

Abstract
Inflammation marks all stages of atherogenesis. DNA hypermethylation in the whole genome or specific genes is associated with inflammation and cardiovascular diseases. Therefore, we aimed to study whether inhibiting DNA methylation by DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine (5-aza-dC) ameliorates atherosclerosis in low-density lipoprotein receptor knockout (Ldlr−/−) mice. Ldlr−/− mice were fed an atherogenic diet and adminisered saline or 5-aza-dC (0.25 mg/kg) for up to 30 weeks. 5-aza-dC treatment markedly decreased atherosclerosis development in Ldlr−/− mice without changes in body weight, plasma lipid profile, macrophage cholesterol levels and plaque lipid content. Instead, this effect was associated with decreased macrophage inflammation. Macrophages with 5-aza-dC treatment had downregulated expression of genes involved in inflammation (TNF-α, IL-6, IL-1β, and inducible nitric oxidase) and chemotaxis (CD62/L-selectin, chemokine [C-C motif] ligand 2/MCP-1 [CCL2/MCP-1], CCL5, CCL9, and CCL2 receptor CCR2). This resulted in attenuated macrophage migration and adhesion to endothelial cells and reduced macrophage infiltration into atherosclerotic plaques. 5-aza-dC also suppressed macrophage endoplasmic reticulum stress, a key upstream signal that activates macrophage inflammation and apoptotic pathways. Finally, 5-aza-dC demethylated liver X receptor α (LXRα) and peroxisome proliferator-activated receptor γ1 (PPARγ1) promoters, which are both enriched with CpG sites. This led to overexpression of LXRα and PPARγ, which may be responsible for 5-aza-dC's anti-inflammatory and atheroprotective effect. Our findings provide strong evidence that DNA methylation may play a significant role in cardiovascular diseases and serve as a therapeutic target for prevention and treatment of atherosclerosis.
Macrophage inflammation plays a significant role in atherogenesis. A hallmark of atherosclerosis is the infiltration of macrophages into the arterial wall, which promotes atherosclerotic plaque formation, progression, and rupture (1, 2). The early development of atherosclerosis involves adhesion of monocytes to activated endothelial cells by vascular cell adhesion molecule 1. The chemokine monocyte chemoattractant protein 1 (MCP-1) and its receptor, chemokine (C-C motif) receptor 2 (CCR2), also play important roles in directing monocyte migration into the intima through chemotaxis (1). Within the intima, monocytes mature into macrophages, which accelerate atherosclerosis by producing proinflammatory cytokines, accumulating cholesterol, and initiating apoptosis (1, 2). Endoplasmic reticulum (ER) stress is one of the pathways that mediate lipid-induced macrophage inflammation during atherogenesis (3, 4). In addition, prolonged ER stress results in apoptotic cell death in macrophages, which may lead to plaque vulnerability and acute cardiac death in later-stage atherosclerosis (4). Experimental interventions that aim to reduce inflammatory processes or ER stress have atheroprotective effects (1). Accumulating evidence strongly supports macrophage inflammation and ER stress as key players in atherosclerosis development.
Although numerous studies have evaluated genetic factors related to atherosclerosis development, much less is known about epigenetic changes that occur without alterations in the DNA sequence. Epigenetic regulation, including DNA methylation, is a molecular link between environmental factors (eg, diets) and complex diseases, including atherosclerosis. DNA methylation of cytosines at primarily CpG dinucleotide sites is the most common epigenetic modification in the genome. CpGs are often enriched in the promoter and the first exon/5′-untranslated region of genes (5). Promoters of transcriptionally active genes are typically hypomethylated, whereas DNA hypermethylation can result in gene silencing by affecting the binding of methylation-sensitive DNA binding proteins and/or by interacting with various histone modifications and corepressors that alter DNA accessibility to transcriptional factors (5). DNA methylation alterations have also been implicated in cardiovascular disease and chronic inflammation. For instance, previous studies showed that global DNA hypermethylation is associated with inflammation and cardiovascular diseases in humans (6, 7). DNA hypermethylation was also found in specific genes such as the promoters of estrogen receptor α and p53, important regulators of smooth muscle cell proliferation, in atherosclerotic plaques from patients undergoing coronary surgery (8). These observations raise an interesting question as to whether inhibiting DNA methylation may have beneficial effects on atherogenesis prevention.
5-Aza-2′-deoxycytidine (5-aza-dC) is a nucleoside-based DNA methyltransferase inhibitor that induces demethylation (9–11). 5-Aza-dC is an anticancer drug and has been widely used in animal models to study the role of DNA methylation in cancer development (12). In this study, we investigated whether inhibiting DNA methylation pharmacologically using 5-aza-dC ameliorates atherosclerosis in low-density lipoprotein (LDL) receptor knockout (Ldlr−/−) mice. We found that 5-aza-dC treatment significantly diminished atherosclerosis development in Ldlr−/− mice without altering lipid and cholesterol metabolism. Instead, 5-aza-dC treatment supressed macrophage inflammation and infiltration into atherosclerotic plaques, reduced ER stress signals, and reduced apoptosis. This was associated with decreased DNA methylation status at the promoters of liver X receptor-α (LXRα) and peroxisome proliferator-activated receptor γ1 (PPARγ1), important transcriptional factors that play key roles during the development of atherosclerosis through regulating macrophage inflammation and/or cholesterol homeostasis (13–15).
Materials and Methods
Mice
Ldlr−/− mice that have been backcrossed to BL/6J background over 10 generations (16) were purchased from The Jackson Laboratory. Eight-week-old mice were put on an atherogenic diet containing 20% calories from fat and 0.2% (wt/wt) cholesterol (17) and were randomly assigned to receive saline or 5-aza-dC injection ip (0.25 mg/kg body weight, 3 times per week) for up to 30 weeks. Body weight was monitored weekly. All animal procedures were approved by the Institutional Animal Care and Use Committee at Wake Forest School of Medicine and Georgia State University.
Plasma lipid and cytokine analysis
Mice were euthanized after food removal for 4 hours. Blood was collected to analyze plasma lipid and cholesterol profiles as described previously (18). Plasma TNFα and IL-6 protein contents were measured using the mouse TNFα and IL6 ELISA kits (R&D Systems).
Quantification of atherosclerosis
Atherosclerosis development was analyzed as described previously (18, 19). Briefly, mice were euthanized 4 hours after food removal. The circulatory system was perfused with PBS before removing the heart and aorta. The upper one-third of the heart was dissected and embedded in optimal cutting temperature compound (Sakura Tissue-Tek), frozen, and stored at −80°C. Blocks were serially cut at 8-μm intervals and stained with hematoxylin and 0.5% Oil Red O (Sigma-Aldrich) to evaluate aortic sinus atherosclerotic intimal area. Atherosclerotic lesion area and Oil Red O-positive area were quantified using Image-Pro Plus software (Media Cybernetics).
Whole aorta (from sinotubular junction to iliac bifurcation) was dissected and fixed in 10% formalin, and the adventitia was cleaned. Aortas were opened along the longitudinal axis and pinned onto black silicon elastomer (Rubber-Cal) for the quantification of atherosclerotic lesion area. The percentage of total aortic surface covered with atherosclerotic lesions was quantified by Image-Pro Plus software (Media Cybernetics).
Immunohistochemistry
Sections of aortic sinus were immunostained with the following primary antibodies: rat monoclonal antibody against CD68 (Clone FA11, 1:75; AbD Serotec), rabbit polyclonal antibodies against phosphor-ER to nucleus signaling 1/inositol-requiring enzyme-1α (ERN1/IRE-1α) (1:500; Abcam) and C/EBP homologous protein (CHOP) (1:50, Santa Cruz Biotechnology), followed by staining with alkaline phosphatase-conjugated mouse antirat (for CD68, 1:50) or donkey antirabbit (for IRE-1α and Chop, 1:500) secondary antibodies (Jackson ImmunoResearch laboratories, West Grove, PA). Control slides contained no primary antibody. The CD68-, IRE-1α-, and Chop-positive areas were analyzed using Image-Pro Plus software (Media Cybernetics).
