

Abstract
The diagnosed incidence of small intestine neuroendocrine tumors (SI-NETs) is increasing, and the underlying genomic mechanisms have not been defined for these tumors. Using exome/genome sequence analysis of SI-NETs, we identified recurrent somatic mutations and deletions in CDKN1B, the cyclin-dependent kinase inhibitor gene, which encodes p27. We observed frameshift mutations of CDKN1B in 14 of 180 SI-NETs, and we detected hemizygous deletions encompassing CDKN1B in 7 out of 50 SI-NETs, nominating p27 as a tumor suppressor and implicating cell cycle dysregulation in the etiology of SI-NET.
Neuroendocrine tumors (NETs) are rare neoplasms (~1 per 100,000) that are thought to arise from endocrine precursor cells and occur most commonly in the lung, pancreas, and small intestine 1. Well-differentiated NETs are typically more indolent than other epithelial malignancies but nevertheless can metastasize 1. Both germline and somatic mutations of the multiple endocrine neoplasia type 1 gene, MEN1, are common in lung and pancreatic NETs 1. Pancreatic NETs are also characterized by recurrent somatic mutations in the DAXX, ATRX, PTEN and TSC2 genes 2. In SI-NETs, by contrast, evidence for focal events indicative of driver alterations has remained inconclusive; hemizygous loss of chromosome 18 is the most frequent known genomic event, followed by arm level gains of chromosomes 4, 5,14 and 20 3-5. Recently, a whole-exome sequencing study of 48 SI-NETs examining somatic single nucleotide variants (SSNVs) identified mutations in several cancer genes although none were recurrently altered 5.
To identify genomic alterations driving tumorigenesis in SI-NETs, we profiled 55 tumors from 50 individuals by a combination of whole-exome and whole-genome sequencing (Fig. 1a and Supplementary Tables 1-5). Mutation analysis of the exome sequencing data with the MuTect algorithm 6,7 revealed a total of 1230 genes with somatic mutations, of which 90% (1113/1230) were mutated in only a single individual. A relatively low non-silent SSNV rate of 0.77/Mb (range 0.13 to 2.51 per Mb) was observed (Figure 1a and Supplementary Fig. 1a and Table 6). Of the 1230 mutated genes in our study, 21 were also found to be mutated in the previous SI-NET study and another 17 in the pancreatic NET study including the cancer census genes ATRX and COL1A1, each in a single individual (Supplementary Fig. 1b-d) 2,5,8. The lack of substantial overlap in recurrently altered genes suggests that many of the mutations are passengers. There are potentially therapeutically targetable mutations in genes including SRC, FYN, KDR and IDH1 (R132H), however each are present within a single individual (Supplementary Table 6).
Figure 1.
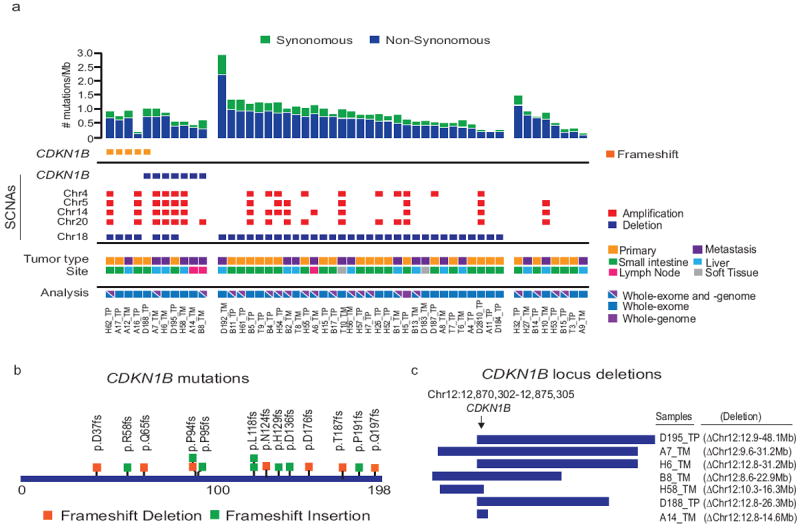
Mutational analysis of 31 small bowel and 19 metastatic SI-NETs.
