

Abstract
Methanogenic archaea are known as human gut inhabitants since more than 30 years ago through the detection of methane in the breath and isolation of two methanogenic species belonging to the order Methanobacteriales, Methanobrevibacter smithii and Methanosphaera stadtmanae. During the last decade, diversity of archaea encountered in the human gastrointestinal tract (GIT) has been extended by sequence identification and culturing of new strains. Here we provide an updated census of the archaeal diversity associated with the human GIT and their possible role in the gut physiology and health. We particularly focus on the still poorly characterized 7th order of methanogens, the Methanomassiliicoccales, associated to aged population. While also largely distributed in non-GIT environments, our actual knowledge on this novel order of methanogens has been mainly revealed through GIT inhabitants. They enlarge the number of final electron acceptors of the gut metabolites to mono- di- and trimethylamine. Trimethylamine is exclusively a microbiota-derived product of nutrients (lecithin, choline, TMAO, L-carnitine) from normal diet, from which seems originate two diseases, trimethylaminuria (or Fish-Odor Syndrome) and cardiovascular disease through the proatherogenic property of its oxidized liver-derived form. This therefore supports interest on these methanogenic species and its use as archaebiotics, a term coined from the notion of archaea-derived probiotics.
Keywords: Human gut microbiota, Methanogens, Methanomassiliicoccus, Methanomethylophilus, Trimethylaminuria, Trimethylamine, Methane, Cardiovascular disease, Archaebiotics, Probiotics
Core tip: Archaea are naturally occurring components of the human gut microbiota, whose biological significance has been recently reevaluated. In this review, an update of the current knowledge about the archaea from the human gut is provided, integrating the new order of methanogens, Methanomassiliicoccales. By its particular metabolism, this lineage is likely a depleting biological agent of a gut microbiota metabolite from diet implied in cardiovascular disease and trimethylaminuria, trimethylamine. The recent provocative proposal of archaea as a new class of probiotics (archaebiotics) should focus the interest on the third domain of life concerning the gut physiopathology and human health.
INTRODUCTION
Archaea are unicellular microorganisms which have long been seen as “extreme bacteria”. However, although some are indeed extremophiles (e.g., hyperthermophiles hyperacidophiles and hyperhalophiles), it is now clear that a large part of them are mesophiles and widespread in the environment[1,2]. They form a separate branch of life, the domain Archaea, alongside of the two other domains: Eukarya and Bacteria[3]. It has recently been suggested that eukaryotes have appeared within the domain Archaea[4], which nevertheless remains controversial[5]. The domain Archaea includes a wide variety of organisms that share properties with both bacteria (various morphologies: coccus, spirillum, bacillus or irregular shapes, presence of a single circular chromosome, lack of introns, similar post-transcriptional modifications) and eukaryotes (similar molecular machinery for DNA replication, RNA transcription and protein translation, presence of histones for chromosomal DNA packaging)[6]. It also presents unique characteristics such as lack of peptidoglycan in the cell wall, when present and membrane formed by L-glycerol ethers/isoprenoids chains instead of D-glycerol esters/fatty acids as in the two other domains[7-9]. Some of these archaea have a unique and particular metabolism, the methanogenesis. Methanogenic archaea are strict anaerobes that occur in a large range of environments, such as freshwater[10] and marine[11] sediments, soils[12,13] and the gut of numerous animal species[14,15], including humans[16-18]. Their physiology and ecology is widely studied for their intrinsic capacity to produce methane which is both an energy source (biogas) in bioreactor[19] and a greenhouse gas emitted from natural and anthropic environments, including livestock[20,21]. As a recurrent component of the human digestive tract, impact of archaea[22] and more particularly methanogens[23,24] on human health have been questioned for many years.
Notewithstanding the fact that archaea are also identified in the oral microbiota of human (see[25] for a recent review), this article mainly focuses on methanogenic and non-methanogenic archaea in the gut. A concise description of methanogenesis and of the two Methanobacteriales, Methanobrevibacter smithii (M. smithii) and Methanosphaera stadtmanae (M. stadtmanae) whose occurrence in the human gut is known since three decades is given. In a second part the current knowledge about the archaeal diversity in our gut is presented from recent studies, and a more detailed focus is made on a newly described order of methanogens, the Methanomassiliicoccales, for which physiological and genomics data from human-retrieved strains are now available. Finally we display a census of the investigated relationships between methanogens and the physiological state of their human host, with a focus on the age relationships, and we discuss the putative implications of the representatives of the Methanomassiliicoccales for human health.
METHANOGENS, METHANOGENESIS AND THE DIGESTIVE TRACT
Until recently, methanogens were organized into 6 orders (Methanobacteriales, Methanococcales, Methanomicrobiales, Methanosarcinales, Methanopyrales, Methanocellales) (filled square yellow boxes in Figure 1), all belonging to the phylum Euryarchaeota. A 7th order of methanogens phylogenetically related to the Thermoplasmatales was recently proposed on the basis of sequences retrieved from human gut[26,27]. Two names were subsequently proposed, Methanoplasmatales[28] and Methanomassiliicoccales[29], the latter being now validated by the International Committee on Systematics of Prokaryotes (ICSP)[30] (Figure 1). Methanogenesis is coupled to different energy conservation systems and represents the sole energetic metabolism of methanogens. For this metabolism, methanogens could only use a limited number of substrates originating from the anaerobic degradation of the organic matter by hydrolytic and fermentative bacteria[31]. Methanogens have therefore a terminal position in the microbial trophic chains. According to the metabolic classification used today, one can define the hydrogenotrophic, methylotrophic and acetotrophic (or acetoclastic) class of methanogenesis[31]. Most of the methanogens are hydrogenotrophs that use H2 (and often formate) to reduce CO2 into methane[32]. The methylotrophic methanogens use methylated compounds such as methanol, methylamines and methyl-sulfides, converting the methyl group of these compounds into CH4 with substrate-specific methyltransferases[33]. Reducing equivalents for this methanogenesis are obtained by an additional oxidation of a methyl group into CO2 through the methyl-oxidation pathway, corresponding to the first steps of the hydrogenotrophic pathway operating in reverse. A variant of this pathway consists in the direct use of H2 present in the environment as an electron donor, instead of the reducing equivalents produced by the methyl-oxidation pathway. Interestingly, the methanogens restricted to this variant were found to be associated to gut environments (see below). Finally, few archaea affiliated to the Methanosarcinales are able to use acetate as substrate for methanogenesis[34]. The methyl-coenzyme M reductase (MCRI or MCRII isoenzyme), responsible for the formation of CH4 from CH3-S-CoM and H-S-CoM, is shared by all methanogens. The alpha-subunit of this enzyme, encoded by mcrA (or mrtA for the isoenzyme MCRII), is considered as a functional marker of methanogens and is widely used for their specific detection by molecular approaches[35]. Owning to the congruence between mcrA and 16S rRNA genes based phylogenies[36], mcrA is also used to investigate the affiliation and phylogenetic diversity of methanogens in various environments, including the human GIT[26,27,37-39]. The digestive tract of animals and especially of ruminants harbors a large variety of methanogens[40] with the different types of methanogenesis (hydrogenotroph, methylotroph and acetotroph), even though acetoclastic methanogenesis is considered to be a minor pathway in rumen[41] and acetoclastic methanogens were never detected in the human distal gut.