Terminal deoxynucleotidyl transferase dUTP nick end labeling assay
Terminal deoxynucleotidyl transferase dUTP nick end labeling assays were used to detect apoptotic cells in atherosclerotic plaques with an in situ cell death detection kit (Roche) according to the manufacturer's instructions.
Analysis of blood and aortic immune cell composition and gene expression
For analysis of aortic immune cell composition, the aortas were harvested, cleaned of adventitia, and digested to release the aortic cells as previously described (20). The cells were incubated with the following antibodies: allophycocyanin (APC)-F4/80 (AbD Serotec) and Pacific blue-B220, Percp-Cy5-CD4, and APC-Cy7-CD8 (BD Pharmingen). Peripheral blood was collected for leukocytes composition analysis (19). Red blood cells were lysed using Ammonium-Chloride-Potassium (ACK) lysing buffer (Lonza). The remaining white blood cells were incubated with the following antibodies: APC-F4/80 (AbD Serotec), Pacific blue-B220, Percp-cy5-CD4, APC-Cy7-CD8, phycoerythrin-CD11b and fluorescein isothiocyanate-7/4 (BD Pharmingen). Data were acquired with fluorescence-activated cell sorting (FACS) using BD FACSCanto II and were analyzed using Cell Quest or FACSDiva software (BD Pharmingen) as we described previously (21).
For gene expression in aortic macrophages, the digested aortas were used to isolate macrophages with rat antimouse F4/80 antibodies (AbD Serotec), followed by a pulldown of F4/80-positive cells with sheep antirat microbeads using magnetic-activated cell sorting system (Miltenyi Biotec) as we described previously (21). The isolated F4/80-positive aortic macrophages were used for RNA extraction with Tri Reagent (Molecular Research Center, Inc) and gene expression analysis (21).
Cell culture
Bone marrow-derived macrophages (BMDMs) and Raw264.7 macrophages were cultured as we previously described (22). In some experiments, Raw264.7 macrophages were treated with 5-aza-dC for up to 6 days and then either harvested or stimulated with lipopolysaccharide (LPS) (100 ng/mL) or stearate (C18:0, 250 μM) (Sigma-Aldrich) for the indicated time before cell collection for RNA and protein measurement. Stearate was conjugated with BSA at a 4:1 molar ratio before treatment (21).
Small interfering RNA knockdown
Raw264.7 macrophages were reversely transfected with PPARγ and/or LXRα small interfering RNA (siRNA) (SMARTpool; Dharmacon, Thermo Scientific) for 2 days using Lipofectamine RNAiMAX (Invitrogen).
Cholesterol efflux to apolipoprotein A1 or high-density lipoprotein
Macrophage cholesterol efflux to apolipoprotein A1 (apoA1) or high-density lipoprotein (HDL) was measured as described previously (23). Briefly, BMDMs pretreated with 5-aza-dC were incubated with 1 μCi/mL [3H]cholesterol in the presence of 50 μg/mL acetylated LDL (acLDL; Kalen Biomedical) for 24 hours. Cells were washed and equilibrated for 2 hours in medium with lipid-free BSA. HDL- or apoA-I–mediated cholesterol efflux was performed by adding HDL or apoA-I for an additional 24 or 4 hours, respectively. Radioactivity was measured in both medium and cell lysates. Cholesterol efflux was calculated as radioactivity in the medium divided by the sum of radioactivity in medium and cell lysate.
Cholesterol content in peritoneal macrophages
Ldlr−/− mice were treated with saline or 5-aza-dC for 12 weeks. Peritoneal macrophages were then isolated from these mice by lavage 3 days after ip injection of 3% thioglycolate (2 mL; Difco, BD Biosciences). Cells were then plated at 1 × 106 per well in 6-well plates for 2 hours, washed, and lipid-extracted with isopropanol. Cholesterol content in the cells was measured by gas-liquid chromatography as previously described (24).
Macrophage migration assay
The macrophage migration assay was performed using a CytoSelect 96-well cell migration assay kit (Cell Biolabs) according to the manufacturer's instructions. Briefly, BMDMs (2.5 × 105 per well) were pretreated with or without 5-aza-dC for 4 days. Cells were washed and plated in the membrane chamber, which was placed on top of the feeder tray. Cells were incubated with 10 ng/mL MCP-1 at 37°C for 20 hours. Migrated cells were dissociated from the membrane, lysed, and quantified using CyQuant GR fluorescent dye with a Victor 3 plate reader (PerkinElmer).
Macrophage adhesion assay
BMDM adhesion assays were conducted with a CytoSelect leukocyte-endothelium adhesion assay kit (Cell Biolabs) according to the manufacturer's instructions. Briefly, endothelial cells (6 × 104 per well, human microvascular endothelial cells, adult dermis; Invitrogen) were seeded in a 96-well plate and incubated overnight. BMDMs were pretreated with or without 5-aza-dC for 4 days and then labeled with LeukoTracker dye for 60 minutes at 37°C. BMDMs were washed with serum-free medium twice, and 4 × 105 cells per well were added into the 96-well plate containing endothelial cells. After 60 minutes incubation, wells were washed and adherent macrophages were lysed. Fluorescent signal from each well was detected with a Victor 3 plate reader (PerkinElmer).
Bisulfite conversion and pyrosequencing of LXRα and PPARγ1 promoter
Raw264.7 macrophages were treated with 0.5μM 5-aza-dC for up to 6 days, and genomic DNA was isolated by phenol/chloroform extraction. DNA (1 μg) was converted and purified using the EpiTect bisulfite kit (QIAGEN). The converted DNA was used as template to amplify the DNA sequence covering putative CpG sites at the LXRα or PPARγ1 promoter. Pyrosequencing primers for LXRα were designed by EpiGenDx, whereas those for PPARγ1 were designed using PyroMark Assay Design version 2.0 software (QIAGEN) (Supplemental Table 1). Pyrosequencing for LXRα was carried out by EpiGenDx, whereas that for PPARγ was carried out using PyroMark Q96ID machine (QIAGEN) according to manufacturer's instructions. Results were analyzed using Pyro-Q-CpG software version 1.0.9 (QIAGEN).
Cloning of the mouse LXRα and PPARγ1 promoters
Mouse LXRα promoter was amplified from bacterial artificial chromosome clones with the primers listed in Supplemental Table 2. The PCR products were then inserted into pGL3-Basic to generate pGL3-LXRα.
Mouse PPARγ1 promoter was amplified from bacterial artificial chromosome clones. Two primer sets (listed in Supplemental Table 2) were used to amplify 2 overlapping fragments on the PPARγ1 promoter. The 2 PCR fragments were digested with KpnI/ApaLI and ApaLI/BglII, respectively, and were then ligated to pGL3-Basic at KpnI/BglII sites to generate pGL3-PPARγ1. The constructs were confirmed by sequencing.
Luciferase reporter assay
To obtain unmethylated promoter, the reporter constructs were transformed into the dam−/dcm− Escherichia coli strain (New England Biolabs). To obtain fully methylated promoter, constructs were incubated with 3 U/μg SssI methylase in the presence of 160μM S-adenosylmethionine (New England Biolabs) at 37°C for 3 hours (25, 26). Methylation of pGL3-LXRα and pGL3-PPARγ1 was confirmed by checking the resistance of reporter constructs to HpaII and HpyCH4IV digestion, respectively. The unmethylated or fully methylated reporter constructs were transfected into Raw264.7 cells, and luciferase activity was measured using the dual-luciferase reporter assay system (Promega).
