle. Cortices were triturated in 1 mL triturating solution (0.05% soybean trypsin inhibitor, 0.001% DNAse1, 1% Albumax in HBSS) and diluted in neuronal culture medium (neurobasal medium, 1% B-27, 50 units/mL penicillin, 50 μg/mL streptomycin). Neurons were plated on glass coverslips or Nunc culture dishes coated with 3 μg/mL poly-d-lysine (30–70 kDa) at a density of 2.5 × 104 cells/cm2 for imaging. Fifty percent of the culture medium was changed first at 5 days in vitro (DIV) and then every 3 days. The synaptic compartment was analyzed at 13 to 16 DIV. Lentiviral infections on neuronal cultures were done at 1 DIV for the analysis of neurite outgrowth and at 7 DIV for the analysis of synaptic effects.
Protein Extraction and Western Blotting
Two hundred milligrams of tissue was homogenized in a reaction tube on ice using micropistilles and 1-mL pipette tips in 5 volumes of RIPA lysis buffer (25 mmol/L Tris-HCl pH 7.4, 150 mmol/L NaCl, 5 mmol/L EDTA, 0.1% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS], 1% NP-40, 1 mmol/L NaF, 1× complete protease inhibitor [Roche, Burgess Hill, West Sussex, UK], 1 mmol/L phenylmethylsulfonyl fluoride, 1 mmol/L Na3VO4). Protein from cell cultures was extracted in RIPA lysis buffer and homogenized by 10 strokes with a 200-μL pipette tip. Samples were centrifuged at 15,000 × g for 15 minutes at 4°C. The supernatant was removed, and the protein concentration was measured by Bradford assay. Protein extracts were snap frozen in liquid nitrogen and stored at −80°C. For SDS–polyacrylamide gel electrophoresis (PAGE), protein samples were thawed on ice and mixed with 4× protein sample buffer (106 mmol/L Tris-HCl, 141 mmol/L tris-base, 4% SDS, 40% glycerol, 0.51 mmol/L EDTA, 4% dithiothreitol, 0.05 mg/mL bromophenol blue, pH 8.5) and Milli-Q water and denatured at 95°C for 5 minutes.
Twenty-five micrograms of protein was separated on 10% or 12.5% SDS-PAGE for 10 minutes at 120 V followed by 1 hour at 160 V and transferred to 0.2 μm supported nitrocellulose or polyvinylidene fluoride membranes (GE Healthcare, Little Chalfont, Buckinghamshire, UK) for 60 to 90 minutes at 25 V. Membranes were rinsed in tris-buffered saline (TBS) and blocked in TBS/0.1% Tween 20 (TBS-T) containing 1:5 diluted LI-COR blocking buffer (LI-COR Biosciences, Cambridge, UK) or 5% bovine serum albumin. Primary antibodies diluted in 1:5 diluted LI-COR blocking buffer were goat anti-EFhd2 (ab24368, 1:1000; Abcam, Cambridge, UK), mouse anti–β-actin (A1978, 1:10,000; Sigma), mouse anti–glyceraldehyde 3-phosphate dehydrogenase (GAPDH, G8795, 1:20,000; Sigma), goat anti-synapsin (C-20, sc-8295, 1:1000; Insight Biotechnology, Middlesex, UK), and mouse anti–β3-tubulin (G7121, 1:2000; Promega, Southampton, UK). Antibodies diluted in 5% bovine serum albumin/TBS-T or TBS-T were rabbit anti–PSD-95 (EP2652Y, ab76115, 1:3000; Abcam), rabbit anti–microtubule-associated protein 2 ([MAP2] ab32454, 1:500; Abcam), and anti–Tau DA9, RZ3, PHF-1, and CP-13 (1:1–1:50; kind gifts from Prof. Peter Davies, Albert-Einstein College of Medicine, Bronx, NY). All were incubated overnight at 4°C. Membranes were washed 3 × 5 minutes in TBS-T and incubated with infrared dye–coupled antibodies in TBS-T (IRDye 800CW or IRDye 680RD goat anti-mouse/anti-rabbit [LI-COR Biosciences] or donkey anti-goat Alexa Fluor 790 [Jackson Immunoresearch, Suffolk, UK]) for 1 hour at room temperature. Bands were detected using the LI-COR Odyssey detection system. Densitometry of stained bands was done in NIH ImageJ software, and values were normalized to the loading control.
Synaptoneurosome Preparation
Crude synaptoneurosomes were prepared according to Tai et al (25). Two hundred milligrams of fresh-frozen human brain tissue was homogenized in a glass dounce homogenizer with 1 mL ice-cold buffer A (25 mmol/L HEPES pH 7.5, 120 mmol/L NaCl, 5 mmol/L KCl, 1 mmol/L MgCl2, and 2 mmol/L CaCl2), supplemented with 2 mmol/L dithiothreitol, protease inhibitors, and phosphatase inhibitors. The homogenate was passed through 2 layers of 80-μm nylon filters (Millipore, Watford, UK), and a 200-μL aliquot of the filtered homogenate was saved. The saved aliquot was mixed with 200 μL water and 70 μL 10% SDS, passed through a 27-gauge needle, and boiled for 5 minutes to prepare the total extract. To prepare filtered synaptoneurosomes, the remainder of the homogenate was passed through a 5-μm Durapor membrane filter (Millipore) to remove large organelles and nuclei and centrifuged at 1,000 × g for 5 minutes. The nonsynaptic supernatant containing cytoplasmic proteins was removed, and the pellet was washed once with buffer A and centrifuged again, yielding the synaptoneurosome pellet.
Quantitative Real-time Polymerase Chain Reaction
Total RNA was extracted from 50- to 100-mg tissue samples using 1 mL Tri-reagent according to the manufacturer’s protocol. Three micrograms of RNA was digested with 3 units DNAse 1 (Promega) in a 10-μL reaction at 37°C for 45 minutes. The reaction was terminated by addition of 1 μL RQI DNAse Stop solution (Promega) and incubation for 10 minutes at 65°C. Complementary DNA was produced from the DNAse-digested RNA using random hexamer primers and Revert Aid Reverse Transcriptase (Fermentas, Fisher Scientific, Loughborough, UK), according to the manufacturer’s instructions. Ten microliters of qPCR reactions using 1 μL 1:20 diluted cDNA and 200 nmol/L primers (EFhd2 forward: TCGACCTGATGGAGCTAAAACTCA, EFhd2 reverse: ACACGTTGATGGCCTGTACCTT; actin forward: CTCGCCTTTGCCGATCC, actin reverse: AACATGATCTGGGTCATCTTCTC; GAPDH forward: CAGTCAGCCGCATCTTCTTT, GAPDH reverse: CCCAATACGACCAAATCCGT) and Brilliant III SYBR green reaction mix (Agilent, Santa Clara, CA) were prepared using a QIAgility robot and run on a Rotor-Gene cycler using a rotor disc 100 (Qiagen, Manchester, UK). Data of 3 runs with samples run in duplicate were analyzed with the Rotor-Gene software based on the ΔΔ-Ct method, with both actin and GAPDH used as reference genes.
Immunocytochemistry
Neurons grown on coverslips were fixed with prewarmed 4% paraformaldehyde solution for 15 minutes at room temperature. Cells were permeabilized with PBS/0.05% Triton X-100 for 5 minutes, and nonspecific antibody binding sites were blocked by incubation in PBS/10% horse serum for 30 minutes. Primary antibodies (rabbit anti-tubulin, T3526, 1:500; Sigma), mouse anti–β3-tubulin (1:2000), goat anti–synapsin 1a/b (1:500), rabbit anti-PSD95 (1:500), and rabbit anti-EFhd2 (ab87006, 1:100; Abcam) were diluted in PBS and incubated overnight at 4°C, followed by 3 × 5-minute washes with PBS/0.02% Tween 20. Fluorochrome-conjugated secondary antibodies (donkey anti-mouse Alexa Fluor 594/488, bovine anti-goat Alexa Fluor 488 [Jackson Immunoresearch, Suffolk, UK], donkey anti-rabbit Alexa Fluor 594 [Invitrogen, Paisley, UK], Alexa Fluor 594 Phalloidin [A1238, 1:1000; Invitrogen, Paisley, UK]) were diluted in PBS and incubated for 1 hour at room temperature in the dark followed by 3 × 5-minute washes with PBS/0.02% Tween 20. One microgram per milliliter 4′,6-diamidino-2-phenylindole was added to the secondary antibody solution as a nuclear counterstain. Coverslips were mounted on microscope slides in Mowiol mounting medium (10% Mowiol-488, 2.5% glycerol, 1.5% propylgallate, 100 mmol/L Tris, pH 8.5).