Figure 1.
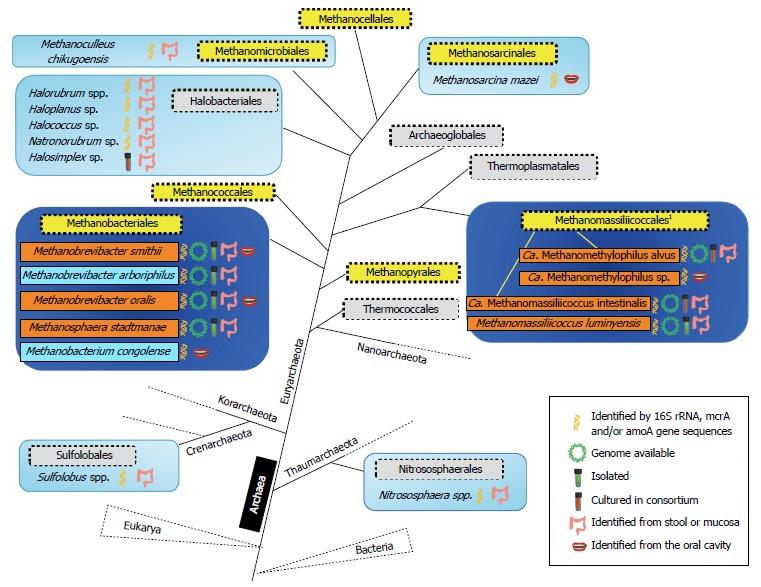
Archaeal representatives associated to the human digestive tract. A simplified phylogeny of the Archaea (adapted from[89,148]) is shown. Taxonomic orders are drawn in filled square boxes, those composed of methanogens in yellow and others in light grey. Orders in which some members have been found in the human digestive tract are indicated in light blue boxes when identified by sequences derived from nested-PCR (or deep sequencing on low yield amplifications for Nitrososphaera spp.) or a limited number of studies, and other are indicated in dark blue boxes in which abundant and recurrently reported species are mentioned in orange boxes or light blue when observed from limited number of studies. For the Methanomassiliicoccales (1), also referred as Methanoplasmatales in the literature, the repartition of Methanomassiliicoccus spp. and Ca. Methanomethylophilus spp. in two different clades is highlighted by yellow branches (see the text for details).
In humans, methanogenesis is mainly H2-dependent, for the reduction of CO2 and for the reduction of methyl-compounds: this H2 depletion optimizes the fermentation and modifies the metabolic pathways of fermentative bacteria[17]. This role is shared with two other types of hydrogenotrophic microorganisms, the reductive acetogenic bacteria (e.g., Ruminococcus spp.[42]) and the sulfate-reducing bacteria (e.g., Desulfovibrio spp.[43]). Hydrogenotrophic methanogenesis from CO2 efficiently decreases the gas partial pressure in the colon by consuming 4 moles of H2 and 1 mole of CO2 to produce 1 mole of CH4. The Figure 2 shows the role of the gut methanogens (white filled box) in the overall gut metabolism of nutrients (blue circle).
Figure 2.
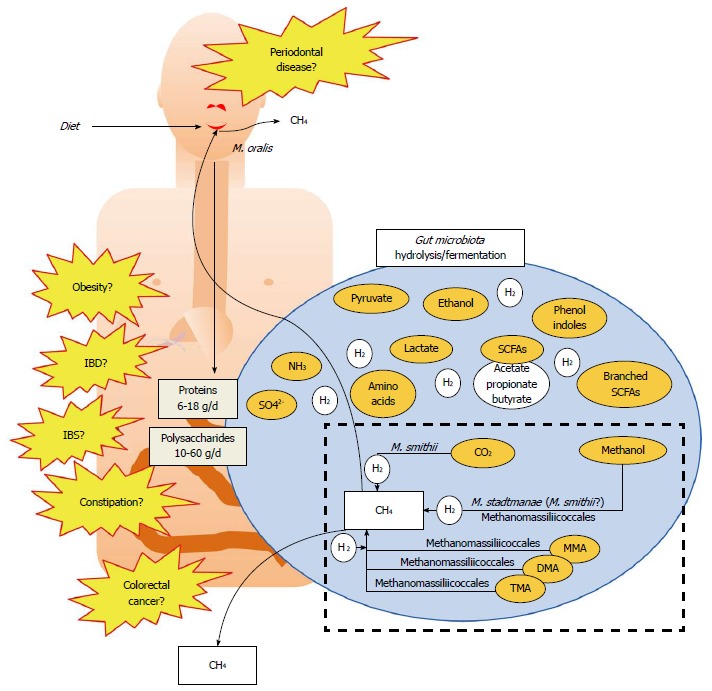
Simplified representation of the role of methanogens in the gut microbiota metabolism and possible association with diseases. Diet leads to proteins and polysaccharides that enter the gut where they are metabolized by the hydrolytic and fermentative bacteria present in the gut (large blue circle). This produces various compounds (some important being indicated in orange circles), and large amounts of H2, which in turn inhibit the fermentation processes. H2 partial pressure is lowered by hydrogenotrophs, three different types being present in the gut (sulfate-reducing bacteria, acetogens and methanogens). Here, only the methanogens are shown, which are either able to use CO2, methanol, or methyl-compounds [monomethylamine (MMA); dimethylamine (DMA) and trimethylamine (TMA)] reduced by hydrogen to form methane, found in breath and flatulence. A highlight on the TMA use is given in Figure 3. The presence of methanogens in the mouth is also indicated. Altogether, the presence of methanogens has been linked to several diseases (in yellow) but these remains to be clarified (see Table 2 for further explanations).