a) Top panel shows the somatic mutation rate per megabase (Mb) of covered target sequence in the 50 cases. Middle panel shows the recurrent somatic mutation of CDKN1B and prominent somatic copy number alterations found in each tumor. Primary (TP) and metastatic (TM) tumors, sites and type of sequencing performed are indicated by colored boxes. b) Schematic representation of frameshift mutations identified in CDKN1B. c) Schematic of the hemizygous deletions identified targeting CDKN1B.
Significantly mutated genes were identified by measuring the nucleotide-specific and sample-specific mutation rates in the SI-NET sequence data, computing an expected gene-specific mutation frequency for the SI-NETs based on the size and nucleotide composition of each gene, and then comparing the actual mutation frequency for each gene to the calculated expected number 9. This analysis of the 50 SI-NET cases identified statistically significant mutations in only one gene, the cell cycle regulator CDKN1B (p=6.5e-10). In total, we found small insertions and deletions within CDKN1B in 10% (5/50) of cases (Fig. 1a and Supplementary Table 7), leading to frameshift mutations (Fig. 1b). These mutations were validated by independent PCR and sequencing. Furthermore, copy number analysis identified hemizygous deletions encompassing CDKN1B in 7 cases (Fig. 1c). Four out of these seven SI-NETs with CDKN1B deletions retained both copies of chromosome 18 compared to 8 out of 35 SI-NETs without CDKN1B deletion (P=0.048, two-tailed Fisher’s exact test. The region encompassing CDKN1B, 12p13, is frequently hemizygously deleted in ovarian, prostate, non-small cell lung cancer and multiple hematological malignancies 10-16.
To confirm the incidence of CDKN1B mutations in SI-NETs, we analyzed two independent cohorts; 48 SI-NETs reported by Banck et al. 5 and an extension set of 81 SI-NETs sequenced to a mean 800-fold coverage at CDKN1B. Two previously unreported somatic deletions within CDKN1B were detected in the Banck et al. cohort that was not previously analyzed for indels 5, resulting in frameshift mutations. The extension cohort revealed 7 small indels within CDKN1B leading to frameshifts; the extension set did not have paired germline DNA so we cannot exclude the possibility that some of these inactivating alterations are germline. Overall, heterozygous frameshift CDKN1B mutations were detected in 8% (14/180) of SI-NETs analyzed.
The presence of heterozygous inactivating mutations in CDKN1B is consistent with the possibility that CDKN1B acts as a haploinsufficient tumor suppressor gene in SI-NETs. One possible explanation is that some p27 expression is necessary for cell proliferation, as has been described in certain oncogenic models 17,18, thus making bi-allelic deletion disfavored. Several recurrently cancer-mutated genes, including FBXW7, PTEN, DICER1 and CREBBP have recently been reported to be haploinsufficient tumor suppressors in mouse genetic models of cancer 19-22. The increased susceptibility to tumors following DNA damage observed in Cdkn1b heterozygous knockout mouse models along with elevated cellular proliferation 23-26 is consistent with the hypothesis that CDKN1B is haploinsufficient for tumor suppression.
Hemizygous loss of chromosome 18 (log2 (copy number/2)<-0.1) was found in ~78% (43/55) of SI-NETs, but was associated with only a slight increase in mutation rate genome wide (Supplementary Fig. 2 and 3). Two genes, including BCL10, a gene mutated in colorectal cancer 27, were found to be altered exclusively in the 12 cases with diploid chr. 18 (Supplementary Fig. 3d). Because of the high frequency of hemizygous deletion of chr.18, we examined the cohort for somatic mutations to growth inhibitory or “STOP” 28 genes within the three frequently deleted regions of this chromosome. While we observed no somatic mutations, it is possible that hemizygous loss of these genes may contribute to SI-NET tumorigenesis through altered gene dosage (Supplementary Table 8). In addition, comparison with genes mutated in small cell lung cancer (SCLC) 29,30, a tumor type that shares neuroendocrine characteristics with SI-NETs, showed 199 genes with mutation in common in both studies; however, this overlap may be due to the high overall mutation rate in SCLC rather than a shared mechanism of tumorigenesis (Supplementary Fig. 4 and Tables 9 and 10).
