

Abstract
Hepatitis C virus (HCV) infection represents an important public health problem worldwide. Reduction of HCV morbidity and mortality is a current challenge owned to several viral and host factors. Virus molecular evolution plays an important role in HCV transmission, disease progression and therapy outcome. The high degree of genetic heterogeneity characteristic of HCV is a key element for the rapid adaptation of the intrahost viral population to different selection pressures (e.g., host immune responses and antiviral therapy). HCV molecular evolution is shaped by different mechanisms including a high mutation rate, genetic bottlenecks, genetic drift, recombination, temporal variations and compartmentalization. These evolutionary processes constantly rearrange the composition of the HCV intrahost population in a staging manner. Remarkable advances in the understanding of the molecular mechanism controlling HCV replication have facilitated the development of a plethora of direct-acting antiviral agents against HCV. As a result, superior sustained viral responses have been attained. The rapidly evolving field of anti-HCV therapy is expected to broad its landscape even further with newer, more potent antivirals, bringing us one step closer to the interferon-free era.
Keywords: Hepatitis C virus, Evolution, Phylogenetics, Drug resistance, Clinical outcome
Core tip: Hepatitis C virus (HCV) infection remains as an important public health problem worldwide. Viral molecular evolution determines, in many ways, the outcome of HCV infection. Here, we present up-to-date information about the role of HCV molecular evolution in virus ransmission, disease progression and antiviral therapy.
INTRODUCTION
Globally, hepatitis C virus (HCV) infection affects approximately 180 million people[1], in addition to three million new infections occurring annually[2,3]. The prevalence of HCV infection varies greatly from region to region, and as a result, the burden of disease exhibits important differences worldwide[2]. HCV is one of the leading causes of chronic liver disease associated with end-stage cirrhosis and hepatocellular carcinoma[4], with approximate 20% of chronically infected patients developing cirrhosis, and about 10% progressing to cancer[5].
HCV is a small single-stranded, positive polarity, enveloped virus belonging to the Hepacivirus genus within the Flaviviridae family. The RNA genome (about 9.6 kb in length) contains a single open reading frame (ORF) encoding for a polyprotein which is flanked by a 5’- and 3’-untranslated regions (UTR). The 5’-UTR contains an internal ribosomal entry site (IRES) which is essential for viral replication. The polyprotein is processed into three structural proteins (C, E1, E2) and seven nonstructural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, NS5B) (Figure 1)[6,7]. The HCV core is a highly conserved protein that makes up the viral nucleocapsid and plays role in pathogenesis[8]. E1 and E2 are highly glycosylated proteins that participate in cell entry (Figure 2)[9]. E2 contains three hypervariable regions (HVR), known as HVR1-3[10], which are under continuous selection pressure exerted by the host immune system. P7 is a 63-amino acid polypeptide that serves as a signal sequence for the translocation of NS2 into the lumen of the endoplasmic reticulum for further cleavage. P7 is also essential for particle assembly and release of infectious virions[11,12]. NS2 is a transmembrane protein required for viral replication while NS3 is the HCV protease and NTPase/helicase[13,14]. The HCV protease disrupts the interferon (IFN) and toll-like receptor-3 (TLR3) signaling pathways by cleaving host proteins including the caspase recruitment domain of the mitochondrial antiviral signaling protein (MAVS)[15,16], and TIR-domain-containing adapter-inducing interferon-β (TRIF) (Figure 2)[17]. NS4A acts as a cofactor for the NS3 protease and NS4B is a small hydrophobic protein required for recruitment of other viral proteins[18,19]. NS5A is a hydrophilic phosphoprotein needed for viral replication[20,21]. Lastly, NS5B is the HCV RNA dependent RNA polymerase (RdRp)[22], which lacks proofreading and error correction mechanisms, resulting in a highly error prone replication process[23].
Figure 1.
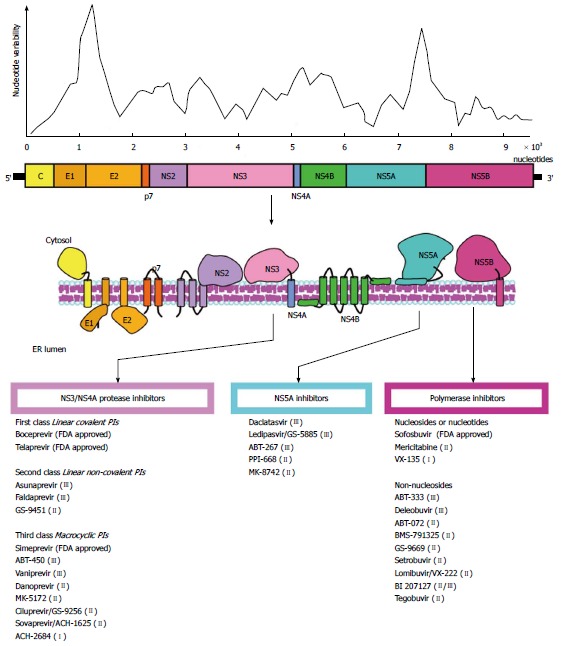
Hepatitis C virus genome and nucleotide variability. A schematic representation of the viral genome is depicted. The degree of nucleotide variability along the viral genome is also shown. The target molecules for anti-HCV therapy are noted, and the antiviral agents are indicated. A selection of FDA approved and in development compounds are shown. Roman numerals in brackets indicate the current clinical phase of development. HCV: Hepatitis C virus; FDA: Food and Drug Administration; PI: Protease inhibitors.
Figure 2.
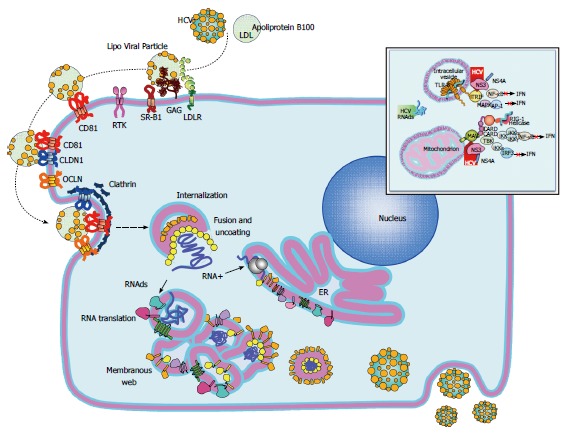
Hepatitis C virus replication cycle. The replicative cycle of hepatitis C virus (HCV) is displayed. HCV interaction with its cell receptors is shown. Upon entry, the HCV genome is released into the cytoplasm and subsequently translated and translocate into the RE. The membranous web is used as scaffold for viral replication. Interferon and TLR3 signaling pathways are disrupted by the HCV NS3/4A protease by cleaving MAVS and TRIF (upper right window). Assembled virions are released via the constitutive secretory pathway. HCV: Hepatitis C virus; MAVS: Mitochondrial antiviral signaling protein; TRIF: Toll-like receptor-domain-containing adapter-inducing interferon-β.
HCV virions bind to the host cellular receptors via E2[6]. The initial viral attachment is mediated by heparin sulfate proteoglycans on the hepatocyte surface[24]. Multiple cellular receptors such as the scavenger receptor class B type I[25], CD81[26,27], claudin-1[28], and occludin[29], in addition to several entry factors including the receptor tyrosine kinases, the epidermal growth factor receptor[30], the ephrin receptor A2[30] and the Niemann-Pick C1-like 1 cholesterol absorption receptor[31] have been identified (Figure 2). Once bound to the cell, HCV particles are then internalized by pH-dependent and clathrin mediated endocytosis[32,33]. Upon entry, the viral genome is released from the nucleocapsid into the cytoplasm and subsequently translated. The NS4B then induces the formation of membranous webs that serve as scaffolds for viral replication. After genome amplification and protein expression, progeny virions are assembled and released by the constitutive secretory pathway (Figure 2)[7,23].
The HCV mutation rate generates a high degree of intrahost genetic diversity[34], allowing for rapid adaptation[35]. This characteristic molecular plasticity of HCV is a key biological property that enables rearrangement of the intrahost viral population under different selection pressures, such as the immune response and antiviral therapy, warranting viral persistence[36,37]. HCV molecular evolution plays a pivotal role in virus transmission, progression of disease and outcome of therapy. Therefore, understanding the mechanisms driving the molecular evolution of HCV is likely to help to implement measures aimed to control HCV-related disease.
GENETIC HETEROGENEITY AND MOLECULAR EVOLUTION
Viral genotypes
Seven major HCV genotypes and several subtypes have been identified (Figure 3)[38]. HCV exhibits a complex taxonomic structure[38]. Genetic diversity between HCV genotypes is about 30%, while subtypes differ by about 15%[39,40]. HCV genotypes show a characteristic distribution in different geographical regions[41]. Genotypes 1, 2 and 3 exhibit a worldwide distribution. Genotypes 1 and 2 are endemic in West Africa while genotype 3 is endemic to the Indian subcontinent. Genotypes 4 and 5 are primarily found in Africa, and genotype 4 is particularly endemic in Egypt and Central Africa. Genotype 6 is endemic of Asia whereas the distribution of genotype 7 has not been fully assessed[41-45]. Genotype 2 is the oldest lineage, followed by genotypes 3, 5 and 6, while genotypes 1 and 4 emerged more recently. Globally distributed genotypes are referred as epidemic, and their rapid dissemination over the last century is primarily attributed to modes of transmission including the use of intravenous drugs, nosocomial transmission and blood transfusions[46]. Endemic genotypes are usually highly divergent and limited to well defined geographic regions[47]. These characteristic distribution patterns facilitate the identification of the site of origin and tracking of the genetic history of different HCV lineages. In general, high degree of genetic variability among HCV strains evolving in relatively small geographical regions suggests long-term evolution. Conversely, strains exhibiting lower genetic heterogeneity have shorter genetic histories and are often associated with recent introduction and higher transmission rates[47].
Figure 3.
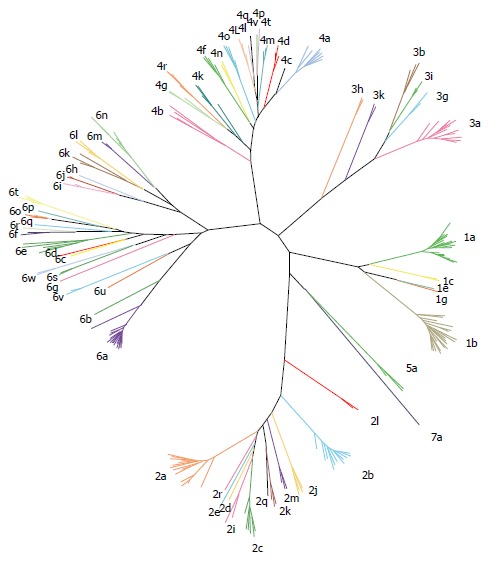
Hepatitis C virus genotypes and subtypes. Representative strains belonging to all seven genotype and all different subtypes are presented.
Molecular evolution
Different molecular mechanisms including mutation, genetic drift, recombination and natural selection shape the molecular evolution of HCV.
Insertion of point mutations by the RdRp is the primary element contributing to the high genetic variability of HCV. The HCV mutation rate in vivo is about 2.5 × 10-5 per nucleotide per genome replication[48]; however, higher estimates have been obtained[49]. Selection determines if a given mutation would be fixed in the viral population. Extrinsic selective pressures, such as the host immune response and antiviral treatment, and intrinsic functional constraints determine the accumulation of mutations in specific subgenomic regions[50]. These constraints define the tolerance to mutations in different regions resulting in an uneven distribution of genetic variability along the genome (Figure 1)[51,52]. Despite the high degree of genetic variability, the HCV antigenic diversity is significantly convergent[51], implying a large but restricted genetic space. This reduction in antigenic diversity is also likely due to structural and functional constrains[53] that restrict the sequence space and favors homoplasy[51]. The homoplastic nature of HCV antigenicity contrasts with the extensive nucleotide variability and represents an important venue for vaccine development[51].
Amino acid variability plays an important role in HCV infection. For instance, amino acid variability in the NS3/4 protease coding region affects its catalytic efficiency[54], allowing HCV to explore a broad range of protease genetic configurations[55]. Interestingly, minor HCV variants can display improved catalytic activities when compared to the master sequence, including those bearing resistant mutations to antiviral drugs[55]. Thus, these functional differences between variants can affect various aspects of the HCV replication cycle.
Genetic bottlenecks are an important force driving the molecular evolution and transmission of HCV[56,57], by inflicting a strong selective pressure during the acute phase of infection[57]. The intensity of the selective pressure imposed by genetic bottlenecks is such that only a handful of HCV variants manage to establish infection[58,59], commonly resulting in a profound founder effect (Figure 4). Upon transmission, HCV nucleotide diversity is reduced by purifying selection and subsequent bottlenecks[57]. Interestingly, the antibody response does not seem to significantly affect the composition of the transmitted/founder HCV population[58]. Rapid evolution after the occurrence of genetic bottlenecks usually results in the emergence of rare fit variants that quickly dominate the next population (Figure 4)[57,59].
Figure 4.
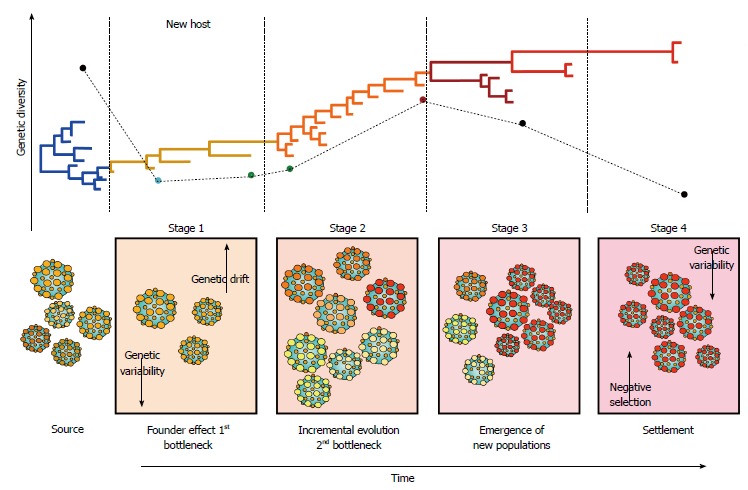
Hepatitis C virus molecular evolution. The staging of molecular evolution intrahost is illustrated. All four stages of infection are portrayed. The phylogenetic relatedness between different populations throughout time is also represented. The main molecular processed driving the molecular evolution are shown in their respective stage.
Genetic drift is also a contributor to the molecular evolution of HCV[60-62]. Genetic drift refers to the stochastic fluctuations in frequencies of variants in the viral population. Overall, large populations are less stochastic and undergo less genetic drift than small populations. While virus populations are often large, genetic drift still has been shown to be an important factor in viral evolution, suggesting that the genetic processes are mostly deterministic. Viral populations undergoing severe genetic bottleneck events frequently experience founder effects, which significantly reduce population size, allowing a small number of variants to establish infection, favoring genetic drift. In HCV infection, the extent of genetic drift is reduced as the time of infection progresses[60-63]. This might be explained by the large population size frequently observed in chronically infected patients in comparison to acute cases. Additionally, occurrence of genetic drift might be affected by fluctuations in the viral load throughout the infection[64]. Anti-HCV therapy might also prompt genetic drift upon relapsing since viral populations are reduced considerably prior to reestablishment as a result of treatment failure.
Genetic recombination is another mechanism that participates in HCV genetic heterogeneity. Recombination generates genetic variability by rearranging genomic molecules during RNA elongation when the polymerase switches from donor to acceptor molecules, resulting in a nascent RNA with traits from both parental viruses[65]. HCV recombination also occurs via endoribonuclease digestion of parental molecules within base-unpaired regions, followed by ligation[66]. For recombination to occur, co-infection or superinfection in the same cell by two parental viruses is required[67]. HCV genetic recombination is considered a rare event[68], and therefore, while co-infection is possible, it remains relatively infrequent. Superinfection exclusion during HCV infection has been reported[69-71]. Thus, simultaneous infection is feasible but sequential infection is severely impaired[72], suggesting that infected cells become refractory to succeeding infections; and therefore, limiting recombination. Nonetheless, naturally occurring inter-genotype, intra-genotype (inter-subtype) and intra-strain HCV recombinants have been reported[73-76]. HCV recombination has important clinical and epidemiological implications that affect molecular epidemiology, surveillance of emergent new lineages and drug resistance strains[67].