Immunohistochemistry
For 3,3′-diaminobenzidine (DAB) staining, 7-μm paraffin sections were rehydrated as follows: 3 × 5 minutes 100% xylene, 2 × 2 minutes 100% ethanol, 2 minutes 80% ethanol, 2 minutes 50% ethanol, 2-minute rinse in tap water. Heat-induced antigen retrieval was done in boiling 100 mmol/L citrate buffer, pH 6.0 for 10 minutes in a microwave pressure cooker, followed by 2 washes of the cooled sections in PBS/0.1% Tween 20 for 5 minutes. Endogenous peroxidases were blocked with 3% H2O2 for 10 minutes. Nonspecific antibody binding sites were blocked with 2.5% horse serum supplied with ImmPRESS Anti-Rabbit Ig (peroxidase) Polymer Detection Kit (Vector Laboratories, Peterborough, UK) for 30 minutes, and rabbit anti-EFhd2 was diluted in 2.5% horse serum in PBS/0.4% Tx-100 and incubated overnight at 4°C. Sections were washed 2 × 5 minutes in TBS/0.02% Tween 20 and incubated with ImmPRESS anti-Rabbit Ig (peroxidase) (Vector Laboratories) for 1 hour. Staining was visualized using DAB Peroxidase Substrate Kit (Vector Laboratories) according to the manufacturer’s instructions. Immunofluorescence staining was done on 13-μm sections from fresh-frozen adult mouse brain tissue fixed in 100% acetone at −20°C for 5 minutes. Nonspecific antibody binding sites were blocked with 2.5% horse serum in PBS/0.1% Tx-100 for 30 minutes and rabbit or goat anti-EFhd2 (Abcam) diluted in 2.5% horse serum in PBS/0.1% Tx-100 and incubated overnight at 4°C. Sections were washed 2 × 5 minutes in PBS/0.02% Tween 20 and incubated with monoclonal rabbit anti-NeuN (ab177487; Abcam) or mouse anti-NeuN (MAB377; Millipore) for 2 hours at room temperature. Sections were washed 2 × 5 minutes in PBS/0.02% Tween 20 and incubated with appropriate combinations of donkey anti-rabbit Alexa Fluor 594 (Invitrogen, Paisley, UK), bovine anti-goat Alexa Fluor 488, and donkey anti-mouse Alexa Fluor 488 (Jackson Immunoresearch, Suffolk, UK) for 1 hour at room temperature. Coverslips were mounted using Mowiol mounting medium (without propylgallate for DAB-stained sections).
Fluorescence Microscopy and Image Analysis
Stained cells were analyzed at a Deltavision imaging system using an Olympus 60X/1.42, Plan Apo N oil immersion objective. Image stacks with a 0.2-μm-step size were acquired and deconvolved using SoftWorX Acquisition and Analysis software. Images were imported into ImageJ using the Bioformats plugin, and colocalization analysis was performed using the Colocalization analysis plugin on image stacks. Total numbers of synapsin puncta were determined as counts of puncta of 0.05- to 1.5-μm2 size and normalized to the length of the tubulin skeleton. Mature synapses were quantified as puncta with colocalization of synapsin 1a/b and PSD-95 and expressed as a ratio of colocalizing puncta compared with the total number of synapsin puncta. Total neurite development was quantified as the area covered by skeletonized β3-tubulin–stained neurites in 3 DIV cultures, normalized to the number of cells per field of view.
Data Analysis
Statistical analysis was done in Prism statistical software (GraphPad, La Jolla, CA). Normality of data sets was confirmed using D’Agostino-Pearson normality test, and one-way analysis of variance (ANOVA) with Dunnett post hoc test or 2-tailed t test was applied depending on the comparisons made and indicated in figure legends.
RESULTS
EFhd2 Protein and mRNA Levels Are Reduced in the Frontal Cortex in Dementia
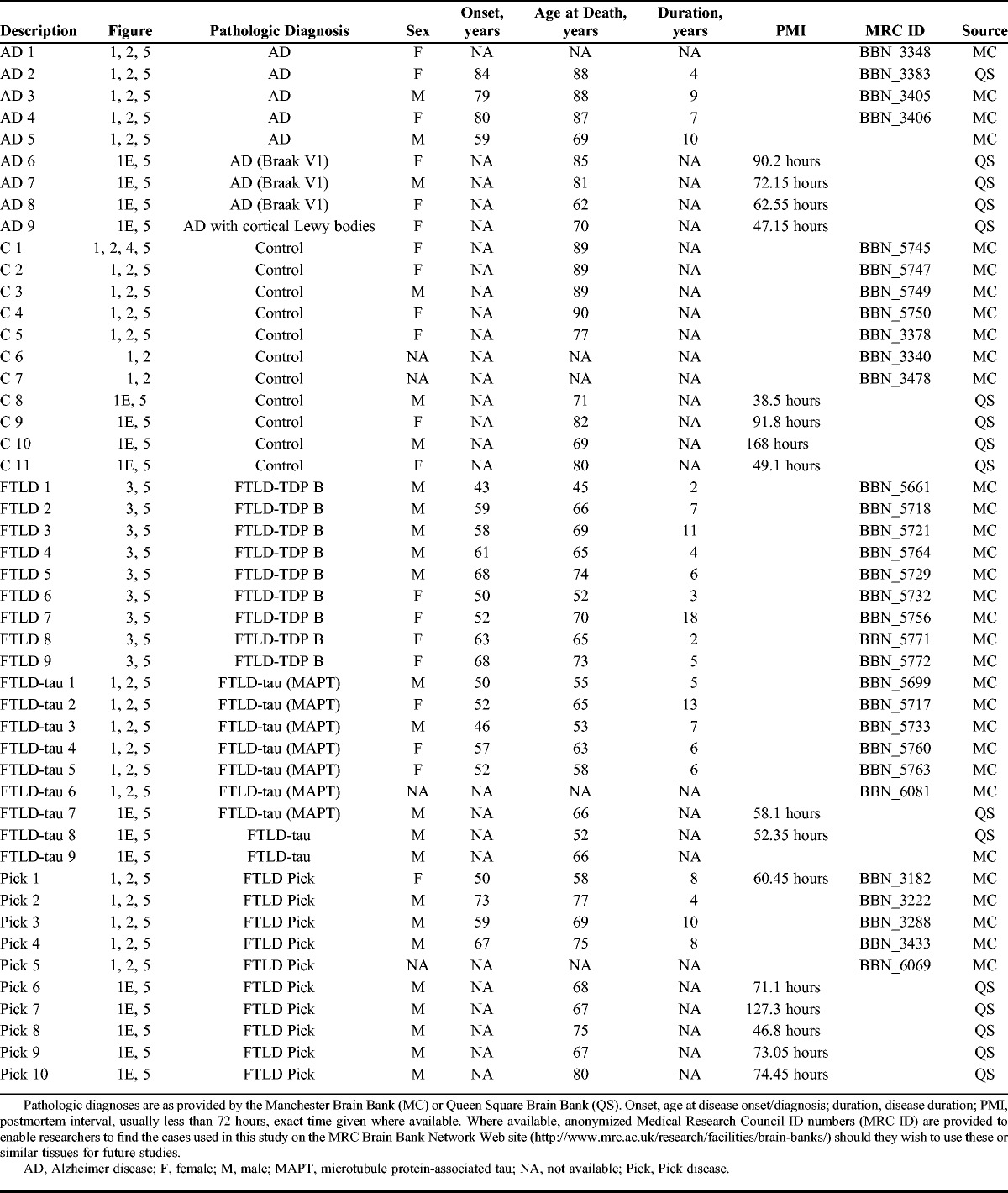
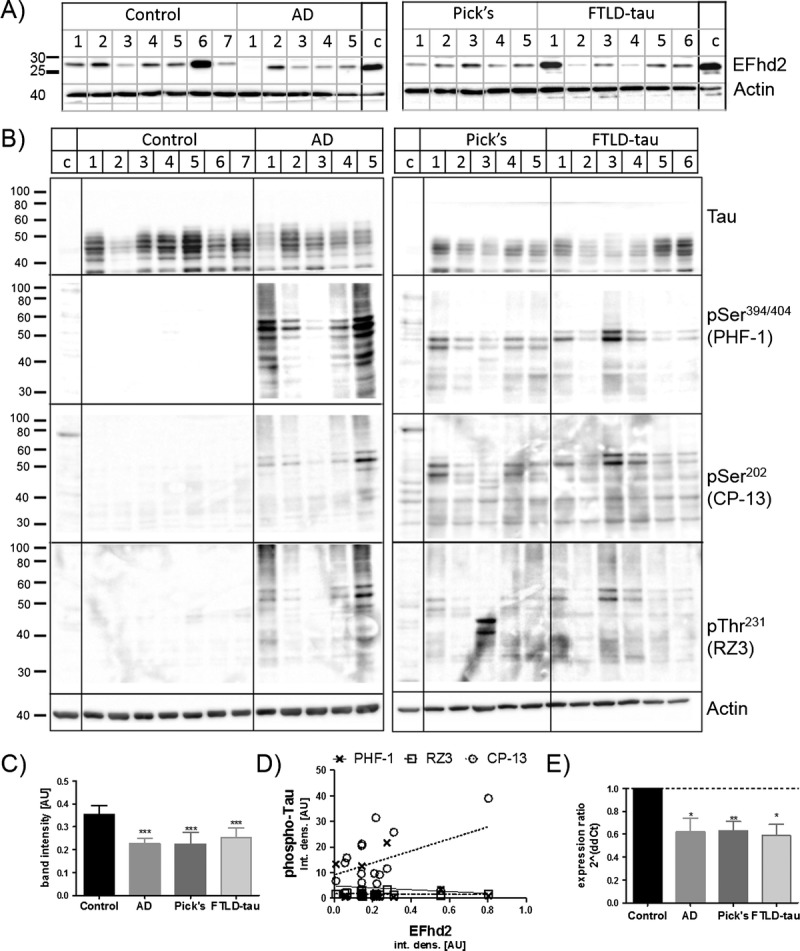
EFhd2 had previously been linked to tau pathology in dementia in a mouse model for FTLD as well as in AD (11, 12). Because AD and FTLD are dementias with different etiologies, these previous findings warranted further investigation into the role played by EFhd2 in these diseases. Therefore, we compared the protein expression of EFhd2 in human brain tissues from individuals with different types of dementia with tau pathology, namely, AD and FTLD-tau and Pick disease, with that in nondemented controls. Protein levels of EFhd2 were analyzed by Western blotting in both frontal cortex as well as hippocampus because both brain regions are affected by their respective neurodegenerative diseases. The analysis revealed a reduction in EFhd2 protein levels in the frontal cortex from AD and both types of FTLD-tau compared with controls (Fig. 1A, C); these changes were confirmed by quantitative real-time PCR (Fig. 1E), suggesting a specific reduction of EFhd2 expression in these tissues. Notably, however, no changes in EFhd2 protein levels were observed in hippocampi (Fig. 2).