To survey for genomic rearrangements, we performed whole genome sequencing on 24 tumor/normal sample pairs. The number of somatic rearrangements detected by paired-end and split-read mapping 31,32 ranged from 0 to 45 per case, with a median of 7 (Supplementary Fig. 5 and Table 11). Of those, 20% (33/163) of rearrangements involved genes or promoter regions, leading to five potential fusion proteins and 2 in-frame deletions, however none were recurrent (Supplementary Table 12). The concordance of SSNVs identified in whole-genome and whole-exome was on average ~95% when sufficient coverage was available 6.
Tumor heterogeneity within epithelial tumors can be exceptionally complex and cells shed from the primary can form distant metastases 33. Approximately 25% of SI-NETs are multifocal tumors at time of resection and 10-15% of neuroendocrine metastases are diagnosed as being of unknown primary origin 34,35. When we compared exomic mutations and copy number data for the paired primary and metastatic tumors, we observed no overlap in SSNVs or SCNAs between the primary and metastasis in 2 out of 5 primary/metastatic/normal trios (Fig. 2 and Supplementary Table 13). We confirmed that germline SNPs were concordant in the trios to exclude sample mix-up. In one particular case (A16), the primary tumor contained a CDKN1B frameshift mutation while the metastasis did not, a phenomenon also reported for PIK3CA and EGFR in breast and non-small cell lung cancer, respectively 36,37. It is hypothesized that the metastases in these two cases may have been derived from either an undiagnosed independent primary lesion, a subclonal population that was not detected by sequencing, or a clone that was shed from the primary tumor early in progression prior to the acquisition of major genomic events. In contrast, Banck et al., assessed the overlap of 35 gene mutations in paired primary and metastasis samples and observed an 83% concordance in 5 cases 5. Given the small number of primary/metastatic/normal SI-NET trios (five cases each) in our two cohorts, the difference between these datasets is consistent with statistical fluctuation. A Fisher’s exact test comparing a case series with two primary/metastatic discordances and three concordances, to a case series with five primary/metastatic somatic mutation concordances and no discordances, yields p = 0.22, consistent with the null hypothesis that the two datasets are identical. This suggests that SI-NETs are observed in a population wherein a subset of patients may harbor multifocal tumors and a subset may harbor unifocal tumors. The discordance amongst primary and metastasis along with the multifocal nature of SI-NETs highlights a challenge in identifying underlying driving events in these tumors.
Figure 2.
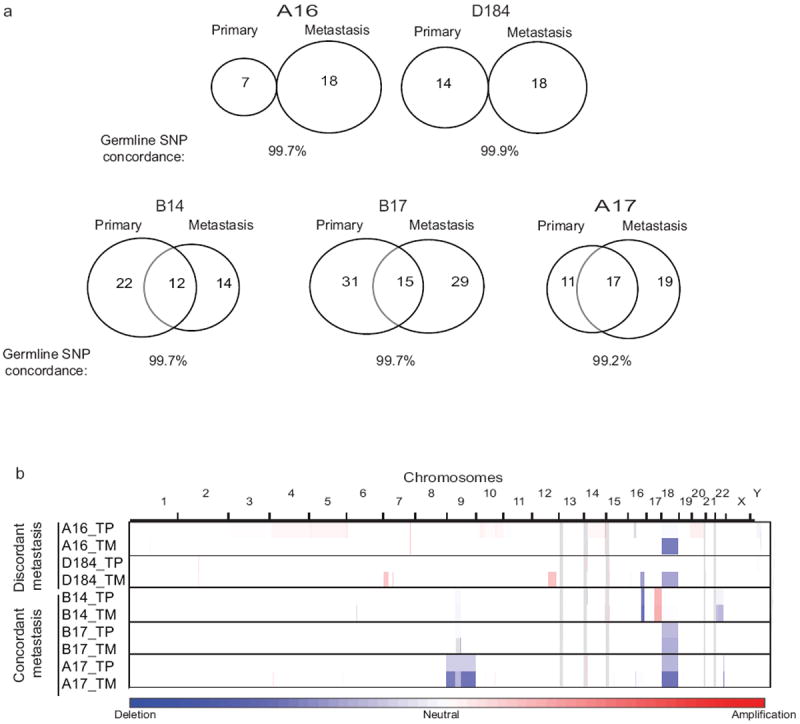
Somatic mutation and copy number discordance between primary and metastatic cases.