HCV exists as an ensemble of closely related but genetically divergent variants, commonly referred as “quasispecies”[77]. Nevertheless, the existence of viral quasispecies has been extensively debated[77-79]. In HCV molecular epidemiology, the term quasispecies has become a synonymous of intrahost genetic variation[79]. Analysis of the HCV intrahost genetic variation is important for genetic relatedness, drug resistance, molecular evolution and epidemiological studies[80,81]. Rapid[34,82], and slow[83,84] HCV divergence has been reported in different settings. Rapid divergence of HCV represents an important challenge for genetic relatedness studies since molecular epidemiological links can be lost between related cases in relatively short periods of time[34].
HCV molecular evolution involves a series of complex processes characterized by temporal variations that constantly reshape the architecture of the viral population[81,83,85,86]. HCV evolves in an specific staging fashion through incremental changes between communities and random mutations accompanied by fluctuation in the frequency of coexisting viral subpopulations (Figure 4)[85]. In stage 1, HCV infection is established in the new host after occurrence of the initial genetic bottleneck, resulting in a strong founder effect before the onset of the adaptive immune response. Stage 2 is characterized by small incremental evolution steps through different communities, due to selective pressures imposed by the immune response. Stage 3 features a genetic diversification leading to the emergence of new subpopulations owned to the decline of previously dominant populations. In stage 4, HCV reaches settlement under strong negative selection[85]. These temporal variations are likely to affect HCV transmission since different viral variants are available at different time points during infection (Figure 4). The degree of “transmissibility” may exhibit important differences among different viral variants; thus, determining the capacity of each population to successfully establish infections in the new host[87]. This staging is also likely to play an important role in clinical outcome and assist in therapy management since viral populations may be differentially sensitive to treatment at particular stages[88].
Compartmentalization
While the liver is the main site for HCV replication, compartmentalization affecting other organs has been suggested to occur in both, natural infections in humans and experimentally infected chimpanzees[81,89-93]. However, limited information on HCV variability in extrahepatic sites is available. Different HCV populations have been found in a variety of tissues and cell types, suggesting the existence of extrahepatic reservoirs[94-96]. The concept of compartmentalization assumes that the distribution of viral variants in a non-random manner varies between tissues with an specific evolution rate for each compartment[97,98]. Compartmentalization in the same organ has also been observed as nontumor and tumor hepatic cells exhibit different populations, and are likely subjected to different selective pressures[99]. Compartmentalization has been shown in immunocompromised[97,100], immunocompetent[101], and transplanted individuals[102,103], further supporting its role in HCV persistence and pathogenesis.
Compartmentalization has also been proposed to play role in HCV molecular evolution[86]. In this evolutionary model, co-existing lineages represent genetically distinct subpopulations of infected liver cells (Figure 5). However, only one subset of viral subpopulations commonly circulates in serum at any given time. This may be explained by the fluctuation of neutralizing antibodies over time which in turn modify the relative frequency of lineages and differences in replication and shedding rates of virus populations[86].
Figure 5.
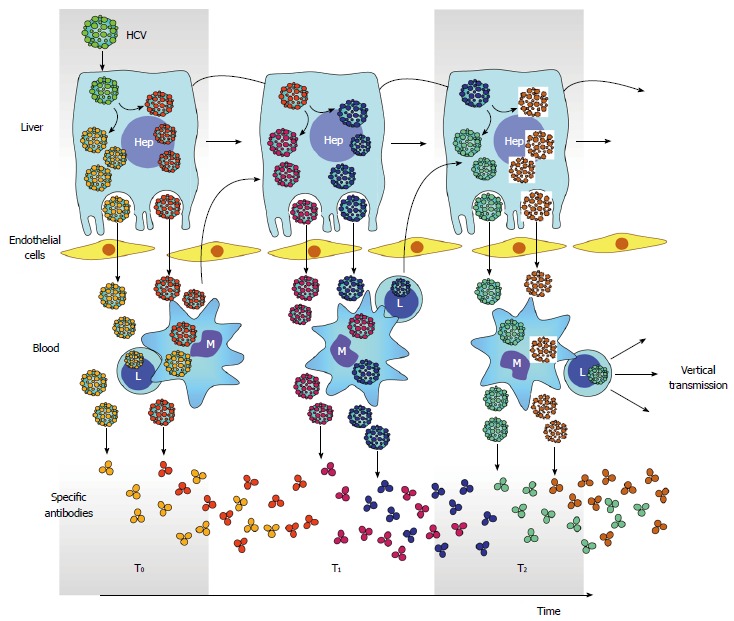
Hepatitis C virus compartmentalization. Generation of multiple HCV subpopulations is color coded. Releasing of different subpopulation to circulation and infection of PBMC is also depicted. Antibodies elicited against different HCV subpopulation is also shown throughout time. However, the generation of neutralizing antibodies is delayed and gives opportunity for arisen of new viral subpopulations and subsequent infection of PBMC. In turn, infection in PBMC facilitates vertical transmission of HCV. PBMC: Peripheral blood mononuclear cells; HCV: Hepatitis C virus.
Different mutation patterns in the HCV 5’UTR have been reported to occur in compartmentalized populations. Importantly, minor nucleotide changes occurring in this region can directly affect HCV translation efficiency. Thus, the effect of mutations in the HCV IRES might be cell type dependent[93,104], resulting in different replication rates. Therefore, HCV compartmentalization is not only determined by cell entry but also replication. NS5A and NS5B compartmentalized variants with different functional properties have also been observed[105,106]. These findings highlight the importance of compartmentalization in functionality of specific viral proteins.
Detection of HCV negative RNA strands has been reported in peripheral blood mononuclear cells (PBMC), lymph nodes, bone narrow and brain. Genetic differences between plasma and PBMC HCV variants have been found to persist for long periods of time[101,102,107]. Compartmentalization can be explained by the distribution of tissue specific receptor on different cell types[95]. Compartmentalization in PBMC is facilitated by high levels of CD81 expression[108]. The existence of HCV variants with different CD81 binding capacities has also been suggested[108]. Higher conservation in the CD81-2 HCV binding region has been observed in PBMC-derived variants in comparison to serum-derived strains[108]. Additionally, PBMC- associated HCV variants have also been detected after resolution of HCV infection, suggesting that compartmentalization can also play role in occult HCV infection[108]. In B cells, the interaction between E2 and CD81, as a result of compartmentalization, leads to cell activation, which in turns protects lymphocytes from activation-induced cell death and regulates their function[94,109]. Therefore, E2-CD81 engagement may contribute to HCV associated B cell lymphoproliferative disorders and insufficient neutralizing antibody production[109]. In human T lymphocytes, HCV infection is mediated by the CD5 receptor, a member of the scavenger receptor cysteine-rich family[110]. However, the factors determining lympho- and hepatotropism are not well known. While compartmentalization remains controversial[111-113], the potential implications in persistence, cell tropism, drug resistance, and vaccine development warrant further research[52].
VIRUS TRANSMISSION
HCV transmission primarily occurs via parenteral routes. Epidemic of recreational injection drugs and unsafe injections resulted in a large number of HCV infections during the 20th century[114,115]. Importantly, differences in transmission rates have resulted in distinctive HCV prevalence and genotype distribution worldwide[116-118].
HCV viral transmission is a dynamic process that has been subjected to significant changes during the last century. Certain risk factors, such as use of illegal intravenous drugs and risk behaviors among homosexual men, significantly facilitate virus transmission and create optimal conditions for rapid HCV molecular evolution[34]. The modes of transmission in a given epidemiological settings affect the intra and interhost genetic variability within the transmission network[34,83,119]. In high risk population groups, higher transmission rates occur among acute cases leading to spread of more infectious variants[87]. However, the patterns of HCV transmission can change over time[120], and this in turn affects HCV transmissibility.
HCV transmission networks are difficult to identify for several reasons. The long incubation period makes it difficult to link related cases to a common source of infection[34]. Additionally, acute HCV infections are usually asymptomatic, making identification of cases challenging[121]. The lack of laboratory methods and appropriate molecular surveillance systems capable of distinguishing acute from chronic infections further complicates identification of incident cases[122]. Recognition of HCV transmission is of critical importance in implementing measures aimed to prevent virus spread. Transmissions of fast evolving viruses, such as HCV, are difficult to trace since strains from epidemiological linked cases are genetically related but rarely identical. Thus, interpretation of viral phylogenies is inherently uncertain and should be used cautiously[123]. Phylogenetic branching not always correspond to directly linked transmission events, and available sequences may not represent all individuals belonging to the transmission network. In consequence, local epidemic sequences can group together in the absence of direct transmission[123], and therefore, phylogenetic linkage among strains cannot show, beyond any doubt, direct transmission between possibly linked cases, despite of inclusion of local unrelated controls[124]. However, local control sequences do help in those instances where direct transmission did not occur by showing sufficient phylogenetic separation between suspected cases[124,125]. Hence, proper epidemiological investigations should be performed in conjunction to adequately assess relatedness between HCV cases. Additionally, in high risk groups re-infection with closely related strains may preclude the use of paraphyletic clustering to relate cases[124].
Estimating the time of infection based purely on genetic data has been reported by using evolutionary molecular clock models[125-127]. However, confidence limits associated with molecular clock estimates can vary significantly, making interpretation difficult. Different factors can affect the accuracy and reliability of estimates of molecular divergence including sampling, temporal and anatomical distribution of sampling, genome region sequenced, super- or re-infection, and evolutionary models and algorithms used[124]. Thus, interpretation and reliability of such approaches requires further validation.
HCV evolution is also affected by other instances such as co-infection with other viruses or pregnancy[35]. This might be associated with the implicit alteration of the immune response in the mother. During pregnancy, viral levels are commonly increased and CD8+ T cell cytotoxicity reduced along with loss of HLA escape mutants, with the consequently emergence of enhanced fitness strains, and further reversion during the inter-pregnancy periods[102]. Thus, maternal cellular immunity impairment and emergence of more fit viruses might facilitate HCV perinatal transmission. Compartmentalization of HCV in PBMC has been proposed to play role in perinatal transmission, acting as Trojan horses for viral entry[128,129]. Moreover, PBMC-infected cells have been shown to be a risk factor for HCV vertical transmission[130,131]. In this setting, children infected perinatally bear infection with more fit viruses[101,132]. Transmission of more fit wild-type or revertant viruses could favor persistent infection in infants[128].
Tracking of HCV infection depends on sequence information originated from different subgenomic regions. The 5’-UTR region has been widely used for detection owned to its degree of conservation across genotypes while the NS5B region is the target of election for HCV genotyping[133,134]. However, these two regions do not contain sufficient sequence information to establish genetic relatedness between clinical isolates. Genetic relatedness studies primarily rely on information obtained from the HVR1[34,81,83,119]. Importantly, the rapid divergence in this region represents a challenge for molecular epidemiological studies, which can lead to loss of genetic links between related isolates[34]. Sequencing of multiple and longer subgenomic regions has been proposed as an alternative to overcome the limitations imposed by the rapid molecular evolution of HCV[34]. The NS5A has also been used to establish relatedness among HCV cases[85], which can aid to restore links between isolates owned to a lower nucleotide substitution rate. Despite the usefulness of different subgenomic regions for the characterization of clinical isolates, whole genome sequencing should be the ultimate goal for HCV molecular characterization.
The selective forces that shape the HCV molecular evolution are complex in nature and required sophisticated methods to be assessed. Until recently, molecular approaches used to detect low frequencies variants and assess the intrahost viral genetic variation, such as standard cloning techniques[57], limiting dilution[135] and single genome amplification[59], were cumbersome, time-consuming, and expensive[136]. Moreover, these conventional methods are difficult to implement and provide limited insights in the composition of the intrahost population[133]. The relatively recent advent of next generation sequencing platforms has allowed the detection of rare variants present at much lower frequencies[136-139], facilitating the characterization of the HCV intrahost viral population in remarkable detail[136,140,141].
DISEASE PROGRESSION
HCV molecular evolution is intimately associated with disease outcome and progression[142,143]. HCV-related liver disease gradually advances from chronic hepatitis to liver cirrhosis and to hepatocellular carcinoma (HCC)[144]. The rate of disease progression differs considerably among HCV cases, and the cause for these differences remains poorly understood. However, viral genotypes have been associated with pathogenesis of HCV-related infection, in addition to dictating the path of treatment[145]. Impairment of different pathways has also been associated with the infecting genotypes, which in turns leads to distinct pathological settings. Overall, HCV genotype 1 is associated with more aggressive disease, increased insulin resistance, poor response to therapy, higher risk of cirrhosis and hepatocellular carcinoma development, while genotype 3 is associated with increased steatosis and fibrosis[145,146]. Thus, the identification and characterization of HCV types and subtypes provides insight into the different outcome of HCV infection and responsiveness to therapy[145].
HCV core has been associated with disease progression by inducing apoptosis through the extrinsic and intrinsic pathways[147]. The signaling pathway converges into a common apoptotic pathway where the caspase enzyme cascade is triggered. As a consequence, intracellular components and DNA are degraded, leading to cells death and ultimately causing liver damage[148]. Apoptosis of liver cells plays an important role in the pathogenesis of end stage liver disease[149]. Interestingly, HCV core induces both pro- and anti-apoptotic effects[150-154]. HCV suppresses apoptosis by preventing the release of cytochrome C in the mitochondria, resulting in inactivation of caspase-9, -3 and -7[155]. When HCV core binds to the downstream death domain of the Fas-associated death domain (FADD) protein and FLICE (FADD-like interleukin-1beta-converting enzyme)-like inhibitory protein results in anti-apoptotic state[156]. HCV core also binds to p53 resulting in either inhibition or activation of apoptosis[157,158]. This dual behavior of HCV core in regulation of apoptosis is of the utmost importance in progression to HCC. The association between different amino acid residues in the HCV core region and disease progression has been reported[159]. Particularly, amino acid 70 in the HCV core has been closely associated with progression to HCC[160-163]. A detailed analysis of the quasispecies nature of the HCV core region showed that mutants bearing this residue is linked to advance of liver disease. Sequence variations in the core region have also been reported in tumor cells[164]. Analyses of the full-length of the core gene from patients with HCC showed that mutations in the core protein may affect the course of HCV infection[165-168]. HCV infection is associated with an increased risk of developing diabetes mellitus[169,170]. Additionally, amino acid substitutions in the HCV core from genotype 1b isolates have been suggested as predictor markers for insulin resistance in diabetic patients without signs of cirrhosis[171]. Isolates from patients with severe insulin resistance bear higher proportions of Glu70 (His70) and/or Met91. Therefore, genetic variability occurring in the HCV core is likely to affect its interaction with host proteins and thereby disease progression.
Steatosis is commonly observed in HCV cases and contributes to progressive hepatic injury. Development of hepatocellular steatosis is more frequently observed in cases infected with HCV genotype 3[172,173]. However, not all patients infected with genotype 3 develop steatosis[174]. Therefore, infection with genotype 3 is only one of the elements in the multi factorial nature of steatosis. Other studies have also implicated HCV genotype 3 in rapid progression to fibrosis[175], further supporting the role of genotyping in disease progression.
The complexity of the viral population in the NS5A region has been shown to be associated with HCC[176]. However, the specific mechanisms by which NS5A promotes disease progression are not understood. The NS5A inhibits the activated protein kinase (PKR), an important mediator of the IFN anti-viral response, apoptosis and cell proliferation[177-179], which can lead to liver damage. Additionally, NS5A can act as transcriptional activators affecting cellular signaling[180]. The NS5A encompasses the interferon-sensitivity determining region (ISDR) which is associated with response to antiviral therapy[181]. Thus, the NS5A interaction with host proteins is pleiotropic in nature, making difficult the characterization of the participant elements in pathogenesis.