Specific tests have been developed, such as the “breath- test”[16], in which the presence of methane in breath is synonymous to the presence of methanogenic archaea in the gut (usually after a lactose challenge test). It was determined that the detection of more than 1 ppm of methane in breath vs atmospheric concentration (lowest detection capacity) reflects the minimum presence of 107-108 methanogens per gram of stool[44,45]. Using this test, the human population may be divided into two different groups: the methane-producers and the non-methane-producers. A review of the studies based on the methane breath test has stressed the high variability of the prevalence of methane producers in different geographic and ethnic populations[23], ranging from 34% to 87%, with level above 70% observed in rural African populations[46-48].
M. SMITHII, THE DOMINANT HUMAN GUT METHANOGEN
Metabolism and adaptations
From the 80s to very recently, only 2 different archaeal species belonging to Methanobacteriales have been identified and isolated from the human gut microbiota, M. smithii[49] and M. stadtmanae[50] (formerly named M. stadtmaniae). These archaea have a different metabolism of methanogenesis, M. smithii using H2 (or formate) to reduce CO2 and M. stadtmanae using H2 to reduce methanol. M. smithii is able to colonized the human GIT at least from the cecum to the rectum[39,51,52]. Pyxigraphic sampling on 6 individuals and quantification by culture-based approaches has suggested that M. smithii preferentially colonizes the distal colon[51]. A recent qPCR-based quantification of the hydrogenotrophic microbes associated to the colonic mucosa did not report significant changes in the abundance of methanogens along the colon[39]. However, this study also reports that the methanogens would represent a larger proportion of the hydrogenotrophic microbes in the rectum than in the left and right colon[39]. Several important adaptations of M. smithii to the human gut environment have been described, using genomics and transcriptomics[53-57]. In a humanized gnotobiotic mouse model, M. smithii was shown to adapt its gene expression in presence of a dominant gut microbe, Bacteroides thetaiotaomicron (B. thetaiotaomicron), and to modify the metabolic pathways of this bacteria[53]. M. smithii strains have also the property to produce glycans mimicking those found in the gut[54] and possess a large diversity of adhesion-like proteins whose substrate-related regulation suggests adaptive answers to different niches in the gut[55]. M. smithii and M. stadtmanae have gene to cope with the presence of bile acid, notably the bile acid salts hydrolase gene (BSH)[54,58], with likely a bacterial origin[59]. Over 15% of the coding genes of these two species might have been transferred from bacteria, with a large contribution of Firmicutes, a dominant bacterial lineage in the gut[56,57]. The putative gut origin of these genes is supported by the observation that 20 of the 22 bacterial OTUs found to be positively associated to M. smithii in 136 subjects were affiliated to the Firmicutes[55]. Several of these genes are involved in transport and surface function and could have facilitated the adaptation of M. smithii and M. stadtmanae to human GIT environment. M. smithii was also shown to be efficient in competition for the nitrogenous nutrient pool[54] and have the capacity to use several end-products derived from organic matter degradation in the gut[54]. In regard to this last aspect, it is interesting to quote that almost all sequenced human GIT associated methanogens possess the genes mtaABC encoding methyl-transferases required for methanol utilization (this study, see Table 1 and below), highlighting the importance of this metabolism for gut methanogens. In the case of M. stadtmanae, it is clear that these genes are involved in methanogenesis[58], but their role remains less clear for Methanobrevibacter spp..
Table 1.
Available genomics data (GenBank, March 2014) from methanogens of the human gut
| Species | Strain (DSMZ collection number) | Status1 | Size (Mb) | GC% | mta | BSH | Accession | Genome reference |
| ABC | gene | number | ||||||
| Methanobacteriales | ||||||||
| Methanobrevibacter smithii | PS (DSM 861) | Chromosome | 1.85 | 31.0 | 1 | 1 | CP000678.1 | [54] |
| F1 (DSM 2374) | Scaffolds | 1.73 | 31.3 | 0 | 1 | ABYV00000000 | [126] | |
| ALI (DSM 2375) | Scaffolds | 1.71 | 31.3 | 1 | 1 | ABYW00000000 | [126] | |
| TS145A | Contigs | 1.78 | 31.1 | 12 | 12 | AEKU00000000 | [55] | |
| TS145B | Contigs | 1.80 | 31.1 | 12 | 12 | AELL00000000 | ||
| TS146A | Contigs | 1.79 | 31.2 | 12 | 12 | AELM00000000 | ||
| TS146B | Contigs | 1.79 | 31.1 | 12 | 12 | AELN00000000 | ||
| TS146C | Contigs | 1.95 | 31.2 | 12 | 12 | AELO00000000 | ||
| TS146D | Contigs | 1.71 | 31.1 | 12 | 12 | AELP00000000 | ||
| TS146E | Contigs | 1.95 | 30.4 | 12 | 12 | AELQ00000000 | ||
| TS147A | Contigs | 2.01 | 30.5 | 12 | 12 | AELR00000000 | ||
| TS147B | Contigs | 1.97 | 30.4 | 12 | 12 | AELS00000000 | ||
| TS147C | Contigs | 1.97 | 30.4 | 12 | 12 | AELT00000000 | ||
| TS94A | Contigs | 1.89 | 30.2 | 12 | 12 | AELU00000000 | ||
| TS94B | Contigs | 1.89 | 30.2 | 12 | 12 | AELV00000000 | ||
| TS94C | Contigs | 1.91 | 30.3 | 12 | 12 | AELW00000000 | ||
| TS95A | Contigs | 1.99 | 30.2 | 12 | 12 | AELX00000000 | ||
| TS95B | Contigs | 1.97 | 30.2 | 12 | 12 | AELY00000000 | ||
| TS95C | Contigs | 1.98 | 30.2 | 12 | 12 | AELZ00000000 | ||
| TS95D | Contigs | 2.01 | 30.2 | 12 | 12 | AEMA00000000 | ||
| TS96A | Contigs | 1.98 | 30.3 | 12 | 12 | AEMB00000000 | ||
| TS96B | Contigs | 1.87 | 30.2 | 12 | 12 | AEMC00000000 | ||
| TS96C | Contigs | 1.82 | 31.1 | 12 | 12 | AEMD00000000 | ||
| Methanobrevibacter arboriphilus | ANOR1 | Scaffolds | 2.22 | 25.5 | 12 | 22 | CBVX000000000 | [83] |
| Methanobrevibacter oralis | JMR01 | Scaffolds | 2.11 | 27.8 | 2 | 2 | CBWS000000000 | [63] |
| Methanosphaera stadtmanae | (DSM 3091) | Chromosome | 1.77 | 27.6 | 4 | 1 | CP000102 | [58] |
| Methanomassiliicoccales | ||||||||
| Methanomassiliicoccus luminyensis | B10 (DSM 25720) | Contigs | 2.62 | 60.5 | 33 | 0 | CAJE00000000 | [88] |
| Ca. Methanomassiliicoccus intestinalis | Issoire-Mx1 | Chromosome | 1.93 | 41.3 | 33 | 0 | CP005934 | [87] |
| Ca. Methanomethylophilus alvus | Mx1201 | Chromosome | 1.67 | 55.6 | 13 | 1 | CP004049 | [86] |
As given by genbank, March 2014;
Original analysis from this work: a tblasn of the protein sequence of mtaA (YP_447810), mtaB (YP_447248), mtaC (YP_447249) and the BSH (Bile Salt Hydrolase) gene (YP_447796) from Methanosphaera stadtmanae on the draft genome sequence of Methanobrevibacter arboriphilus ANOR1, Methanobrevibacter oralis JMR01 and Methanobrevibacter smithii strains from[55].