a) Venn diagram depicting the concordance and discordance in somatic mutation calls in the primary or metastatic lesion analyzed in 5 cases. b) Copy number profiles for concordant and discordant primary and metastatic tumors.
Somatic mutations targeting the cell-cycle regulatory gene CDKN1B were the most frequent gene-specific events in SI-NETs. CDKN1B encodes a cyclin-dependent kinase inhibitor that binds to and inhibits Cdk2 and Cdk4 38,39. Mouse models of Cdkn1b haploinsufficiency have larger body and organ size and enhanced sensitivity to mitogenic stimulation owing to greater Cdk2 activity 23-25. In contrast to CDKN2A and related genes encoding Cdk4/Cdk6 inhibitors, somatic mutations in CDKN1B have been recently reported at low frequency in breast and prostate cancers 16,40,41. CDKN1B is also known as MEN4, a gene mutated in the germline of families with a phenotype of MEN-1 syndrome without an identifiable MEN1 gene mutation 42. Furthermore, menin, the MEN1 gene product, associates with promoter regions to mediate expression of CDKN1B and CDKN2C through epigenetic regulation 43,44.
In summary, this study presents a comprehensive genomic analysis of somatic variants and whole-genomes of SI-NETs. SI-NETs are dominated by large, arm-level copy number gains and losses but there were strikingly few recurrent somatic gene alterations. The discovery of recurrent CDKN1B mutations raises the possibility that the p21/p27/p57 family may represent haploinsufficient tumor suppressors, and suggests a focus on cell cycle regulation in understanding the pathogenesis of SI-NETs.
Supplementary Material
Supplementary Figure 1: Comparison of somatic mutation rates in our study and the published neuroendocrine tumor literature
a) Plot comparing the number of mutations per megabase (Mb) in primary and metastatic SI-NETs, the bar represents the mean. b) Venn diagram of the concordant gene mutations identified previously in SI-NETs 5 and the current study of SI-NETs. c) Plot comparing the total number of mutations per case in pancreatic NETs 2 and the current study, the bar represents the mean. d) Venn diagram of the concordant gene mutations identified in both studies (bold indicates membership in the Cancer Gene Census) 8
Supplementary Figure 2: SCNA and GISTIC analysis of 55 small intestine and metastatic NETs.
a) Somatic copy number alterations in 55 primary and metastatic small intestine NETS. Upper panel, summary plot of SCNAs across all chromosomes. Lower panel, heat map of copy number increases (red) and decreases (blue). b) GISTIC 2.0 q-values for amplifications (B) and deletions (C) are plotted across the genome (y-axis). The significantly deleted peaks are labeled with the size of the region and putative gene targets (Supplementary Tables 14 and 15).