FIGURE 1.
EFhd2 protein levels in the frontal cortex are reduced in tauopathies. (A) Western blot images of EFhD2 in human frontal cortex tissue RIPA buffer extracts from nondemented controls, individuals with Alzheimer disease (AD), Pick disease (Pick’s), and frontotemporal dementia with tau mutations (FTLD-tau). Western blots run with a 293T-cell line lysate loaded as a calibrator on each gel. Beta-actin was used as a loading control. (B) Western blot analysis of total and phosphorylated Tau protein in human frontal cortex samples. (C) Quantification of EFhd2 protein bands by densitometry, n = 3 for each sample. Error bars = SEM. *** p < 0.001 compared with control with 1-way ANOVA and Dunnett post hoc test. (D) Scatter plots of densitometry values for EFhd2 against values for each of the phosphorylated Tau antigens (PHF-1, CP-13, RZ3). Linear regression was done in GraphPad prism and showed no correlation between EFhd2 and any of the phospho-tau antigens. (E) Quantitative real-time PCR for EFhd2, quantified by the ΔΔ-Ct method. Actin and glyceraldehyde 3-phosphate dehydrogenase were used as housekeeping genes. * p < 0.05, ** p < 0.01 compared with control with 1-way ANOVA and Dunnett post hoc test.
FIGURE 2.
EFhd2 protein levels are not altered in the hippocampi of human tauopathy patients. (A) Western blots showing EFhd2 protein levels in human hippocampal tissue RIPA buffer extracts from nondemented controls and individuals with Alzheimer disease (AD), Pick disease (Pick’s), and frontotemporal dementia with tau mutations (FTLD-tau). Western blot were repeated 3 times with a 293T-cell line lysate loaded as a calibrator on each gel. Beta-actin was used as a loading control. (B) Western blot analysis of total and phosphorylated tau protein in human frontal cortex samples. (C) Quantification of EFhd2 protein bands by densitometry, n = 3 for each sample. Error bars = SEM. (D) Scatter plots of densitometry values for EFhd2 against values for each of the phosphorylated Tau antigens (PHF-1, CP-13, RZ3). Linear regression was done in GraphPad prism and showed no correlation between EFhd2 and any of the phospho-tau antigens.
Because of its link to hyperphosphorylated tau (12), we next tested if changes in EFhd2 expression levels correlated with the amount of tau pathology in these brain tissues. The abundance of hyperphosphorylated tau was detected by antibody clones CP-13 (phosphorylated Ser-202), RZ3 (phosphorylated Thr-231), and PHF-1 (phosphorylated Ser-394/404) in the previously studied samples. As expected, extensive tau hyperphosphorylation could be detected at all phosphorylation sites in the frontal cortex as well as hippocampal tissues from samples from AD and both forms of FTLD-tau (Figs. 1B, 2B). However, the amount of tau phosphorylation detected by Western blotting did not correlate with the abundance of EFhd2 protein in individual samples (Figs. 1D, 2D).
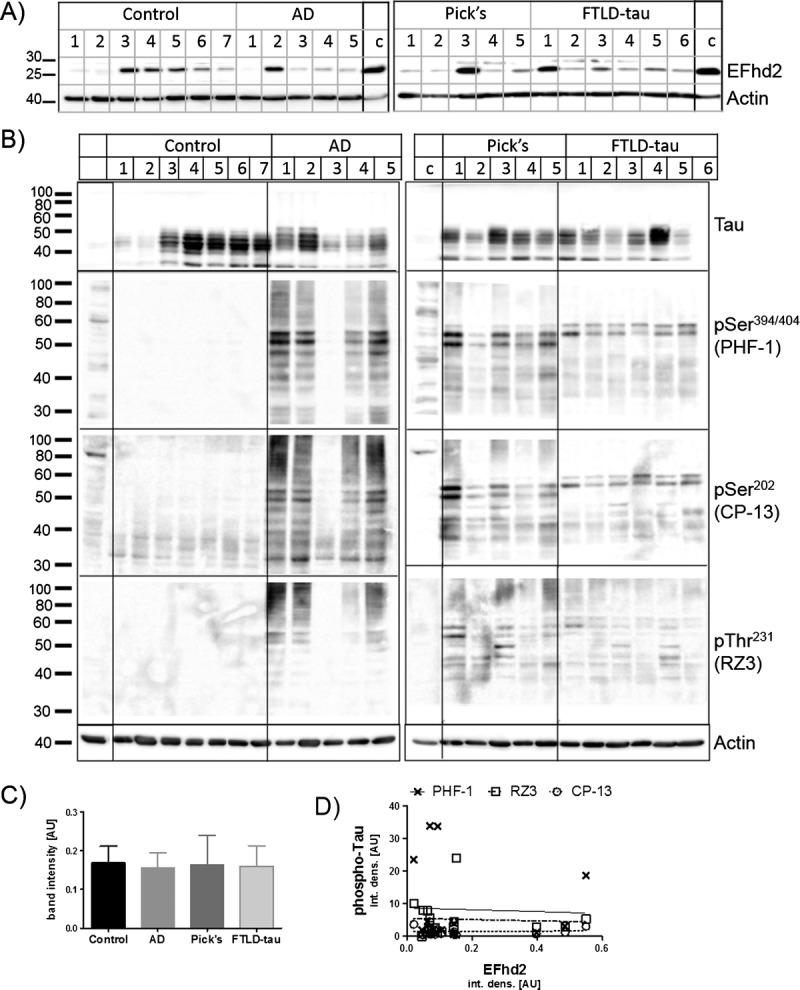
We therefore asked if changes in EFhd2 protein expression are limited to dementias with tau pathology. To answer this question, EFhd2 protein abundance was compared between nondemented controls and cases with tau-negative FTLD (FTLD-TDP Type B). As in the previously studied tau-positive cases, significantly lower levels of EFhd2 protein were detected in the frontal cortex tissues in FTLD-TDB Type B cases when compared with nondemented controls (Fig. 3A, B). Again, no significant changes in EFhd2 levels could be detected in hippocampal tissues from the same cases (Fig. 3A, C). The absence of tau pathology was confirmed by Western blotting for tau phosphorylated at Ser-394/404 (PHF-1 epitope), which had clearly been detected in AD and FTLD-tau (Fig. 3A, bottom).
FIGURE 3.