Numbers of mtaBC genes detected, homologues of mtaA genes are also present in the 3 Methanomassiliicoccales genomes but their specific implication in either methanol and/or methylamines utilization pathways has not yet been determined.
M. smithii does not only occur in the distal gut, but also sparsely in the oral cavity[60] and the vagina[61]. In the oral cavity, methanogenic archaea are dominated by a third species, Methanobrevibacter oralis (M. oralis), isolated in the mid 90’[62] and whose genome was recently sequenced[63]. This species have attracted a great attention because its occurrence is strongly related to periodontitis[24]. While M. smithii and M. oralis are phylogenetically close (their 16S rRNA gene share 98% identity), they are clearly specialized on gut and oral cavity, respectively. One aspect of this specialization is supported by the presence of a bile salt hydrolase gene in all sequenced M. smithii species but not in the genome of M. oralis (this study, see Table 1). Another aspect might rely on the versatility of these two species, M. oralis lacking the mtaABC genes present in almost all other M. smithii strains. Moreover, M. oralis is able to reduce CO2 with H2 but not formate for methanogenesis, a difference compared to M. smithii[62]. Obviously their niche partitioning is probably under dependence of other properties, notably those favoring M. oralis over M. smithii in the mouth, which will require further investigations.
Prevalence in the population
In a large analysis of three different individuals gut bacteria and archaea, with samples from different segments of the intestine and feces (cecum; ascending, transverse, distal and sigmoid colon), the cloning/sequencing of 16S rRNA genes[52] revealed the presence of archaea in 2 of the 3 individuals. Of the 1534 archaeal clones, all the sequences corresponded to M. smithii. This agrees with previous culture-dependent studies[45,47] and subsequent culture-independent studies indicating M. smithii is the most prevalent and abundant methanogen in the human gut[26,27,37,44,64]. Recent metagenomics studies support also this predominance[65-68], even if this kind of studies is prone to difficulties and limitations to correctly attribute nucleic sequences to species: it necessitates more particularly referenced genomes, data that are not always available at the species level for archaea from the gut. The gut microbiota shotgun metagenomics analysis performed on 96 healthy Russian adults indicated that there were variations among individuals, and among populations[69], therefore confirming results from methane breath-tests mentioned above[23,48,70,71]: M. smithii was the second and third most prevalent prokaryotic species in the microbiota in two of the 96 Russian adults (representing between 11% and 14%), and more globally, Russian adults showed generally a higher level of M. smithii than Chinese, Danish and US people, while being lower compared to the Amerindian group[69]. Based on a modified method of DNA extraction, Dridi et al[64] reported that 95.7% of 700 individuals tested by qPCR were positive for M. smithii. However, subsequent studies performed with the same method reported lower levels with 75%-89%[72] and 64%[73] of individuals positive to M. smithii.
Therefore, it seems that M. smithii is found in more than 50% of the adult population, forming a highly prevalent microbe of the gut, but remains undetected in some people. Whether the lack of detection is due to a real absence or of a technical threshold remains still unclear, but it is conceivable that the methanogens present at low abundance in some individuals have likely a very low physiological incidence. M. stadtmanae shows a lower prevalence: it has been found approximately in 1 people on 3 to 5 (32.6%[74] and 17%-25%[27] of the population).
RECENT EXPANSION OF THE ARCHAEAL DIVERSITY ASSOCIATED WITH THE HUMAN GASTROINTESTINAL TRACT
The advent of molecular methods[75,76] has greatly improved our knowledge of the gut microbiota composition. As for bacterial populations, studies based on culture-independent approaches have largely participated to the identification of novel archaeal phylotypes associated to the human GIT. During the last decade, gene sequences retrieved from human oral cavity, colonic mucosa or feces have been affiliated to several taxonomic orders shown in light and dark blue boxes in Figure 1: they represent 1 order within Crenarchaeota and Thaumarchaeota, and 5 within the Euryarchaeota, showing that 3 of the 5 archaeal phyla are represented in the human GIT.
Large diversity of archaea occurring likely at a low abundance
A set of sequences closely related to Sulfolobus species of the phylum Crenarchaeota was obtained by nested PCR on feces of 4 adults[77]. Colonization of the gut by these archaea would be particularly surprising considering the acido-thermophilic nature of other known representatives of Crenarchaeota. Sequences affiliated to this phylum were never retrieved in other studies, and therefore their presence may be attributed to an unusual case or to a microorganism in transit. However, some of the “Uncultured Crenarchaeote” sequences of this study (AY887071; AY887074; AY887077; AY887078) are closely related to “Candidatus Nitrososphaera gargensis”[78], a moderately thermophilic ammonium oxidizer of the phylum Thaumarchaeota[2], not yet proposed in 2005. More recently, an investigation on the associations of diet with fungal and archaeal populations by 16S pyrosequencing also revealed the presence of sequences affiliated to the genus Nitrososphaera, in low yield amplifications and only in samples without M. smithii[79]. These sequences were detected in 16 samples from the 96 analyzed. A nested PCR assay targeting the amoA gene (encoding the ammonia mono-oxygenase) confirmed this result. The report of sequences affiliated to Nitrososphaera in two independent studies argues for their presence, even at low abundance, in the human gut microbiota[79]. Their low abundance might be due, for a part to their oxygen requirement for ammonium-oxidation, as reported for other ammonium-oxidizing archaea so far[80].