Supplementary Figure 3: Chromosome 18 centric specific analysis a) Plot comparing the total number of somatic variants in chromosome 18 as detected by whole-genome sequencing (WGS) for cases with or without chr.18 LOH, the bar represents the mean. b) Plot comparing the total genome wide somatic variants detected by WGS for cases with or without chr.18 LOH, the bar represents the mean. c) Plot comparing the number of mutations genome-wide detected by whole-exome sequencing (WES) for cases with or without chr.18 LOH, the bar represents the mean. d) Venn diagram of gene mutations found in more than 1 case and relation to chr.18 status. Mutations in four genes were exclusive to the chr.18 diploid cases, while mutations in five genes were present in both chr. 18 diploid and LOH cases. The greater number of gene mutations found in the chr.18 LOH cases is a reflection of the greater sample size. Genes in bold are present in the Cancer Gene Census 8.
Supplementary Figure 4: Comparison of somatic variants with the published small cell lung cancer literature
a) Plot comparing the total number of mutations per case in SCLC 29 and the current study of SI-NETs. b) Venn diagram of the concordant gene mutations identified in both studies (see Supplemental Table 9 for genes). c) Plot comparing the total number of mutations per case in SCLC 30 and the current study. b) Venn diagram of the concordant gene mutations identified in both studies (see Supplemental Table 10 for genes).
Supplementary Figure 5: CIRCOS plots detailing chromosomal rearrangements in 24 tumors identified by whole-genome sequencing.
Table 1: Cohort overview and platforms used
Table 2: Cohort information file with site of tumor, age and gender
Table 3: Sequencing metrics for whole-exome sequencing
Table 4: Sequencing metrics for whole-genome sequencing
Table 5: Mutation type breakdown
Table 6: Complete list of somatic mutations identified by exome sequencing
Table 7: Frameshift mutations identified in CDKN1B
Table 8: Chromosome 18 STOP genes deleted in SI-NETs
Table 9: Concordance of SI-NET mutation data with SCLC mutations from Rudin et al.
Table 10: Concordance of SI-NET mutation data with SCLC mutations from Peifer et al.
Table 11: Predicted chromosomal rearrangements identified by whole-genome sequencing
Table 12: Predicted fusion proteins identified by whole-genome sequencing
Table 13: Somatic mutations identified in tumor/metastasis/normal trios
Table 14: GISTIC 2.0 arm-level SCNA analysis results
Table 15: GISTIC 2.0 focal deletion analysis results
Acknowledgments
This work was supported by grants from the Caring for Carcinoid Foundation Sciences (S.L.A, and M.M.), the Raymond and Beverly Sackler Foundation for the Arts and Sciences (C.T., A.K., S.L.A, and M.M.) and Cancer Research UK (C.T., and A.K.).
References
- 1.Vinik AI, et al. Pancreas. 2010;39:713–34. doi: 10.1097/MPA.0b013e3181ebaffd. [DOI] [PubMed] [Google Scholar]
- 2.Jiao Y, et al. Science. 2011;331:1199–203. doi: 10.1126/science.1200609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kulke MH, et al. Genes Chromosomes Cancer. 2008;47:591–603. doi: 10.1002/gcc.20561. [DOI] [PubMed] [Google Scholar]
- 4.Cunningham JL, et al. Genes Chromosomes Cancer. 2011;50:82–94. doi: 10.1002/gcc.20834. [DOI] [PubMed] [Google Scholar]
- 5.Banck MS, et al. J Clin Invest. 2013 [Google Scholar]
- 6.Cibulskis K, et al. Nat Biotechnol. 2013;31:213–9. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lawrence MS, et al. Nature. 2013 [Google Scholar]
- 8.Futreal PA, et al. Nat Rev Cancer. 2004;4:177–83. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Getz G, et al. Science. 2007;317:1500. doi: 10.1126/science.1138764. [DOI] [PubMed] [Google Scholar]
- 10.Hatta Y, Takeuchi S, Yokota J, Koeffler HP. Br J Cancer. 1997;75:1256–62. doi: 10.1038/bjc.1997.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kibel AS, Freije D, Isaacs WB, Bova GS. Genes Chromosomes Cancer. 1999;25:270–6. [PubMed] [Google Scholar]
- 12.Takeuchi S, et al. Cancer Res. 1996;56:738–40. [PubMed] [Google Scholar]
- 13.Pietenpol JA, et al. Cancer Res. 1995;55:1206–10. [PubMed] [Google Scholar]
- 14.Hetet G, et al. Hematol J. 2000;1:42–7. doi: 10.1038/sj.thj.6200008. [DOI] [PubMed] [Google Scholar]
