

Abstract
Objective
Atherosclerosis is the primary driver of cardiovascular disease, the leading cause of death worldwide. Identification of naturally occurring atheroprotective genes has become a major goal for development of interventions that will limit atheroma progression and associated adverse events. To this end, we have identified SPRR3 (small proline-rich repeat protein 3) as selectively upregulated in vascular smooth muscle cells (VSMCs) of atheroma-bearing arterial tissue versus healthy arterial tissue. In this study, we sought to determine the role of SPRR3 in atheroma pathophysiology.
Approach and Results
We found that atheroprone ApoE-null mice lacking SPRR3 developed significantly greater atheroma burden. To determine the cellular driver(s) of this increase, we evaluated SPRR3-dependent changes in bone-marrow-derived cells, endothelial cells (ECs), and VSMCs. Bone marrow transplant of SPRR3-expressing cells into SPRR3−/− ApoE−/− recipients failed to rescue atheroma burden. Similarly, ECs did not exhibit a response to SPRR3 loss. However, atheromas from SPRR3-deficient mice exhibited increased TUNEL-positive VSMCs compared with control. Cell death in SPRR3-deficient VSMCs was significantly increased in vitro. Conversely, SPRR3-overexpressing VSMCs exhibited reduced apoptosis compared with control. We also observed a PI3K/Akt-dependent positive association between SPRR3 expression and levels of active Akt in VSMCs. The survival advantage seen in SPRR3-overexpressing VSMCs was abrogated following the addition of a PI3K/Akt pathway inhibitor.
Conclusions
These results indicate that SPRR3 protects the lesion from VSMC loss by promoting survival signaling in plaque VSMCs, thereby significantly decreasing atherosclerosis progression. As the first identified atheroma-specific VSMC pro-survival factor, SPRR3 represents a potential target for lesion-specific modulation of VSMC survival.
Keywords: vascular smooth muscle, atherosclerosis, Akt
Introduction
Cardiovascular disease, the leading cause of death worldwide1, accounts for one in three deaths in the United States1 and is primarily driven by atherosclerosis. Initiation of atherosclerosis is believed to stem from a confluence of injury to the endothelium along with lipid deposition in the vascular wall, which then triggers an ongoing cycle of endothelial cell (EC) activation, inflammatory cell infiltration, hyperplastic vascular smooth muscle cell (VSMC) proliferation, and further lipid accumulation2. Atherosclerosis progression ultimately results in a toxic microenvironment in which dead or dying lipid-laden foam cells are sequestered from the circulation by a VSMC-derived fibrous cap2–3. Atheroprotective genes or factors that delay the initiation or progression of the atheroma are of great interest for their potential to delay adverse events4.
We sought to identify novel, potentially atheroprotective genes in a previous study, in which we conducted a transcript screen for alternately regulated genes in human arteries versus veins and other tissues5. The most strongly upregulated of the significantly altered genes was the small proline-rich repeat protein 3 (SPRR3), a member of the small proline-rich repeat protein family. Interestingly, immunofluorescence staining of human thoracic aorta showed that, while expression was high in atheroma, no SPRR3 expression was detected in healthy arterial tissue5. SPRRs are primarily expressed in cornified envelope and stratified epithelium such as skin and, in the case of SPRR3, foregut and esophagus6. Interestingly, some studies have shown that SPRR3 is also associated with pro-survival functions in colorectal cancer7. However, the mechanism by which SPRR3 promotes cell survival remains undetermined. We have previously investigated the mechanism by which SPRR3 is expressed specifically in VSMCs within atheromas. The mechanism of SPRR3 expression in atherosclerotic plaques requires both cyclic strain and the expression of integrin α1β1, a major receptor for collagen type I; VSMCs isolated from integrin α1β1 knockout mice failed to upregulate SPRR3 transcripts under cyclic strain.8 We confirmed that VSMCs within atheromas expressed integrin α1β18–9. SPRR3 may, therefore, represent a VSMC-derived response to the atheroma microenvironment.
To elucidate the pathophysiological function of SPRR3 in the atheroma, we developed an SPRR3-deficient mouse bred onto an atherogenic ApoE−/− background. Here, we have provided evidence for SPRR3 as an atheroprotective factor that operates specifically in the atheroma and promotes VSMC survival. We also uncovered a novel role for SPRR3 as an atheroprotective protein that regulates lesion development via interaction with the Akt signaling pathway in atheroma VSMCs.
Methods
Materials and Methods are available in the online-only Data Supplement.
Results
Atherosclerosis is increased in ApoE-null mice lacking SPRR3
We have shown previously that arterial SPRR3 is expressed in humans and mice at sites of lesion development5, 8. We verified the atheroma expression pattern of SPRR3 in aortic root atheroma sections from 6 month old ApoE-null mice fed normal diet immunostained for both SPRR3 and a VSMC-specific marker (smooth muscle α-actin, α-SMA). SPRR3 colocalized with α-SMA, indicating that expression was primarily found in VSMCs (Fig. 1, A). A comparison of SPRR3 transcript levels in atheroma-bearing arterial tissue versus normal arterial tissue indicated a 19-fold increase (p=0.02) in SPRR3 mRNA expression (Fig. 1, B).
Figure 1. SPRR3 was enriched in atheroma VSMCs but not in normal arteries.
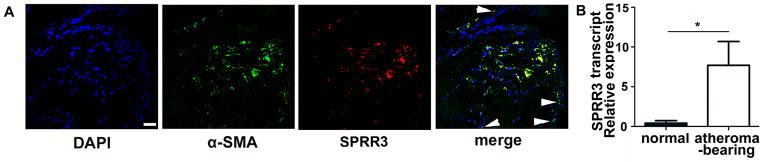
(A) Fluorescent images were acquired by confocal fluorescent microscopy with x40 lens. A representative Z plane shows a high degree of colocalization of α-SMA (green) and SPRR3 (red) in aortic root lesions of 6 month normal diet-fed SPRR3+/+ApoE−/− mice. Scale bar, 50 μm. Arrowheads indicate VSMCs not positive for SPRR3 expression. (B) mRNA isolated from atheroma-bearing artery and normal artery (of SPRR3+/+ApoE−/− mice) was assessed for SPRR3 expression using real time qRT-PCR (N=3/group). Bars represent mean ±SD. * p < 0.05
To investigate the role of SPRR3 in atherosclerotic lesions, we generated SPRR3 knockout mice (Figure I, A–C in the online-only Data Supplement). SPRR3 knockout mice were overtly phenotypically normal and reproduced normally compared with WT C57Bl/6 mice. Serum cholesterol and triglyceride levels (Table I in the online-only Data Supplement; N=6), as well as body weight measurements, were comparable to WT. The lack of an associated phenotype in SPRR3 knockout mice was expected, due to the near total absence of atheroma formation in wild type mice with normal lipid profiles. Therefore, we crossed SPRR3 knockout mice onto the atherogenic ApoE-null background. We assessed atherosclerosis severity in 6 month old ApoE-null (SPRR3+/+ApoE−/−) and DKO (SPRR3−/−ApoE−/−) mice fed normal chow (4.5% fat by weight with 13% of calories derived from fat, diet 5001; PMI, St Louis, Missouri, USA). Serum triglyceride and cholesterol levels did not differ between groups (Table I in the online-only Data Supplement; N=6).