EFhd2 protein levels in the frontal cortex are reduced in patients with dementia without tauopathy. (A) Western blots showing EFhd2 protein levels in human frontal cortex (left) and hippocampal (right) tissue RIPA extracts from nondemented controls and individuals with frontotemporal lobar degeneration FTLD-TDP Type B. A sample from AD frontal cortex was run as a calibrator and phospho-tau–positive control on each gel. No calibrator sample was run for EFhd2 and actin detection in hippocampal samples because all samples were run on the same SDS-PAGE. Beta-actin was used as a loading control. (B) Quantification of EFhd2 protein bands by densitometry in frontal cortex samples. * p < 0.05 with all data points (left), *** p < 0.001 without encircled outlier (right), compared with control by t test. (C) Quantification of EFhd2 protein bands in samples from human hippocampus from nondemented controls and individuals with FTLD-TDP Type B.
Together, these results demonstrate that EFhd2 expression levels are altered in the frontal cortex in patients with different types of dementia involving this brain region. Thus, this finding fits with the idea of a frontal cortex–related role of EFhd2 in cognitive processes (12–15). Whereas tau pathology is a common feature of some of the dementia cases studied here, dysfunction and aggregation of tau itself caused by its hyperphosphorylation per se were not linked to these changes.
EFhd2 Is Expressed in Neurons in Brain Tissues
EFhd2 protein expression in neurons of the brain has previously been described (11, 24, 26), but its expression and function have also been linked to cells of the immune system (26, 27), muscle (28), and epithelial cells (22, 23). Therefore, we wanted to verify that EFhd2 expression was exclusively neuronal. As expected, immunohistochemistry revealed EFhd2-labeled cells with neuronal morphology in human brain samples (Fig. 4-I); its neuronal location could be confirmed by double immunofluorescence for EFhd2 and the neuronal marker NeuN in adult mouse brain tissue (Fig. 4-II). In agreement with recent mRNA expression data (24), abundant EFhd2 labeling was present in the forebrain (hippocampus, cortex, and subcortical structures; Fig. 4-IIA–C) and 2 different antibodies against EFhd2 exclusively labeled cells that were colabeled with NeuN in the frontal cortex (Fig. 4-IID–F, J, K). At the very least, therefore, the vast majority of EFhd2 in the adult mouse brain, and probably the human brain, is present in neurons.
FIGURE 4.
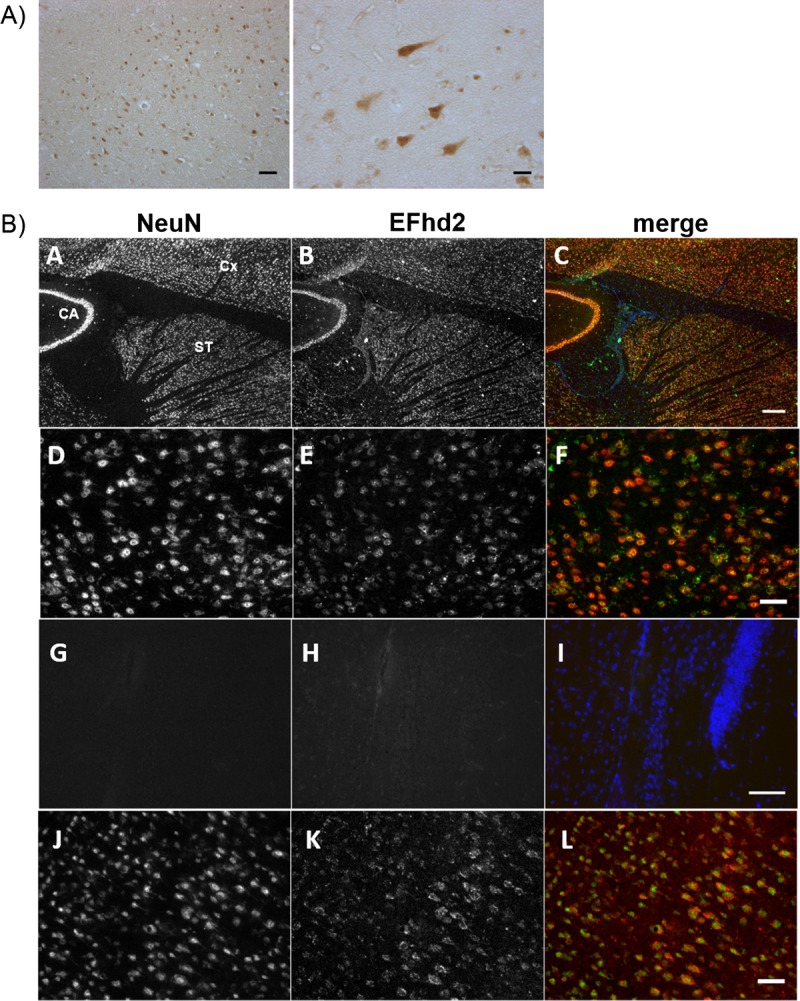
(I) Immunohistochemistry (IHC) with a rabbit polyclonal antibody (RbpAb) on human brain tissues for EFhd2. Scale bars = 100 μm (left); 50 μm (right). (II) IHC on mouse brain for NeuN (red: A, D, G, J), EFhd2 with a goat polyclonal antibody (green: B, E, H), and merge (C, F, I). (A–C) EFhd2 is present in neuronal layers of the hippocampus (cornu ammonis [CA]), cortex (Cx), and subcortical structures (striatum [ST]). Scale bar = 250 μm. (D–F) Expression in the frontal cortex is restricted to anti-NeuN–labeled neurons. Scale bar = 50 μm. (G–I) No primary antibody, merge with 4′,6-diamidino-2-phenylindole. Scale bar = 100 μm. (J–L) Similar results were obtained using an RbpAb against an N-terminal epitope of EFhd2 stained in the frontal cortex (red). Scale bar = 50 μm.
Compartment-Specific Changes in EFhd2 Protein Abundance
Having established its neuronal localization, we tested whether changes in EFhd2 protein abundance are associated with specific compartments in human brain tissue. To this end, filtered synaptoneurosomes were prepared from human brain frontal cortex tissues from dementia and nondemented control patients. Success of the fractionation procedure was verified by Western blotting for PSD-95 as a marker for the postsynaptic density and NeuN as a marker for neuronal cell nuclei/cell bodies. We confirmed that there was an absence, or at least a significant reduction, of NeuN in the synaptoneurosome and nonsynaptic supernatant fractions and the accumulation of PSD-95 in the synaptoneurosome fraction compared with the nonsynaptic supernatant (Figure, Supplemental Digital Content 1, http://links.lww.com/NEN/A673). Samples with a significant amount of NeuN in the synaptoneurosome fraction (Figure, Supplemental Digital Content 1, http://links.lww.com/NEN/A673) were excluded from the analysis because this indicated insufficient fractionation of the tissue. We then compared EFhd2 protein levels by Western blot analysis in each of the fractions (total tissue homogenates, pelleted synaptoneurosomes, and nonsynaptic cytoplasmic supernatant). Note that we did not assess an enrichment of EFhd2 in the synaptoneurosome fraction because we expected EFhd2 to only partially associate with this compartment based on earlier results obtained by Purohit et al (24). Comparison of EFhd2 protein levels between groups within each of the fractions again revealed significantly less EFhd2 in tissue homogenates from FLTD-tau, Pick disease, and FTD-TDP Type B cases as compared with controls (nonsignificant reduction in AD samples; Fig. 5A, B, left), a finding which is in line with our earlier results (Fig. 1). Whereas there was a tendency toward lower levels of EFhd2 in dementia in synaptoneurosomes (Fig. 5B, middle) as well as nonsynaptic cytoplasmic supernatants in cases of Pick disease and AD (Fig. 5A, bottom; 5B, right), changes in these fractions were not significant. There were no changes in the levels PSD-95 in tissue homogenates between groups (Figure, Supplemental Digital Content 2A, B, left, http://links.lww.com/NEN/A674), which is consistent with previous reports of variable effects of dementia on the postsynaptic density (2); also, there were no significant changes of NeuN (Figure, Supplemental Digital Content 2A, B, right, http://links.lww.com/NEN/A674). These findings do not indicate the absence of synapse and neuronal loss in the dementia tissues studied here. Extensive atrophy is generally observed at the later stages of dementia (29), which translates into a net loss of neurons and synapses but is no longer reflected by a relative loss of neuronal and synaptic proteins compared with whole tissue markers. Importantly, adjustment of EFhd2 densitometry results to levels of NeuN in each sample, as a measure of potential neuronal cell loss in dementia (30, 31), did not alter the overall outcome (Figure, Supplemental Digital Content 2C, http://links.lww.com/NEN/A674). These results indicate that there are global changes in EFhd2 protein abundance that are mainly associated with cell bodies and neurites. We also observe EFhd2 protein in the synaptic compartment, indicating that it could be playing a role in synaptic processes in the human brain.
FIGURE 5.