- 15.Le Toriellec E, et al. Blood. 2008;111:2321–8. doi: 10.1182/blood-2007-06-095570. [DOI] [PubMed] [Google Scholar]
- 16.Barbieri CE, et al. Nat Genet. 2012;44:685–9. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muraoka RS, et al. Mol Cell Biol. 2002;22:2204–19. doi: 10.1128/MCB.22.7.2204-2219.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao H, et al. Proc Natl Acad Sci U S A. 2004;101:17204–9. doi: 10.1073/pnas.0407693101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sancho R, et al. Gastroenterology. 2010;139:929–41. doi: 10.1053/j.gastro.2010.05.078. [DOI] [PubMed] [Google Scholar]
- 20.Ying H, et al. Cancer discovery. 2011;1:158–69. doi: 10.1158/2159-8290.CD-11-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar MS, et al. Genes Dev. 2009;23:2700–4. doi: 10.1101/gad.1848209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zimmer SN, et al. Blood. 2011;118:69–79. doi: 10.1182/blood-2010-09-307942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kiyokawa H, et al. Cell. 1996;85:721–32. doi: 10.1016/s0092-8674(00)81238-6. [DOI] [PubMed] [Google Scholar]
- 24.Nakayama K, et al. Cell. 1996;85:707–20. doi: 10.1016/s0092-8674(00)81237-4. [DOI] [PubMed] [Google Scholar]
- 25.Fero ML, et al. Cell. 1996;85:733–44. doi: 10.1016/s0092-8674(00)81239-8. [DOI] [PubMed] [Google Scholar]
- 26.Fero ML, Randel E, Gurley KE, Roberts JM, Kemp CJ. Nature. 1998;396:177–80. doi: 10.1038/24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cancer Genome Atlas, N. Nature. 2012;487:330–7. [Google Scholar]
- 28.Solimini NL, et al. Science. 2012;337:104–9. doi: 10.1126/science.1219580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rudin CM, et al. Nat Genet. 2012;44:1111–6. doi: 10.1038/ng.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peifer M, et al. Nat Genet. 2012;44:1104–10. doi: 10.1038/ng.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Banerji S, et al. Nature. 2012;486:405–9. doi: 10.1038/nature11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Imielinski M, et al. Cell. 2012;150:1107–20. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gerlinger M, et al. N Engl J Med. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Habbe N, Fendrich V, Heverhagen A, Ramaswamy A, Bartsch DK. Surgery today. 2012 doi: 10.1007/s00595-012-0408-1. [DOI] [PubMed] [Google Scholar]
- 35.Polish A, Vergo MT, Agulnik M. J Natl Compr Canc Netw. 2011;9:1397–402. doi: 10.6004/jnccn.2011.0118. [DOI] [PubMed] [Google Scholar]
- 36.Curtit E, et al. Oncologist. 2013 doi: 10.1634/theoncologist.2012-0350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park S, et al. J Thorac Oncol. 2009;4:809–15. doi: 10.1097/JTO.0b013e3181a94af4. [DOI] [PubMed] [Google Scholar]
- 38.Polyak K, et al. Cell. 1994;78:59–66. doi: 10.1016/0092-8674(94)90572-x. [DOI] [PubMed] [Google Scholar]
- 39.Toyoshima H, Hunter T. Cell. 1994;78:67–74. doi: 10.1016/0092-8674(94)90573-8. [DOI] [PubMed] [Google Scholar]
- 40.TCGA. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stephens PJ, et al. Nature. 2012;486:400–4. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pellegata NS, et al. Proc Natl Acad Sci U S A. 2006;103:15558–63. doi: 10.1073/pnas.0603877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Milne TA, et al. Proc Natl Acad Sci U S A. 2005;102:749–54. doi: 10.1073/pnas.0408836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karnik SK, et al. Proc Natl Acad Sci U S A. 2005;102:14659–64. doi: 10.1073/pnas.0503484102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Comparison of somatic mutation rates in our study and the published neuroendocrine tumor literature
a) Plot comparing the number of mutations per megabase (Mb) in primary and metastatic SI-NETs, the bar represents the mean. b) Venn diagram of the concordant gene mutations identified previously in SI-NETs 5 and the current study of SI-NETs. c) Plot comparing the total number of mutations per case in pancreatic NETs 2 and the current study, the bar represents the mean. d) Venn diagram of the concordant gene mutations identified in both studies (bold indicates membership in the Cancer Gene Census) 8
Supplementary Figure 2: SCNA and GISTIC analysis of 55 small intestine and metastatic NETs.