Total RNA extraction and quantitative RT-PCR
Total RNA was extracted using the Tri Reagent kit (Molecular Research Center), and gene expression was assessed by quantitative RT-PCR as we previously described (22).
Immunoblotting
Immunoblotting was performed as we previously described (22). The information for antibodies used in the study is in Table 1.
Table 1.
Antibodies
| Peptide/Protein Target | Name of Antibody | Manufacturer, Catalog No., and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|
| For IHC | ||||
| CD68 | CD68 | AbD Serotec | Rat; monoclonal | 75 |
| IRE-1α | IRE-1α | Abcam | Rabbit; polyclonal | 500 |
| Chop | Chop | Santa Cruz | Rabbit; polyclonal | 50 |
| For Western blotting | ||||
| IRE-1α | IRE-1α | Cell Signaling | Rabbit; polyclonal | 1000 |
| β-Actin | β-Actin | Cell Signaling | Rabbit; polyclonal | 1000 |
Abbreviation: IHC, immunohistochemistry.
Statistics
All data are expressed as mean ± SE. Differences between groups were analyzed for statistical significance by t test or one-way or two-way ANOVA as appropriate.
Results
5-Aza-dC ameliorates the development of atherosclerosis
To study whether inhibiting DNA methylation by 5-aza-dC affects atherosclerosis development, we fed 8-week-old male Ldlr−/− mice an atherogenic diet and treated them with either saline or low-dose 5-aza-dC (0.25 mg/kg) for up to 30 weeks. This low dose of 5-aza-dC has been shown to effectively reduce tumor genesis without any visible adverse effects over a period of 18 weeks (27). As expected, 5-aza-dC treatment did not alter body weight, liver weight, or epididymal fat pad mass (Supplemental Figure 1, A and B), nor did it change food intake (data not shown). However, 5-aza-dC treatment significantly decreased aortic root atherosclerotic lesion intimal area by 35% (Figure 1, A and B) and atherosclerotic surface lesion area in whole aorta by 35% (Figure 1, C and D) compared with control mice receiving saline injection.
Figure 1.
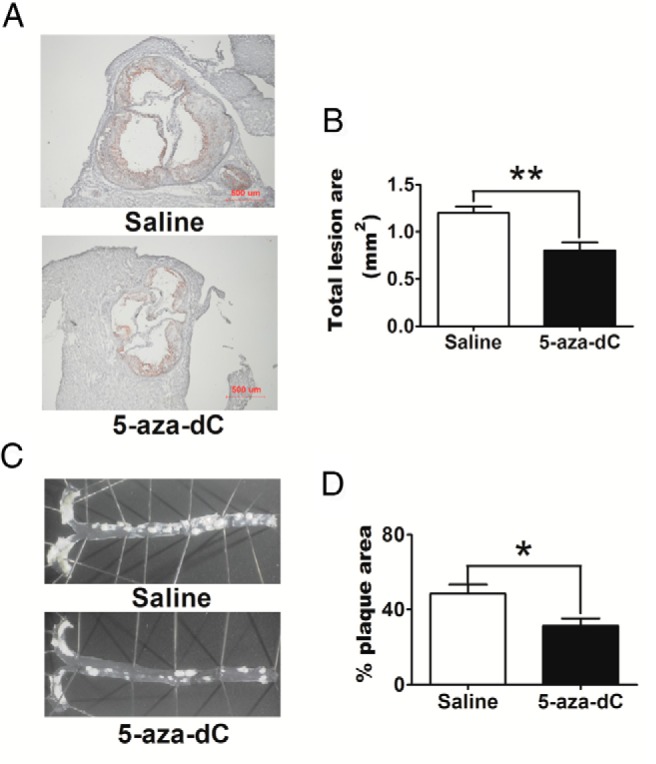
5-Aza-dC ameliorates atherosclerosis development in Ldlr−/− mice. A, Representative cross-sectional image of aortic sinus stained with Oil Red O. B, Quantification of aortic lesion area. C, Representative images of aortic surface lesions. D, Aortic surface lesion area normalized to total aortic surface area. Eight-week-old male Ldlr−/− mice (n = 6 per group) were fed an atherogenic diet and were ip injected with either saline or 5-aza-dC (0.25 mg/kg body weight) 3 times a week. Quantification of atherosclerosis was conducted as described in Materials and Methods. For B and D, data are expressed as mean ± SEM; n = 6 per group. *, P < .05; **, P < .01.
5-Aza-dC does not affect plasma lipid profiles and foam cell formation
The development of atherosclerosis is affected by various risk factors, one of which is elevated plasma cholesterol concentrations (28). Surprisingly, 5-aza-dC treatment did not change plasma triglyceride, total cholesterol and cholesterol ester levels, and only slightly reduced free cholesterol levels (Supplemental Figure 1C) relative to saline-injected control mice. In addition, there was also no difference in plasma very-low-density lipoprotein, LDL, or HDL cholesterol concentrations between 5-aza-dC- and saline-treated groups (Supplemental Figure 1, D and E).
Macrophages play a critical role in the development of atherosclerosis by accumulating lipid and cholesterol to form lipid-laden foam cells (28). However, we found that 5-aza-dC did not change the amount of total cholesterol, FC, and CE accumulated in peritoneal macrophages isolated from 5-aza-dC-treated Ldlr−/− mice (Supplemental Figure 1F). In addition, 5-aza-dC had no effect on cholesterol efflux to HDL and even slightly decreased cholesterol efflux to apoA1 (Supplemental Figure 1, G and H). Finally, 5-aza-dC treatment did not alter neutral lipid deposition in aortic root atherosclerotic plaques (Supplemental Figure 1I). These data suggest that inhibiting DNA methylation by 5-aza-dC reduces atherosclerosis independent of lipid and cholesterol metabolism in atherogenic Ldlr−/− mice.
5-Aza-dC reduces macrophage content and inflammation in atherosclerotic plaques
Macrophage inflammation is another risk factor that plays an important role in the development of atherosclerosis (2). We therefore explored whether 5-aza-dC ameliorates atherosclerosis by inhibiting macrophage inflammation. Using immunohistochemistry, we stained macrophages in aortic sinus sections with anti-CD68 antibodies and found that atherosclerotic plaque macrophage content was significantly lower in 5-aza-dC-treated mice (Figure 2, A and B). Using FACS analysis, we found that the percentage of F4/80+ macrophages was significantly decreased in total live cells isolated from whole aorta in 5-aza-dC-treated mice compared with that of saline-treated controls (Figure 2C). CD8+ T lymphocytes and B220+ B lymphocytes were also decreased in the 5-aza-dC-treated group, whereas there was no significant difference in CD4+ T lymphocytes (Supplemental Figure 2A). These data suggested that 5-aza-dC treatment in Ldlr−/− mice resulted in reduced infiltration of immune cell populations into atherosclerotic lesions.
Figure 2.