In individuals progressing to end-stage liver disease, complexity of the quasispecies population at baseline is greater and tends to reduce over time, while non-progressors course with gradual increase in complexity[182]. Patients progressing to end stage liver disease, who likely exert less immune pressure on the HCV population, exhibit reduction in the complexity of the viral population[182]. High genetic complexity of the intrahost HCV population and liver injury has also been observed in immune-competent children associated with vertical transmission in comparison to immune-suppressed patients[183]. The dynamics of the HCV quasispecies are also associated with progression to fibrosis after liver transplantation[184-187]. Recurrence of HCV infection is universal after transplantation; however, clinical outcome is variable[188,189]. Several factors affect the outcome of recurrent HCV disease upon transplantation including HCV genotype[190,191], virus load[192], and co-infection with cytomegalovirus[193,194]. Co-infection with human immunodeficiency (HIV) virus has also been associated with progression to severe liver disease[195]. Co-infected patients with unfavorable prognosis are presented with a relatively stable quasispecies population which can be a reflection of the limited immune pressure commonly observed in these patients[184].
MOLECULAR ASPECTS OF THERAPY AND DRUG RESISTANCE
The “ideal” outcome of the anti-HCV treatment is a sustained virologic response (SVR), defined as undetectable viral RNA for six months after completion of treatment[196]. Until recently, the only option for HCV therapy was a combination of pegylated interferon-α (PEG-IFNα) and ribavirin (RBV). The efficacy of this regime ranged between 20%-80%, depending on race, disease stage, infecting genotype and distinct single nucleotide polymorphisms located in the IFN-λ3 promoter gene[197,198]. Poor outcome was the common feature among patients infected with HCV genotype 1 undergoing IFNα-RBV therapy[196]; thus, the infecting genotype was an important determinant for response to treatment[199]. Therefore, efforts aimed to find predictive markers for SVR were pursued by several groups. Besides infecting genotype and viral titer, other viral molecular factors used to predict IFN therapy outcome rendered controversial results. For instance, the 39 amino acids located in the interferon sensitivity determining region (ISDR) in the N5A gene were proposed to be predictive of SVR[181,200,201]. Thus, four or more substitutions in this small region correlated with responsiveness in Asian patients infected with HCV genotype 1b. However, the association between ISDR and SVR in other races rendered controversial results[202,203].
Major advances in the anti-HCV therapy have been made during the past few years[204]. This has resulted in significant improvement in SVR. The landscape of treatments for HCV is expected to change drastically with more efficient IFN-free antiviral therapies being approved in the near future[205]. Insights into the molecular structure of different HCV proteins have greatly fostered the development of new drugs, commonly referred as direct-acting antiviral agents (DAA). Experience with anti-retrovirals has provided a valuable framework for the development of DAA against HCV. In theory, every step of the HCV replication cycle is a potential target for antiviral therapy; however, good results with HIV protease inhibitors suggested the HCV protease as an ideal candidate. Nonetheless, both NS3 and NS5B have been targets for anti-HCV therapy; with two-thirds of drugs directed against these proteins currently in Phase II and III trials, and few already approved by the United States Food and Drug Administration (FDA). In addition, DAA directed to the NS5A protein have also been developed (Figure 1, Table 1).
Table 1.
Amino acid substitutions within the direct-acting antiviral agents target associated with resistance to different direct-acting antiviral agents agents
| Drug target | DAA agents | RAV |
| NS3/4A protease | ||
| Telaprevir | V36M/A1, T54A/S1, R155K/T1, A156S/T/V1 | |
| Boceprevir | V36M/A1, T54A/S1, V55A1, R155K/T1, A156S/T/V1, V158I, V170A, M175L | |
| Faldaprevir | R155K1, A156T/V, D168V/E1 | |
| Simeprevir | V36M, F43S, Q80K1, S122A/R, R155K1, A156T/V, D168V/E1, I170T | |
| ABT-450 | V36M/A/G, V55I, R155K1, A156T/V, D168A/V1 | |
| Danoprevir | V36M/A, V55I, R155K1, D168E/T1 | |
| Vaniprevir | R155G1/K/N/S/T, A156V, D168V1/G/A | |
| NS5B polymerase | ||
| Mericitabine | S282T, L159F, L320F | |
| BI 207127 | P495L1/S/A/T/Q, P496S, V499A | |
| Lomibuvir/VX-222 | L419S1, R422K1, M423TV | |
| Setrobuvir | M414T/L, G554D, D559G | |
| NS5A | ||
| Daclatasvir | M28V/A/T, Q30R/E/H, L31V/M1, Q54H/N/Y, H58D, Q62R/E, A92K/T, Y93H/N/C1 |
Amino acid position number represents the key mutations that are clearly associated with virologic failure. A: Alanine; C: Cysteine; D: Aspartate; E: Glutamate; F: Phenylalanine; G: Glycine; H: Histidine; L: Leucine; I: Isoleucine; K: Lysine; M: Methionine; N: Asparagine; P: Proline; Q: Glutamine; R: Arginine; S: Serine; T: Threonine; V: Valine; Y: Tyrosine; DAA: Direct-acting antiviral agents; RAV: Resistant associated variants.
HCV NS3 protease inhibitors
From a chemical point of view, HCV NS3/4A protease inhibitors can be divided into three main categories: (1) linear peptidomimetics with an alpha-ketoamide group that binds the active site, covalently blocking the enzymatic activity (first class); (2) linear non-covalent peptidomimetic inhibitors (second class); and (3) macrocyclic non-covalent peptidomimetic inhibitors (third class)[206,207].
In May 2011, telaprevir and boceprevir, both belonging to the first class of protease inhibitors, were approved by the FDA for treatment of patients chronically infected with HCV genotype 1. In large Phase III studies, SVR rates of 67%-75% among treatment-naive and 59%-64% in treatment-experienced patients were achieved with triple regimens (PEG-IFNα, RBV plus either telaprevir or boceprevir) in comparison to the standard therapy with IFNα/RBV[109,208-210]. SVR rates depend on the patient previous response to dual therapy and fibrosis stage[211], being considerably lower in patients with null response and liver cirrhosis[210,212]. Additionally, these drugs still require the leading phase with IFNα/RBV; and therefore, are prone to develop drug resistance[207]. Two features contribute to this observation: (1) the low-fidelity of the HCV RdRp and large progeny generated per day; and (2) the complex structure of the HCV protease active site. The estimated error rate of the HCV RpRd is about 10-fold higher than the HIV reverse transcriptase[213], and the virion production rate about 100-fold higher, originating resistant mutants as minor populations in treatment naïve patients[133,214,215]. While some studies have reported frequencies of HCV resistance mutants < 1%[133,214,215], others have shown higher proportions[216]. These resistant associated variants (RAV) can rapidly emerge and come to prominence after few days of treatment and are responsible for treatment failure in many patients[217-219].
Boceprevir and telaprevir share extensive cross-resistance. RAV most frequently associated with telaprevir monotherapy include R155K, A156S/T/V, V36M, and T54A/S (Table 1). Similar variants are observed with boceprevir monotherapy, in addition to V170A and V55A (Table 1). Similar profiles of resistance in vitro have been observed for each substitution resulting in low-to-moderate resistance for V36M, T54A/S, V55A, R155K, A156S, and V170A, and high resistance for A156T/V and double mutants R155K/V36M[218,220]. Substitutions associated with resistance to protease inhibitors generally reduce the catalytic efficiency of the HCV protease. For this reason, these mutants are rarely detected as the dominant variant in the intrahost HCV population in the absence of the antiviral drug. Recent studies have shown that non-responders to protease inhibitors carried a dominant strain of RAV at the time of virus breakthrough or relapse, but the wild-type strain re-emerged as the dominant variant after completion of therapy[221,222]. RAV have been reported for most antiviral agents in development, with widespread cross-resistance for both linear and macrocyclic groups[218,223]. For example, certain substitutions involving residues such as R155 as well as R155/A156 often confer resistance against most protease inhibitors at a moderate fitness cost to the virus. Thus, in the absence of antiviral drugs, RAV replicate with moderate efficiency compared to wild-type viruses[222,224].
Telaprevir and boceprevir have restricted spectrum of action over HCV genotypes. While telaprevir has some clinical effect against genotype 2, and boceprevir seems to be effective against genotype 3, their use is prescribed off-label for these genotypes[223]. Drug resistance to protease inhibitors displays a low genetic barrier (i.e., the number of nucleotide changes required to generate RAV) which represents a particular problem in HCV subgenotype 1a infection, as the genetic barrier is lower compared to subgenotype 1b[225].
Second and third classes of NS3 protease inhibitors, which do not form covalent bounds with their targets, have several advantages over the first-class compounds[226]. These NS3 protease inhibitors include linear non-covalent molecules such as faldaprevir, asunaprevir, sovaprevir, GS-9451 and macrocyclic inhibitors such as simeprevir, danoprevir, ABT-450, GS-9256; and vaniprevir (Table 1)[207,227-234]. Most of these new DAA are currently in Phase II or III clinical trials with the exception of simeprevir which has been already approved by FDA. These antiviral agents have showed high rates of SVR, comparable or higher than boceprevir or telaprevir triple-combination regimens, in HCV genotype 1 patients when used in combination with PEG-IFN and RBV. These NS3 HCV protease inhibitors tend to have a broader spectrum of action over different HCV genotypes; however, these DAA are not pan genotypic antivirals owned to different inhibitory efficacies across genotypes[227,235,236]. Simeprevir exhibits high antiviral activity against genotypes 2, 4, 5, and 6 whereas no effect has been observed with genotype 3[236]. First class protease inhibitors genetic barrier is low and extensive cross-resistance in comparison to second and third class protease inhibitors[218]. In particular, mutations R155K/T/Q have been shown to confer broad cross-resistance while mutations D168/E/G/H/T/Y confer resistance specifically to non-covalent peptidomimetic inhibitors, regardless of linear or macrocyclic structure[218,233,237-239]. Notably, D168 is one of the few active site residues not entirely conserved among HCV genotypes, which is replaced by glutamine in isolates belonging to genotype 3, partly explaining why HCV genotype 3 is “naturally resistant”[240]. Moreover, substitution Q80K is present in about 20% of genotype 1a sequences and confers resistance to Simeprevir[237]. This mutant has been associated with breakthrough infection in treatment-naïve subjects[220].
NS5B polymerase inhibitors
Another strategy to inhibit viral replication is the development of polymerase inhibitors which interfere with viral replication by binding to the NS5B RdRp. NS5B inhibitors can be divided into two distinct categories: nucleosides inhibitors and non- nucleoside inhibitors.
Nucleoside analogue inhibitors mimic the natural substrates of the polymerase causing direct chain termination[241,242]. The NS5B active site is well-conserved across HCV genotypes as amino acid substitutions in this location are generally poorly tolerated and result in remarkable loss of fitness[223,243]. Among nucleoside analogue, mericitabine has been shown to increase SVR rates in patients infected with genotype 1 and 4[244]. Sofosbuvir, recently approved by the FDA, has demonstrated extraordinary efficacy for treatment of adults infected with genotypes 1 and 4 in combination with IFN/RBV, and for the treatment of adults infected with genotypes 2 and 3 in IFN-free regimens[106,245,246]. Importantly, several factors usually associated with poor outcomes in HCV infected patients had no effect on the efficacy of sofosbuvir; thus, only male gender and the presence of cirrhosis are predictive of unfavorable outcome. Substitution S282T conferring resistance to sofosbuvir has been observed in vitro but not in vivo[223]. However, mutants L159F/L320F in the NS5B polymerase conferring low-level resistance to mericitabine and sofosbuvir has been reported recently[223,247]. Overall, nucleoside polymerase inhibitors tend to exhibit good activity against a broad range of genotypes and have a high genetic barrier to resistance which makes them a promising class of anti-HCV agents.
Non-nucleoside inhibitors are chemically and functionally much more diverse than nucleoside inhibitors. Non-nucleoside inhibitors usually bind to several discrete sites on the HCV polymerase, which results in conformational protein changes before the elongation complex is formed[241,242]. A limitation of this mechanism of action is that binding sites are less conserved among genotypes compared to the active site. As a consequence, lower cross-genotypic activity and higher probability of RAV development is observed. Despite efficacy against HCV genotype 1, resistance occurs at a low fitness cost[218].
Several non-nucleoside inhibitors targeting at least four distinct allosteric binding sites are in development. Two of these binding sites, named “thumb I” and “thumb II”, are located on the polymerase thumb domain, whereas the other two sites, “palm I” and “palm II” are close to the active site cavity and involve primarily amino acids from the palm domain. Certain non-nucleoside inhibitors develop drug resistant variants carrying mutations at different positions, e.g., BI 207127 targeting thumb I at P495, P496 and V499; lomibuvir targeting thumb II at L419, R422 and M423 as well as setrobuvir affecting palm I at M411, G554 and D559[220].
NS5A inhibitors
Disruption of the recruiting capabilities of NS5A can also be targeted for antiviral drug development. Recently, identification of lead compounds capable of inducing a rapid decline in viral load and emergence of RAV, characterized by amino acid substitutions located in the NS5A protein, confirmed their specificity[248,249]. These agents feature broadly genotypic coverage but low genetic barrier to resistance[207,250].
Daclatasvir is a replication complex inhibitor currently in Phase III. Daclatasvir is active at picomolar concentrations in vitro displaying activity over a broad range of HCV genotypes[249,251]. The resistance profile of daclatasvir has been linked to the N-terminus of NS5A[249]. The most remarkable RAV associated to daclatasvir are Y93H/C/N, which also confer cross-resistance to other NS5A inhibitors[249]. Interestingly, Y93 is found near the protein dimer interface, leading to speculate that NS5A inhibitors might affect the monomer/dimer equilibrium[252]. Monotherapy with these drugs rapidly selects for the outgrowth of resistant variants, similar to other HCV protease inhibitors[220,251,253,254].
Interferon-free therapy regimens
IFN-free combination trials are ongoing with different DAA. The current challenge is the development of an oral regimen including compounds with different mechanisms of action, synergistic interactions and pan-genotypic activity. Drugs in an IFN-free therapy should not display overlapping resistance profiles. In principle, this is achievable since most DAA target different viral proteins or binding sites in the same protein. Several DAA are expected to successfully complete phase III trials in anticipation of licensing. Initially, different IFN-based regimens (sofosbuvir, faldaprevir and simeprevir) will be readily available for treatment of HCV genotype 1. In the near future, combination of antiviral agents lacking cross-resistance with good safety profile will be the new recommended therapy regime. Recent clinical studies have shown SVR > 90% in patients administered with DAA cocktails including protease and polymerase inhibitor[207]. For example, Sofosbuvir is currently being evaluated in IFN-free combinations with simeprevir and daclatasvir. In turn daclatasvir efficacy is being assessed in combination with asunaprevir and/or non-nucleoside polymerase inhibitor BMS791325[207,255].
The rapid development of DAA has broadened the landscape of anti-HCV drugs which will probably include extensive number of possible ingredients for an effective combinatory regimen. Results from recent clinical trials have firmly established the concept that a permanent cure can be achieved with IFN-free combinations of DAA[96,256].
CONCLUSION
HCV molecular evolution in many ways dictates virus transmission, progression to severe liver disease and therapy outcome. The molecular mechanisms exploited by the virus to achieve persistence are numerous and complex in nature. The sophisticated evolutionary process to which HCV is subjected during transmission and upon infection further limits our understanding of the participating elements of HCV pathogenesis. The remarkable HCV mutation rate represents a challenging task for vaccine development and molecular epidemiology. In this new era of advance sequencing technologies, the implementation of enhanced molecular surveillance is of the utmost importance to accurately monitor circulation of viral strains. Comprehensive molecular studies are also required to uncover the participant elements responsible for virulence.
The rapidly evolving field of DAA is heading to the development of IFN-free regimens with superior SVR and pan genotypic activity and less prone to side effects. However, emergence of RAV remains an important concern. Importantly, and despite the emergence of HCV RAV, which are frequently developed upon anti-HCV treatment, high SVR rates are attainable. Detection of minor variants should be conducted before and during treatment with new drugs to monitor development of resistance and better manage patients.
The major advances on therapy have been the results of many efforts focused on discovering the nuances associated with HCV replication. However, the development of successful vaccines imposed a more daunting task. Thus, the path to HCV vaccination should be as exciting, if not more, than the road to efficacious treatment.