To determine whether SPRR3 loss alters atherosclerosis progression, we compared the extent of atherosclerosis in 6-month-old gender-matched DKO mice with ApoE-null control mice fed a normal chow diet. We used Sudan IV to quantify atherosclerosis by en face analysis of aortas, in which DKO mice exhibited a 3-fold increase in lesion area over control (0.7 ± 0.14% in SPRR3+/+ApoE−/− vs 2.2±0.6 in SPRR3−/−ApoE−/−; Fig. 2, A–B; p=0.02; N=13). Similar results were observed in aortic root lesions stained for lipid levels with Oil Red O (Figure II, A–B in the online-only Data Supplement; DKO increased 51% over ApoE-null, p=0.03). To determine whether the increased atheroma burden observed on normal chow would be maintained on an atherogenic diet, we fed ApoE-null and DKO mice a high fat diet (9% fat by weight with 21% of calories derived from fat, diet 5021; PMI, St Louis, Missouri, USA) for 6 months. Following the atherogenic diet, a 3 fold greater lesion area was observed in DKO aortas than in ApoE-null aortas (2.3±0.8 in SPRR3+/+ApoE−/− vs 6.8±1.9 in SPRR3−/−ApoE−/−; Figure III, A–B in the online-only Data Supplement; p=0.048; N=8). The significant increase in atheroma burden in DKO mice indicated that loss of SPRR3 resulted in increased plaque burden.
Figure 2. Atherosclerosis progression was accelerated in ApoE-null mice lacking SPRR3.
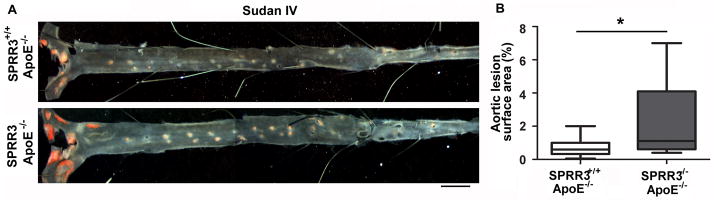
(A) Representative en face images of Sudan IV (red) stained aortas from 6 mo. old gender-matched SPRR3+/+ApoE−/− and SPRR3−/−ApoE−/− mice (normal chow). Scale bar, 2mm. (B) Quantification of aortic lesion surface area. (N=13 mice/group). Bars represent mean ±SD. * p<0.05
VSMC survival is selectively reduced in atheromas of SPRR3-deficient mice
Since SPRR3 expression was primarily observed in atheroma VSMCs, we sought to determine whether VSMC localization or content was altered in mice lacking SPRR3. An initial stain with anti-α-SMA of aortic root sections from mice fed normal chow suggested DKO mice had lower α-SMA-positive VSMC content than age-matched ApoE-null mice (Figure 3, A). To quantify the change in lesion VSMC content, we collected confocal images of anti-α-SMA-stained and anti-smooth muscle myosin heavy chain (SM-MHC)-stained aortic root sections from DKO and ApoE-null mice fed normal chow (Figure 3, B–C). VSMC content was quantified as the number of α-SMA-positive or SM-MHC-positive cells normalized to plaque cellularity (cells/mm2). α-SMA-positive staining indicated that DKO (N=8) lesions had a 37.3% (p<0.001) reduction in lesion VSMC content compared with control (N=8; Fig. 3, D). Similar results were observed in SM-MHC-stained sections (Figure IV in the online-only Data Supplement, p=0.0001; N=3/group). When VSMC content was quantified in DKO and ApoE-null mice fed high fat diet for 6 months, lesion VSMC content was reduced by 48.1% (p=0.002) in mice lacking SPRR3.
Figure 3. Increased VSMC apoptosis in SPRR3-null atheroma but not in disease-free arteries.

(A) Low magnification photomicrographs of aortic root sections from 6 mo. SPRR3+/+ApoE−/− and SPRR3−/−ApoE−/− mice fed normal chow stained with α-SMA (green). Scale bar, 100 μm. (B) H&E stained aortic root sections from 6 mo. SPRR3+/+ApoE−/− and SPRR3−/−ApoE−/− mice (normal chow). Scale bar, 100 μm. Boxed area magnified in C. (C) Representative Z plane from confocal fluorescent microscopy images collected with x40 lens. Sections stained with α-SMA and DAPI (blue). Scale bar, 50 μm. (D) Quantification of %α-SMA positive cap cells in aortic root sections from 6 month old SPRR3+/+ApoE−/− or SPRR3−/−ApoE−/− mice fed normal diet (N=8). (E) Quantification of % α-SMA positive cap cells in aortic root sections from SPRR3+/+ApoE−/− or SPRR3−/−ApoE−/− mice fed high fat diet for 6 months (N=6). (F) Representative Z plane from confocal image of α-SMA-TUNEL (red) showed a high degree of colocalization in aortic root of SPRR3+/+ApoE−/− and SPRR3−/−ApoE−/− mice fed high fat diet for 6 months. Scale bar, 40 μm. (G) Quantification of %fibrous cap α-SMA/-TUNEL colocalization in (F) assessed in 10 fields (N=5 mice/group). Bars represent mean ±SD. * < 0.05.