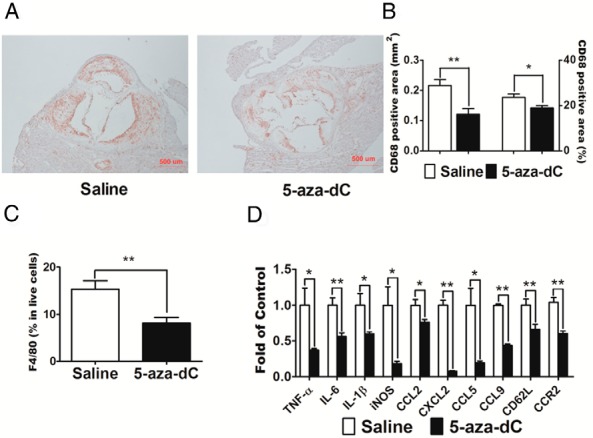
5-aza-dC reduces macrophage content and inflammation in intima of artery wall in Ldlr−/− mice. A, Representative cross-sectional image of aortic sinus stained with CD68 antibody. B, Quantification of CD68-positive lesion area (square millimeters) and percentage in total lesion area. C, 5-Aza-dC decreases the percentage of F4/80+ cells in live aortic cells. Eight-week-old male Ldlr−/− mice (n = 6 per group) were treated with 5-aza-dC as described in Materials and Methods. For A and 2, the upper one-third of the heart was dissected and embedded in Optimum cutting temperature (OCT) medium, cryosectioned at 8-μm intervals, and stained with anti-CD68 antibodies. For C, aortas were digested to release aortic cells. The cells were stained with APC-conjugated anti-F4/80 antibodies and were analyzed using BD FACSCanto II as described in Materials and Methods. D, Expression of inflammatory genes in aortic macrophages. Aortic macrophages were isolated using a magnetic-activated cell sorting system as described in Materials and Methods. The expression of inflammatory genes was measured by real-time RT-PCR and normalized to cyclophilin; n = 8 per group. Data are expressed as mean ± SEM. *, P < .05; **, P < .01.
5-Aza-dC (decitabine) has been successfully used in treating acute and chronic myelodysplastic leukemia (9, 11), and B and T cell lymphomas (29–31). To study whether 5-aza-dC treatment affects hematopoiesis, we analyzed circulating leukocyte populations in mice treated with saline or 5-aza-dC using FACS analysis. We have gated blood monocytes based on their surface expression of CD11b and the monocyte/macrophage-specific marker F4/80 (32). We found that there was no difference in circulating CD11b+F4/80+ cells between saline and 5-aza-dC-treated mice (Supplemental Figure 2B), nor is there any difference in circulating CD11b+F4/80+7/4low resident and CD11b+F4/80+7/4high inflammatory monocyte subset composition (Supplemental Figure 2C), the latter being the monocyte subset that infiltrates into inflammatory sites, including atherosclerotic lesions (33, 34). In addition, we did not find any differences in circulating CD4+ and CD8+ T lymphocytes between saline- and 5-aza-dC-treated mice (Supplemental Figure 2, D and E). However, 5-aza-dC treatment did reduce circulating B220+ B lymphocyte populations (Supplemental Figure 2F). Our data suggested that with the dose and length used in this study, 5-aza-dC treatment in Ldlr−/− mice did not change circulating leukocyte populations of monocyte and resident and inflammatory monocyte subset, CD4+ and CD8+ T lymphocytes, but reduced B220+ B lymphocyte populations. The role of B lymphocytes in atherosclerosis is not fully elucidated (35), whereas it is well-documented that monocyte/macrophage infiltration into the arterial wall significantly promotes atherosclerotic plaque formation and progression (1, 2). We therefore further studied the effect of 5-aza-dC on monocyte/macrophage infiltration and inflammation.
We found that the expression of proinflammatory cytokines, such as TNF-α, IL-6, IL-1β, and inducible nitric oxide synthase (iNOS), was significantly reduced in macrophages isolated from atherosclerotic plaques from 5-aza-dC-treated mice compared with that of saline-treated mice (Figure 2D). Moreover, 5-aza-dC also suppressed the expression of chemokines and adhesion molecules such as chemokine (C-C motif) ligand 2 (CCL2)/MCP-1, chemokine C-X-C motif ligand 2 (CXCL2), CCL5, CCL9, CD62L/L-selectin, and the CCL2 receptor CCR2, in macrophages isolated from atherosclerotic plaques (Figure 2D). These data suggest that 5-aza-dC inhibits the expression of genes involved in inflammatory and chemotactic processes, which may contribute to the decreased macrophage infiltration into atherosclerotic plaques observed in our 5-aza-dC-treated animal models. Consistent with reduced macrophage inflammatory gene expression, plasma levels of IL-6 and TNF-α were also reduced in 5-aza-dC-treated Ldlr−/− mice (Supplemental Figure 3, A and B).
5-Aza-dC suppresses macrophage inflammation, migration, and adhesion
To determine whether 5-aza-dC suppression of plaque macrophage inflammation is a direct effect of 5-aza-dC or is secondary to a systemic effect on other tissues, we tested whether 5-aza-dC suppressed macrophage inflammation in vitro. It has been reported that 5-aza-dC led to rapid loss of DNA methyltransferase activity in cell culture at a dose low enough to avoid triggering cell death (36). Using a similar dose range, we found that there was no difference in DNA amount in macrophages treated with 5-aza-dC at up to 2μM compared with saline-treated cells (Supplemental Figure 4). We therefore chose a low dose of 5-aza-dC (0.5μM) that did not affect macrophage proliferation and viability. We found that pretreating Raw264.7 macrophages with 0.5μM 5-aza-dC substantially inhibited basal and LPS-stimulated expression of proinflammatory genes, such as TNF-α, IL-6, and iNOS, and chemokines CCL2, CCL5, and CCL9 (Figure 3, A–F). Similar results were observed on 5-aza-dC inhibition of the saturated fatty acid stearate-stimulated gene expression in macrophages (Figure 3G).
Figure 3.

5-aza-dC suppresses macrophage inflammation, migration, and adhesion. A–G, 5-Aza-dC inhibits LPS-stimulated (A–F) and stearate-stimulated (G) expression of proinflammatory genes in macrophages. Raw264.7 macrophages were pretreated with 5-aza-dC (0.5μM) for 4 days and then treated with LPS (100 ng/mL) (A–F) or stearate (C18:0, 250μM) (G) for 4 hours. Gene expression was measured by real-time RT-PCR and normalized to cyclophilin; n = 4 per group. H–I, 5-Aza-dC reduces macrophage adhesion to endothelial cells (H) and migration (I). BMDMs were pretreated with 0.5μM 5-Aza-dC for 4 days, and BMDM-endothelium adhesion or migration assays were performed as described in Materials and Methods; n = 5 per group. All data are expressed as mean ± SEM. *, P < .05; **, P < .01.
To test whether 5-aza-dC's anti-inflammatory effect affects macrophage function, we performed macrophage adhesion and chemotaxis assays in BMDMs. We found that 5-aza-dC pretreatment markedly reduced macrophage adhesion to endothelial cells and suppressed its ability to migrate in an MCP-1-induced chemotaxis assay (Figure 3, H and I).
5-Aza-dC inhibits macrophage ER stress
ER stress has emerged as a key upstream signal that can activate macrophage inflammation, leading to the production of inflammatory mediators (3). In addition, prolonged ER stress results in macrophage apoptosis, which may lead to plaque vulnerability and acute cardiac death in advanced atherosclerosis (37). We found that 5-aza-dC treatment markedly downregulated ERN1/IRE-1α and CHOP mRNA levels in a time course ranging from 2 to 6 days (Figure 4, A and B). 5-Aza-dC treatment also dramatically downregulated the total protein and phosphorylation of ERN1/IRE-1α in macrophages treated with stearate, a physiological inducer of macrophage ER stress (Figure 4, C–E) (3). 5-Aza-dC inhibition of macrophage ER stress might be physiologically significant, as our immunohistochemical data revealed that 5-aza-dC injection significantly reduced phospho-ERN1/IRE-1α and CHOP levels in atherosclerotic plaques of Ldlr−/− mice fed an atherogenic diet compared with saline-treated mice (Figure 5, A–D). The downregulation of ER stress is associated with decreased number of apoptotic cells in the plaques of Ldlr−/− mice receiving 5-aza-dC treatment (Figure 5, E and F). Our data indicate that 5-aza-dC suppresses macrophage ER stress, which may contribute to its inhibition of inflammation and its protective effect against atherosclerosis (3, 4).