Footnotes
Supported by Project Salud 2012-C01-181585, CONACYT and PAPIIT TA200112, Dirección General de Asuntos del Personal Academico, Universidad Nacional Autónoma de México (in part); Argentine National Agency for Scientific and Technology Promotion (PICT 2012 Nº804) and National Research Council (CONICET, PIP 2010 Nº51)
P- Reviewer: Waheed Y, Pazienza V, Song M S- Editor: Ma YJ L- Editor: A E- Editor: Liu XM
References
- 1.Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology. 2013;57:1333–1342. doi: 10.1002/hep.26141. [DOI] [PubMed] [Google Scholar]
- 2.Alter MJ. Epidemiology of hepatitis C virus infection. World J Gastroenterol. 2007;13:2436–2441. doi: 10.3748/wjg.v13.i17.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lavanchy D. The global burden of hepatitis C. Liver Int. 2009;29 Suppl 1:74–81. doi: 10.1111/j.1478-3231.2008.01934.x. [DOI] [PubMed] [Google Scholar]
- 4.Lauer GM, Walker BD. Hepatitis C virus infection. N Engl J Med. 2001;345:41–52. doi: 10.1056/NEJM200107053450107. [DOI] [PubMed] [Google Scholar]
- 5.McHutchison JG, Bacon BR. Chronic hepatitis C: an age wave of disease burden. Am J Manag Care. 2005;11:S286–S295; quiz S307-S311. [PubMed] [Google Scholar]
- 6.Stanley ML, Walker C, Alter MJ, Yi M. Hepatitis C Virus. In: Knipe DM, Howley PM, editors. Fields Virology. Philadelphia: Lippincott Williams & Wilkins; 2007. p. 1280. [Google Scholar]
- 7.Chevaliez S, Pawlotsky JM. HCV Genome and Life Cycle. In: Tan SL, editor. Hepatitis C Viruses: Genomes and Molecular Biology. UK: Norfolk; 2006. [Google Scholar]
- 8.Khaliq S, Jahan S, Pervaiz A. Sequence variability of HCV Core region: important predictors of HCV induced pathogenesis and viral production. Infect Genet Evol. 2011;11:543–556. doi: 10.1016/j.meegid.2011.01.017. [DOI] [PubMed] [Google Scholar]
- 9.Drummer HE, Maerz A, Poumbourios P. Cell surface expression of functional hepatitis C virus E1 and E2 glycoproteins. FEBS Lett. 2003;546:385–390. doi: 10.1016/s0014-5793(03)00635-5. [DOI] [PubMed] [Google Scholar]
- 10.Troesch M, Meunier I, Lapierre P, Lapointe N, Alvarez F, Boucher M, Soudeyns H. Study of a novel hypervariable region in hepatitis C virus (HCV) E2 envelope glycoprotein. Virology. 2006;352:357–367. doi: 10.1016/j.virol.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 11.Steinmann E, Penin F, Kallis S, Patel AH, Bartenschlager R, Pietschmann T. Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions. PLoS Pathog. 2007;3:e103. doi: 10.1371/journal.ppat.0030103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steinmann E, Pietschmann T. Hepatitis C virus p7-a viroporin crucial for virus assembly and an emerging target for antiviral therapy. Viruses. 2010;2:2078–2095. doi: 10.3390/v2092078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gallinari P, Brennan D, Nardi C, Brunetti M, Tomei L, Steinkühler C, De Francesco R. Multiple enzymatic activities associated with recombinant NS3 protein of hepatitis C virus. J Virol. 1998;72:6758–6769. doi: 10.1128/jvi.72.8.6758-6769.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stapleford KA, Lindenbach BD. Hepatitis C virus NS2 coordinates virus particle assembly through physical interactions with the E1-E2 glycoprotein and NS3-NS4A enzyme complexes. J Virol. 2011;85:1706–1717. doi: 10.1128/JVI.02268-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci USA. 2005;102:17717–17722. doi: 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 17.Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M, Ray SC, Gale M, Lemon SM. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci USA. 2005;102:2992–2997. doi: 10.1073/pnas.0408824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morikawa K, Lange CM, Gouttenoire J, Meylan E, Brass V, Penin F, Moradpour D. Nonstructural protein 3-4A: the Swiss army knife of hepatitis C virus. J Viral Hepat. 2011;18:305–315. doi: 10.1111/j.1365-2893.2011.01451.x. [DOI] [PubMed] [Google Scholar]
- 19.Gouttenoire J, Penin F, Moradpour D. Hepatitis C virus nonstructural protein 4B: a journey into unexplored territory. Rev Med Virol. 2010;20:117–129. doi: 10.1002/rmv.640. [DOI] [PubMed] [Google Scholar]
- 20.Macdonald A, Crowder K, Street A, McCormick C, Harris M. The hepatitis C virus NS5A protein binds to members of the Src family of tyrosine kinases and regulates kinase activity. J Gen Virol. 2004;85:721–729. doi: 10.1099/vir.0.19691-0. [DOI] [PubMed] [Google Scholar]
- 21.Reed KE, Xu J, Rice CM. Phosphorylation of the hepatitis C virus NS5A protein in vitro and in vivo: properties of the NS5A-associated kinase. J Virol. 1997;71:7187–7197. doi: 10.1128/jvi.71.10.7187-7197.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Behrens SE, Tomei L, De Francesco R. Identification and properties of the RNA-dependent RNA polymerase of hepatitis C virus. EMBO J. 1996;15:12–22. [PMC free article] [PubMed] [Google Scholar]
- 23.Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus. Nat Rev Microbiol. 2007;5:453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 24.Barth H, Schafer C, Adah MI, Zhang F, Linhardt RJ, Toyoda H, Kinoshita-Toyoda A, Toida T, Van Kuppevelt TH, Depla E, et al. Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J Biol Chem. 2003;278:41003–41012. doi: 10.1074/jbc.M302267200. [DOI] [PubMed] [Google Scholar]
- 25.Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, Filocamo G, Traboni C, Nicosia A, Cortese R, Vitelli A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002;21:5017–5025. doi: 10.1093/emboj/cdf529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hadlock KG, Lanford RE, Perkins S, Rowe J, Yang Q, Levy S, Pileri P, Abrignani S, Foung SK. Human monoclonal antibodies that inhibit binding of hepatitis C virus E2 protein to CD81 and recognize conserved conformational epitopes. J Virol. 2000;74:10407–10416. doi: 10.1128/jvi.74.22.10407-10416.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G, et al. Binding of hepatitis C virus to CD81. Science. 1998;282:938–941. doi: 10.1126/science.282.5390.938. [DOI] [PubMed] [Google Scholar]
- 28.Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wölk B, Hatziioannou T, McKeating JA, Bieniasz PD, Rice CM. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature. 2007;446:801–805. doi: 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- 29.Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, de Jong YP, Rice CM. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature. 2009;457:882–886. doi: 10.1038/nature07684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, Zona L, Davis C, Mee CJ, Turek M, Gorke S, et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med. 2011;17:589–595. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sainz B, Barretto N, Martin DN, Hiraga N, Imamura M, Hussain S, Marsh KA, Yu X, Chayama K, Alrefai WA, et al. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat Med. 2012;18:281–285. doi: 10.1038/nm.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blanchard E, Belouzard S, Goueslain L, Wakita T, Dubuisson J, Wychowski C, Rouillé Y. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J Virol. 2006;80:6964–6972. doi: 10.1128/JVI.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hsu M, Zhang J, Flint M, Logvinoff C, Cheng-Mayer C, Rice CM, McKeating JA. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc Natl Acad Sci USA. 2003;100:7271–7276. doi: 10.1073/pnas.0832180100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cruz-Rivera M, Carpio-Pedroza JC, Escobar-Gutiérrez A, Lozano D, Vergara-Castaneda A, Rivera-Osorio P, Martinez-Guarneros A, Chacon CA, Fonseca-Coronado S, Vaughan G. Rapid hepatitis C virus divergence among chronically infected individuals. J Clin Microbiol. 2013;51:629–632. doi: 10.1128/JCM.03042-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Honegger JR, Kim S, Price AA, Kohout JA, McKnight KL, Prasad MR, Lemon SM, Grakoui A, Walker CM. Loss of immune escape mutations during persistent HCV infection in pregnancy enhances replication of vertically transmitted viruses. Nat Med. 2013;19:1529–1533. doi: 10.1038/nm.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.von Hahn T, Yoon JC, Alter H, Rice CM, Rehermann B, Balfe P, McKeating JA. Hepatitis C virus continuously escapes from neutralizing antibody and T-cell responses during chronic infection in vivo. Gastroenterology. 2007;132:667–678. doi: 10.1053/j.gastro.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 37.Ralston R, Jacobson I, Scull M. The conundrum of relapse in STAT-C therapy: does HCV play the Red Queen or Rip Van Winkle? Semin Liver Dis. 2011;31:410–419. doi: 10.1055/s-0031-1297929. [DOI] [PubMed] [Google Scholar]
- 38.Smith DB, Bukh J, Kuiken C, Muerhoff AS, Rice CM, Stapleton JT, Simmonds P. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology. 2014;59:318–327. doi: 10.1002/hep.26744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bukh J, Miller RH, Purcell RH. Genetic heterogeneity of hepatitis C virus: quasispecies and genotypes. Semin Liver Dis. 1995;15:41–63. doi: 10.1055/s-2007-1007262. [DOI] [PubMed] [Google Scholar]
- 40.Simmonds P, Holmes EC, Cha TA, Chan SW, McOmish F, Irvine B, Beall E, Yap PL, Kolberg J, Urdea MS. Classification of hepatitis C virus into six major genotypes and a series of subtypes by phylogenetic analysis of the NS-5 region. J Gen Virol. 1993;74(Pt 11):2391–2399. doi: 10.1099/0022-1317-74-11-2391. [DOI] [PubMed] [Google Scholar]
- 41.Lavanchy D. Evolving epidemiology of hepatitis C virus. Clin Microbiol Infect. 2011;17:107–115. doi: 10.1111/j.1469-0691.2010.03432.x. [DOI] [PubMed] [Google Scholar]
- 42.Agha S, Tanaka Y, Saudy N, Kurbanov F, Abo-Zeid M, El-Malky M, Khalaf M, Ohta N, Yoshizawa H, Mizokami M. Reliability of hepatitis C virus core antigen assay for detection of viremia in HCV genotypes 1, 2, 3, and 4 infected blood donors: a collaborative study between Japan, Egypt, and Uzbekistan. J Med Virol. 2004;73:216–222. doi: 10.1002/jmv.20078. [DOI] [PubMed] [Google Scholar]
- 43.McOmish F, Yap PL, Dow BC, Follett EA, Seed C, Keller AJ, Cobain TJ, Krusius T, Kolho E, Naukkarinen R. Geographical distribution of hepatitis C virus genotypes in blood donors: an international collaborative survey. J Clin Microbiol. 1994;32:884–892. doi: 10.1128/jcm.32.4.884-892.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kao JH, Chen PJ, Lai MY, Yang PM, Sheu JC, Wang TH, Chen DS. Genotypes of hepatitis C virus in Taiwan and the progression of liver disease. J Clin Gastroenterol. 1995;21:233–237. doi: 10.1097/00004836-199510000-00014. [DOI] [PubMed] [Google Scholar]
- 45.Simmonds P. The origin of hepatitis C virus. Curr Top Microbiol Immunol. 2013;369:1–15. doi: 10.1007/978-3-642-27340-7_1. [DOI] [PubMed] [Google Scholar]
- 46.Magiorkinis G, Magiorkinis E, Paraskevis D, Ho SY, Shapiro B, Pybus OG, Allain JP, Hatzakis A. The global spread of hepatitis C virus 1a and 1b: a phylodynamic and phylogeographic analysis. PLoS Med. 2009;6:e1000198. doi: 10.1371/journal.pmed.1000198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jackowiak P, Kuls K, Budzko L, Mania A, Figlerowicz M, Figlerowicz M. Phylogeny and molecular evolution of the hepatitis C virus. Infect Genet Evol. 2014;21:67–82. doi: 10.1016/j.meegid.2013.10.021. [DOI] [PubMed] [Google Scholar]
- 48.Ribeiro RM, Li H, Wang S, Stoddard MB, Learn GH, Korber BT, Bhattacharya T, Guedj J, Parrish EH, Hahn BH, et al. Quantifying the diversification of hepatitis C virus (HCV) during primary infection: estimates of the in vivo mutation rate. PLoS Pathog. 2012;8:e1002881. doi: 10.1371/journal.ppat.1002881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cuevas JM, González-Candelas F, Moya A, Sanjuán R. Effect of ribavirin on the mutation rate and spectrum of hepatitis C virus in vivo. J Virol. 2009;83:5760–5764. doi: 10.1128/JVI.00201-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lohmann V, Körner F, Dobierzewska A, Bartenschlager R. Mutations in hepatitis C virus RNAs conferring cell culture adaptation. J Virol. 2001;75:1437–1449. doi: 10.1128/JVI.75.3.1437-1449.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Campo DS, Dimitrova Z, Yokosawa J, Hoang D, Perez NO, Ramachandran S, Khudyakov Y. Hepatitis C virus antigenic convergence. Sci Rep. 2012;2:267. doi: 10.1038/srep00267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Farci P. New insights into the HCV quasispecies and compartmentalization. Semin Liver Dis. 2011;31:356–374. doi: 10.1055/s-0031-1297925. [DOI] [PubMed] [Google Scholar]
- 53.Penin F, Combet C, Germanidis G, Frainais PO, Deléage G, Pawlotsky JM. Conservation of the conformation and positive charges of hepatitis C virus E2 envelope glycoprotein hypervariable region 1 points to a role in cell attachment. J Virol. 2001;75:5703–5710. doi: 10.1128/JVI.75.12.5703-5710.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Quer J, Esteban JI, Cos J, Sauleda S, Ocaña L, Martell M, Otero T, Cubero M, Palou E, Murillo P, et al. Effect of bottlenecking on evolution of the nonstructural protein 3 gene of hepatitis C virus during sexually transmitted acute resolving infection. J Virol. 2005;79:15131–15141. doi: 10.1128/JVI.79.24.15131-15141.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Franco S, Parera M, Aparicio E, Clotet B, Martinez MA. Genetic and catalytic efficiency structure of an HCV protease quasispecies. Hepatology. 2007;45:899–910. doi: 10.1002/hep.21623. [DOI] [PubMed] [Google Scholar]