The reduced atheroma VSMC content in mice lacking SPRR3 could be due to an increase in lesion VSMC cell death or a reduction in proliferation. To determine whether the reduced VSMC content was related to VSMC survival, we compared VSMC death in aortic root lesions of ApoE-null and DKO mice following 6 months on a high-fat diet. In the aortic root, the number of fibrous cap cells co-staining for TUNEL and α-SMA was increased 70.6% in mice lacking SPRR3 (Fig. 3, E–F; p=0.0316). However, there was no significant difference between groups in basal cell death levels in arterial tissue lacking atheroma, as measured by the percent of cells co-stained for TUNEL and α-SMA (4.2±2.3% in SPRR3+/+ApoE−/−, N=7; 4.32±3.2% in SPRR3−/−ApoE−/−, N=10; p=0.67). There was also no significant difference in the number of cells costaining for MOMA-2 and TUNEL or vWF and TUNEL (Figure VI in the online-only Data Supplement, MOMA-2-TUNEL p=0.91) or in the number of Ki67-positive cells (see Supplemental Text and Figure V in the online-only Data Supplement).
Increased atherosclerosis in ApoE-null mice lacking SPRR3 is not attributable to bone-marrow derived cells
Since SPRR3 expression was primarily but not exclusively detected in VSMCs8 (Figure 1, A), we sought to identify the affected cell type(s) driving increased atheroma burden in mice with global SPRR3 deficiency. As macrophages are the primary source of foam cells in murine atherosclerosis10–11, assessments of plaque burden that rely on lipid staining may reflect changes in macrophage recruitment or foam cell formation. We therefore evaluated changes in macrophage content relative to lesion size in aortic root lesions of ApoE-null and DKO mice fed high fat diet for 4 months. Macrophage content was quantified by MOMA-2 staining, which binds an intracellular target in mouse monocytes and macrophages. Although absolute MOMA-2-positive area was increased in DKO mice, after controlling for lesion size, there was no significant difference in MOMA-2-positive staining between ApoE-null (47.9±6.2%) and DKO (34.3±2.1%) aortic roots (Fig. 4, A–B; N=8; p=0.14). Similarly, there was no significant difference in CD3-positive staining, an indicator of T cell content, between ApoE-null (8.07±3.38%) and DKO (11.4±0.94%) aortic roots (p=0.06) after controlling for lesion size. These data suggested the SPRR3-dependent increase in atheroma burden observed in DKO mice was not mediated by bone marrow (BM) -derived cells.
Figure 4. Bone-marrow derived cells did not contribute to SPRR3-dependent effects on atherosclerosis development.

(A) Representative photomicrographs of MOMA-2-stained aortic root sections of 6 mo. mice (normal chow; N=6–8). Scale bar, 100 μm. (B) Quantification of MOMA-2 staining normalized to lesion size (i.e. MOMA-2 positive area/lesion area). (C–G) Donor bone marrow from SPRR3+/+ApoE−/−, SPRR3−/−ApoE+/+, or SPRR3−/−ApoE−/− (N=6) were transplanted into SPRR3−/−ApoE−/− recipients following lethal irradiation. Mice were allowed 2 months for engraftment followed by 4 months high fat diet prior to evaluation. (C) Schematic depicting BMT experiments and anticipated results (D) Images (scale bar, 100 μm) of ORO-stained (red) lesions in aortic root sections and (E) quantification of lesion area in ORO-stained aortic root sections. (F) Images (scale bar, 2mm) of Sudan IV (red) positive staining in en face and (G) quantification of %Sudan IV positive surface area in en face. Bars represent mean ±SD. * < 0.05
To further confirm that the effects of SPRR3 loss were mediated through arterial, rather than BM-derived cells, we performed bone marrow transplantation (BMT) experiments. BM from ApoE-null (SPRR3+/+ApoE−/−), SPRR3 knockout (SPRR3−/−ApoE+/+), or DKO (SPRR3−/−ApoE−/−) donor mice was transplanted into 2-week old lethally irradiated DKO recipient mice. The experimental approach and anticipated results are explained in Figure 4, C. After 4 months of high fat diet, plaque burden was assessed in aortic root sections and in dissected aortas of each group. Previous studies have demonstrated that rescue of ApoE expression in the BM compartment of ApoE-null mice is sufficient to reduce lesion burden12. We therefore expected that DKO recipients of SPRR3−/−ApoE+/+ donor cells (positive control) would exhibit significantly reduced atheroma burden compared with DKO recipients of DKO donor cells (negative control). Should the effects of SPRR3 on atherosclerosis be mediated through the bone marrow compartment, we expected that transplant of SPRR3+/+ApoE−/− donor cells into DKO recipients would rescue atheroma burden. Conversely, should the effects of SPRR3 be mediated through arterial cells, we expected to see no significant difference between DKO recipients of SPRR3+/+ApoE−/− donor cells and the negative control group. Assessment of plaque burden in the aortic root of DKO recipients of SPRR3+/+ bone marrow cells showed that SPRR3+/+ bone marrow cells failed to rescue plaque formation, as recipient mice experienced no significant change in lesion size when compared with recipients of DKO bone marrow. In contrast, DKO recipients of ApoE+/+ bone marrow experienced a 58.3% reduction (p<0.05) in aortic root lesion size when compared with control recipients (Fig. 4, D–E). En face analysis of dissected aortas showed an 81.3% (p<0.01) reduction in aortic surface area covered by Sudan IV lipid stain (Fig. 4, F–G) following ApoE+/+ BMT and no significant difference in SPRR3+/+ BMT recipients. These data demonstrated that increased atherosclerosis observed in DKO mice was not mediated by BM-derived cells but instead by arterial cells.
EC survival and proliferation are not altered following SPRR3 loss
Two arterial cell types, endothelial cells (ECs) and VSMCs, are commonly involved in atheroma development. To determine whether SPRR3 loss affected EC function, we isolated primary ECs from WT C57Bl/6 mice or from SPRR3 knockout mice (Figure VII, A in the online-only Data Supplement). Cell morphology assessment by brightfield microscopy and vWF staining confirmed the purity of our samples (Figure VII, A in the online-only Data Supplement). We compared pooled groups of cells for differences in survival, proliferation, or SPRR3 mRNA expression (Figure VII, B, C, and D, respectively in the online-only Data Supplement). Quantification of TdT-labeled WT ECs and SPRR3-KO ECs did not show significant survival differences between groups (25.1±7.3% WT ECs; 25.6±8.6% SPRR3-KO ECs; p=0.89). Similarly, BrdU labeling of SPRR3-KO ECs was not significantly different from that of WT (0.08±0.01 WT ECs; 0.02±0.04 SPRR3-KO ECs; p=0.04). Quantitative RT-PCR analysis of SPRR3 transcript expression in ECs isolated from WT C57Bl/6 mice showed little to no SPRR3 transcript expression. These data suggested that altered atheroma pathophysiology in SPRR3-deficient mice was not due to changes in EC function.