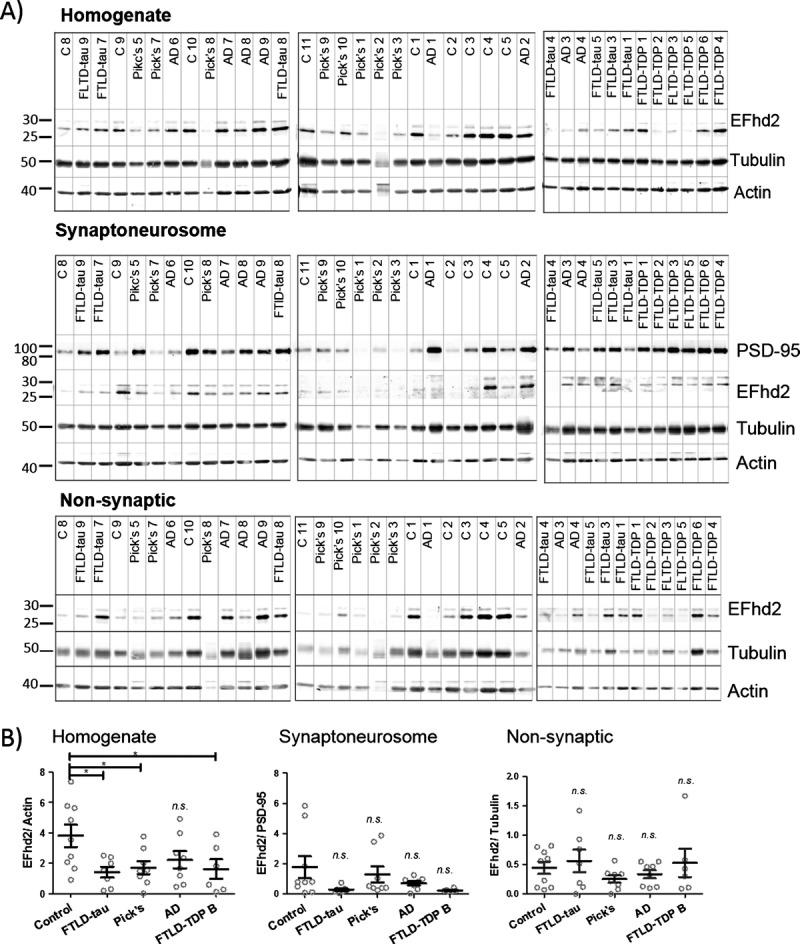
EFhd2 protein levels in the frontal cortex are reduced in total homogenates from patients with dementia but not in synaptoneurosomes. (A) Western blots showing EFhd2 protein levels in human frontal cortex tissue extracts from nondemented controls and individuals with Alzheimer disease (AD), frontotemporal lobar degeneration (FTLD)-tau (FTLD-tau), Pick disease, or FTLD-TDP Type B (FTLD-TDP). Beta-actin was used as a loading control for homogenates and nonsynaptic fractions; PSD-95 was used as a loading control for synaptoneurosome fractions. (B) Quantification of EFhd2 protein bands by densitometry. Error bars = SEM. Homogenate: 1-way ANOVA, p = 0.0301; * p < 0.05 compared with control by using Dunnett post hoc test. Synaptoneurosomes: 1-way ANOVA, p = 0.1108. Nonsynaptic: 1-way ANOVA, p = 0.4848.
EFhd2 Is Present in Neuronal Growth Cones and Nascent Presynaptic Structures in Developing Neurons In Vitro and Affects Synapse Formation
The role of EFhd2 in neuronal function has not been investigated in detail. A recent report revealed that a loss of EFhd2 does not have a direct impact on synaptic function, but that it does affect transport along microtubules in neuronal cultures (24). Studies in other cell types have found EFhd2 at the leading edge of cells where it influences lamellipodia formation (22, 23). Therefore, we investigated the potential association of EFhd2 with growth cones, which represent the leading edge of neurons, and synapses in developing neuronal cultures. As predicted, coimmunocytochemistry for EFhd2 with tau, tubulin, or (filamentous) actin in cortical neuronal cultures clearly showed an accumulation of EFhd2 at the tip of neuronal growth cones (Fig. 6A). A recent study demonstrated that EFhd2 can also be associated with the synaptic compartment in the rodent brain and in mature neuronal cultures (24). In addition to this, coimmunofluorescence staining with the presynaptic marker synapsin 1a/b in neuronal cultures revealed that EFhd2 can be present at nascent synaptic structures as early as 8 DIV—at a time before the recruitment of postsynaptic elements in vitro (Fig. 6B, arrows).
FIGURE 6.
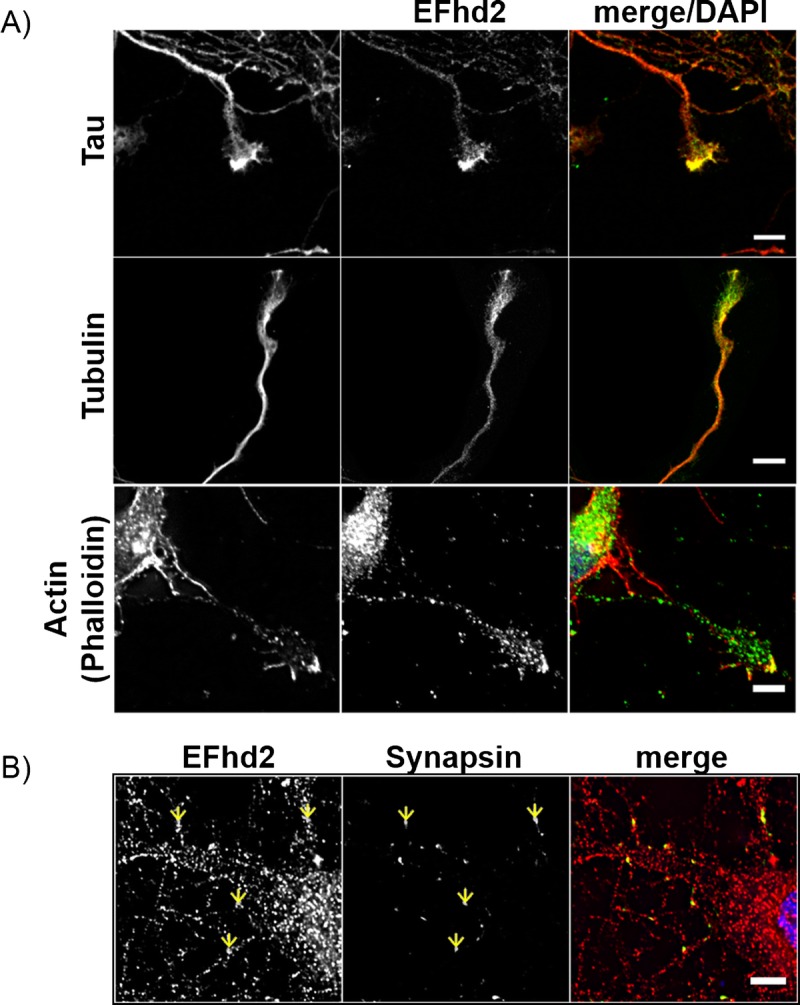
EFhd2 is present in neuronal growth cones and nascent presynaptic structures in developing neurons. (A) Coimmunofluorescence of EFhd2 with cytoskeletal proteins. Top: Confocal images of EFhd2 (green) and Tau (red) in 10 days in vitro (DIV) neurons. Scale bar = 5 μm. Middle: epifluorescence images of EFhd2 (green) and tubulin (red) in 3 DIV neurons. Scale bar = 10 μm. Bottom: confocal images of EFhd2 (green) and Phalloidin (red) in 7 DIV neurons. Scale bar = 5 μm (top, bottom). (B) Immunocytochemistry of EFhd2 (rabbit polyclonal antibody, red) and synapsin (green) on 8 DIV neurons. Yellow arrows indicate foci of colocalization. Scale bar = 5 μm (bottom).
The recruitment of EFhd2 to neuronal growth cones raised the possibility that EFhd2 could have a function in neurite development. To test this hypothesis, overexpression of EGFP-EFhd2 or EGFP alone or knockdown of EFhd2 was induced in primary cortical neurons by lentiviral infection at 1 DIV. Successful expression of EGFP-EFhd2 or EGFP and EFhd2 knockdown were confirmed in 3 DIV cultures by fluorescence imaging (Fig. 7A) and Western blotting (Fig. 7C), respectively. The effect of this modulation of EFhd2 expression on the outgrowth neurites in cortical neurons was then tested by quantification of the length of MAP2-labeled neurites per cell at 3 DIV. Interestingly, EGFP-EFhd2 overexpression or EFhd2 knockdown did not have any effect on the length of neurites developed until 3 DIV (Fig. 7B) or at a later stage at 8 DIV (Figure, Supplemental Digital Content 3, http://links.lww.com/NEN/A675).
FIGURE 7.