Figure 4.

5-Aza-dC inhibits macrophage ER stress. A and B, 5-Aza-dC inhibits ERN1/IRE-1α (A) and Chop (B) mRNA expression in macrophages. Raw264.7 macrophages were treated with 5-aza-dC (0.5μM) for up to 6 days. ERN1/IRE-1α (A) and Chop (B) mRNA expression was measured by real-time RT-PCR and normalized to cyclophilin; n = 4 per group. C–E, 5-Aza-dC inhibits phosphorylated (p-) and total ERN1/IRE-1α in macrophages. Raw264.7 macrophages were pretreated with 5-aza-dC (0.5μM) for 2 days and then were stimulated with stearate (C18:0, 250μM) for 1 day. Phosphorylated and total ERN1/IRE-1α was measured by immunoblotting. A representative blot is shown in C. The blots were quantitated for total (D) and phosphorylated (E) ERN1/IRE-1α expression with a Li-COR Odyssey infrared imager system and normalized to that of β-actin; n = 3 per group. All data are expressed as mean ± SEM. **, P < .01 (A and B); for D and E, groups labeled with different superscripts are statistically different from each other, P < .05.
Figure 5.

5-Aza-dC reduces ER stress and apoptosis in atherosclerotic plaques of Ldlr−/− mice. A and B, Representative cross-sectional image of aortic sinus stained with phospho-ERN1/IRE-1α (A) or Chop (B) antibodies. C and D, Quantification of phospho-ERN1/IRE-1α (C) or Chop (D) positive areas. E and F, 5-Aza-dC reduces apoptosis in atherosclerotic plaques of Ldlr−/− mice: E, representative images of apoptotic cells in aortic sinus; F, quantification of apoptotic cell numbers per section. All data are expressed as mean ± SEM; n = 6 per group. *, P < .05; **, P < .01. Abbreviation: TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
Demethylation of LXRα and PPARγ1 promoters are potential targets for 5-aza-dC's atheroprotective effect
LXRα plays a key role in the regulation of atherogenesis through its abilities to 1) modulate cholesterol efflux and 2) antagonize macrophage inflammation (14, 38). Interestingly, we found that 5-aza-dC markedly upregulated LXRα mRNA levels in Raw264.7 macrophages in a time-dependent manner (Figure 6A). Moreover, LXRα mRNA expression was significantly upregulated in macrophages isolated from atherosclerotic plaques of 5-aza-dC-treated mice compared with that of saline-treated mice (Figure 6B).
Figure 6.

5-Aza-dC induces LXRα and PPARγ1 expression via regulating their promoter DNA methylation. A and B, 5-Aza-dC stimulates LXRα mRNA in Raw26.47 macrophages (A) and aortic macrophages (B). C and D, 5-Aza-dC stimulates PPARγ mRNA in Raw264.7 macrophages (C) and aortic macrophages (D). In A and C, Raw264.7 macrophages were treated with 5-aza-dC (0.5μM) for up to 6 days; n = 6 per group. In B and D, aortic macrophages were isolated from saline- or 5-aza-dC-treated Ldlr−/− mice; n = 6 per group. LXRα and PPARγ mRNA were measured by real-time RT-PCR and normalized to cyclophilin. E and F, Promoter activities of LXRα (E) and PPARγ1 (F) were regulated by DNA methylation. LXRα and PPARγ1 promoters (Supplemental Figure 5, A and B) were cloned into pGL3-luciferase expression vector. Either unmethylated or fully methylated reporter constructs were transfected into Raw264.7 cells, and luciferase activity was measured; n = 3 per group. G and H, 5-Aza-dC demethylates LXRα promoter. Raw264.7 macrophages were treated with 5-aza-dC (0.5μM) for up to 6 days. CpG methylation was measured by pyrosequencing; n = 3 per group. In G, the average DNA methylation level at the LXRα promoter from CpG sites 9 to 25 (according to Supplemental Figure 6A) was calculated and expressed as a function of treatment time. H, DNA methylation levels at individual CpG sites on LXRα promoter (according to Supplemental Figure 5A) in Raw264.7 macrophages treated with 5-aza-dC for 6 days. I, DNA methylation levels at PPARγ1 promoter (according to Supplemental Figure 5B) in Raw264.7 macrophages treated with 5-aza-dC for 4 days; n = 3 per group. All data are expressed as mean ± SEM. For E and F, groups labeled with different superscripts are statistically different from each other, P < .05; for the rest of the graphs: *, P < .05; **, P < .01.
PPARγ also has potent anti-inflammatory and cardioprotective effects during the development of atherosclerosis (15, 39–41). There are 2 isoforms of PPARγ, namely PPARγ1 and PPARγ2 (42–44). Whereas PPARγ1 is more broadly expressed in various tissues including adipocytes and macrophages, PPARγ2 is expressed only in adipocytes (45). Using a real-time RT-PCR primer/probe set recognizing both isoforms, we found that PPARγ mRNA levels were markedly upregulated in both 5-aza-dC-treated Raw264.7 macrophages (Figure 6C) and in macrophages isolated from atherosclerotic plaques of 5-aza-dC-treated Ldlr−/− mice compared with saline-treated controls (Figure 6D). However, we could not detect PPARγ2 expression in macrophages using a primer/probe set specific for the PPARγ2 isoform (data not shown), similar to a previous report (45). This indicates that PPARγ1 is the major form in macrophages and that 5-aza-dC regulates its expression.
The proximal promoters and 5′-untranslated regions of both LXRα and PPARγ1 are enriched with CpG islands (Supplemental Figure 5, A and B). This raises the possibility that LXRα and PPARγ1 promoter DNA methylation status may be regulated by 5-aza-dC. Indeed, luciferase activity of unmethylated LXRα promoter was ∼200-fold higher than that of fully methylated promoter constructs (Figure 6E), suggesting that the LXRα promoter activity was regulated by DNA methylation. Similar results were obtained using unmethylated and fully methylated PPARγ1 promoter luciferase constructs (Figure 6F). In addition, treatment of macrophages with 5-aza-dC time-dependently decreased average DNA methylation levels at the LXRα promoter, starting after 1 day of 5-aza-dC treatment, with maximum reduction at 6 days of treatment (Figure 6G). Supplemental Figure 5C and Figure 6H show that 5-aza-dC inhibited DNA methylation levels at individual CpG sites on the LXRα promoter in macrophages after 4 and 6 days of treatment, respectively. Similarly, 5-aza-dC significantly reduced DNA methylation levels of PPARγ1 at the promoter/5-untranslated region after 4 days of treatment (Figure 6I). These data suggest that the promoters of LXRα and PPARγ1 can be regulated by DNA methylation; 5-aza-dC may induce LXRα and PPARγ1 expression through reducing DNA methylation on their promoters, which may be responsible for 5-aza-dC's anti-inflammatory and atheroprotective effects.
To further study whether LXRα and/or PPARγ is necessary in mediating 5-aza-dC's effect on inhibiting macrophage inflammation and ER stress, we deleted LXRα and/or PPARγ in Raw264.7 macrophages using siRNAs targeting the 2 genes. As expected, 5-aza-dC significantly reduced basal and LPS-induced expression of genes involved in inflammation and chemotaxis, including TNF-α, IL-6, CCL2, and CCL5 in Raw264.7 macrophages transfected with scrambled siRNAs (Figure 7, A–D). Deletion of LXRα or PPARγ partially abolished 5-aza-dC's effect, whereas deleting both LXRα and PPARγ exerted a synergistic effect on abolishing 5-aza-dC's inhibitory effects on LPS-induced inflammatory and chemotactic gene expression (Figure 7, A–D). Similarly, 5-aza-dC significantly reduced ER stress gene expression, including IRE-1α and CHOP in Raw264.7 macrophages transfected with scrambled siRNAs (Figure 7, E and F). Deletion of LXRα or PPARγ partially abolished 5-aza-dC's effect, whereas deleting both LXRα and PPARγ exerted an additive effect on abolishing 5-aza-dC's inhibitory effects on ER stress gene expression (Figure 7, E and F). Interestingly, knocking down PPARγ was more potent in abolishing 5-aza-dC's inhibitory effects on inflammatory and chemotactic gene expression, whereas knocking down LXRα was more potent in abolishing 5-aza-dC's inhibitory effects on ER stress gene expression.