- 56.Wang GP, Sherrill-Mix SA, Chang KM, Quince C, Bushman FD. Hepatitis C virus transmission bottlenecks analyzed by deep sequencing. J Virol. 2010;84:6218–6228. doi: 10.1128/JVI.02271-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bull RA, Luciani F, McElroy K, Gaudieri S, Pham ST, Chopra A, Cameron B, Maher L, Dore GJ, White PA, et al. Sequential bottlenecks drive viral evolution in early acute hepatitis C virus infection. PLoS Pathog. 2011;7:e1002243. doi: 10.1371/journal.ppat.1002243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.D’Arienzo V, Moreau A, D’Alteroche L, Gissot V, Blanchard E, Gaudy-Graffin C, Roch E, Dubois F, Giraudeau B, Plantier JC, et al. Sequence and functional analysis of the envelope glycoproteins of hepatitis C virus variants selectively transmitted to a new host. J Virol. 2013;87:13609–13618. doi: 10.1128/JVI.02119-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li H, Stoddard MB, Wang S, Blair LM, Giorgi EE, Parrish EH, Learn GH, Hraber P, Goepfert PA, Saag MS, et al. Elucidation of hepatitis C virus transmission and early diversification by single genome sequencing. PLoS Pathog. 2012;8:e1002880. doi: 10.1371/journal.ppat.1002880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kato N, Ootsuyama Y, Sekiya H, Ohkoshi S, Nakazawa T, Hijikata M, Shimotohno K. Genetic drift in hypervariable region 1 of the viral genome in persistent hepatitis C virus infection. J Virol. 1994;68:4776–4784. doi: 10.1128/jvi.68.8.4776-4784.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van Doorn LJ, Quint W, Tsiquaye K, Voermans J, Paelinck D, Kos T, Maertens G, Schellekens H, Murray K. Longitudinal analysis of hepatitis C virus infection and genetic drift of the hypervariable region. J Infect Dis. 1994;169:1226–1235. doi: 10.1093/infdis/169.6.1226. [DOI] [PubMed] [Google Scholar]
- 62.Okamoto H, Kojima M, Okada S, Yoshizawa H, Iizuka H, Tanaka T, Muchmore EE, Peterson DA, Ito Y, Mishiro S. Genetic drift of hepatitis C virus during an 8.2-year infection in a chimpanzee: variability and stability. Virology. 1992;190:894–899. doi: 10.1016/0042-6822(92)90933-g. [DOI] [PubMed] [Google Scholar]
- 63.Allain JP, Dong Y, Vandamme AM, Moulton V, Salemi M. Evolutionary rate and genetic drift of hepatitis C virus are not correlated with the host immune response: studies of infected donor-recipient clusters. J Virol. 2000;74:2541–2549. doi: 10.1128/jvi.74.6.2541-2549.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kao JH, Chen PJ, Lai MY, Wang TH, Chen DS. Quasispecies of hepatitis C virus and genetic drift of the hypervariable region in chronic type C hepatitis. J Infect Dis. 1995;172:261–264. doi: 10.1093/infdis/172.1.261. [DOI] [PubMed] [Google Scholar]
- 65.Worobey M, Holmes EC. Evolutionary aspects of recombination in RNA viruses. J Gen Virol. 1999;80(Pt 10):2535–2543. doi: 10.1099/0022-1317-80-10-2535. [DOI] [PubMed] [Google Scholar]
- 66.Scheel TK, Galli A, Li YP, Mikkelsen LS, Gottwein JM, Bukh J. Productive homologous and non-homologous recombination of hepatitis C virus in cell culture. PLoS Pathog. 2013;9:e1003228. doi: 10.1371/journal.ppat.1003228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.González-Candelas F, López-Labrador FX, Bracho MA. Recombination in hepatitis C virus. Viruses. 2011;3:2006–2024. doi: 10.3390/v3102006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shi W, Freitas IT, Zhu C, Zheng W, Hall WW, Higgins DG. Recombination in hepatitis C virus: identification of four novel naturally occurring inter-subtype recombinants. PLoS One. 2012;7:e41997. doi: 10.1371/journal.pone.0041997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tscherne DM, Evans MJ, von Hahn T, Jones CT, Stamataki Z, McKeating JA, Lindenbach BD, Rice CM. Superinfection exclusion in cells infected with hepatitis C virus. J Virol. 2007;81:3693–3703. doi: 10.1128/JVI.01748-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Webster B, Ott M, Greene WC. Evasion of superinfection exclusion and elimination of primary viral RNA by an adapted strain of hepatitis C virus. J Virol. 2013;87:13354–13369. doi: 10.1128/JVI.02465-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Webster B, Wissing S, Herker E, Ott M, Greene WC. Rapid intracellular competition between hepatitis C viral genomes as a result of mitosis. J Virol. 2013;87:581–596. doi: 10.1128/JVI.01047-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schaller T, Appel N, Koutsoudakis G, Kallis S, Lohmann V, Pietschmann T, Bartenschlager R. Analysis of hepatitis C virus superinfection exclusion by using novel fluorochrome gene-tagged viral genomes. J Virol. 2007;81:4591–4603. doi: 10.1128/JVI.02144-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kalinina O, Norder H, Mukomolov S, Magnius LO. A natural intergenotypic recombinant of hepatitis C virus identified in St. Petersburg. J Virol. 2002;76:4034–4043. doi: 10.1128/JVI.76.8.4034-4043.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sentandreu V, Jiménez-Hernández N, Torres-Puente M, Bracho MA, Valero A, Gosalbes MJ, Ortega E, Moya A, González-Candelas F. Evidence of recombination in intrapatient populations of hepatitis C virus. PLoS One. 2008;3:e3239. doi: 10.1371/journal.pone.0003239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Colina R, Casane D, Vasquez S, García-Aguirre L, Chunga A, Romero H, Khan B, Cristina J. Evidence of intratypic recombination in natural populations of hepatitis C virus. J Gen Virol. 2004;85:31–37. doi: 10.1099/vir.0.19472-0. [DOI] [PubMed] [Google Scholar]
- 76.Kalinina O, Norder H, Magnius LO. Full-length open reading frame of a recombinant hepatitis C virus strain from St Petersburg: proposed mechanism for its formation. J Gen Virol. 2004;85:1853–1857. doi: 10.1099/vir.0.79984-0. [DOI] [PubMed] [Google Scholar]
- 77.Wilke CO. Quasispecies theory in the context of population genetics. BMC Evol Biol. 2005;5:44. doi: 10.1186/1471-2148-5-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Holmes EC. The RNA virus quasispecies: fact or fiction? J Mol Biol. 2010;400:271–273. doi: 10.1016/j.jmb.2010.05.032. [DOI] [PubMed] [Google Scholar]
- 79.Holmes EC. Does hepatitis C virus really form quasispecies? Infect Genet Evol. 2010;10:431–432. doi: 10.1016/j.meegid.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 80.Gismondi MI, Becker PD, Díaz Carrasco JM, Guzmán CA, Campos RH, Preciado MV. Evolution of hepatitis C virus hypervariable region 1 in immunocompetent children born to HCV-infected mothers. J Viral Hepat. 2009;16:332–339. doi: 10.1111/j.1365-2893.2009.01071.x. [DOI] [PubMed] [Google Scholar]
- 81.Gismondi MI, Díaz Carrasco JM, Valva P, Becker PD, Guzmán CA, Campos RH, Preciado MV. Dynamic changes in viral population structure and compartmentalization during chronic hepatitis C virus infection in children. Virology. 2013;447:187–196. doi: 10.1016/j.virol.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 82.Ray SC, Fanning L, Wang XH, Netski DM, Kenny-Walsh E, Thomas DL. Divergent and convergent evolution after a common-source outbreak of hepatitis C virus. J Exp Med. 2005;201:1753–1759. doi: 10.1084/jem.20050122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Escobar-Gutiérrez A, Soudeyns H, Larouche A, Carpio-Pedroza JC, Martinez-Guarneros A, Vazquez-Chacon CA, Fonseca-Coronado S, Yamasaki LH, Ruiz-Tovar K, Cruz-Rivera M. Vertical transmission of hepatitis C virus: a tale of multiple outcomes. Infect Genet Evol. 2013;20:465–470. doi: 10.1016/j.meegid.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Larouche A, Gaëtan G, El-Bilali N, Quesnel-Vallières M, Martin SR, Alvarez F, Shoukry NH, Soudeyns H. Seronegative hepatitis C virus infection in a child infected via mother-to-child transmission. J Clin Microbiol. 2012;50:2515–2519. doi: 10.1128/JCM.00622-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ramachandran S, Campo DS, Dimitrova ZE, Xia GL, Purdy MA, Khudyakov YE. Temporal variations in the hepatitis C virus intrahost population during chronic infection. J Virol. 2011;85:6369–6380. doi: 10.1128/JVI.02204-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gray RR, Salemi M, Klenerman P, Pybus OG. A new evolutionary model for hepatitis C virus chronic infection. PLoS Pathog. 2012;8:e1002656. doi: 10.1371/journal.ppat.1002656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Khudyakov Y. Molecular surveillance of hepatitis C. Antivir Ther. 2012;17:1465–1470. doi: 10.3851/IMP2476. [DOI] [PubMed] [Google Scholar]
- 88.Gerlach JT, Diepolder HM, Zachoval R, Gruener NH, Jung MC, Ulsenheimer A, Schraut WW, Schirren CA, Waechtler M, Backmund M, et al. Acute hepatitis C: high rate of both spontaneous and treatment-induced viral clearance. Gastroenterology. 2003;125:80–88. doi: 10.1016/s0016-5085(03)00668-1. [DOI] [PubMed] [Google Scholar]
- 89.Shimizu YK, Igarashi H, Kanematu T, Fujiwara K, Wong DC, Purcell RH, Yoshikura H. Sequence analysis of the hepatitis C virus genome recovered from serum, liver, and peripheral blood mononuclear cells of infected chimpanzees. J Virol. 1997;71:5769–5773. doi: 10.1128/jvi.71.8.5769-5773.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cabot B, Esteban JI, Martell M, Genescà J, Vargas V, Esteban R, Guardia J, Gómez J. Structure of replicating hepatitis C virus (HCV) quasispecies in the liver may not be reflected by analysis of circulating HCV virions. J Virol. 1997;71:1732–1734. doi: 10.1128/jvi.71.2.1732-1734.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Navas S, Martín J, Quiroga JA, Castillo I, Carreño V. Genetic diversity and tissue compartmentalization of the hepatitis C virus genome in blood mononuclear cells, liver, and serum from chronic hepatitis C patients. J Virol. 1998;72:1640–1646. doi: 10.1128/jvi.72.2.1640-1646.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ducoulombier D, Roque-Afonso AM, Di Liberto G, Penin F, Kara R, Richard Y, Dussaix E, Féray C. Frequent compartmentalization of hepatitis C virus variants in circulating B cells and monocytes. Hepatology. 2004;39:817–825. doi: 10.1002/hep.20087. [DOI] [PubMed] [Google Scholar]
- 93.Forton DM, Karayiannis P, Mahmud N, Taylor-Robinson SD, Thomas HC. Identification of unique hepatitis C virus quasispecies in the central nervous system and comparative analysis of internal translational efficiency of brain, liver, and serum variants. J Virol. 2004;78:5170–5183. doi: 10.1128/JVI.78.10.5170-5183.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zignego AL, Giannini C, Monti M, Gragnani L. Hepatitis C virus lymphotropism: lessons from a decade of studies. Dig Liver Dis. 2007;39 Suppl 1:S38–S45. doi: 10.1016/s1590-8658(07)80009-0. [DOI] [PubMed] [Google Scholar]
- 95.Baré P. Hepatitis C virus and peripheral blood mononuclear cell reservoirs Patricia Baré. World J Hepatol. 2009;1:67–71. doi: 10.4254/wjh.v1.i1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bürgel B, Friesland M, Koch A, Manns MP, Wedemeyer H, Weissenborn K, Schulz-Schaeffer WJ, Pietschmann T, Steinmann E, Ciesek S. Hepatitis C virus enters human peripheral neuroblastoma cells - evidence for extra-hepatic cells sustaining hepatitis C virus penetration. J Viral Hepat. 2011;18:562–570. doi: 10.1111/j.1365-2893.2010.01339.x. [DOI] [PubMed] [Google Scholar]
- 97.Blackard JT, Hiasa Y, Smeaton L, Jamieson DJ, Rodriguez I, Mayer KH, Chung RT. Compartmentalization of hepatitis C virus (HCV) during HCV/HIV coinfection. J Infect Dis. 2007;195:1765–1773. doi: 10.1086/518251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Blackard JT, Kemmer N, Sherman KE. Extrahepatic replication of HCV: insights into clinical manifestations and biological consequences. Hepatology. 2006;44:15–22. doi: 10.1002/hep.21283. [DOI] [PubMed] [Google Scholar]
- 99.Sobesky R, Feray C, Rimlinger F, Derian N, Dos Santos A, Roque-Afonso AM, Samuel D, Bréchot C, Thiers V. Distinct hepatitis C virus core and F protein quasispecies in tumoral and nontumoral hepatocytes isolated via microdissection. Hepatology. 2007;46:1704–1712. doi: 10.1002/hep.21898. [DOI] [PubMed] [Google Scholar]
- 100.Jouvencel AC, Neau D, Faure M, Neau M, Martinaud C, Legrand E, Trimoulet P, Garrigue I, Le Bail B, Bioulac-Sage P, et al. Plasma and liver hepatitis C virus variability in patients coinfected with human immunodeficiency virus. J Clin Microbiol. 2006;44:1877–1880. doi: 10.1128/JCM.44.5.1877-1880.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vera-Otarola J, Barría MI, León U, Marsac D, Carvallo P, Soza A, López-Lastra M. Hepatitis C virus quasispecies in plasma and peripheral blood mononuclear cells of treatment naïve chronically infected patients. J Viral Hepat. 2009;16:633–643. doi: 10.1111/j.1365-2893.2009.01112.x. [DOI] [PubMed] [Google Scholar]
- 102.Roque-Afonso AM, Ducoulombier D, Di Liberto G, Kara R, Gigou M, Dussaix E, Samuel D, Féray C. Compartmentalization of hepatitis C virus genotypes between plasma and peripheral blood mononuclear cells. J Virol. 2005;79:6349–6357. doi: 10.1128/JVI.79.10.6349-6357.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schramm F, Soulier E, Royer C, Weitten T, Fafi-Kremer S, Brignon N, Meyer N, Ellero B, Woehl-Jaegle ML, Meyer C, et al. Frequent compartmentalization of hepatitis C virus with leukocyte-related amino acids in the setting of liver transplantation. J Infect Dis. 2008;198:1656–1666. doi: 10.1086/592986. [DOI] [PubMed] [Google Scholar]
- 104.Laporte J, Bain C, Maurel P, Inchauspe G, Agut H, Cahour A. Differential distribution and internal translation efficiency of hepatitis C virus quasispecies present in dendritic and liver cells. Blood. 2003;101:52–57. doi: 10.1182/blood-2002-03-0818. [DOI] [PubMed] [Google Scholar]
- 105.Pellerin M, Lopez-Aguirre Y, Penin F, Dhumeaux D, Pawlotsky JM. Hepatitis C virus quasispecies variability modulates nonstructural protein 5A transcriptional activation, pointing to cellular compartmentalization of virus-host interactions. J Virol. 2004;78:4617–4627. doi: 10.1128/JVI.78.9.4617-4627.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Blackard JT, Ma G, Welge JA, Martin CM, Sherman KE, Taylor LE, Mayer KH, Jamieson DJ. Analysis of a non-structural gene reveals evidence of possible hepatitis C virus (HCV) compartmentalization. J Med Virol. 2012;84:242–252. doi: 10.1002/jmv.22269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Di Liberto G, Roque-Afonso AM, Kara R, Ducoulombier D, Fallot G, Samuel D, Feray C. Clinical and therapeutic implications of hepatitis C virus compartmentalization. Gastroenterology. 2006;131:76–84. doi: 10.1053/j.gastro.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 108.Welker MW, Welsch C, Ochs D, Hofmann WP, Herrmann E, Piiper A, Hartmann RW, Zeuzem S, Sarrazin C, Kronenberger B. Comparison of envelope 2 CD81 binding regions in PBMC-derived versus serum-derived hepatitis C virus isolates: higher conservation of CD81 region 2 in PBMC isolates. J Viral Hepat. 2011;18:181–192. doi: 10.1111/j.1365-2893.2010.01296.x. [DOI] [PubMed] [Google Scholar]