To investigate whether increased atheroma burden was associated with an effect of SPRR3 loss on VSMC function, we generated conditionally immortalized VSMCs that were WT or null for SPRR3 expression. Although SPRR3 protein was undetectable in VSMCs at baseline consistent with our previous publications 8, we detected mRNA transcript expression in VSMCs with wild type SPRR3 (Figure VII, D in the online-only Data Supplement).
Loss of SPRR3 reduces VSMC survival
To determine the role of SPRR3 in VSMC survival, we isolated primary VSMCs from SPRR3-KO (SPRR3−/−ApoE+/+) and WT (C57Bl/6) mice for culture under cyclic strain. As described in our previous work, cyclic strain causes upregulation of SPRR3 expression in VSMCs in vitro8. VSMCs were transferred to collagen-I treated elastomer membranes and subjected to 72 hours of cyclic strain to promote SPRR3 expression. Treatment with 0.5 mM H2O2 for 2 hours resulted in a 54.4±0.23% increase in TdT-labeling in primary SPRR3-KO VSMCs as measured by TUNEL staining compared with primary WT VSMCs (Fig. 5, A; p=0.02) while no difference was observed in unchallenged groups under strain or between WT VSMCs and SPRR3-KO VSMCs not exposed to cyclic strain (Figure VIII, A–B in the online-only Data Supplement). This finding demonstrated a protective role for SPRR3 in VSMCs exposed to oxidative stress under cyclic strain.
Figure 5. SPRR3 modulates VSMC survival.

(A) Primary VSMCs isolated from SPRR3−/− ApoE+/+ or WT mice exposed to 2 hours of oxidative stress (0.5mM H2O2) under cyclic strain were stained with TUNEL. %TUNEL-positive VSMCs were calculated among >200 cells/treatment in 3 replicates. (B) Viability in 24 hour serum-starved VSMCs overexpressing vector or SPRR3 measured by MTT. (C–E) SPRR3 overexpression enhances VSMC survival under 4 hours oxidative stress (0.25mM H2O2). (C) Western blot and analysis of active caspase-3 expression. (D) Photomicrograph showing IF staining of VSMCs expressing active caspase-3 (red); DAPI (blue) stains nuclei. Scale bar, 50 μm. Arrows indicate examples of caspase-3 positive cells. (E) Quantification of active caspase-3 IF in >200 cells/treatment (N=3). Bars represent mean ±SD. * < 0.05
VSMCs overexpressing SPRR3 exhibit reduced apoptosis and proliferation
If loss of SPRR3 leads to increased VSMC apoptosis, we hypothesized a pro-survival phenotype would be observed with SPRR3 overexpression. To generate SPRR3-overexpressing VSMCs, we isolated VSMCs from transgenic H-2Kb-tsA58 mice that we then retrovirally transfected with LZRS plasmid expressing GFP alone (GFP-VSMCs) or expressing human SPRR3 and GFP (SPRR3-VSMCs). Cells from the H-2Kb-tsA58 mouse expressing heat-labile T-antigen behind the mouse major histocompatibility complex H-2Kb are immortalized at 33°C and return to primary phenotype after a week at 37°C8.
We evaluated the effect of SPRR3 overexpression on VSMC survival using MTT dye reduction assays in serum-starved cells and both immunoblot and immunocytochemical staining analysis of active caspase-3 expression in cells exposed to oxidative stress. MTT dye reduction assays conducted under serum deprivation conditions indicated a 20% increase (p=0.03) in survival of SPRR3-VSMCs compared with control GFP-VSMCs (Fig. 5, B). This increase in VSMC viability observed with SPRR3 overexpression was not due to increased proliferation. WT-VSMCs and SPRR3-KO VSMCs treated with bromodioxyuridine (BrdU) stain revealed no significant difference in proliferation of VSMCs lacking SPRR3 when compared with control VSMCs (Figure IX in the online-only Data Supplement, p=0.43). To determine whether the increase in VSMC number observed in SPRR3-VSMCs was due to reduced apoptosis, we evaluated active caspase-3 levels by immunoblotting (Fig. 5, C) or immunofluorescence (Fig. 5, D–E) following oxidative stress (via 0.25mM H2O2), a pro-apoptotic trigger frequently observed in atherosclerotic lesions. Both methods demonstrated that SPRR3-VSMCs expressed less active caspase-3 in response to oxidative stress.
Akt phosphorylation is increased in VSMCs expressing SPRR3
A myriad of signaling changes takes place in VSMCs associated with plaque development and progression. Many of these signaling changes are associated with the functional changes observed in VSMCs during plaque progression (e.g. proliferation, migration, synthesis of cap material) and with responses to novel interactions and stressors in the plaque microenvironment (e.g. altered mechanical strain, pro-inflammatory signals, ROS, growth factors). Signaling pathways with pro-survival functions frequently observed to be activated in plaque VSMCs include p38, ERK1/2, and Akt13–17. We investigated the activity of each of these kinases by immunoblot analysis of whole cell lysates from control GFP-VSMCs and SPRR3-VSMCs. Western blot analysis indicated that Akt phosphorylation at Ser473 was increased in SPRR3-VSMCs relative to GFP-VSMCs (Fig. 6, A) and in WT-VSMCs relative to SPRR3-KO VSMCs (Fig. 6, B). However, there was no difference in phosphorylated p38 or phosphorylated ERK1/2 levels in GFP-VSMCs or SPRR3-VSMCs, indicating that SPRR3 was not a downstream effector of these pathways (Figure X in the online-only Data Supplement).
Figure 6. SPRR3 mediated a PI3K-dependent increase in Akt activation required for SPRR3-dependent VSMC survival benefits.

(A) Immunoblot analysis of pAkt in cell lysate from 24 hour serum-starved VSMCs overexpressing vector or SPRR3. (B) Immunoblot analysis of pAkt in cell lysate from 24-hour serum-starved VSMCs isolated from wildtype or SPRR3 KO mice. (C) Addition of IGF-1 (20ng/mL) increased pAkt levels proportionally in VSMCs overexpressing vector or SPRR3. The SPRR3-dependent pAkt increase was ablated with addition of Ly294002 (25μM). (D–E) Quantification of TUNEL-positive VSMCs overexpressing vector (D) and SPRR3 (E) with and without Ly294002 applied at the same time as oxidative stress challenge. Apoptosis was measured following 4 hours of oxidative stress (0.25mM H2O2). %TUNEL positive VSMCs calculated among >100 cells/treatment in 4 replicates. Bars represent mean ±SD. * < 0.05; ** < 0.01.