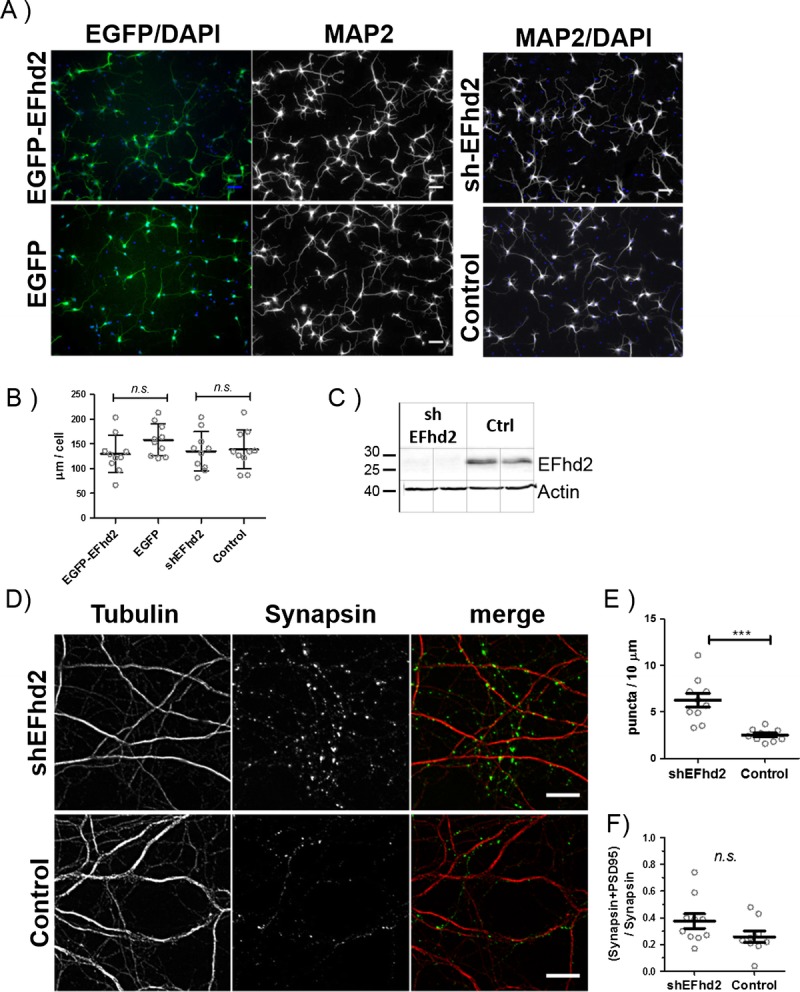
Neurite development in expression short hairpin RNA (shRNA) targeting EFhd2 (shEFhd2) and enhanced green fluorescent protein (EGFP)-EFhd2 transgenic neuronal cultures. (A) Representative images of primary cortical neurons infected with lentiviral supernatants for shEFhd2 or a scrambled control (shScr) overexpression of EGFP-EFhd2 or EGFP alone. Cells were infected at 1 day in vitro (DIV) and fixed and stained for microtubule-associated protein 2 (MAP2) at 3 DIV. Scale bar = 50 μm. (B) Quantification of anti-MAP2–immunostained neurites from 10 fluorescence images (20× magnifications) per group. n.s., nonsignificant by 2-tailed t test. Results are representative of 3 independent experiments. (C) Verification of EFhd2 knockdown by Western blotting. (D) Immunocytochemistry for synapsin and tubulin in neurons with EFhd2 knockdown (shEFhd2, top) or expression scrambled control shRNA (bottom). Scale bar = 10 μm. (E) Quantification of the number of synapsin puncta per 10 μm, labeled for tubulin in 3 independent experiments. ** p < 0.01 by 2-tailed t test, error bars = SEM, n = 10 fields of view for each experiment. (F) Quantification of the number of mature synapses labeled with synapsin and PDS-95 colocalization compared with the total number of synapsin puncta in the same cultures analyzed in E. Error bars = SEM, n = 10. Nonsignificant by 2-tailed t test.
Because of its partial association with nascent synapses in addition to neuronal growth cones in vitro (Fig. 6B), a potential effect of the loss of EFhd2 on the formation of synapses in cortical neuronal cultures was tested. Presynaptic (synapsin 1a/b) puncta in total per micrometer of β3-tubulin–labeled neurites were counted in mature neuronal cultures at 15 to 16 DIV after lentiviral knockdown of EFhd2 or infection with a control shRNA (Fig. 7D). Indeed, this analysis revealed that knockdown of EFhd2 facilitates the development of a larger number of synapses in vitro (Fig. 7E). At the same time, there was no difference in the ratio comparing mature synapses identified by the colocalization of synapsin and PSD95 with nascent structures labeled by synapsin alone (Fig. 7F). These results suggests that a downregulation of EFhd2 in neurons enhances their capacity to induce the formation of new functional synapses but does not affect their ability to convert these to mature synapses by recruiting postsynaptic densities.
DISCUSSION
EFhd2 is a novel calcium-binding protein; its biologic roles in the CNS have only recently started to emerge. Here we show that EFhd2 protein and mRNA levels are reduced in the frontal cortex in AD, Pick disease, and FTLD with tau pathology and that protein levels are also reduced in FTLD-TDP Type B (without tauopathy) compared with nondemented controls (Figs. 1, 3). We confirmed these findings in separate sets of experiments using different methods for protein extraction; EFhd2 protein levels were also found to be reduced in synaptoneurosome preparations from dementia compared with control patients (Fig. 5).
Interestingly, no significant changes in EFhd2 protein abundance could be observed in the hippocampus of either FTLD or AD cases (Figs. 2, 3), although this brain region is markedly affected in AD alongside the neocortex (32, 33). We showed by immunohistochemistry that EFhd2 is strongly expressed in the hippocampus (Fig. 4-IIA-C), which is in agreement with earlier studies (12, 24). However, it is possible that EFhd2 has functions relevant to cognitive processes in neuronal subtypes that are present in the neocortex but not the hippocampal allocortex, and it is also worth noting that its role in the hippocampus has not been investigated to date. Most studies involving EFhd2 in the CNS have analyzed frontal cortex tissues and have investigated neuropathologic processes that specifically affect the frontal cortex, such as FTLD (12), schizophrenia (14), and psychiatric conditions linked to suicide (15). Our present data support the notion that EFhd2 might play a significant role in the function of the frontal cortex, and that changes in its expression or posttranslational modification could be related to common processes underlying (pre)frontal cortex pathology.
The observations that protein and mRNA expression changes are present in tauopathies as well as tau-negative FTLD-TDP Type B and that there is no correlation between the amount of tau hyperphosphorylation and levels of EFhd2 in tauopathies (Figs. 1, 2) argue against a causative relationship between the two. Studies by Vega et al (12) and Ferrer-Acosta et al (11) identified EFhd2 as a protein coaggregating with pathologic tau in AD; a mouse model for FTLD also suggested an association with tau by coimmunoprecipitation from nondemented control brain tissue (12). Its interaction with nonpathologic tau species under physiologic conditions has, however, not been studied. Our data of both EFhd2 and tau proteins at the tip of neuronal growth cones in cultured neurons (Fig. 6A, top) support the idea of a functional interaction between them. We saw a similar relationship between EFhd2 and tubulin as well as actin (Fig. 6A, middle, bottom). Indeed, Purohit et al (24) demonstrated a direct effect of EFhd2 on microtubule transport in the absence of tau, and previous studies in non-neuronal cells established EFhd2 as an actin-binding protein involved in the regulation of actin bundling (22, 23, 34). Together, these findings imply that a functional or physical interaction between EFhd2 and tau might also be caused by their common interaction with tubulin and actin and put the role of EFhd2 in the regulation of cytoskeletal function into the center of interest.
EFhd2 is partially located in synapses, but it is most strongly associated with neuronal cell bodies and neurites (24). EFhd2 protein abundance are global and affect not only cell bodies and neurites in brain tissues affected by dementia but also synaptic compartment (Fig. 5). The observed reduction of EFhd2 mRNA levels supports this view (Fig. 1E). Because changes tended toward a reduction of EFhd2 in dementia in all fractions studied (Fig. 5B), our results also imply that there is no major redistribution of EFhd2 to or from the synaptoneurosomal compartment associated with dementia. It has to be acknowledged, however, that these findings are based on crude synaptoneurosome preparations by filtration from frozen tissues (as applied by Tai et al [25]) and merely indicate overall changes. Purohit et al (24) showed that EFhd2 is associated with the cytosolic as well as the plasma membrane fraction in synaptosomes isolated by differential centrifugation. In light of this, future more extensive and precise analyses of EFhd2 distribution and its potential changes in localization in neuropathologic conditions by immunoelectron microscopy and the isolation of synaptosomes and their subfractionation by density gradient centrifugation from fresh tissues would address these issues.