Non-methanogenic euryarchaeota affiliated to the Halobacteriales were reported by two studies based on nested-PCR. The analysis of fecal samples using the PCR fingerprinting method DGGE (denaturing gradient gel electrophoresis) showed the presence of several sequences related to halophilic archaea in healthy Koreans. DNA sequences analyses revealed from 93% to 99% of identity to known Halobacteriales, Halorubrum koreense, Halorubrum alimentarium and Halococcus morrhuae[81]. These euryarchaeota are also found in salt-fermented seafood, therefore arguing that they may be related to Korean eating habits[81]. The presence of a large diversity (15 phylotypes with a 97.5% similarity cut off on 16S rRNA gene sequences) of haloarchaea was also detected by nested-PCR approaches in colonic mucosa biopsies and, in a lower extent, from feces obtained from German subjects with IBD[82]. Parallel detection of archaea in table salts has revealed a high and underestimated diversity of haloarchaea suggesting the possible origin of these archaea, regardless of their transient or persistent status in the human gut[82]. Several phylotypes were also detected in pre-endoscopic lavage solutions but were different from those retrieved from the biopsies. Interestingly one haloarchaea related to Halosimplex sp. was enriched in a low salt concentration medium (2.5% w/v) inoculated with microbes associated to a biopsy, showing that some of them are viable.
The few studies performed on archaea associated to mucosa have also detected methanogens not previously retrieved from feces samples[39,82], suggesting that mucosa displays an overall larger diversity of archaea than feces, possibly reflecting greater niche heterogeneity and refuges when less adapted to gut conditions compared to M. smithii and M. stadtmanae. In addition to the haloarchaea, Oxley et al[82] have also reported sequences closely related to Methanobrevibacter arboriphilus from biopsies of two individuals. A strain of this species was subsequently isolated from a human stool sample and its genome was sequenced[83] (Table 1). Clone sequences of mcrA closely related to Methanoculleus chikugoensis, a hydrogenotrophic methanogen of the order Methanomicrobiales were found associated to the intestinal mucosa[39]. As reviewed by Nguyen-Hieu et al[84], several other archaea such as M. mazei and M. congolense were also sparsely retrieved in the oral cavity.
Overall, this larger diversity suggests new functionalities and putative new archaea-host interactions. However most of these lineages of methanogenic and non-methanogenic archaea were identified from nested-PCR based experiments, suggesting there low abundance and possibly transient status in the human GIT[39,77,79,81,82]. This is not the case of sequences affiliated to the recently proposed 7th order of methanogens[26,27,37,38].
Seventh order of methanogens: From an uncultured Thermoplasmatales-related lineage to the Methanomassiliicoccales
In 2008, two studies on the human gut methanogens revealed, in addition to sequences attributed to M. smithii and M. stadtmanae, mcrA sequences extremely distant to any other cultured methanogens[26,37]. These sequences were associated to the Methanosarcinales order in the study of Scanlan et al[37] due to a weak, but higher homology with sequences of this order than of all others[37]. The other study concluded to a clearly distinct and distant evolutionary branch compared to all other methanogens, and postulated for a new order of methanogens[26]. In parallel, the investigation of the co-occurring archaeal 16S rRNA genes provided a sequence related to the Thermoplasmatales, only in the sample positive for the atypical mcrA sequence[26]. The almost strict co-occurrence of Thermoplasmatales-related 16S rRNA sequences and atypical mcrA sequences was extended to a larger range of human-associated samples, highlighting also the diversity of this putative new order[27]. The final evidences of the existence of these new methanogens phylogenetically related to the Thermoplasmatales were subsequently provided by the culturing of several representatives of this order from human feces samples, either in pure culture (Methanomassiliicoccus luminyensis[85]) or in consortia (Ca. Methanomethylophilus alvus[86] and Ca. Methanomassiliicoccus intestinalis[87]), and the sequencing of their genome[86-88] (Table 1). The phylogenetic positioning of these methanogens was confirmed by a phylogenomic approach including 7472 marker positions from 84 Euryarchaeota genomes[89]. On the basis of enrichment cultures from hindgut of termites the name of Methanoplasmatales was proposed for this order[28]. However, according to the rules of the International Code of Nomenclature of Bacteria[90], the name of Methanomassiliicoccales was then claimed by Iino et al[29] in 2013 and subsequently validated by the International Committee on Systematics of Prokaryotes[30]. In the human digestive tract, the occurrence of the Methanomassiliicoccales representatives is not restricted to the colon as the 16S rRNA gene sequences of Ca. M. alvus shares 97%-98% identity with two 16S rRNA gene sequences retrieved in the oral cavity (FJ458322[91]; JQ433705[38]). These two sequences derived from oral cavity were almost similar to each other (2 differences on 357 aligned sites), suggesting that they belong to two strains of a same Ca. Methanomethylophilus species. In their study, Horz et al[38] retrieved it in 10% of the subjects where it was estimated to represent 0.5% of the overall prokaryotic community[84]. It is thus likely that some species of Methanomassiliicoccales are more adapted to the oral cavity while others are more adapted to the colonic environment, similarly to the niche partitioning of M. smithii and M. oralis between the colon and the oral cavity, respectively[92].