Figure 7.

PPARγ and LXRα knockdowns substantially reverse the inhibitory effects of 5-aza-dC on inflammation and ER stress. A–F, PPARγ and LXRα knockdowns additively reverse the inhibitory effects of 5-aza-dC on TNF-α (A), IL-6 (B), CCL2 (C), CCL5 (D), IRE-1α (E), and Chop (F) mRNA expression in Raw264.7 macrophages. Raw264.7 macrophages were reversely transfected with PPARγ1 and/or LXRα siRNAs. Cells were then pretreated with 5-aza-dC (0.5μM) for 4 days. In A–D, cells were further stimulated with LPS (100 ng/mL) for 4 hours. Gene expression was measured by real-time RT-PCR and normalized to cyclophilin. All data are expressed as mean ± SE; n = 6 per group. Groups labeled with different letters are statistically significantly different, P < .05.
Discussion
Most complex diseases, including cardiovascular disease, are the result of gene and environment interactions. One mechanism by which environmental factors affect gene expression patterns involves reprograming the epigenome (46, 47). Growing evidence converges to suggest that epigenetic events, such as DNA methylation, figure prominently in the development of atherosclerosis (8). During the preparation of this manuscript, Dunn et al (48) reported that 5-aza-dC treatment reduced aortic lesions in apoE−/− mice through suppression of endothelial inflammation. In addition, 5-aza-dC can also demethylate FOXP3, leading to increased regulatory T cell populations (49, 50) and thereby reducing atherosclerosis (51). Consistent with these findings, we demonstrate the atheroprotective effect of 5-aza-dC in Ldlr−/− mice, indicating that DNA methylation may play an important role in the development of atherosclerosis. However, we demonstrate for the first time that the atheroprotective effect of 5-aza-dC may be due to its ability to suppress the macrophage inflammation and ER stress, 2 key events involved in the development of atherosclerosis and plaque vulnerability, by demethylating LXRα and PPARγ1 promoters.
When we used a low dose of 5-aza-dC in the current study that did not affect body weight, fat mass, and food intake, we found that 5-aza-dC had minimal effects on hematopoiesis, because it did not change circulating CD4+ and CD8+ T lymphocyte populations, nor did it change the circulating monocyte population. Although 5-aza-dC reduced the blood B220+ B lymphocyte population in drug-treated animals, the role of B lymphocytes in atherosclerosis is not fully elucidated (35). On the other hand, it is well documented that macrophages play an important role in the development of atherosclerosis (2), and we found that 5-aza-dC specifically reduced macrophage infiltration into atherosclerotic plaques without altering the circulating monocyte population as well as circulating CD11b+F4/80+7/4low resident and CD11b+F4/80+7/4High inflammatory monocyte subset composition. We also found that 5-aza-dC treatment inhibited inflammatory and chemotactic gene expression in aortic macrophages isolated from 5-aza-dC-treated atherogenic Ldlr−/− mice. Similarly, treating macrophages with 5-aza-dC at a low dose that did not cause cytotoxicity significantly suppressed LPS- and stearate-induced inflammatory and chemotactic gene expression and suppressed macrophage adhesion and migration in in vitro assays. Our in vitro studies may also partly explain the in vivo phenotype we observed, that low-dose 5-aza-dC suppressed macrophage inflammation and infiltration into atherosclerotic plaques without altering circulating monocyte populations.
ER stress is a key upstream signal that can activate macrophage inflammation and apoptotic pathways, thus playing an important role in the development and progression of atherosclerosis (3, 4). We found that 5-aza-dC potently inhibited macrophage ER stress, leading to fewer apoptotic cells in atherosclerotic plaques than that of saline-treated mice. This may contribute partly to the anti-inflammatory and atheroprotective effects of 5-aza-dC. Therefore, our data suggest that inhibiting DNA methylation with low-dose 5-aza-dC in atherogenic Ldlr−/− mice may have atheroprotective benefits primarily by inhibiting monocyte/macrophage infiltration into atherosclerotic plaques and suppressing macrophage inflammation and ER stress without affecting monocyte/macrophage lineage development and cell viability.
LXRα and PPARγ are important transcriptional factors that play key roles during the development of atherosclerosis through regulating macrophage inflammation and/or cholesterol homeostasis (13–15). LXR family members, including LXRα and LXRβ, are cholesterol sensors that lower cholesterol levels by regulating the expression of genes involved in lipid metabolism and reverse cholesterol transport in response to specific oxysterol ligands in macrophages (52). Recent data suggest that LXRs also exert potent anti-inflammatory effects. LXR activation significantly suppresses bacteria- and LPS-induced proinflammatory cytokine expression in vitro and inhibits inflammatory gene expression in the aortas of atherosclerotic mice in vivo (14). The anti-inflammatory function of LXR is likely mediated through its ability to inhibit nuclear factor-κB (NF-κB) signaling (14). Consequently, LXR activation is atheroprotective, whereas LXR deletion exacerbates atherosclerosis development (13). Similarly, in addition to its role in lipid homeostasis, PPARγ also has potent anti-inflammatory effects (38). Numerous data demonstrate that PPARγ activation ameliorates inflammatory responses in various tissues, including pancreas, lungs, joints, nervous system, and gastrointestinal tract (38). Activation of PPARγ inhibits inflammatory gene expression by several mechanisms including suppression of NF-κB signaling (14), a key transcriptional factor that directly regulates the expression of many proinflammatory cytokines linked to atherosclerosis (53). PPARγ can inhibit the ability of NF-κB to drive inflammatory gene expression by physically interacting with NF-κB to reduce its DNA binding to target gene promoters, increasing cytoplasmic redistribution of the NF-κB subunit p65, and increasing the stability of the NF-κB corepressors (14). In addition, mice with macrophage-specific deletion of PPARγ display obesity-induced insulin resistance, primarily due to exacerbated macrophage inflammation (54), whereas conditional deletion of PPARγ in macrophages increases atherosclerosis in Ldlr−/− mice (39). Our present study shows that 5-aza-dC strongly antagonizes inflammatory gene expression by demethylating LXRα and PPARγ1 promoters and inducing their gene expression. Although we did not directly examine the NF-κB signaling in the present study, the inhibitory effect of 5-aza-dC on inflammatory gene expression may be mediated by LXRα and PPARγ1 suppression of NF-κB signaling, because the inflammatory genes inhibited by 5-aza-dC treatment are mostly NF-κB downstream target genes such as TNF-α, IL-6, IL-1β, etc. Indeed, 5-aza-dC has been shown to inhibit NF-κB signaling in several previous reports (55–57). These data demonstrate that both LXRα and PPARγ are molecular targets for 5-aza-dC to antagonize macrophage inflammation and ameliorate atherosclerosis development possibly through their abilities to suppress NF-κB signaling.