a) Somatic copy number alterations in 55 primary and metastatic small intestine NETS. Upper panel, summary plot of SCNAs across all chromosomes. Lower panel, heat map of copy number increases (red) and decreases (blue). b) GISTIC 2.0 q-values for amplifications (B) and deletions (C) are plotted across the genome (y-axis). The significantly deleted peaks are labeled with the size of the region and putative gene targets (Supplementary Tables 14 and 15).
Supplementary Figure 3: Chromosome 18 centric specific analysis a) Plot comparing the total number of somatic variants in chromosome 18 as detected by whole-genome sequencing (WGS) for cases with or without chr.18 LOH, the bar represents the mean. b) Plot comparing the total genome wide somatic variants detected by WGS for cases with or without chr.18 LOH, the bar represents the mean. c) Plot comparing the number of mutations genome-wide detected by whole-exome sequencing (WES) for cases with or without chr.18 LOH, the bar represents the mean. d) Venn diagram of gene mutations found in more than 1 case and relation to chr.18 status. Mutations in four genes were exclusive to the chr.18 diploid cases, while mutations in five genes were present in both chr. 18 diploid and LOH cases. The greater number of gene mutations found in the chr.18 LOH cases is a reflection of the greater sample size. Genes in bold are present in the Cancer Gene Census 8.
Supplementary Figure 4: Comparison of somatic variants with the published small cell lung cancer literature
a) Plot comparing the total number of mutations per case in SCLC 29 and the current study of SI-NETs. b) Venn diagram of the concordant gene mutations identified in both studies (see Supplemental Table 9 for genes). c) Plot comparing the total number of mutations per case in SCLC 30 and the current study. b) Venn diagram of the concordant gene mutations identified in both studies (see Supplemental Table 10 for genes).
Supplementary Figure 5: CIRCOS plots detailing chromosomal rearrangements in 24 tumors identified by whole-genome sequencing.
Table 1: Cohort overview and platforms used
Table 2: Cohort information file with site of tumor, age and gender
Table 3: Sequencing metrics for whole-exome sequencing
Table 4: Sequencing metrics for whole-genome sequencing
Table 5: Mutation type breakdown
Table 6: Complete list of somatic mutations identified by exome sequencing
Table 7: Frameshift mutations identified in CDKN1B
Table 8: Chromosome 18 STOP genes deleted in SI-NETs
Table 9: Concordance of SI-NET mutation data with SCLC mutations from Rudin et al.
Table 10: Concordance of SI-NET mutation data with SCLC mutations from Peifer et al.
Table 11: Predicted chromosomal rearrangements identified by whole-genome sequencing
Table 12: Predicted fusion proteins identified by whole-genome sequencing
Table 13: Somatic mutations identified in tumor/metastasis/normal trios
Table 14: GISTIC 2.0 arm-level SCNA analysis results
Table 15: GISTIC 2.0 focal deletion analysis results