UNIQUE PROPERTIES OF THE METHANOMASSILIICOCCALES IN THE HUMAN GUT
The only isolated strain of this order, Methanomassiliicoccus luminyensis B10, was obtained from human feces[85]. This strain is H2 and methanol dependent for its methanogenesis[85], like M. stadtmanae[58]. This is also the case for Ca. Methanomethylophilus alvus[86] and Ca. Methanomassiliicoccus intestinalis[87]. Other members of the Methanomassiliicoccales cultured in consortia from the GIT of higher termites and millipedes[28] and from an anaerobic digester sludge[29] share the same characteristic. The first genome analyses of Ca. M. alvus, Ca. M. intestinalis and M. luminyensis revealed several fundamental aspects and putative generalized characteristics of this order[87-89]. First of all, the genes involved in the CO2-reduction/methyl-oxydation pathways are completely absent[89], while the other methanol-using methanogen under dependence of H2, M. stadtmanae, still possesses most of them. Moreover, the three genomes show the genetic potential to encode an unusual amino acid, pyrrolysine (Pyl) discovered 10 years earlier in Methanosarcinaceae[93-95]. This 22nd proteinogenic amino acid is derived from lysine which is then incorporated into proteins by a specific genetic code during the translation, and relies on the amber stop codon suppression though specific tRNAs[94]. This strange event in the evolution of the genetic code is restricted to a few bacteria and methanogenic archaea from the Methanosarcinaceae family. In the Methanomassiliicoccales, this seems followed by a specific evolution, with consequences for the entire genome (very low use of amber codon UAG as translation stop signal, regardless of GC%) (Borrel et al[95]).
Adding methylamines to the list of valuable electron acceptors in the human gut
Pyrrolysine is an essential component of the catalytic site of the methyltransferase which is involved in the methylotrophic methanogenesis from methylated amines [monomethylamine (MMA), dimethylamine (DMA) and trimethylamine (TMA)]. The genes for these methyltransferases (respectively mtmB, mtbB and mttB) are present in the three genomes, their open reading frame being interrupted by an amber stop codon[86,87,95,96], as observed in the Methanosarcinaceae[94]. The detection of mttB transcripts, having an amber stop codon presumably coding for Pyl has also been demonstrated in inhabitants of the cattle rumen belonging to the Methanomassiliicoccales (the Rumen Cluster C, RCC cluster[97]). In this study, this gene was overexpressed in ruminal microbiota cultures in the presence of TMA, coupled to enhanced methanogenesis, providing strong clues that TMA is a substrate for methanogenesis in the representatives of the Methanomassiliicoccales. Using the isolated strain M. luminyensis B10 in pure culture, we definitively demonstrated that TMA, in presence of H2, is one of its methanogenic substrates, as are also MMA and DMA, in addition to methanol[96]. Therefore, this leads to reconsider the electron acceptors in the human gut by including methylamines, when representatives of Methanomassiliicoccales are present (Figure 3). This could have likely important consequences for the digestive physiology as well as for human pathologies.
Figure 3.
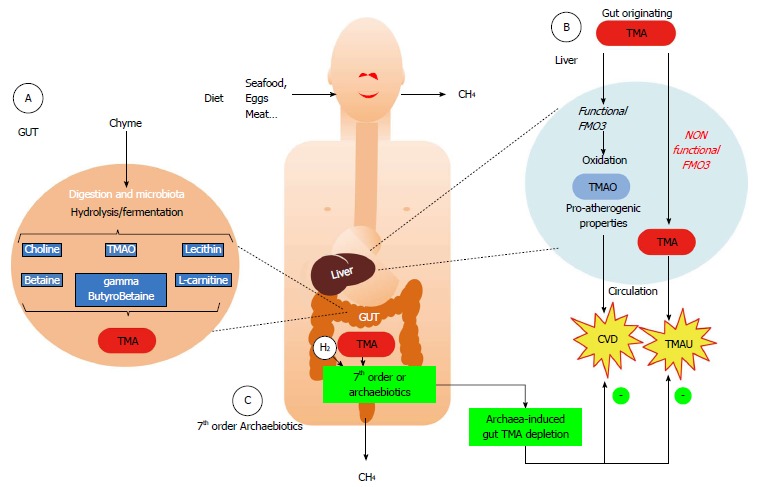
Faith of trimethylamine originating from the gut microbiota metabolism and putative role of representatives of Methanomassiliicoccales. A: Several common foods (containing lecithin/choline, L-carnitine, trimethylamine oxide,…) are metabolized into trimethylamine (TMA) by some bacterial components of the microbiota; B: When the microbiota does not contain any members of the 7th order of methanogens, the gut-originating TMA reaches the liver, where it is oxidized by the flavin-monoxygenase FMO3 into trimethylamine oxide (TMAO) (Blue arrows in B). This odorless molecule has recently been shown to be associated to cardiovascular diseases (CVD) and has pro-atherogenic properties. In some people however, with a deficiency in liver oxidation, the TMA is not or less metabolized (orange arrows in B) and reaches various body fluids, leading to trimethylaminuria (TMAU) or “fish-odor syndrome”; C: When present, either naturally or by supplementation (Archaebiotics), it is hypothesized that members of the 7th order of methanogens, through their up to now unique metabolism in the gut, might decrease the concentration of TMA by converting it into methane, therefore leading to an archaea-mediated TMA depletion before it can reach the liver. This could therefore be a convenient way to prevent CVD or limit TMAU.
Large environmental distribution with a clade evolution dedicated to the GIT?
Sequence database mining associated to phylogenetic studies have now shown that this order of methanogens has a large environmental distribution[28,29,89]. It seems that this order may be divided in at least 3 different evolutionary clades[89], two of them being represented in Figure 1. One is almost only composed of GIT members[28] and is proposed to form a distinct family among this order to which we will refer therein as Ca. Methanomethylophilaceae. A second clade, more distantly related, is exclusively composed of sequences retrieved from other environments and is to date, without any genome sequences and/or isolated representative. At last, there is a third clade, with an intermediary phylogenetic position between these two formers containing sequences from both GIT and non-GIT environments[89]: even if retrieved from human feces, M. luminyensis and Ca. M. intestinalis belong to this latter clade (family Methanomassiliicoccaceae among the Methanomassiliicoccales order), mostly composed of sequences derived from sediments and soils[89].
Ca. M. alvus belongs to the clade mainly composed of sequences from GIT, and therefore its genome could be of particular interest to understand the role of the Methanomassiliicoccales in the human (and other animals) GIT. It reveals typical adaptations to the GIT which are not present in the others. For example, the genome of Ca. M. alvus encodes a bile salts hydrolase (BSH) that confers resistance to the antimicrobial properties of bile salts in the digestive environment[96] (Table 1), absent from M. luminyensis and Ca. M. intestinalis. We report here the presence of the BSH genes in the genomes of the 20 strains of M. smithii isolated and sequenced by Hansen et al[55] and in the genome of M. arboriphilus[83] (Table 1). This gene is poorly represented in other archaeal lineages with the notable exception of a distant homolog present in numerous Halobacteriales genomes[98].