- 109.Chen Z, Zhu Y, Ren Y, Tong Y, Hua X, Zhu F, Huang L, Liu Y, Luo Y, Lu W, et al. Hepatitis C virus protects human B lymphocytes from Fas-mediated apoptosis via E2-CD81 engagement. PLoS One. 2011;6:e18933. doi: 10.1371/journal.pone.0018933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sarhan MA, Pham TN, Chen AY, Michalak TI. Hepatitis C virus infection of human T lymphocytes is mediated by CD5. J Virol. 2012;86:3723–3735. doi: 10.1128/JVI.06956-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.McGuinness PH, Bishop GA, McCaughan GW, Trowbridge R, Gowans EJ. False detection of negative-strand hepatitis C virus RNA. Lancet. 1994;343:551–552. [PubMed] [Google Scholar]
- 112.Lanford RE, Chavez D, Chisari FV, Sureau C. Lack of detection of negative-strand hepatitis C virus RNA in peripheral blood mononuclear cells and other extrahepatic tissues by the highly strand-specific rTth reverse transcriptase PCR. J Virol. 1995;69:8079–8083. doi: 10.1128/jvi.69.12.8079-8083.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Laskus T, Radkowski M, Wang LF, Cianciara J, Vargas H, Rakela J. Hepatitis C virus negative strand RNA is not detected in peripheral blood mononuclear cells and viral sequences are identical to those in serum: a case against extrahepatic replication. J Gen Virol. 1997;78(Pt 11):2747–2750. doi: 10.1099/0022-1317-78-11-2747. [DOI] [PubMed] [Google Scholar]
- 114.Armstrong GL. Commentary: Modelling the epidemiology of hepatitis C and its complications. Int J Epidemiol. 2003;32:725–726. doi: 10.1093/ije/dyg266. [DOI] [PubMed] [Google Scholar]
- 115.Pawlotsky JM, Tsakiris L, Roudot-Thoraval F, Pellet C, Stuyver L, Duval J, Dhumeaux D. Relationship between hepatitis C virus genotypes and sources of infection in patients with chronic hepatitis C. J Infect Dis. 1995;171:1607–1610. doi: 10.1093/infdis/171.6.1607. [DOI] [PubMed] [Google Scholar]
- 116.Pybus OG, Cochrane A, Holmes EC, Simmonds P. The hepatitis C virus epidemic among injecting drug users. Infect Genet Evol. 2005;5:131–139. doi: 10.1016/j.meegid.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 117.Pybus OG, Charleston MA, Gupta S, Rambaut A, Holmes EC, Harvey PH. The epidemic behavior of the hepatitis C virus. Science. 2001;292:2323–2325. doi: 10.1126/science.1058321. [DOI] [PubMed] [Google Scholar]
- 118.Thomson BJ, Finch RG. Hepatitis C virus infection. Clin Microbiol Infect. 2005;11:86–94. doi: 10.1111/j.1469-0691.2004.01061.x. [DOI] [PubMed] [Google Scholar]
- 119.Escobar-Gutiérrez A, Vazquez-Pichardo M, Cruz-Rivera M, Rivera-Osorio P, Carpio-Pedroza JC, Ruíz-Pacheco JA, Ruiz-Tovar K, Vaughan G. Identification of hepatitis C virus transmission using a next-generation sequencing approach. J Clin Microbiol. 2012;50:1461–1463. doi: 10.1128/JCM.00005-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Williams IT, Bell BP, Kuhnert W, Alter MJ. Incidence and transmission patterns of acute hepatitis C in the United States, 1982-2006. Arch Intern Med. 2011;171:242–248. doi: 10.1001/archinternmed.2010.511. [DOI] [PubMed] [Google Scholar]
- 121.Recommendations for prevention and control of hepatitis C virus (HCV) infection and HCV-related chronic disease. Centers for Disease Control and Prevention. MMWR Recomm Rep. 1998;47:1–39. [PubMed] [Google Scholar]
- 122.Araujo AC. Antibody- and genome-based identification of recent HCV infection. Antivir Ther. 2012;17:1459–1464. doi: 10.3851/IMP2464. [DOI] [PubMed] [Google Scholar]
- 123.Abecasis AB, Geretti AM, Albert J, Power L, Weait M, Vandamme AM. Science in court: the myth of HIV fingerprinting. Lancet Infect Dis. 2011;11:78–79. doi: 10.1016/S1473-3099(10)70283-8. [DOI] [PubMed] [Google Scholar]
- 124.Vandamme AM, Pybus OG. Viral phylogeny in court: the unusual case of the Valencian anesthetist. BMC Biol. 2013;11:83. doi: 10.1186/1741-7007-11-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.González-Candelas F, Bracho MA, Wróbel B, Moya A. Molecular evolution in court: analysis of a large hepatitis C virus outbreak from an evolving source. BMC Biol. 2013;11:76. doi: 10.1186/1741-7007-11-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wróbel B, Torres-Puente M, Jiménez N, Bracho MA, García-Robles I, Moya A, González-Candelas F. Analysis of the overdispersed clock in the short-term evolution of hepatitis C virus: Using the E1/E2 gene sequences to infer infection dates in a single source outbreak. Mol Biol Evol. 2006;23:1242–1253. doi: 10.1093/molbev/msk012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.de Oliveira T, Pybus OG, Rambaut A, Salemi M, Cassol S, Ciccozzi M, Rezza G, Gattinara GC, D’Arrigo R, Amicosante M, et al. Molecular epidemiology: HIV-1 and HCV sequences from Libyan outbreak. Nature. 2006;444:836–837. doi: 10.1038/444836a. [DOI] [PubMed] [Google Scholar]
- 128.Indolfi G, Azzari C, Resti M. Hepatitis: Immunoregulation in pregnancy and perinatal transmission of HCV. Nat Rev Gastroenterol Hepatol. 2014;11:6–7. doi: 10.1038/nrgastro.2013.230. [DOI] [PubMed] [Google Scholar]
- 129.Indolfi G, Azzari C, Resti M. Perinatal transmission of hepatitis C virus. J Pediatr. 2013;163:1549–1552.e1. doi: 10.1016/j.jpeds.2013.06.077. [DOI] [PubMed] [Google Scholar]
- 130.Azzari C, Moriondo M, Indolfi G, Betti L, Gambineri E, de Martino M, Resti M. Higher risk of hepatitis C virus perinatal transmission from drug user mothers is mediated by peripheral blood mononuclear cell infection. J Med Virol. 2008;80:65–71. doi: 10.1002/jmv.21023. [DOI] [PubMed] [Google Scholar]
- 131.Azzari C, Resti M, Moriondo M, Ferrari R, Lionetti P, Vierucci A. Vertical transmission of HCV is related to maternal peripheral blood mononuclear cell infection. Blood. 2000;96:2045–2048. [PubMed] [Google Scholar]
- 132.Kudo T, Yanase Y, Ohshiro M, Yamamoto M, Morita M, Shibata M, Morishima T. Analysis of mother-to-infant transmission of hepatitis C virus: quasispecies nature and buoyant densities of maternal virus populations. J Med Virol. 1997;51:225–230. [PubMed] [Google Scholar]
- 133.Fonseca-Coronado S, Escobar-Gutiérrez A, Ruiz-Tovar K, Cruz-Rivera MY, Rivera-Osorio P, Vazquez-Pichardo M, Carpio-Pedroza JC, Ruíz-Pacheco JA, Cazares F, Vaughan G. Specific detection of naturally occurring hepatitis C virus mutants with resistance to telaprevir and boceprevir (protease inhibitors) among treatment-naïve infected individuals. J Clin Microbiol. 2012;50:281–287. doi: 10.1128/JCM.05842-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Fonseca-Coronado S, Vaughan G, Cruz-Rivera MY, Carpio-Pedroza JC, Ruiz-Tovar K, Ruiz-Pacheco JA, Escobar-Gutiérrez A. Interleukin-28B genotyping by melt-mismatch amplification mutation assay PCR analysis using single nucleotide polymorphisms rs12979860 and rs8099917, a useful tool for prediction of therapy response in hepatitis C patients. J Clin Microbiol. 2011;49:2706–2710. doi: 10.1128/JCM.00877-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ramachandran S, Xia GL, Ganova-Raeva LM, Nainan OV, Khudyakov Y. End-point limiting-dilution real-time PCR assay for evaluation of hepatitis C virus quasispecies in serum: performance under optimal and suboptimal conditions. J Virol Methods. 2008;151:217–224. doi: 10.1016/j.jviromet.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 136.Cruz-Rivera M, Forbi JC, Yamasaki LH, Vazquez-Chacon CA, Martinez-Guarneros A, Carpio-Pedroza JC, Escobar-Gutiérrez A, Ruiz-Tovar K, Fonseca-Coronado S, Vaughan G. Molecular epidemiology of viral diseases in the era of next generation sequencing. J Clin Virol. 2013;57:378–380. doi: 10.1016/j.jcv.2013.04.021. [DOI] [PubMed] [Google Scholar]
- 137.Forbi JC, Purdy MA, Campo DS, Vaughan G, Dimitrova ZE, Ganova-Raeva LM, Xia GL, Khudyakov YE. Epidemic history of hepatitis C virus infection in two remote communities in Nigeria, West Africa. J Gen Virol. 2012;93:1410–1421. doi: 10.1099/vir.0.042184-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Miura M, Maekawa S, Takano S, Komatsu N, Tatsumi A, Asakawa Y, Shindo K, Amemiya F, Nakayama Y, Inoue T, et al. Deep-sequencing analysis of the association between the quasispecies nature of the hepatitis C virus core region and disease progression. J Virol. 2013;87:12541–12551. doi: 10.1128/JVI.00826-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lauck M, Alvarado-Mora MV, Becker EA, Bhattacharya D, Striker R, Hughes AL, Carrilho FJ, O’Connor DH, Pinho JR. Analysis of hepatitis C virus intrahost diversity across the coding region by ultradeep pyrosequencing. J Virol. 2012;86:3952–3960. doi: 10.1128/JVI.06627-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Beerenwinkel N, Günthard HF, Roth V, Metzner KJ. Challenges and opportunities in estimating viral genetic diversity from next-generation sequencing data. Front Microbiol. 2012;3:329. doi: 10.3389/fmicb.2012.00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Caraballo Cortés K, Zagordi O, Laskus T, Płoski R, Bukowska-Ośko I, Pawełczyk A, Berak H, Radkowski M. Ultradeep pyrosequencing of hepatitis C virus hypervariable region 1 in quasispecies analysis. Biomed Res Int. 2013;2013:626083. doi: 10.1155/2013/626083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Farci P, Shimoda A, Coiana A, Diaz G, Peddis G, Melpolder JC, Strazzera A, Chien DY, Munoz SJ, Balestrieri A, et al. The outcome of acute hepatitis C predicted by the evolution of the viral quasispecies. Science. 2000;288:339–344. doi: 10.1126/science.288.5464.339. [DOI] [PubMed] [Google Scholar]
- 143.Farci P, Strazzera R, Alter HJ, Farci S, Degioannis D, Coiana A, Peddis G, Usai F, Serra G, Chessa L, et al. Early changes in hepatitis C viral quasispecies during interferon therapy predict the therapeutic outcome. Proc Natl Acad Sci USA. 2002;99:3081–3086. doi: 10.1073/pnas.052712599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Niederau C, Lange S, Heintges T, Erhardt A, Buschkamp M, Hürter D, Nawrocki M, Kruska L, Hensel F, Petry W, et al. Prognosis of chronic hepatitis C: results of a large, prospective cohort study. Hepatology. 1998;28:1687–1695. doi: 10.1002/hep.510280632. [DOI] [PubMed] [Google Scholar]
- 145.Ripoli M, Pazienza V. Impact of HCV genetic differences on pathobiology of disease. Expert Rev Anti Infect Ther. 2011;9:747–759. doi: 10.1586/eri.11.94. [DOI] [PubMed] [Google Scholar]
- 146.Amoroso P, Rapicetta M, Tosti ME, Mele A, Spada E, Buonocore S, Lettieri G, Pierri P, Chionne P, Ciccaglione AR, et al. Correlation between virus genotype and chronicity rate in acute hepatitis C. J Hepatol. 1998;28:939–944. doi: 10.1016/s0168-8278(98)80340-1. [DOI] [PubMed] [Google Scholar]
- 147.Bantel H, Lügering A, Poremba C, Lügering N, Held J, Domschke W, Schulze-Osthoff K. Caspase activation correlates with the degree of inflammatory liver injury in chronic hepatitis C virus infection. Hepatology. 2001;34:758–767. doi: 10.1053/jhep.2001.28229. [DOI] [PubMed] [Google Scholar]
- 148.Aweya JJ, Tan YJ. Modulation of programmed cell death pathways by the hepatitis C virus. Front Biosci (Landmark Ed) 2011;16:608–618. doi: 10.2741/3709. [DOI] [PubMed] [Google Scholar]
- 149.Malhi H, Gores GJ. Cellular and molecular mechanisms of liver injury. Gastroenterology. 2008;134:1641–1654. doi: 10.1053/j.gastro.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Shiu TY, Huang SM, Shih YL, Chu HC, Chang WK, Hsieh TY. Hepatitis C virus core protein down-regulates p21(Waf1/Cip1) and inhibits curcumin-induced apoptosis through microRNA-345 targeting in human hepatoma cells. PLoS One. 2013;8:e61089. doi: 10.1371/journal.pone.0061089. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 151.Jahan S, Khaliq S, Siddiqi MH, Ijaz B, Ahmad W, Ashfaq UA, Hassan S. Anti-apoptotic effect of HCV core gene of genotype 3a in Huh-7 cell line. Virol J. 2011;8:522. doi: 10.1186/1743-422X-8-522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Nakamura H, Aoki H, Hino O, Moriyama M. HCV core protein promotes heparin binding EGF-like growth factor expression and activates Akt. Hepatol Res. 2011;41:455–462. doi: 10.1111/j.1872-034X.2011.00792.x. [DOI] [PubMed] [Google Scholar]
- 153.Berg CP, Schlosser SF, Neukirchen DK, Papadakis C, Gregor M, Wesselborg S, Stein GM. Hepatitis C virus core protein induces apoptosis-like caspase independent cell death. Virol J. 2009;6:213. doi: 10.1186/1743-422X-6-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Siavoshian S, Abraham JD, Thumann C, Kieny MP, Schuster C. Hepatitis C virus core, NS3, NS5A, NS5B proteins induce apoptosis in mature dendritic cells. J Med Virol. 2005;75:402–411. doi: 10.1002/jmv.20283. [DOI] [PubMed] [Google Scholar]
- 155.Machida K, Tsukiyama-Kohara K, Seike E, Toné S, Shibasaki F, Shimizu M, Takahashi H, Hayashi Y, Funata N, Taya C, et al. Inhibition of cytochrome c release in Fas-mediated signaling pathway in transgenic mice induced to express hepatitis C viral proteins. J Biol Chem. 2001;276:12140–12146. doi: 10.1074/jbc.M010137200. [DOI] [PubMed] [Google Scholar]
- 156.Saito K, Meyer K, Warner R, Basu A, Ray RB, Ray R. Hepatitis C virus core protein inhibits tumor necrosis factor alpha-mediated apoptosis by a protective effect involving cellular FLICE inhibitory protein. J Virol. 2006;80:4372–4379. doi: 10.1128/JVI.80.9.4372-4379.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kao CF, Chen SY, Chen JY, Wu Lee YH. Modulation of p53 transcription regulatory activity and post-translational modification by hepatitis C virus core protein. Oncogene. 2004;23:2472–2483. doi: 10.1038/sj.onc.1207368. [DOI] [PubMed] [Google Scholar]
- 158.Otsuka M, Kato N, Lan K, Yoshida H, Kato J, Goto T, Shiratori Y, Omata M. Hepatitis C virus core protein enhances p53 function through augmentation of DNA binding affinity and transcriptional ability. J Biol Chem. 2000;275:34122–34130. doi: 10.1074/jbc.M000578200. [DOI] [PubMed] [Google Scholar]
- 159.Koike K. Molecular basis of hepatitis C virus-associated hepatocarcinogenesis: lessons from animal model studies. Clin Gastroenterol Hepatol. 2005;3:S132–S135. doi: 10.1016/s1542-3565(05)00700-7. [DOI] [PubMed] [Google Scholar]