An initial study analyzing the effect of EFhd2 on synaptic function found no impact on the turnover of synaptic vesicles in cultured neurons (24). We show here that knockdown of EFhd2 does, however, boost the formation of new synaptic connections in cortical neuronal cultures (Fig. 7E, F), whereas it does not affect the growth of neurites (Fig. 7). Interestingly, EFhd2 has been shown to affect vesicular transport along microtubules (24); thus, it is conceivable that this function is underlying its effect on synapse development, which strongly depends on cytoskeletal rearrangements (including tau [35]) and is modulated by signaling cascades that EFhd2 has been linked to in other cell types, such as PI3K signaling (36, 37). Importantly, the formation of new synaptic connections is not restricted to embryonic development but is also important in the adult brain, and failure to maintain current and develop new synapses is strongly linked to dementia (1). Possible functions of EFhd2 could be in synapse formation (e.g. pruning) during development and/or synapse maintenance in the adult brain. In this context, it is interesting to note that Purohit et al (24) found higher levels of EFhd2 expression in the adult mouse brain compared with embryonic brain tissues, which our own preliminary RT-PCR data could confirm (not shown). This suggests that the function of EFhd2 might primarily be in the postnatal or adult brain. Synaptic plasticity is also increasingly being studied in the frontal cortex with regard to dementia and other psychiatric disorders, for example, schizophrenia and mood disorders (38). Our data together with the earlier studies raise the possibility that EFhd2 expression levels impact on these processes in the brain, specifically in the frontal cortex, and that its own regulation of expression or posttranslational modification (13, 21, 39) could therefore be important. A recent study suggests that such posttranslational modification could indeed be relevant for neurodegenerative diseases, that is, phosphorylation of EFhd2 by Cdk5 was found to occur in AD and to affect its calcium-binding activity (13). Cdk5 itself has previously been linked to tauopathy (40), specifically in AD (41–43), and to other neurodegenerative diseases such as Parkinson disease (44, 45). The positive impact of EFhd2 knockdown on microtubule transport (24) and synapse numbers (Fig. 7E, F) implies that, when present, EFhd2 might enable the integration of signaling cues that decide the fate of nascent synapses; this spatiotemporal regulation is likely to be of pivotal importance for correct neuronal network function and hence cognitive processes. A hypothesis based on current data on the role of EFhd2 in neurons in the brain and how it might influence neuropathologic processes (or be influenced by them) is depicted in Figure 8.
FIGURE 8.
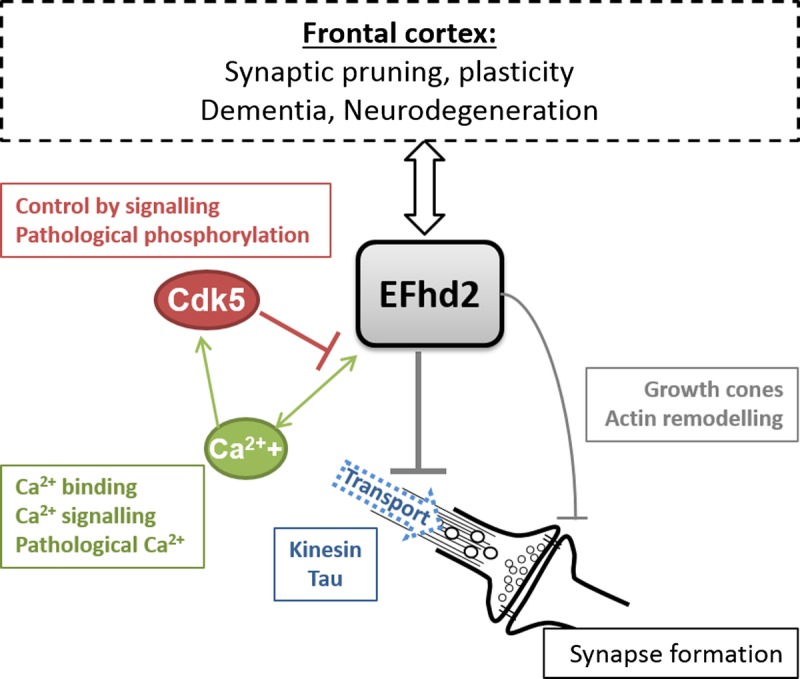
Hypothesis on the function of EFhd2 in the brain. EFhd2 inhibits kinesin-mediated anterograde microtubule transport in neurons. This could also involve physical or functional interaction with the microtubule binding protein tau. It also negatively regulates the formation of synapses either directly through its effect on actin remodeling and a potential function in growth cones other than neurite outgrowth or indirectly through its negative impact on microtubule transport. EFhd2 is a Ca2+-binding protein, and its function is regulated by intracellular Ca2+. Pathologically elevated Ca2+ might therefore impact on EFhd2 function. Ca2+ is furthermore involved in signaling mechanisms activating the Cdk5 protein kinase, which inhibits EFhd2 by phosphorylation of residues relevant for its Ca2+-binding activity. Pathologically elevated Ca2+ levels and aberrant activation of Cdk5 have previously been described in neurodegenerative diseases. Changes in EFhd2 expression as well as synapse loss are associated with dementia and other neurodegenerative diseases affecting the frontal cortex. EFhd2 might be involved in processes such as synaptic pruning or morphologic plasticity in this brain region, and its loss might have a profound impact on the balance of synapse maintenance and exacerbate or be a sign of synaptic pathology.
The mechanisms underlying the reduction of EFhd2 expression in the brain have not been elucidated so far. It has been reported that EFhd2 expression is controlled by pathways involving protein kinase C (β/ε/ ζ), as well as nuclear factor-κB in immune cells (17, 18). Therefore, it is plausible that similar mechanisms would exist in neuronal cells. Interestingly, neurotrophic signaling, which activates signaling cascades involving protein kinase C and nuclear factor-κB, is known to be disturbed in dementia (46–48), and this might contribute to the observed changes in EFhd2 expression. Further studies delineating the pathways involved in the regulation of EFhd2 expression in the brain are needed and might elucidate new pathways that orchestrate the maintenance of synapses in the frontal cortex. Consequently, these studies will help to understand the context of EFhd2 activity in neurons and determine whether its dysregulation could be an indicator of synaptic pathology in the human frontal cortex.
Supplementary Material
Footnotes
This work was funded by a research grant from Alzheimer’s Research UK (Eva Borger, Tara Spires-Jones, Frank Gunn-Moore) and the 600th University of St. Andrews anniversary BRAINS appeal.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s Web site (www.jneuropath.com).