A comparative genome analysis has been realized for these human digestive representatives of Methanomassiliicoccales and revealed other significant differences between the Methanomassiliicoccus spp. and Ca. M. alvus[99]. Thus, Methanomassiliicoccus spp. could be more recent human GIT inhabitants, and/or accidental ones, giving them a different role in the human GIT, positive or negative. Therefore, obtaining other human and environmental strains coupled with epidemiological studies will allow learning more about them.
RELATIONSHIPS OF METHANOGENS WITH PHYSIOLOGICAL STATES OF THE HUMAN HOST
During the last three decades, studies have tried to determine if any relationship existed between the presence of methanogens and some human diseases. These possible diseases are indicated in the Figure 2, while the Table 2 summarizes some of the putative pros and cons of the role of archaea from available data on humans concerning various digestive pathologies (colorectal cancers CRC, inflammatory bowel disease IBD, irritable bowel syndrome IBS, obesity, constipation). To date, no direct link with mechanisms has been established between these diseases and methanogens, and contradictory results were found for example concerning CRC. However, it was proposed that the presence of M. smithii promotes calories intake from diet: in an animal model, M. smithii was shown to influence the metabolism of one of the most important saccharolytic microbiota species, B. thetaiotaomicron[53]. This phenomenon was observed in germ-free mice inoculated with M. smithii and B. thetaiotaomicron, together or separately[53]: the co-implementation led to a mutual gain with an increase in energy retrieval (hydrolysis and fermentation), and importantly, an increased lipogenesis and host fat. Very recently, M. stadtmanae has been shown to be more prevalent in IBD patients[100]. Interestingly, M. stadtmanae stimulates in vitro TNF production, an inflammatory cytokine, from peripheral blood mononuclear cells of healthy patients, while patients affected by IBD have a higher circulating IgG response to M. stadtmanae[100]. At last, methanogens seem also linked to a pathology of the oral cavity, the periodontitis, in which the severity corroborates to the abundance of methanogens[24] (Table 2).
Table 2.
Possible association between several human diseases and methanogens
| Pathology | Pros for a role or an association | Cons |
| Colorectal cancer | 80% of colorectal cancer patients are methane producers[127] | No significant relation with the amount of breath methane[48,71,129-131] |
| Methane production increases with the severity of colorectal cancer[128] | Native Africans with high level of methanogenic archaea are less susceptible to sporadic colorectal cancer[48] | |
| Obesity | Higher levels of methanogens in obese[132] | Decreased proportion of M. smithii in obese[72,73,133] |
| Anorexia | Higher levels of M. smithii in anorexic people (adaption to diet restriction?)[134] | |
| IBD | Lower proportion of methane-producers/methanogen carriers in patients with IBD[104,135] | |
| IBS | Correlation with higher production of methane (also compared to IBD patients)[136] | No significant difference in methane production with controls[137] |
| Diverticulosis | Subjects with diverticulosis often with high level of methanogens[45] | |
| Constipation | Association with chronic constipation/transit time[138,139] | No significant difference between constipated children and controls[144] |
| Level of methane significantly associated with the severity of constipation[136,140] | ||
| Association with IBS constipation/increase of transit time[141-143] | ||
| Periodontitis | Possible pathogenic role and increased proportions of hydrogenotrophs and methanogens in severe cases[24,145,146] | |
| After therapy, decrease in the prevalence of archaea[147] |
IBD: Inflammatory bowel disease; IBS: Irritable bowel disease.
Higher prevalence and diversity of methanogens with age
Among all the various relationships sought between the occurrence of methanogens and the physiological state of human host cohorts, the age relationship was the most consistently observed. As highlighted by the literature review of Levitt et al[23] (2006) on studies based on the methane breath test, the proportion of people excreting methane is higher among adults than among children with virtually no methane producers under 5-year-old[16,101,102]. Such age-related increase of the proportion of methane producers was less clearly observed during adulthood with more studies reporting no statistical change (see for example[23,103,104]). Recently, an association between age (and also sex) and breath methane was described in the German population (428 subjects, age range: 4-95)[105]. Studies based on molecular approaches support this relationship and provide supplemental information through the lower detection limit of the methanogens and the access to their diversity. Significantly lower proportion of methanogens among babies[27] and children[44] than among adults were also reported based on PCR approaches. Time monitoring of the gut microbiota composition in babies over 2 years have shown that gut associated methanogens can be present in the early life, with 7 on 14 babies positive for methanogens[106]. However, their abundance was low (103-106 copies/g of stool), likely below the necessary threshold for CH4 detection in the breath and they were only temporarily present during the first weeks of life[107]. This transient state contrast with reports of stable populations of methanogens in adults sampled at several time points[55,107,108]. The prevalence of M. smithii has been linked to bacterial genera[79] and gut enterotypes[65] which are also considered stables in adults, suggesting the very specific competitive and symbiotic interactions of methanogens with other member of the microbiota. Due to the fast evolving composition of the microbiota of babies, the occurrence of the favorable ecological conditions for the colonization of M. smithii could only be temporary. Another non-exclusive reason of this low prevalence and transient state could rely on the high transit time in babies.
One study from our group[27] has reported a not significant trend of increased prevalence among elderly adults (79.4 ± 6.7 years) in comparison to younger adults (33.0 ± 6.4 years) which agrees with previous reports based on the methane breath test[27]. Importantly this study has pointed an increase of the diversity of the methanogens. Indeed, several distant phylotypes affiliated to the Methanomassiliicoccales were more frequently observed in the elderlies (40% of those tested on a small panel of 20 patients) and far less in adults (10%), and almost absent in the newborns (only 1 positive out of 17 tested)[27]. Accordingly, a similar age-related increase of M. luminyensis and possibly Ca. M. intestinalis was subsequently observed based on qPCR[74]. The increase of diversity between adults and elderlies remains unexplained, even if it is now well known that the gut microbiota of the elderlies is different from the adults[109,110] with variations due to diet/nutrition, residence location and the general health of the elderlies[66]. Several hypotheses can be made: first, methanogenic archaea are known to be insensitive to most of the antibiotics used in human health[9,111], mainly due to the composition of their membrane and cell wall, but also to their specificity in their DNA/RNA metabolism. It is therefore tempting to hypothesize that they could be selected by treatments accumulation during the life. Second, methanogenic archaea have relatively long doubling time, and the slower transit time observed with aging may be also a factor of this over representation in the elderlies compared to the adult, and even more compared to newborns and infants under two years[109,112]. Collectively these studies suggest that the prevalence of methanogens in the population increases rapidly in the first years of life, mainly by the colonization of M. smithii, and that the last part of life is characterized by an increase of the diversity through the colonization by various representatives of the Methanomassiliicoccales. Another hypothesis relies not directly to aging, but to the different generations between elderlies compared to present adults: exposure to farm animals, germs carried by food and diet habits, etc… have evolved over the years, and it may be possible that present adults have less chance to be inoculated than have their parents. Studies on various ethnical/geographical populations would provide interesting complementary data.