LXRα and PPARγ may have atheroprotective effects through their regulation of lipid/cholesterol metabolism and/or inflammation (13–15). However, we found that 5-aza-dC did not affect cholesterol and lipid metabolism, because there was no difference in plasma lipid and cholesterol concentrations or lipoprotein cholesterol distribution, macrophage cholesterol levels, and atherosclerotic plaque lipid content between saline- and 5-aza-dC-treated mice. Nor did we find that 5-aza-dC affected cholesterol efflux from macrophages. The mechanisms underlying the discrepancy between 5-aza-dC's anti-inflammatory effect and its inability to regulate cholesterol/lipid metabolism is not clear, although it appears that 5-aza-dC induces LXRα and PPARγ1 expression, which may affect both pathways. Possibly other pathways, which may be regulated by 5-aza-dC, may act in concert with 5-aza-dC's effects on LXRα and PPARγ1 expression to regulate inflammation, but not lipid/cholesterol metabolism. Nonetheless, 5-aza-dC's anti-inflammatory function appears to play a dominant role in its protection against atherosclerosis.
We found that inhibiting DNA methylation by 5-aza-dC significantly reduced ER stress and apoptosis in atherosclerotic plaques, which may contribute to its atheroprotective effects. The mechanism by which 5-aza-dC suppresses ER stress is not clear. Inducing ER stress suppresses PPARγ expression (58), whereas the PPARγ agonist pioglitazone reduces ER stress in liver (59). However, it is not known whether the effect of pioglitazone on reducing ER stress is dependent on PPARγ and whether a similar effect takes place in macrophages. On the other hand, activating LXR signaling suppressed ER stress by modulating membrane phospholipid composition (60). Interestingly, our data suggested that knocking down PPARγ was more potent in abolishing 5-aza-dC's inhibitory effects on inflammatory and chemotactic gene expression, whereas knocking down LXRα was more potent in abolishing 5-aza-dC's inhibitory effects on ER stress gene expression. Further study is needed to elucidate the role of PPARγ and LXRα in mediating 5-aza-dC's inhibition of ER stress.
In summary, our data demonstrate that inhibiting DNA methylation by 5-aza-dC protects against atherosclerosis development. 5-Aza-dC may exert its atheroprotective effects by antagonizing macrophage inflammation, infiltration, and ER stress without affecting plasma lipid/cholesterol profiles, macrophage cholesterol traffic, and circulating monocyte/macrophage lineage development. This may be due to its ability to demethylate LXRα and PPARγ1 promoters in macrophages. These findings provide strong evidence that DNA methylation plays a significant role in cardiovascular diseases and may serve as a therapeutic target for the prevention and treatment of atherosclerosis.
Acknowledgments
This work was supported by National Institutes of Health Grants R01HL107500 (to B.X.) and R01DK084172 (to H.S.), American Heart Association Grants 10SDG3900046 (to B.X.) and 11GRNT7370080 (to H.S.), and American Diabetes Association Grant 7–13-BS-159 (to H.S.).
Disclosure Summary: The authors have no conflict of interests to disclose.
Footnotes
- APC
- allophycocyanin
- apoA1
- apolipoprotein A1
- 5-aza-dC
- 5-aza-2′-deoxycytidine
- BMDM
- bone marrow-derived macrophage
- CCL2
- chemokine (C-C motif) ligand 2
- CCR2
- chemokine (C-C motif) receptor 2
- ER
- endoplasmic reticulum
- ERN1/IRE-1α
- ER to nucleus signaling 1/inositol-requiring enzyme-1α
- FACS
- fluorescence-activated cell sorting
- HDL
- high-density lipoprotein
- iNOS
- inducible nitric oxide synthase
- LDL
- low-density lipoprotein
- LPS
- lipopolysaccharide
- LXRα
- liver X receptor-α
- MCP-1
- monocyte chemoattractant protein 1
- NF-κB
- nuclear factor-κB
- PPARγ1
- peroxisome proliferator-activated receptor γ1
- siRNA
- small interfering RNA.
References
- 1. Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis (*). Annu Rev Immunol. 2009;27:165–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Libby P, Ridker PM, Hansson GK. Inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol. 2009;54:2129–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Erbay E, Babaev VR, Mayers JR, et al. Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat Med. 2009;15:1383–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res. 2010;107:839–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Luczak MW, Jagodzinski PP. The role of DNA methylation in cancer development. Folia Histochem Cytobiol. 2006;44:143–154. [PubMed] [Google Scholar]
- 6. Stenvinkel P, Karimi M, Johansson S, et al. Impact of inflammation on epigenetic DNA methylation - a novel risk factor for cardiovascular disease? J Intern Med. 2007;261:488–499. [DOI] [PubMed] [Google Scholar]
- 7. Sharma P, Kumar J, Garg G, et al. Detection of altered global DNA methylation in coronary artery disease patients. DNA Cell Biol. 2008;27:357–365. [DOI] [PubMed] [Google Scholar]
- 8. Dong C, Yoon W, Goldschmidt-Clermont PJ. DNA methylation and atherosclerosis. J Nutr. 2002;132:2406S–2409S. [DOI] [PubMed] [Google Scholar]
- 9. Christman JK. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene. 2002;21:5483–5495. [DOI] [PubMed] [Google Scholar]
- 10. Patra SK, Bettuzzi S. Epigenetic DNA-(cytosine-5-carbon) modifications: 5-aza-2′-deoxycytidine and DNA-demethylation. Biochemistry (Mosc). 2009;74:613–619. [DOI] [PubMed] [Google Scholar]
- 11. Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer. 2008;123:8–13. [DOI] [PubMed] [Google Scholar]
- 12. Li Q, Bartlett DL, Gorry MC, O'Malley ME, Guo ZS. Three epigenetic drugs up-regulate homeobox gene Rhox5 in cancer cells through overlapping and distinct molecular mechanisms. Mol Pharmacol. 2009;76:1072–1081. [DOI] [PubMed] [Google Scholar]
- 13. Calkin AC, Tontonoz P. Liver X receptor signaling pathways and atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30:1513–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med. 2003;9:213–219. [DOI] [PubMed] [Google Scholar]
- 15. Gerry JM, Pascual G. Narrowing in on cardiovascular disease: the atheroprotective role of peroxisome proliferator-activated receptor gamma. Trends Cardiovasc Med. 2008;18:39–44. [DOI] [PubMed] [Google Scholar]
- 16. Ishibashi S, Brown MS, Goldstein JL, Gerard RD, Hammer RE, Herz J. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J Clin Invest. 1993;92:883–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Temel RE, Lee RG, Kelley KL, et al. Intestinal cholesterol absorption is substantially reduced in mice deficient in both ABCA1 and ACAT2. J Lipid Res. 2005;46:2423–2431. [DOI] [PubMed] [Google Scholar]
- 18. Rong S, Cao Q, Liu M, et al. Macrophage 12/15 lipoxygenase expression increases plasma and hepatic lipid levels and exacerbates atherosclerosis. J Lipid Res. 2012;53:686–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brown AL, Zhu X, Rong S, et al. Omega-3 fatty acids ameliorate atherosclerosis by favorably altering monocyte subsets and limiting monocyte recruitment to aortic lesions. Arterioscler Thromb Vasc Biol. 2012;32:2122–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Galkina E, Kadl A, Sanders J, Varughese D, Sarembock IJ, Ley K. Lymphocyte recruitment into the aortic wall before and during development of atherosclerosis is partially L-selectin dependent. J Exp Med. 2006;203:1273–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang X, Yang Z, Xue B, Shi H. Activation of the cholinergic antiinflammatory pathway ameliorates obesity-induced inflammation and insulin resistance. Endocrinology. 152:836–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang X, Wang X, Liu D, Yu L, Xue B, Shi H. Epigenetic regulation of macrophage polarization by DNA methyltransferase 3b. Mol Endocrinol. 2014;28:565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhu X, Lee JY, Timmins JM, et al. Increased cellular free cholesterol in macrophage-specific Abca1 knock-out mice enhances pro-inflammatory response of macrophages. J Biol Chem. 2008;283:22930–22941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Furbee JW, Jr, Sawyer JK, Parks JS. Lecithin:cholesterol acyltransferase deficiency increases atherosclerosis in the low density lipoprotein receptor and apolipoprotein E knockout mice. J Biol Chem. 2002;277:3511–3519. [DOI] [PubMed] [Google Scholar]