Several growth factors have been implicated in phosphorylated Akt (pAkt) activation in lesion VSMCs, including IGF-118–19. We investigated whether IGF-1 may enhance SPRR3-mediated increases in pAkt levels. Treatment of GFP- or SPRR3-VSMCs with IGF-1 triggered a proportionally equal increase in pAkt levels, indicating that SPRR3 was not a downstream effector of IGF-1 (Fig. 6, C). Addition of a PI3K/Akt inhibitor (Ly294002) abolishes pAkt in both GFP-VSMCs and SPRR3-VSMCs, indicating that SPRR3 increases pAkt levels in a canonical, PI3K/Akt-dependent manner (Fig. 6, C). To assess whether the observed SPRR3-mediated survival benefits were dependent on Akt activity, we evaluated GFP- and SPRR3-VSMCs with and without Ly294002 and assessed cell death using TUNEL staining. The pro-survival effect of SPRR3-overexpression was abolished in cells treated with Ly294002, in which we detected a 49% increase (p=0.005) in TdT-labeling compared to untreated cells (Fig. 6, D–E).
Discussion
We have identified SPRR3 as an atheroprotective factor in VSMCs that is uniquely upregulated in mouse and human atherosclerotic lesions. Global knockout of SPRR3 in the mouse led to significantly increased atheroma burden along with reduced survival of VSMCs, but not of endothelial cells (ECs). Additionally, transplantation of SPRR3+/+ bone marrow cells into mice lacking SPRR3 did not alter atherosclerosis development. Intriguingly, SPRR3 expression in cultured VSMCs was positively associated with Akt activity. In vitro studies in VSMCs challenged with oxidative stress indicated an SPRR3-mediated survival benefit. When these studies were conducted in the presence of a PI3K/Akt inhibitor, the SPRR3-mediated survival benefit was lost. Our findings indicated that SPRR3 expression in VSMCs played a critical role in Akt-mediated survival signaling in atherosclerotic lesions, protecting the atheroma from excessive VSMC apoptosis and leading to higher VSMC content in the fibrous cap.
Most studies intent on identification of atheroprotective genes have investigated ECs or immune cells. In ECs, studies have focused on key differences between gene expression in arterial tissue exposed to laminar versus turbulent flow, as exposure to turbulent flow is associated with increased risk of atheroma initiation20–25. In immune cells, studies have investigated the role of genes that either reduce inflammation or improve cholesterol homeostasis, both interventions leading to reduced lesion size and complexity26–29. Less is understood, however, about VSMC-specific atheroprotective genes. A few studies have investigated regulation of VSMC proliferation and intima-media thickness, mechanisms most critical at atheroma initiation30–32. More work is needed in investigating later stages of atheroma development where VSMC survival and maintenance of a synthetic phenotype may be more important, as these characteristics are critical for regulation of atheroma progression and fibrous cap stability33.
Previously, Bennett’s group demonstrated accelerated atheroma progression and a vulnerable phenotype in an inducible diphtheria toxin-driven mouse model of chronic, low level VSMC apoptosis34. A potential explanation for this effect may be that an increase in VSMC apoptosis within the lesion will, in turn, increase cellular debris and other pro-inflammatory stimuli, recruiting macrophages and other immune cells to the lesion and accelerating foam cell formation. The effect may also suggest a protective role of VSMCs in the lesion, either through an unidentified secreted factor(s) or through delay of lipid deposition or macrophage entry. While the mechanism(s) driving atheroma progression in lesions experiencing chronic VSMC apoptosis remains uncharacterized, Bennett’s model emphasizes the importance of reducing VSMC apoptosis in regulation of atheroma size and composition. Despite the potential for use in delaying atheroma progression, no one has previously identified a lesion-specific regulator of VSMC survival.
SPRR3 knockout mice demonstrated altered VSMC survival in atheromas, while VSMCs in normal arterial tissue remained unaffected. Cells residing in the atheroma microenvironment encounter a myriad of stressors, putting them at increased risk of cell death. Stressors in the atheroma milieu include extracellular cholesterol and oxidized fatty acids, hypoxic conditions, rising ROS levels, and an array of inflammatory cytokines released by both activated ECs and macrophages35–39. The critical role VSMCs play in synthesis and subsequent maintenance of the barrier between these atheroma components and the circulation makes them an indispensible part of a stable plaque. Our data suggested that SPRR3 activity specifically in the atheroma may be part of an adaptive response to the injurious atheroma microenvironment by protecting VSMCs from death.
The PI3K/Akt pathway is an important regulator of cell survival. We showed that upregulation of SPRR3 in VSMCs was positively associated with increased Akt phosphorylation; and the reverse was also true, as VSMCs lacking SPRR3 had reduced pAkt levels. The SPRR family has, until recently, been recognized solely as a family of structural proteins providing support against high levels of mechanical strain. Some evidence has been published showing an association between increased SPRR3 levels and increased colorectal or breast cancer cell proliferation and Akt activity7, 40, however the mechanism for this phenotype or whether SPRR3 plays a role in oncogenesis has not yet been studied. Previous work has not identified a kinase-interacting role for SPRR family members. Thus, our current study uncovered a novel function for SPRR3 through activation of Akt to promote VSMC survival. Consistent with our hypothesis, other groups have identified pro-survival activity for SPRR family members. One investigation suggested that SPRR2a may modulate p53 transcription and p300 acetylation status in cholangiocarcinoma cells leading to reduced apoptosis41. Another group demonstrated that SPRR1a is upregulated downstream of the gp130 pathway in cardiomyocytes and protected against ischemic stress-induced apoptosis42. Future studies will focus on elucidation of the precise molecular mechanism by which SPRR3 may activate Akt specifically in plaque VSMCs, allowing a targeted intervention aimed at VSMC protection within the lesion.
Our survival studies, in which cultured VSMCs under oxidative stress were treated with a PI3K/Akt inhibitor, showed that pAkt upregulation was required for an SPRR3-mediated survival benefit. The SPRR3-dependent increase in Akt activity occurs independently of the insulin-like growth factor signaling pathway, as treatment with IGF-1 results in a proportional increase in pAkt across cell types. Tissue-specific activators of Akt are rare. To our knowledge, the only other protein that promotes Akt activity in a tissue-specific manner is T-cell leukemia/lymphoma 1 (TCL1). Expressed only in lymphoid tissue, TCL1 was discovered due to its proto-oncogene activity in human T cell malignancies43. TCL1 increases Akt activation by binding the PH domain of Akt and causing a conformational change to promote Ser473 phosphorylation44. SPRR3 represents, to our knowledge, the first VSMC-specific activator of Akt and one that has the potential to mediate the VSMC atherosclerosis response.