- 160.Akuta N, Suzuki F, Hirakawa M, Kawamura Y, Sezaki H, Suzuki Y, Hosaka T, Kobayashi M, Kobayashi M, Saitoh S, et al. Amino acid substitutions in hepatitis C virus core region predict hepatocarcinogenesis following eradication of HCV RNA by antiviral therapy. J Med Virol. 2011;83:1016–1022. doi: 10.1002/jmv.22094. [DOI] [PubMed] [Google Scholar]
- 161.Akuta N, Suzuki F, Kawamura Y, Yatsuji H, Sezaki H, Suzuki Y, Hosaka T, Kobayashi M, Kobayashi M, Arase Y, et al. Amino acid substitutions in the hepatitis C virus core region are the important predictor of hepatocarcinogenesis. Hepatology. 2007;46:1357–1364. doi: 10.1002/hep.21836. [DOI] [PubMed] [Google Scholar]
- 162.Akuta N, Suzuki F, Kawamura Y, Yatsuji H, Sezaki H, Suzuki Y, Hosaka T, Kobayashi M, Kobayashi M, Arase Y, et al. Substitution of amino acid 70 in the hepatitis C virus core region of genotype 1b is an important predictor of elevated alpha-fetoprotein in patients without hepatocellular carcinoma. J Med Virol. 2008;80:1354–1362. doi: 10.1002/jmv.21202. [DOI] [PubMed] [Google Scholar]
- 163.Fishman SL, Factor SH, Balestrieri C, Fan X, Dibisceglie AM, Desai SM, Benson G, Branch AD. Mutations in the hepatitis C virus core gene are associated with advanced liver disease and hepatocellular carcinoma. Clin Cancer Res. 2009;15:3205–3213. doi: 10.1158/1078-0432.CCR-08-2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Rüster B, Zeuzem S, Krump-Konvalinkova V, Berg T, Jonas S, Severin K, Roth WK. Comparative sequence analysis of the core- and NS5-region of hepatitis C virus from tumor and adjacent non-tumor tissue. J Med Virol. 2001;63:128–134. [PubMed] [Google Scholar]
- 165.Alam SS, Nakamura T, Naganuma A, Nozaki A, Nouso K, Shimomura H, Kato N. Hepatitis C virus quasispecies in cancerous and noncancerous hepatic lesions: the core protein-encoding region. Acta Med Okayama. 2002;56:141–147. doi: 10.18926/AMO/31716. [DOI] [PubMed] [Google Scholar]
- 166.De Mitri MS, Mele L, Chen CH, Piccinini A, Chianese R, D’Errico A, Alberti A, Pisi E. Comparison of serum and liver hepatitis C virus quasispecies in HCV-related hepatocellular carcinoma. J Hepatol. 1998;29:887–892. doi: 10.1016/s0168-8278(98)80115-3. [DOI] [PubMed] [Google Scholar]
- 167.Horie C, Iwahana H, Horie T, Shimizu I, Yoshimoto K, Yogita S, Tashiro S, Ito S, Itakura M. Detection of different quasispecies of hepatitis C virus core region in cancerous and noncancerous lesions. Biochem Biophys Res Commun. 1996;218:674–681. doi: 10.1006/bbrc.1996.0121. [DOI] [PubMed] [Google Scholar]
- 168.Ogata S, Nagano-Fujii M, Ku Y, Yoon S, Hotta H. Comparative sequence analysis of the core protein and its frameshift product, the F protein, of hepatitis C virus subtype 1b strains obtained from patients with and without hepatocellular carcinoma. J Clin Microbiol. 2002;40:3625–3630. doi: 10.1128/JCM.40.10.3625-3630.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Allison ME, Wreghitt T, Palmer CR, Alexander GJ. Evidence for a link between hepatitis C virus infection and diabetes mellitus in a cirrhotic population. J Hepatol. 1994;21:1135–1139. doi: 10.1016/s0168-8278(05)80631-2. [DOI] [PubMed] [Google Scholar]
- 170.Antonelli A, Ferri C, Fallahi P, Pampana A, Ferrari SM, Goglia F, Ferrannini E. Hepatitis C virus infection: evidence for an association with type 2 diabetes. Diabetes Care. 2005;28:2548–2550. doi: 10.2337/diacare.28.10.2548. [DOI] [PubMed] [Google Scholar]
- 171.Akuta N, Suzuki F, Hirakawa M, Kawamura Y, Yatsuji H, Sezaki H, Suzuki Y, Hosaka T, Kobayashi M, Kobayashi M, et al. Amino acid substitutions in the hepatitis C virus core region of genotype 1b are the important predictor of severe insulin resistance in patients without cirrhosis and diabetes mellitus. J Med Virol. 2009;81:1032–1039. doi: 10.1002/jmv.21473. [DOI] [PubMed] [Google Scholar]
- 172.Adinolfi LE, Gambardella M, Andreana A, Tripodi MF, Utili R, Ruggiero G. Steatosis accelerates the progression of liver damage of chronic hepatitis C patients and correlates with specific HCV genotype and visceral obesity. Hepatology. 2001;33:1358–1364. doi: 10.1053/jhep.2001.24432. [DOI] [PubMed] [Google Scholar]
- 173.Rubbia-Brandt L, Leandro G, Spahr L, Giostra E, Quadri R, Malé PJ, Negro F. Liver steatosis in chronic hepatitis C: a morphological sign suggesting infection with HCV genotype 3. Histopathology. 2001;39:119–124. doi: 10.1046/j.1365-2559.2001.01208.x. [DOI] [PubMed] [Google Scholar]
- 174.Tachi Y, Katano Y, Honda T, Hayashi K, Ishigami M, Itoh A, Hirooka Y, Nakano I, Samejima Y, Goto H. Impact of amino acid substitutions in the hepatitis C virus genotype 1b core region on liver steatosis and hepatic oxidative stress in patients with chronic hepatitis C. Liver Int. 2010;30:554–559. doi: 10.1111/j.1478-3231.2009.02164.x. [DOI] [PubMed] [Google Scholar]
- 175.Bochud PY, Cai T, Overbeck K, Bochud M, Dufour JF, Müllhaupt B, Borovicka J, Heim M, Moradpour D, Cerny A, et al. Genotype 3 is associated with accelerated fibrosis progression in chronic hepatitis C. J Hepatol. 2009;51:655–666. doi: 10.1016/j.jhep.2009.05.016. [DOI] [PubMed] [Google Scholar]
- 176.De Mitri MS, Cassini R, Bagaglio S, Morsica G, Andreone P, Marino N, Bernardi M. Evolution of hepatitis C virus non-structural 5A gene in the progression of liver disease to hepatocellular carcinoma. Liver Int. 2007;27:1126–1133. doi: 10.1111/j.1478-3231.2007.01537.x. [DOI] [PubMed] [Google Scholar]
- 177.Ghosh AK, Majumder M, Steele R, Meyer K, Ray R, Ray RB. Hepatitis C virus NS5A protein protects against TNF-alpha mediated apoptotic cell death. Virus Res. 2000;67:173–178. doi: 10.1016/s0168-1702(00)00141-6. [DOI] [PubMed] [Google Scholar]
- 178.Ghosh AK, Steele R, Meyer K, Ray R, Ray RB. Hepatitis C virus NS5A protein modulates cell cycle regulatory genes and promotes cell growth. J Gen Virol. 1999;80(Pt 5):1179–1183. doi: 10.1099/0022-1317-80-5-1179. [DOI] [PubMed] [Google Scholar]
- 179.Gale M, Blakely CM, Kwieciszewski B, Tan SL, Dossett M, Tang NM, Korth MJ, Polyak SJ, Gretch DR, Katze MG. Control of PKR protein kinase by hepatitis C virus nonstructural 5A protein: molecular mechanisms of kinase regulation. Mol Cell Biol. 1998;18:5208–5218. doi: 10.1128/mcb.18.9.5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Chung KM, Song OK, Jang SK. Hepatitis C virus nonstructural protein 5A contains potential transcriptional activator domains. Mol Cells. 1997;7:661–667. [PubMed] [Google Scholar]
- 181.Enomoto N, Sakuma I, Asahina Y, Kurosaki M, Murakami T, Yamamoto C, Ogura Y, Izumi N, Marumo F, Sato C. Mutations in the nonstructural protein 5A gene and response to interferon in patients with chronic hepatitis C virus 1b infection. N Engl J Med. 1996;334:77–81. doi: 10.1056/NEJM199601113340203. [DOI] [PubMed] [Google Scholar]
- 182.Qin H, Shire NJ, Keenan ED, Rouster SD, Eyster ME, Goedert JJ, Koziel MJ, Sherman KE. HCV quasispecies evolution: association with progression to end-stage liver disease in hemophiliacs infected with HCV or HCV/HIV. Blood. 2005;105:533–541. doi: 10.1182/blood-2004-04-1452. [DOI] [PubMed] [Google Scholar]
- 183.Canobio S, Guilbert CM, Troesch M, Samson J, Lemay M, Pelletier VA, Bernard-Bonnin AC, Kozielski R, Lapointe N, Martin SR, et al. Differing patterns of liver disease progression and hepatitis C virus (HCV) quasispecies evolution in children vertically coinfected with HCV and human immunodeficiency virus type 1. J Clin Microbiol. 2004;42:4365–4369. doi: 10.1128/JCM.42.9.4365-4369.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Arenas JI, Gallegos-Orozco JF, Laskus T, Wilkinson J, Khatib A, Fasola C, Adair D, Radkowski M, Kibler KV, Nowicki M, et al. Hepatitis C virus quasi-species dynamics predict progression of fibrosis after liver transplantation. J Infect Dis. 2004;189:2037–2046. doi: 10.1086/386338. [DOI] [PubMed] [Google Scholar]
- 185.Martell M, Esteban JI, Quer J, Vargas V, Esteban R, Guardia J, Gómez J. Dynamic behavior of hepatitis C virus quasispecies in patients undergoing orthotopic liver transplantation. J Virol. 1994;68:3425–3436. doi: 10.1128/jvi.68.5.3425-3436.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Gretch DR, Polyak SJ, Wilson JJ, Carithers RL, Perkins JD, Corey L. Tracking hepatitis C virus quasispecies major and minor variants in symptomatic and asymptomatic liver transplant recipients. J Virol. 1996;70:7622–7631. doi: 10.1128/jvi.70.11.7622-7631.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Pessoa MG, Bzowej N, Berenguer M, Phung Y, Kim M, Ferrell L, Hassoba H, Wright TL. Evolution of hepatitis C virus quasispecies in patients with severe cholestatic hepatitis after liver transplantation. Hepatology. 1999;30:1513–1520. doi: 10.1002/hep.510300610. [DOI] [PubMed] [Google Scholar]
- 188.Chopra KB, Demetris AJ, Blakolmer K, Dvorchik I, Laskus T, Wang LF, Araya VR, Dodson F, Fung JJ, Rakela J, et al. Progression of liver fibrosis in patients with chronic hepatitis C after orthotopic liver transplantation. Transplantation. 2003;76:1487–1491. doi: 10.1097/01.TP.0000088668.28950.7C. [DOI] [PubMed] [Google Scholar]
- 189.Araya V, Rakela J, Wright T. Hepatitis C after orthotopic liver transplantation. Gastroenterology. 1997;112:575–582. doi: 10.1053/gast.1997.v112.agast970575. [DOI] [PubMed] [Google Scholar]
- 190.Vargas HE, Laskus T, Wang LF, Radkowski M, Poutous A, Lee R, Demetris JA, Gayowski T, Marino IR, Singh N, et al. The influence of hepatitis C virus genotypes on the outcome of liver transplantation. Liver Transpl Surg. 1998;4:22–27. doi: 10.1002/lt.500040103. [DOI] [PubMed] [Google Scholar]
- 191.Féray C, Gigou M, Samuel D, Paradis V, Mishiro S, Maertens G, Reynés M, Okamoto H, Bismuth H, Bréchot C. Influence of the genotypes of hepatitis C virus on the severity of recurrent liver disease after liver transplantation. Gastroenterology. 1995;108:1088–1096. doi: 10.1016/0016-5085(95)90207-4. [DOI] [PubMed] [Google Scholar]
- 192.Charlton M, Seaberg E, Wiesner R, Everhart J, Zetterman R, Lake J, Detre K, Hoofnagle J. Predictors of patient and graft survival following liver transplantation for hepatitis C. Hepatology. 1998;28:823–830. doi: 10.1002/hep.510280333. [DOI] [PubMed] [Google Scholar]
- 193.Burak KW, Kremers WK, Batts KP, Wiesner RH, Rosen CB, Razonable RR, Paya CV, Charlton MR. Impact of cytomegalovirus infection, year of transplantation, and donor age on outcomes after liver transplantation for hepatitis C. Liver Transpl. 2002;8:362–369. doi: 10.1053/jlts.2002.32282. [DOI] [PubMed] [Google Scholar]
- 194.Rosen HR, Chou S, Corless CL, Gretch DR, Flora KD, Boudousquie A, Orloff SL, Rabkin JM, Benner KG. Cytomegalovirus viremia: risk factor for allograft cirrhosis after liver transplantation for hepatitis C. Transplantation. 1997;64:721–726. doi: 10.1097/00007890-199709150-00010. [DOI] [PubMed] [Google Scholar]
- 195.Bica I, McGovern B, Dhar R, Stone D, McGowan K, Scheib R, Snydman DR. Increasing mortality due to end-stage liver disease in patients with human immunodeficiency virus infection. Clin Infect Dis. 2001;32:492–497. doi: 10.1086/318501. [DOI] [PubMed] [Google Scholar]
- 196.Zeuzem S, Hultcrantz R, Bourliere M, Goeser T, Marcellin P, Sanchez-Tapias J, Sarrazin C, Harvey J, Brass C, Albrecht J. Peginterferon alfa-2b plus ribavirin for treatment of chronic hepatitis C in previously untreated patients infected with HCV genotypes 2 or 3. J Hepatol. 2004;40:993–999. doi: 10.1016/j.jhep.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 197.Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, Heinzen EL, Qiu P, Bertelsen AH, Muir AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461:399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 198.Pearlman BL, Ehleben C, Saifee S. Treatment extension to 72 weeks of peginterferon and ribavirin in hepatitis c genotype 1-infected slow responders. Hepatology. 2007;46:1688–1694. doi: 10.1002/hep.21919. [DOI] [PubMed] [Google Scholar]
- 199.Webster G, Barnes E, Brown D, Dusheiko G. HCV genotypes--role in pathogenesis of disease and response to therapy. Baillieres Best Pract Res Clin Gastroenterol. 2000;14:229–240. doi: 10.1053/bega.1999.0072. [DOI] [PubMed] [Google Scholar]
- 200.Shen C, Hu T, Shen L, Gao L, Xie W, Zhang J. Mutations in ISDR of NS5A gene influence interferon efficacy in Chinese patients with chronic hepatitis C virus genotype 1b infection. J Gastroenterol Hepatol. 2007;22:1898–1903. doi: 10.1111/j.1440-1746.2006.04566.x. [DOI] [PubMed] [Google Scholar]
- 201.Murakami T, Enomoto N, Kurosaki M, Izumi N, Marumo F, Sato C. Mutations in nonstructural protein 5A gene and response to interferon in hepatitis C virus genotype 2 infection. Hepatology. 1999;30:1045–1053. doi: 10.1002/hep.510300405. [DOI] [PubMed] [Google Scholar]
- 202.Zeuzem S, Lee JH, Roth WK. Mutations in the nonstructural 5A gene of European hepatitis C virus isolates and response to interferon alfa. Hepatology. 1997;25:740–744. doi: 10.1002/hep.510250341. [DOI] [PubMed] [Google Scholar]
- 203.Murphy MD, Rosen HR, Marousek GI, Chou S. Analysis of sequence configurations of the ISDR, PKR-binding domain, and V3 region as predictors of response to induction interferon-alpha and ribavirin therapy in chronic hepatitis C infection. Dig Dis Sci. 2002;47:1195–1205. doi: 10.1023/a:1015349924116. [DOI] [PubMed] [Google Scholar]
- 204.Ilyas JA, Vierling JM. An overview of emerging therapies for the treatment of chronic hepatitis C. Med Clin North Am. 2014;98:17–38. doi: 10.1016/j.mcna.2013.10.011. [DOI] [PubMed] [Google Scholar]
- 205.Ploss A, Dubuisson J. New advances in the molecular biology of hepatitis C virus infection: towards the identification of new treatment targets. Gut. 2012;61 Suppl 1:i25–i35. doi: 10.1136/gutjnl-2012-302048. [DOI] [PubMed] [Google Scholar]
- 206.Delang L, Neyts J, Vliegen I, Abrignani S, Neddermann P, De Francesco R. Hepatitis C virus-specific directly acting antiviral drugs. Curr Top Microbiol Immunol. 2013;369:289–320. doi: 10.1007/978-3-642-27340-7_12. [DOI] [PubMed] [Google Scholar]