- 25. Fujiki K, Kano F, Shiota K, Murata M. Expression of the peroxisome proliferator activated receptor gamma gene is repressed by DNA methylation in visceral adipose tissue of mouse models of diabetes. BMC Biol. 2009;7:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shore A, Karamitri A, Kemp P, Speakman JR, Lomax MA. Role of Ucp1 enhancer methylation and chromatin remodelling in the control of Ucp1 expression in murine adipose tissue. Diabetologia. 2010;53:1164–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McCabe MT, Low JA, Daignault S, Imperiale MJ, Wojno KJ, Day ML. Inhibition of DNA methyltransferase activity prevents tumorigenesis in a mouse model of prostate cancer. Cancer Res. 2006;66:385–392. [DOI] [PubMed] [Google Scholar]
- 28. Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Clozel T, Yang S, Elstrom RL, et al. Mechanism-based epigenetic chemosensitization therapy of diffuse large B-cell lymphoma. Cancer Discov. 2013;3:1002–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kalac M, Scotto L, Marchi E, et al. HDAC inhibitors and decitabine are highly synergistic and associated with unique gene-expression and epigenetic profiles in models of DLBCL. Blood. 2011;118:5506–5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hassler MR, Klisaroska A, Kollmann K, et al. Antineoplastic activity of the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine in anaplastic large cell lymphoma. Biochimie. 2012;94:2297–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gordon S, Hamann J, Lin HH, Stacey M. F4/80 and the related adhesion-GPCRs. Euro J Immunol. 2011;41:2472–2476. [DOI] [PubMed] [Google Scholar]
- 33. Rosas M, Thomas B, Stacey M, Gordon S, Taylor PR. The myeloid 7/4-antigen defines recently generated inflammatory macrophages and is synonymous with Ly-6B. J Leukoc Biol. 2010;88:169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Swirski FK, Libby P, Aikawa E, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Perry HM, Bender TP, McNamara CA. B cell subsets in atherosclerosis. Front Immunol. 2012;3:373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Creusot F, Acs G, Christman JK. Inhibition of DNA methyltransferase and induction of Friend erythroleukemia cell differentiation by 5-azacytidine and 5-aza-2′-deoxycytidine. J Biol Chem. 1982;257:2041–2048. [PubMed] [Google Scholar]
- 37. Tabas I. Macrophage apoptosis in atherosclerosis: consequences on plaque progression and the role of endoplasmic reticulum stress. Antioxid Redox Signal. 2009;11:2333–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bensinger SJ, Tontonoz P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature. 2008;454:470–477. [DOI] [PubMed] [Google Scholar]
- 39. Babaev VR, Yancey PG, Ryzhov SV, et al. Conditional knockout of macrophage PPARgamma increases atherosclerosis in C57BL/6 and low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25:1647–1653. [DOI] [PubMed] [Google Scholar]
- 40. Pascual G, Sullivan AL, Ogawa S, et al. Anti-inflammatory and antidiabetic roles of PPARgamma. Novartis Found Symp. 2007;286:183–196; discussion 196–203. [DOI] [PubMed] [Google Scholar]
- 41. Staels B. PPARγ and atherosclerosis. Curr Med Res Opin. 2005;21(Suppl 1):S13–S20. [DOI] [PubMed] [Google Scholar]
- 42. Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM. mPPAR gamma 2: tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994;8:1224–1234. [DOI] [PubMed] [Google Scholar]
- 43. Zhu Y, Alvares K, Huang Q, Rao MS, Reddy JK. Cloning of a new member of the peroxisome proliferator-activated receptor gene family from mouse liver. J Biol Chem. 1993;268:26817–26820. [PubMed] [Google Scholar]
- 44. Zhu Y, Qi C, Korenberg JR, et al. Structural organization of mouse peroxisome proliferator-activated receptor gamma (mPPAR gamma) gene: alternative promoter use and different splicing yield two mPPAR gamma isoforms. Proc Natl Acad Sci U S A. 1995;92:7921–7925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lefterova MI, Steger DJ, Zhuo D, et al. Cell-specific determinants of peroxisome proliferator-activated receptor gamma function in adipocytes and macrophages. Mol Cell Biol. 2010;30:2078–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Edwards TM, Myers JP. Environmental exposures and gene regulation in disease etiology. Environ Health Perspect. 2007;115:1264–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Skinner MK, Manikkam M, Guerrero-Bosagna C. Epigenetic transgenerational actions of environmental factors in disease etiology. Trends Endocrinol Metab. 2010;21:214–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dunn J, Qiu H, Kim S, et al. Flow-dependent epigenetic DNA methylation regulates endothelial gene expression and atherosclerosis. J Clin Invest. 2014;124:3187–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lal G, Zhang N, van der Touw W, et al. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J Immunol. 2009;182:259–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lü CX, Xu RD, Cao M, et al. FOXP3 demethylation as a means of identifying quantitative defects in regulatory T cells in acute coronary syndrome. Atherosclerosis. 2013;229:263–270. [DOI] [PubMed] [Google Scholar]
- 51. Sasaki N, Yamashita T, Takeda M, Hirata K. Regulatory T cells in atherogenesis. J Atheroscler Thromb. 2012;19:503–515. [DOI] [PubMed] [Google Scholar]
- 52. Repa JJ, Mangelsdorf DJ. The liver X receptor gene team: potential new players in atherosclerosis. Nat Med. 2002;8:1243–1248. [DOI] [PubMed] [Google Scholar]
- 53. de Winther MP, Kanters E, Kraal G, Hofker MH. Nuclear factor kappaB signaling in atherogenesis. Arterioscler Thromb Vasc Biol. 2005;25:904–914. [DOI] [PubMed] [Google Scholar]
- 54. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fabre C, Grosjean J, Tailler M, et al. A novel effect of DNA methyltransferase and histone deacetylase inhibitors: NFkappaB inhibition in malignant myeloblasts. Cell Cycle. 2008;7:2139–2145. [DOI] [PubMed] [Google Scholar]
- 56. Wang J, Yang B, Han L, et al. Demethylation of miR-9–3 and miR-193a genes suppresses proliferation and promotes apoptosis in non-small cell lung cancer cell lines. Cell Physiol Biochem. 2013;32:1707–1719. [DOI] [PubMed] [Google Scholar]
- 57. Jiang R, Li Y, Zhang A, et al. The acquisition of cancer stem cell-like properties and neoplastic transformation of human keratinocytes induced by arsenite involves epigenetic silencing of let-7c via Ras/NF-κB. Toxicol Lett. 2014;227:91–98. [DOI] [PubMed] [Google Scholar]
- 58. Nguyen MT, Chen A, Lu WJ, et al. Regulation of chemokine and chemokine receptor expression by PPARgamma in adipocytes and macrophages. PLoS One. 2012;7:e34976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yoshiuchi K, Kaneto H, Matsuoka TA, et al. Pioglitazone reduces ER stress in the liver: direct monitoring of in vivo ER stress using ER stress-activated indicator transgenic mice. Endocr J. 2009;56:1103–1111. [DOI] [PubMed] [Google Scholar]
- 60. Rong X, Albert CJ, Hong C, et al. LXRs regulate ER stress and inflammation through dynamic modulation of membrane phospholipid composition. Cell Metab. 2013;18:685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]