Since chronic VSMC apoptosis leads to accelerated atherosclerosis, and SPRR3-mediated protection was lost following PI3K/Akt inhibition, the increase in atheroma burden in SPRR3 knockout mice may be at least partly attributable to altered Akt activity. Global knockout of Akt1 (the most widely expressed isoform of Akt) or Akt3 in ApoE-null mice leads to accelerated atheroma progression45, 26. Global Akt2 knockout on an LDLR-deficient background, meanwhile, leads to smaller, more complex atheromas with a 6-fold increase in % TUNEL-positive cells and reduced cap VSMC content46. Bone marrow transplantation studies revealed that Akt1 deficiency in the BM alone is insufficient to increase the atherosclerotic phenotype in ApoE-null mice, indicating the increased atherosclerosis is probably due to changes in the arterial cells45. Akt1−/− mice experience reduced survival of ECs, macrophages, and VSMCs, and Akt1 loss is associated with increased inflammatory molecule expression (e.g. IL-6, TNF-α) in the artery wall45, 47. Mice experiencing a global knockout of Akt3 also exhibit increased atherosclerosis on an ApoE-null background26. Loss of Akt3 did not affect macrophage survival, but foam cell formation was increased in Akt3−/− macrophages in vitro and rescue of Akt3 in the BM of Akt3-null mice was atheroprotective. While both the Akt1 and Akt3 studies examined the specific role of BM-derived cell expression of Akt relative to arterial expression, neither study attempted to separate the role of VSMC expression of Akt from that of ECs. In vitro studies have indicated that VSMCs isolated from atheroma in humans and mice have downregulated activity of Akt pathway members (e.g. FoxO3, GSK3-β) and are more prone to apoptosis13. In our study, upregulation of Akt activity by SPRR3 may be stabilizing atheroma VSMCs and thereby slowing plaque progression.
Altogether, we show loss of SPRR3 in the plaque accelerates atheroma progression while reducing VSMC pro-survival signals. While atheroprotective signaling has been identified in areas of shear stress, these pathways are generally downregulated in atheroma-prone regions experiencing turbulent mechanical stress. Our findings demonstrated that atheroprotective signals also occur in atheroma-resident cells. Moreover, the atheroma-specific expression pattern provided a unique opportunity for targeted intervention in atheroma VSMCs. Our data suggested lesion VSMC expression of SPRR3 provided a defense mechanism in atherosclerotic plaques, in which pro-survival Akt signaling was promoted to stabilize these cells under mechanical strain, slowing plaque progression. Identification of intermediary signaling interactions may provide key targets for future therapy of patients with early onset or advanced stages of CVD.
Supplementary Material
Significance Statement.
Our studies show that SPRR3 modulation of VSMC survival is crucial to reduced atheroma progression. We show that loss of normal atheroma expression of SPRR3 leads to increased atheroma burden and reduced VSMC survival. However, endothelial cells and hematopoietic cells are unaffected by SPRR3 loss. Increased SPRR3 expression, by contrast, protects VSMCs from oxidative stress-induced apoptosis in an Akt-dependent manner. Since arterial SPRR3 expression is limited to atheroma VSMCs, this novel atheroprotective gene provides a unique opportunity for future efforts at development of interventions that delay atheroma progression and vulnerability.
Acknowledgments
En face images were collected with the aid of the VUMC Cell Imaging Shared Resource. Histology was, in part, performed through VUMC Translational Pathology Shared Resource.
Sources of Funding
This work was supported by funding from the Veterans Affairs Merit Award to P.P. Young, and Vanderbilt University Clinical and Translational Science Award Grant (UL1 RR024975-01 from NCRR/NIH) to P.P. Young; HL105375 (VRB and MFL), HL57986 (SF) and DK59637 (Lipid, Lipoprotein and Atherosclerosis Core of the Vanderbilt Mouse Metabolic Phenotype Centers).
Nonstandard Abbreviations and Acronyms
- VSMC
Vascular smooth muscle cell
- EC
Endothelial cell
- SPRR3
Small proline-rich repeat protein 3
- ApoE
Apolipoprotein E
- α-SMA
Smooth muscle α-actin
Footnotes
Disclosures
None.
References
- 1.Roger VL, Go AS, Lloyd-Jones DM, et al. Heart disease and stroke statistics--2012 update: a report from the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zarins CK, Giddens DP, Bharadvaj BK, Sottiurai VS, Mabon RF, Glagov S. Carotid bifurcation atherosclerosis. Quantitative correlation of plaque localization with flow velocity profiles and wall shear stress. Circ Res. 1983;53:502–514. doi: 10.1161/01.res.53.4.502. [DOI] [PubMed] [Google Scholar]
- 3.Kwon GP, Schroeder JL, Amar MJ, Remaley AT, Balaban RS. Contribution of macromolecular structure to the retention of low-density lipoprotein at arterial branch points. Circulation. 2008;117:2919–2927. doi: 10.1161/CIRCULATIONAHA.107.754614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.National Heart L, and Blood Institute. Working Group on Atheroprotective Genes. 2000 Sep, 2000. [Google Scholar]
- 5.Young PP, Modur V, Teleron AA, Ladenson JH. Enrichment of genes in the aortic intima that are associated with stratified epithelium: implications of underlying biomechanical and barrier properties of the arterial intima. Circulation. 2005;111:2382–2390. doi: 10.1161/01.CIR.0000164235.26339.78. [DOI] [PubMed] [Google Scholar]
- 6.Fischer DF, Sark MW, Lehtola MM, Gibbs S, van de Putte P, Backendorf C. Structure and evolution of the human SPRR3 gene: implications for function and regulation. Genomics. 1999;55:88–99. doi: 10.1006/geno.1998.5622. [DOI] [PubMed] [Google Scholar]