REFERENCES
- 1. Crimins JL, Pooler A, Polydoro M, et al. The intersection of amyloid beta and tau in glutamatergic synaptic dysfunction and collapse in Alzheimer’s disease. Ageing Res Rev 2013; 12: 757– 63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gong Y, Lippa CF. Review: Disruption of the postsynaptic density in Alzheimer’s disease and other neurodegenerative dementias. Am J Alzheimers Dis Other Demen 2010; 25: 547– 55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhou L, Miller BL, McDaniel CH, et al. Frontotemporal dementia: Neuropil spheroids and presynaptic terminal degeneration. Ann Neurol 1998; 44: 99– 109 [DOI] [PubMed] [Google Scholar]
- 4. Scheff SW, Neltner JH, Nelson PT. Is synaptic loss a unique hallmark of Alzheimer’s disease? Biochem Pharmacol 2014; 88: 517– 28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Goedert M, Ghetti B, Spillantini MG. Frontotemporal dementia: Implications for understanding Alzheimer disease. Cold Spring Harb Perspect Med 2012; 2: a006254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ittner LM. Dendritic function of tau mediactes amyloid-b toxicity in Alzheimer’s disease mouse models. Cell 2010; 142: 387– 97 [DOI] [PubMed] [Google Scholar]
- 7. Shipton OA, Leitz JR, Dworzak J, et al. Tau protein is required for amyloid β-induced impairment of hippocampal long-term potentiation. J Neurosci 2011; 31: 1688– 92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eckermann K, Mocanu MM, Khlistunova I, et al. The beta-propensity of tau determines aggregation and synaptic loss in inducible mouse models of tauopathy. J Biol Chem 2007; 282: 31755– 65 [DOI] [PubMed] [Google Scholar]
- 9. Hall GF, Lee VM, Lee G, et al. Staging of neurofibrillary degeneration caused by human tau overexpression in a unique cellular model of human tauopathy. Am J Pathol 2001; 158: 235– 46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bennion Callister J, Pickering-Brown SM. doi: 10.1016/j.expneurol.2014.06.001. Pathogenesis/genetics of frontotemporal dementia and how it relates to ALS [epub ahead of print June 8, 2014]. Exp Neurol doi: 10.1016/j.expneurol.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ferrer-Acosta Y, Rodríguez-Cruz EN, Orange F, et al. Efhd2 is a novel amyloid protein associated to pathological tau in Alzheimer’s disease. J Neurochem 2013; 125: 921– 31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vega IE, Traverso EE, Ferrer-Acosta Y, et al. A novel calcium-binding protein is associated with tau proteins in tauopathy. J Neurochem 2008; 106: 96– 106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vázquez-Rosa E, Rodríguez-Cruz EN, Serrano S, et al. Cdk5 phosphorylation of EFhd2 at S74 affects its calcium binding activity. Protein Sci 2014; 23: 1197– 207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Martins-de-Souza D, Gattaz WF, Schmitt A, et al. Prefrontal cortex shotgun proteome analysis reveals altered calcium homeostasis and immune system imbalance in schizophrenia. Eur Arch Psychiatry Clin Neurosci 2009; 259: 151– 63 [DOI] [PubMed] [Google Scholar]
- 15. Kekesi KA, Juhasz G, Simor A, et al. Altered functional protein networks in the prefrontal cortex and amygdala of victims of suicide. PLoS One 2012; 7: e50532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhai J, Strom AL, Kilty R, et al. Proteomic characterization of lipid raft proteins in amyotrophic lateral sclerosis mouse spinal cord. FEBS J 2009; 276: 3308– 23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim YD, Kwon MS, Na BR, et al. Swiprosin-1 expression is up-regulated through protein kinase C-theta and NF-kappaB pathway in T cells. Immune Netw 2013; 13: 55– 62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thylur RP, Kim YD, Kwon MS, et al. Swiprosin-1 is expressed in mast cells and up-regulated through the protein kinase C beta I/eta pathway. J Cell Biochem 2009; 108: 705– 15 [DOI] [PubMed] [Google Scholar]
- 19. Hagen S, Brachs S, Kroczek C, et al. The B-cell receptor–induced calcium flux involves a calcium mediated positive feedback loop. Cell Calcium 2012; 51: 411– 17 [DOI] [PubMed] [Google Scholar]
- 20. Kroczek C, Lang C, Brachs S, et al. Swiprosin-1/EFhd2 controls B cell receptor signaling through the assembly of the B cell receptor, Syk, and phospholipase C gamma2 in membrane rafts. J Immunol 2010; 184: 3665– 76 [DOI] [PubMed] [Google Scholar]
- 21. Blagoev B, Ong S-E, Kratchmarova I, et al. Temporal analysis of phosphotyrosine-dependent signaling networks by quantitative proteomics. Nat Biotech 2004; 22: 1139– 45 [DOI] [PubMed] [Google Scholar]
- 22. Huh YH, Kim SH, Chung KH, et al. Swiprosin-1 modulates actin dynamics by regulating the F-actin accessibility to cofilin. Cell Mol Life Sci 2013; 70: 4841– 54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kwon MS, Park KR, Kim YD, et al. Swiprosin-1 is a novel actin bundling protein that regulates cell spreading and migration. PLoS One 2013; 8: e71626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Purohit P, Perez-Branguli F, Prots I, et al. The Ca2+ sensor protein Swiprosin-1/EFhd2 is present in neurites and involved in kinesin-mediated transport in neurons. PLoS One 2014; 9: e103976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tai HC, Serrano-Pozo A, Hashimoto T, et al. The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am J Pathol 2012; 181: 1426– 35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Avramidou A, Kroczek C, Lang C, et al. The novel adaptor protein Swiprosin-1 enhances BCR signals and contributes to BCR-induced apoptosis. Cell Death Differ 2007; 14: 1936– 47 [DOI] [PubMed] [Google Scholar]
- 27. Vuadens F, Rufer N, Kress A, et al. Identification of swiprosin 1 in human lymphocytes. Proteomics 2004; 4: 2216– 20 [DOI] [PubMed] [Google Scholar]
- 28. Hornbruch-Freitag C, Griemert B, Buttgereit D, et al. Drosophila Swiprosin-1/EFHD2 accumulates at the prefusion complex stage during Drosophila myoblast fusion. J Cell Sci 2011; 124: 3266– 78 [DOI] [PubMed] [Google Scholar]
- 29. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011; 7: 263– 69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Andrade-Moraes CH, Oliveira-Pinto AV, Castro-Fonseca E, et al. Cell number changes in Alzheimer’s disease relate to dementia, not to plaques and tangles. Brain 2013; 136: 3738– 52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Keage HA, Hunter S, Matthews FE, et al. TDP-43 in the population: Prevalence and associations with dementia and age. J Alzheimers Dis 2014; 42: 641– 50 [DOI] [PubMed] [Google Scholar]
- 32. Duyckaerts C, Delatour B, Potier MC. Classification and basic pathology of Alzheimer disease. Acta Neuropathol 2009; 118: 5– 36 [DOI] [PubMed] [Google Scholar]
- 33. Nordberg A. PET imaging of amyloid in Alzheimer’s disease. Lancet Neurol 2004; 3: 519– 27 [DOI] [PubMed] [Google Scholar]
- 34. Meng X, Wilkins JA. Compositional characterization of the cytoskeleton of NK-like cells. J Proteome Res 2005; 4: 2081– 87 [DOI] [PubMed] [Google Scholar]
- 35. Chen Q, Zhou Z, Zhang L, et al. Tau protein is involved in morphological plasticity in hippocampal neurons in response to BDNF. Neurochem Int 2012; 60: 233– 42 [DOI] [PubMed] [Google Scholar]
- 36. Cuesto G, Enriquez-Barreto L, Carames C, et al. Phosphoinositide-3-kinase activation controls synaptogenesis and spinogenesis in hippocampal neurons. J Neurosci 2011; 31: 2721– 33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ramesh TP, Kim YD, Kwon MS, et al. Swiprosin-1 regulates cytokine expression of human mast cell line HMC-1 through actin remodeling. Immune Netw 2009; 9: 274– 84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Goto Y, Yang CR, Otani S. Functional and dysfunctional synaptic plasticity in prefrontal cortex: Roles in psychiatric disorders. Biol Psych 2010; 67: 199– 207 [DOI] [PubMed] [Google Scholar]
- 39. Blethrow JD, Glavy JS, Morgan DO, et al. Covalent capture of kinase-specific phosphopeptides reveals Cdk1-cyclin B substrates. Proc Natl Acad Sci USA 2008; 105: 1442– 47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mandelkow EM, Biernat J, Drewes G, et al. Tau domains, phosphorylation, and interactions with microtubules. Neurobiol Aging 1995; 16: 355– 62 [DOI] [PubMed] [Google Scholar]
- 41. Cole AR, Noble W, van Aalten L, et al. Collapsin response mediator protein-2 hyperphosphorylation is an early event in Alzheimer’s disease progression. J Neurochem 2007; 103: 1132– 44 [DOI] [PubMed] [Google Scholar]
- 42. Cruz JC, Kim D, Moy LY, et al. p25/Cyclin-dependent kinase 5 induces production and intraneuronal accumulation of amyloid beta in vivo. J Neurosci 2006; 26: 10536– 41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pei J-J, Grundke-Iqbal I, Iqbal K, et al. Accumulation of cyclin-dependent kinase 5 (cdk5) in neurons with early stages of Alzheimer’s disease neurofibrillary degeneration. Brain Res 1998; 797: 267– 77 [DOI] [PubMed] [Google Scholar]
- 44. Qu D, Rashidian J, Mount MP, et al. Role of Cdk5-mediated phosphorylation of Prx2 in MPTP toxicity and Parkinson’s disease. Neuron 2007; 55: 37– 52 [DOI] [PubMed] [Google Scholar]
- 45. Smith PD, Crocker SJ, Jackson-Lewis V, et al. Cyclin-dependent kinase 5 is a mediator of dopaminergic neuron loss in a mouse model of Parkinson’s disease. Proc Natl Acad Sci USA 2003; 100: 13650– 55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Capsoni S, Brandi R, Arisi I, et al. A dual mechanism linking NGF/proNGF imbalance and early inflammation to Alzheimer’s disease neurodegeneration in the AD11 anti-NGF mouse model. CNS Neurol Dis Drug Targ 2011; 10: 635– 47 [DOI] [PubMed] [Google Scholar]
- 47. Holscher C. Diabetes as a risk factor for Alzheimer’s disease: Insulin signalling impairment in the brain as an alternative model of Alzheimer’s disease. Biochem Soc Trans 2011; 39: 891– 97 [DOI] [PubMed] [Google Scholar]
- 48. Schindowski K, Belarbi K, Buee L. Neurotrophic factors in Alzheimer’s disease: Role of axonal transport. Genes Brain Behav 2008; 7 (Suppl 1): 43– 56 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.