- 207.Bartenschlager R. Hepatitis C Virus: From Molecular Virology to Antiviral Therapy (Current Topics in Microbiology and Immunology). 2013th. Publisher: Springer; 2013. p. 369. [Google Scholar]
- 208.Bacon BR, Gordon SC, Lawitz E, Marcellin P, Vierling JM, Zeuzem S, Poordad F, Goodman ZD, Sings HL, Boparai N, et al. Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med. 2011;364:1207–1217. doi: 10.1056/NEJMoa1009482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Poordad F, McCone J, Bacon BR, Bruno S, Manns MP, Sulkowski MS, Jacobson IM, Reddy KR, Goodman ZD, Boparai N, et al. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med. 2011;364:1195–1206. doi: 10.1056/NEJMoa1010494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Zeuzem S, Andreone P, Pol S, Lawitz E, Diago M, Roberts S, Focaccia R, Younossi Z, Foster GR, Horban A, et al. Telaprevir for retreatment of HCV infection. N Engl J Med. 2011;364:2417–2428. doi: 10.1056/NEJMoa1013086. [DOI] [PubMed] [Google Scholar]
- 211.Welsch C, Jesudian A, Zeuzem S, Jacobson I. New direct-acting antiviral agents for the treatment of hepatitis C virus infection and perspectives. Gut. 2012;61 Suppl 1:i36–i46. doi: 10.1136/gutjnl-2012-302144. [DOI] [PubMed] [Google Scholar]
- 212.Hézode C, Fontaine H, Dorival C, Larrey D, Zoulim F, Canva V, de Ledinghen V, Poynard T, Samuel D, Bourlière M, et al. Triple therapy in treatment-experienced patients with HCV-cirrhosis in a multicentre cohort of the French Early Access Programme (ANRS CO20-CUPIC) - NCT01514890. J Hepatol. 2013;59:434–441. doi: 10.1016/j.jhep.2013.04.035. [DOI] [PubMed] [Google Scholar]
- 213.Mansky LM, Temin HM. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J Virol. 1995;69:5087–5094. doi: 10.1128/jvi.69.8.5087-5094.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Lu L, Dekhtyar T, Masse S, Pithawalla R, Krishnan P, He W, Ng T, Koev G, Stewart K, Larson D, et al. Identification and characterization of mutations conferring resistance to an HCV RNA-dependent RNA polymerase inhibitor in vitro. Antiviral Res. 2007;76:93–97. doi: 10.1016/j.antiviral.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 215.Cubero M, Esteban JI, Otero T, Sauleda S, Bes M, Esteban R, Guardia J, Quer J. Naturally occurring NS3-protease-inhibitor resistant mutant A156T in the liver of an untreated chronic hepatitis C patient. Virology. 2008;370:237–245. doi: 10.1016/j.virol.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 216.Kuntzen T, Timm J, Berical A, Lennon N, Berlin AM, Young SK, Lee B, Heckerman D, Carlson J, Reyor LL, et al. Naturally occurring dominant resistance mutations to hepatitis C virus protease and polymerase inhibitors in treatment-naïve patients. Hepatology. 2008;48:1769–1778. doi: 10.1002/hep.22549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Kieffer TL, Sarrazin C, Miller JS, Welker MW, Forestier N, Reesink HW, Kwong AD, Zeuzem S. Telaprevir and pegylated interferon-alpha-2a inhibit wild-type and resistant genotype 1 hepatitis C virus replication in patients. Hepatology. 2007;46:631–639. doi: 10.1002/hep.21781. [DOI] [PubMed] [Google Scholar]
- 218.Sarrazin C, Zeuzem S. Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology. 2010;138:447–462. doi: 10.1053/j.gastro.2009.11.055. [DOI] [PubMed] [Google Scholar]
- 219.Zeminian LB, Padovani JL, Corvino SM, Silva GF, Pardini MI, Grotto RM. Variability and resistance mutations in the hepatitis C virus NS3 protease in patients not treated with protease inhibitors. Mem Inst Oswaldo Cruz. 2013;108:13–17. doi: 10.1590/S0074-02762013000100002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Wyles DL. Antiviral resistance and the future landscape of hepatitis C virus infection therapy. J Infect Dis. 2013;207 Suppl 1:S33–S39. doi: 10.1093/infdis/jis761. [DOI] [PubMed] [Google Scholar]
- 221.Susser S, Vermehren J, Forestier N, Welker MW, Grigorian N, Füller C, Perner D, Zeuzem S, Sarrazin C. Analysis of long-term persistence of resistance mutations within the hepatitis C virus NS3 protease after treatment with telaprevir or boceprevir. J Clin Virol. 2011;52:321–327. doi: 10.1016/j.jcv.2011.08.015. [DOI] [PubMed] [Google Scholar]
- 222.Halfon P, Locarnini S. Hepatitis C virus resistance to protease inhibitors. J Hepatol. 2011;55:192–206. doi: 10.1016/j.jhep.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 223.Manns MP, von Hahn T. Novel therapies for hepatitis C - one pill fits all? Nat Rev Drug Discov. 2013;12:595–610. doi: 10.1038/nrd4050. [DOI] [PubMed] [Google Scholar]
- 224.Shah N, Pierce T, Kowdley KV. Review of direct-acting antiviral agents for the treatment of chronic hepatitis C. Expert Opin Investig Drugs. 2013;22:1107–1121. doi: 10.1517/13543784.2013.806482. [DOI] [PubMed] [Google Scholar]
- 225.McCown MF, Rajyaguru S, Kular S, Cammack N, Nájera I. GT-1a or GT-1b subtype-specific resistance profiles for hepatitis C virus inhibitors telaprevir and HCV-796. Antimicrob Agents Chemother. 2009;53:2129–2132. doi: 10.1128/AAC.01598-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Ciesek S, von Hahn T, Manns MP. Second-wave protease inhibitors: choosing an heir. Clin Liver Dis. 2011;15:597–609. doi: 10.1016/j.cld.2011.05.014. [DOI] [PubMed] [Google Scholar]
- 227.White PW, Llinàs-Brunet M, Amad M, Bethell RC, Bolger G, Cordingley MG, Duan J, Garneau M, Lagacé L, Thibeault D, et al. Preclinical characterization of BI 201335, a C-terminal carboxylic acid inhibitor of the hepatitis C virus NS3-NS4A protease. Antimicrob Agents Chemother. 2010;54:4611–4618. doi: 10.1128/AAC.00787-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Lin TI, Lenz O, Fanning G, Verbinnen T, Delouvroy F, Scholliers A, Vermeiren K, Rosenquist A, Edlund M, Samuelsson B, et al. In vitro activity and preclinical profile of TMC435350, a potent hepatitis C virus protease inhibitor. Antimicrob Agents Chemother. 2009;53:1377–1385. doi: 10.1128/AAC.01058-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Seiwert SD, Andrews SW, Jiang Y, Serebryany V, Tan H, Kossen K, Rajagopalan PT, Misialek S, Stevens SK, Stoycheva A, et al. Preclinical characteristics of the hepatitis C virus NS3/4A protease inhibitor ITMN-191 (R7227) Antimicrob Agents Chemother. 2008;52:4432–4441. doi: 10.1128/AAC.00699-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Sheng XC, Appleby T, Butler T, Cai R, Chen X, Cho A, Clarke MO, Cottell J, Delaney WE, Doerffler E, et al. Discovery of GS-9451: an acid inhibitor of the hepatitis C virus NS3/4A protease. Bioorg Med Chem Lett. 2012;22:2629–2634. doi: 10.1016/j.bmcl.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 231.Sheng XC, Casarez A, Cai R, Clarke MO, Chen X, Cho A, Delaney WE, Doerffler E, Ji M, Mertzman M, et al. Discovery of GS-9256: a novel phosphinic acid derived inhibitor of the hepatitis C virus NS3/4A protease with potent clinical activity. Bioorg Med Chem Lett. 2012;22:1394–1396. doi: 10.1016/j.bmcl.2011.12.038. [DOI] [PubMed] [Google Scholar]
- 232.Agarwal A, Zhang B, Olek E, Robison H, Robarge L, Deshpande M. Rapid and sharp decline in HCV upon monotherapy with NS3 protease inhibitor, ACH-1625. Antivir Ther. 2012;17:1533–1539. doi: 10.3851/IMP2359. [DOI] [PubMed] [Google Scholar]
- 233.McPhee F, Sheaffer AK, Friborg J, Hernandez D, Falk P, Zhai G, Levine S, Chaniewski S, Yu F, Barry D, et al. Preclinical Profile and Characterization of the Hepatitis C Virus NS3 Protease Inhibitor Asunaprevir (BMS-650032) Antimicrob Agents Chemother. 2012;56:5387–5396. doi: 10.1128/AAC.01186-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Liverton NJ, Carroll SS, Dimuzio J, Fandozzi C, Graham DJ, Hazuda D, Holloway MK, Ludmerer SW, McCauley JA, McIntyre CJ, et al. MK-7009, a potent and selective inhibitor of hepatitis C virus NS3/4A protease. Antimicrob Agents Chemother. 2010;54:305–311. doi: 10.1128/AAC.00677-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Reesink HW, Fanning GC, Farha KA, Weegink C, Van Vliet A, Van ‘t Klooster G, Lenz O, Aharchi F, Mariën K, Van Remoortere P, et al. Rapid HCV-RNA decline with once daily TMC435: a phase I study in healthy volunteers and hepatitis C patients. Gastroenterology. 2010;138:913–921. doi: 10.1053/j.gastro.2009.10.033. [DOI] [PubMed] [Google Scholar]
- 236.Moreno C, Berg T, Tanwandee T, Thongsawat S, Van Vlierberghe H, Zeuzem S, Lenz O, Peeters M, Sekar V, De Smedt G. Antiviral activity of TMC435 monotherapy in patients infected with HCV genotypes 2-6: TMC435-C202, a phase IIa, open-label study. J Hepatol. 2012;56:1247–1253. doi: 10.1016/j.jhep.2011.12.033. [DOI] [PubMed] [Google Scholar]
- 237.Lenz O, Verbinnen T, Lin TI, Vijgen L, Cummings MD, Lindberg J, Berke JM, Dehertogh P, Fransen E, Scholliers A, et al. In vitro resistance profile of the hepatitis C virus NS3/4A protease inhibitor TMC435. Antimicrob Agents Chemother. 2010;54:1878–1887. doi: 10.1128/AAC.01452-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Dvory-Sobol H, Wong KA, Ku KS, Bae A, Lawitz EJ, Pang PS, Harris J, Miller MD, Mo H. Characterization of resistance to the protease inhibitor GS-9451 in hepatitis C virus-infected patients. Antimicrob Agents Chemother. 2012;56:5289–5295. doi: 10.1128/AAC.00780-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Lagacé L, White PW, Bousquet C, Dansereau N, Dô F, Llinas-Brunet M, Marquis M, Massariol MJ, Maurice R, Spickler C, et al. In vitro resistance profile of the hepatitis C virus NS3 protease inhibitor BI 201335. Antimicrob Agents Chemother. 2012;56:569–572. doi: 10.1128/AAC.05166-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Trozzi C, Bartholomew L, Ceccacci A, Biasiol G, Pacini L, Altamura S, Narjes F, Muraglia E, Paonessa G, Koch U, et al. In vitro selection and characterization of hepatitis C virus serine protease variants resistant to an active-site peptide inhibitor. J Virol. 2003;77:3669–3679. doi: 10.1128/JVI.77.6.3669-3679.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Koch U, Narjes F. Recent progress in the development of inhibitors of the hepatitis C virus RNA-dependent RNA polymerase. Curr Top Med Chem. 2007;7:1302–1329. doi: 10.2174/156802607781212211. [DOI] [PubMed] [Google Scholar]
- 242.Holler TP, Parkinson T, Pryde DC. Targeting the non-structural proteins of hepatitis C virus: beyond hepatitis C virus protease and polymerase. Expert Opin Drug Discov. 2009;4:293–314. doi: 10.1517/17460440902762802. [DOI] [PubMed] [Google Scholar]
- 243.Pawlotsky JM, Najera I, Jacobson I. Resistance to mericitabine, a nucleoside analogue inhibitor of HCV RNA-dependent RNA polymerase. Antivir Ther. 2012;17:411–423. doi: 10.3851/IMP2088. [DOI] [PubMed] [Google Scholar]
- 244.Pockros PJ, Jensen D, Tsai N, Taylor R, Ramji A, Cooper C, Dickson R, Tice A, Kulkarni R, Vierling JM, et al. JUMP-C: a randomized trial of mericitabine plus pegylated interferon alpha-2a/ribavirin for 24 weeks in treatment-naïve HCV genotype 1/4 patients. Hepatology. 2013;58:514–523. doi: 10.1002/hep.26275. [DOI] [PubMed] [Google Scholar]
- 245.Lawitz E, Mangia A, Wyles D, Rodriguez-Torres M, Hassanein T, Gordon SC, Schultz M, Davis MN, Kayali Z, Reddy KR, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med. 2013;368:1878–1887. doi: 10.1056/NEJMoa1214853. [DOI] [PubMed] [Google Scholar]
- 246.Gane EJ, Stedman CA, Hyland RH, Ding X, Svarovskaia E, Symonds WT, Hindes RG, Berrey MM. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med. 2013;368:34–44. doi: 10.1056/NEJMoa1208953. [DOI] [PubMed] [Google Scholar]
- 247.Tong X, Le Pogam S, Li L, Haines K, Piso K, Baronas V, Yan JM, So SS, Klumpp K, Nájera I. In vivo emergence of a novel mutant L159F/L320F in the NS5B polymerase confers low-level resistance to the HCV polymerase inhibitors mericitabine and sofosbuvir. J Infect Dis. 2014;209:668–675. doi: 10.1093/infdis/jit562. [DOI] [PubMed] [Google Scholar]
- 248.Guedj J, Dahari H, Rong L, Sansone ND, Nettles RE, Cotler SJ, Layden TJ, Uprichard SL, Perelson AS. Modeling shows that the NS5A inhibitor daclatasvir has two modes of action and yields a shorter estimate of the hepatitis C virus half-life. Proc Natl Acad Sci USA. 2013;110:3991–3996. doi: 10.1073/pnas.1203110110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Gao M, Nettles RE, Belema M, Snyder LB, Nguyen VN, Fridell RA, Serrano-Wu MH, Langley DR, Sun JH, O’Boyle DR, et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature. 2010;465:96–100. doi: 10.1038/nature08960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Pawlotsky JM. New antiviral agents for hepatitis C. F1000 Biol Rep. 2012;4:5. doi: 10.3410/B4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Asselah T. NS5A inhibitors: a new breakthrough for the treatment of chronic hepatitis C. J Hepatol. 2011;54:1069–1072. doi: 10.1016/j.jhep.2010.11.033. [DOI] [PubMed] [Google Scholar]
- 252.Conte I, Giuliano C, Ercolani C, Narjes F, Koch U, Rowley M, Altamura S, De Francesco R, Neddermann P, Migliaccio G, et al. Synthesis and SAR of piperazinyl-N-phenylbenzamides as inhibitors of hepatitis C virus RNA replication in cell culture. Bioorg Med Chem Lett. 2009;19:1779–1783. doi: 10.1016/j.bmcl.2009.01.066. [DOI] [PubMed] [Google Scholar]
- 253.Asselah T, Marcellin P. Direct acting antivirals for the treatment of chronic hepatitis C: one pill a day for tomorrow. Liver Int. 2012;32 Suppl 1:88–102. doi: 10.1111/j.1478-3231.2011.02699.x. [DOI] [PubMed] [Google Scholar]
- 254.Fridell RA, Wang C, Sun JH, O’Boyle DR, Nower P, Valera L, Qiu D, Roberts S, Huang X, Kienzle B, et al. Genotypic and phenotypic analysis of variants resistant to hepatitis C virus nonstructural protein 5A replication complex inhibitor BMS-790052 in humans: in vitro and in vivo correlations. Hepatology. 2011;54:1924–1935. doi: 10.1002/hep.24594. [DOI] [PubMed] [Google Scholar]
- 255.Schinazi R, Halfon P, Marcellin P, Asselah T. HCV direct-acting antiviral agents: the best interferon-free combinations. Liver Int. 2014;34 Suppl 1:69–78. doi: 10.1111/liv.12423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Ferenci P. Treatment of chronic hepatitis C--are interferons really necessary? Liver Int. 2012;32 Suppl 1:108–112. doi: 10.1111/j.1478-3231.2011.02705.x. [DOI] [PubMed] [Google Scholar]