- 7.Cho DH, Jo YK, Roh SA, Na YS, Kim TW, Jang SJ, Kim YS, Kim JC. Upregulation of SPRR3 promotes colorectal tumorigenesis. Mol Med. 2010;16:271–277. doi: 10.2119/molmed.2009.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pyle AL, Atkinson JB, Pozzi A, Reese J, Eckes B, Davidson JM, Crimmins DL, Young PP. Regulation of the atheroma-enriched protein, SPRR3, in vascular smooth muscle cells through cyclic strain is dependent on integrin alpha1beta1/collagen interaction. Am J Pathol. 2008;173:1577–1588. doi: 10.2353/ajpath.2008.080042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schapira K, Lutgens E, de Fougerolles A, Sprague A, Roemen A, Gardner H, Koteliansky V, Daemen M, Heeneman S. Genetic deletion or antibody blockade of alpha1beta1 integrin induces a stable plaque phenotype in ApoE−/− mice. Arterioscler Thromb Vasc Biol. 2005;25:1917–1924. doi: 10.1161/01.ATV.0000174807.90292.2f. [DOI] [PubMed] [Google Scholar]
- 10.Shi W, Wang X, Wang NJ, McBride WH, Lusis AJ. Effect of macrophage-derived apolipoprotein E on established atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2000;20:2261–2266. doi: 10.1161/01.atv.20.10.2261. [DOI] [PubMed] [Google Scholar]
- 11.Tabas I. Cholesterol in health and disease. J Clin Invest. 2002;110:583–590. doi: 10.1172/JCI16381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Linton MF, Atkinson JB, Fazio S. Prevention of atherosclerosis in apolipoprotein E-deficient mice by bone marrow transplantation. Science. 1995;267:1034–1037. doi: 10.1126/science.7863332. [DOI] [PubMed] [Google Scholar]
- 13.Allard D, Figg N, Bennett MR, Littlewood TD. Akt regulates the survival of vascular smooth muscle cells via inhibition of FoxO3a and GSK3. J Biol Chem. 2008;283:19739–19747. doi: 10.1074/jbc.M710098200. [DOI] [PubMed] [Google Scholar]
- 14.Baas AS, Berk BC. Differential activation of mitogen-activated protein kinases by H2O2 and O2- in vascular smooth muscle cells. Circ Res. 1995;77:29–36. doi: 10.1161/01.res.77.1.29. [DOI] [PubMed] [Google Scholar]
- 15.Lehoux S, Esposito B, Merval R, Loufrani L, Tedgui A. Pulsatile stretch-induced extracellular signal-regulated kinase 1/2 activation in organ culture of rabbit aorta involves reactive oxygen species. Arterioscler Thromb Vasc Biol. 2000;20:2366–2372. doi: 10.1161/01.atv.20.11.2366. [DOI] [PubMed] [Google Scholar]
- 16.Xu Q, Liu Y, Gorospe M, Udelsman R, Holbrook NJ. Acute hypertension activates mitogen-activated protein kinases in arterial wall. J Clin Invest. 1996;97:508–514. doi: 10.1172/JCI118442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ip JH, Fuster V, Badimon L, Badimon J, Taubman MB, Chesebro JH. Syndromes of accelerated atherosclerosis: role of vascular injury and smooth muscle cell proliferation. J Am Coll Cardiol. 1990;15:1667–1687. doi: 10.1016/0735-1097(90)92845-s. [DOI] [PubMed] [Google Scholar]
- 18.Bennett MR, Boyle JJ. Apoptosis of vascular smooth muscle cells in atherosclerosis. Atherosclerosis. 1998;138:3–9. doi: 10.1016/s0021-9150(98)00013-6. [DOI] [PubMed] [Google Scholar]
- 19.Bennett MR, Evan GI, Schwartz SM. Apoptosis of human vascular smooth muscle cells derived from normal vessels and coronary atherosclerotic plaques. J Clin Invest. 1995;95:2266–2274. doi: 10.1172/JCI117917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dai G, Vaughn S, Zhang Y, Wang ET, Garcia-Cardena G, Gimbrone MA., Jr Biomechanical forces in atherosclerosis-resistant vascular regions regulate endothelial redox balance via phosphoinositol 3-kinase/Akt-dependent activation of Nrf2. Circ Res. 2007;101:723–733. doi: 10.1161/CIRCRESAHA.107.152942. [DOI] [PubMed] [Google Scholar]
- 21.Dimmeler S, Zeiher AM. Exercise and cardiovascular health: get active to “AKTivate” your endothelial nitric oxide synthase. Circulation. 2003;107:3118–3120. doi: 10.1161/01.CIR.0000074244.82874.A0. [DOI] [PubMed] [Google Scholar]
- 22.Hahn C, Orr AW, Sanders JM, Jhaveri KA, Schwartz MA. The subendothelial extracellular matrix modulates JNK activation by flow. Circ Res. 2009;104:995–1003. doi: 10.1161/CIRCRESAHA.108.186486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hergenreider E, Heydt S, Treguer K, Boettger T, Horrevoets AJ, Zeiher AM, Scheffer MP, Frangakis AS, Yin X, Mayr M, Braun T, Urbich C, Boon RA, Dimmeler S. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat Cell Biol. 2012;14:249–256. doi: 10.1038/ncb2441. [DOI] [PubMed] [Google Scholar]
- 24.Park JG, Yoo JY, Jeong SJ, Choi JH, Lee MR, Lee MN, Hwa Lee J, Kim HC, Jo H, Yu DY, Kang SW, Rhee SG, Lee MH, Oh GT. Peroxiredoxin 2 deficiency exacerbates atherosclerosis in apolipoprotein E-deficient mice. Circ Res. 2011;109:739–749. doi: 10.1161/CIRCRESAHA.111.245530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Surapisitchat J, Hoefen RJ, Pi X, Yoshizumi M, Yan C, Berk BC. Fluid shear stress inhibits TNF-alpha activation of JNK but not ERK1/2 or p38 in human umbilical vein endothelial cells: Inhibitory crosstalk among MAPK family members. Proc Natl Acad Sci U S A. 2001;98:6476–6481. doi: 10.1073/pnas.101134098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ding L, Biswas S, Morton RE, Smith JD, Hay N, Byzova TV, Febbraio M, Podrez EA. Akt3 deficiency in macrophages promotes foam cell formation and atherosclerosis in mice. Cell Metab. 2012;15:861–872. doi: 10.1016/j.cmet.2012.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kyaw T, Tay C, Krishnamurthi S, Kanellakis P, Agrotis A, Tipping P, Bobik A, Toh BH. B1a B lymphocytes are atheroprotective by secreting natural IgM that increases IgM deposits and reduces necrotic cores in atherosclerotic lesions. Circ Res. 2011;109:830–840. doi: 10.1161/CIRCRESAHA.111.248542. [DOI] [PubMed] [Google Scholar]
- 28.Langlois D, Forcheron F, Li JY, del Carmine P, Neggazi S, Beylot M. Increased atherosclerosis in mice deficient in perilipin1. Lipids Health Dis. 2011;10:169. doi: 10.1186/1476-511X-10-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Navab M, Reddy ST, Van Lenten BJ, Fogelman AM. HDL and cardiovascular disease: atherogenic and atheroprotective mechanisms. Nat Rev Cardiol. 2011;8:222–232. doi: 10.1038/nrcardio.2010.222. [DOI] [PubMed] [Google Scholar]
- 30.Boucher P, Li WP, Matz RL, Takayama Y, Auwerx J, Anderson RG, Herz J. LRP1 functions as an atheroprotective integrator of TGFbeta and PDFG signals in the vascular wall: implications for Marfan syndrome. PLoS One. 2007;2:e448. doi: 10.1371/journal.pone.0000448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doran AC, Lehtinen AB, Meller N, Lipinski MJ, Slayton RP, Oldham SN, Skaflen MD, Yeboah J, Rich SS, Bowden DW, McNamara CA. Id3 is a novel atheroprotective factor containing a functionally significant single-nucleotide polymorphism associated with intima-media thickness in humans. Circ Res. 2010;106:1303–1311. doi: 10.1161/CIRCRESAHA.109.210294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karas RH, van Eickels M, Lydon JP, Roddy S, Kwoun M, Aronovitz M, Baur WE, Conneely O, O’Malley BW, Mendelsohn ME. A complex role for the progesterone receptor in the response to vascular injury. J Clin Invest. 2001;108:611–618. doi: 10.1172/JCI11374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clarke MC, Figg N, Maguire JJ, Davenport AP, Goddard M, Littlewood TD, Bennett MR. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med. 2006;12:1075–1080. doi: 10.1038/nm1459. [DOI] [PubMed] [Google Scholar]
- 34.Clarke MC, Littlewood TD, Figg N, Maguire JJ, Davenport AP, Goddard M, Bennett MR. Chronic apoptosis of vascular smooth muscle cells accelerates atherosclerosis and promotes calcification and medial degeneration. Circ Res. 2008;102:1529–1538. doi: 10.1161/CIRCRESAHA.108.175976. [DOI] [PubMed] [Google Scholar]
- 35.Deguchi JO, Yamazaki H, Aikawa E, Aikawa M. Chronic hypoxia activates the Akt and beta-catenin pathways in human macrophages. Arterioscler Thromb Vasc Biol. 2009;29:1664–1670. doi: 10.1161/ATVBAHA.109.194043. [DOI] [PubMed] [Google Scholar]
- 36.Geng YJ, Henderson LE, Levesque EB, Muszynski M, Libby P. Fas is expressed in human atherosclerotic intima and promotes apoptosis of cytokine-primed human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1997;17:2200–2208. doi: 10.1161/01.atv.17.10.2200. [DOI] [PubMed] [Google Scholar]
- 37.Geng YJ, Wu Q, Muszynski M, Hansson GK, Libby P. Apoptosis of vascular smooth muscle cells induced by in vitro stimulation with interferon-gamma, tumor necrosis factor-alpha, and interleukin-1 beta. Arterioscler Thromb Vasc Biol. 1996;16:19–27. doi: 10.1161/01.atv.16.1.19. [DOI] [PubMed] [Google Scholar]
- 38.Harada-Shiba M, Kinoshita M, Kamido H, Shimokado K. Oxidized low density lipoprotein induces apoptosis in cultured human umbilical vein endothelial cells by common and unique mechanisms. J Biol Chem. 1998;273:9681–9687. doi: 10.1074/jbc.273.16.9681. [DOI] [PubMed] [Google Scholar]
- 39.Reid VC, Brabbs CE, Mitchinson MJ. Cellular damage in mouse peritoneal macrophages exposed to cholesteryl linoleate. Atherosclerosis. 1992;92:251–260. doi: 10.1016/0021-9150(92)90285-o. [DOI] [PubMed] [Google Scholar]
- 40.Kim JC, Yu JH, Cho YK, Jung CS, Ahn SH, Gong G, Kim YS, Cho DH. Expression of SPRR3 is associated with tumor cell proliferation in less advanced stages of breast cancer. Breast Cancer Res Treat. 2012;133:909–916. doi: 10.1007/s10549-011-1868-5. [DOI] [PubMed] [Google Scholar]
- 41.Mizuguchi Y, Specht S, Lunz JG, 3rd, Isse K, Corbitt N, Takizawa T, Demetris AJ. SPRR2A enhances p53 deacetylation through HDAC1 and down regulates p21 promoter activity. BMC Mol Biol. 2012;13:20. doi: 10.1186/1471-2199-13-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pradervand S, Yasukawa H, Muller OG, et al. Small proline-rich protein 1A is a gp130 pathway- and stress-inducible cardioprotective protein. EMBO J. 2004;23:4517–4525. doi: 10.1038/sj.emboj.7600454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laine J, Kunstle G, Obata T, Sha M, Noguchi M. The protooncogene TCL1 is an Akt kinase coactivator. Mol Cell. 2000;6:395–407. doi: 10.1016/s1097-2765(00)00039-3. [DOI] [PubMed] [Google Scholar]
- 44.Laine J, Kunstle G, Obata T, Noguchi M. Differential regulation of Akt kinase isoforms by the members of the TCL1 oncogene family. J Biol Chem. 2002;277:3743–3751. doi: 10.1074/jbc.M107069200. [DOI] [PubMed] [Google Scholar]
- 45.Fernandez-Hernando C, Ackah E, Yu J, Suarez Y, Murata T, Iwakiri Y, Prendergast J, Miao RQ, Birnbaum MJ, Sessa WC. Loss of Akt1 leads to severe atherosclerosis and occlusive coronary artery disease. Cell Metab. 2007;6:446–457. doi: 10.1016/j.cmet.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rensing KL, de Jager SC, Stroes ES, Vos M, Twickler MT, Dallinga-Thie GM, de Vries CJ, Kuiper J, Bot I, von der Thusen JH. Akt2/LDLr double knockout mice display impaired glucose tolerance and develop more complex atherosclerotic plaques than LDLr knockout mice. Cardiovasc Res. 2013 doi: 10.1093/cvr/cvt252. [DOI] [PubMed] [Google Scholar]
- 47.Fernandez-Hernando C, Jozsef L, Jenkins D, Di Lorenzo A, Sessa WC. Absence of Akt1 reduces vascular smooth muscle cell migration and survival and induces features of plaque vulnerability and cardiac dysfunction during atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29:2033–2040. doi: 10.1161/ATVBAHA.109.196394. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.