TMA-based methanogenesis, a beneficial incidence in human health?
Due to their very specific metabolism leading to TMA depletion for methanogenesis, the currently identified representatives of the Methanomassiliicoccales might have a positive effect on health. TMA is responsible of the rotting fish characteristic odor, to which our olfaction is extremely sensitive. This prevents us to ingest spoiled fishes and seafood. TMA is however a product of diet metabolism by the intestinal microbiota, usually associated to the consumption of foods containing either choline and lecithin (such as eggs, beef, dairy products, vegetables…), L-carnitine (meat, energy drinks…) or TMAO (trimethylamine-N-oxide in seafood, fish…), as exemplified in Figure 3A[113-116]. TMA is then absorbed in the intestine, goes to the liver where it is oxidized to the odorless TMAO via a flavin monooxygenase (mainly the FMO3), which reaches the circulation before being eliminated in the urine, leading to a TMAO vs (TMA+TMAO) urine ratio superior to 92% (Figure 3B). It is likely that TMA is also metabolized into methane when archaea from the Methanomassiliicoccales are present, preferentially to methanol for which inter-microbial species competition occurs in the intestine (i.e., with M. stadtmanae, and possibly M. smithii). Therefore, when present, the Methanomassiliicoccales could lower the intestinal TMA concentration, leading to lowered TMA intestinal absorption, and in consequence to a lower TMAO plasmatic level after liver conversion.
The gut origin of TMA is involved at least in two human diseases. The team of the Pr Stanley Hazen in Cleveland recently demonstrated an association between plasma levels of TMAO and the risk of myocardial infarction, stroke, and death within the next three years. Thus, in 2595 patients, plasma levels of L- carnitine, related to high plasmatic TMAO levels is correlated to a high incidence of cardiovascular events, with or without adjustments to the traditional risk factors[115]. This is identical with plasma levels of choline, betaine and TMAO[117], the upper quartile of plasmatic TMAO concentration increasing the major adverse cardiovascular events to 2.5[113]. These prognostic values of choline and betaine are only effective when concomitant to an increase in plasma TMAO level, i.e. from TMA production in the gut[118]. Plasma TMAO seems however much more than a plasma marker: it seems to promote foam cell formation by increasing the expression of receptors involved in macrophage uptake of circulating cholesterol (scavenger receptors CD36 and SR-A1), and conversely by reducing the reverse cholesterol transport and its elimination as bile acids from the intestine[115]. Therefore, limiting TMA production and absorption in the intestine is a mean to reduce plasmatic TMAO level, and its deleterious effects on blood vessels, which is likely to what the Methanomassiliicoccales contributes naturally when present.
There is also another human pathology, the trimethylaminuria[119-121] (TMAU) which involves the gut microbiota TMA production. After intestinal TMA production and absorption, some people are unable to oxidize it into TMAO in their liver, due to a genetic or acquired liver deficiency affecting the FMO3 functionalities. In this case, unmetabolized TMA reaches the general circulation and is distributed throughout the body fluids. It is next excreted through sweat, exhalation and urine [where TMAO vs (TMAO+TMA) ratio is then below 92%, with a decrease depending on the severity of the FMO3 deficiency]. Thus, TMAU causes an unpleasant odor of rotten fish to the affected person, due to its presence in sweat and breath, for which the human olfactory system is very sensitive. This causes an extremely debilitating disease, at the social and psychological level[120], for which no real treatments are proposed: the affected people often empirically adapt their diet in order to limit gut TMA production, by renouncing to eggs, red meat, beans,…. Still poorly recognized and diagnosed, its prevalence remains unknown, but some FMO3 encoding gene mutations could affect about 1% of the population[122]. Here again, the unique metabolic properties of the Methanomassiliicoccales could limit gut TMA concentration, without the bad side effects of limiting choline inputs by diet.
We then suppose the physiological positive impact that may have the Methanomassiliicoccales as a TMA depleting agent in the intestinal microbiota, leading to the conversion of TMA into CH4, an odorless and inert gas before it reaches the liver for eventual conversion. This metabolism is so far only realized by a few methanogens, and among gut-naturally occurring methanogens, only members of the Methanomassiliicoccales. What can be inherently possible in the gut microbiota containing the methanogenic Methanomassiliicoccales could be reproduced by supplementing the gut microbiota by this kind of microorganism. Accordingly, the 7th order of methanogens could therefore be used as a probiotics to prevent cardiovascular disease, acting by reducing the amount of the pro-atherogenic TMAO produced by the liver and therefore its plasma level. A supply with Methanomassiliicoccales could also be a simple way to limit TMAU, by reducing the TMA concentration in the gut and in consequence, its olfactory impact on the body. In both pathology cases, it will be possible to avoid drastic diets (as it is usually done for TMAU patients), thus limiting nutritional deficiencies risks. This concept of probiotics has recently been developed under the name of “archaebiotics” as to show the innovative and the positive aspect that we expect from archaeal strains to human health[96]. However, it remains to be shown that this metabolic activity is effective in the human GIT (as it is already done in the rumen[97]) without causing inconvenience or bad side effects. Preliminary in vitro studies on human microbiota[123,124] are therefore necessary to determine which representatives of the Methanomassiliicoccales could be supplied and further tested for their impact on other gut microbiota components, as it was recently done for the gut methanogen M. smithii (W. Tottey, personal communication). Using exhaustive microbiota analysis tools applicable to in-vitro systems such as Next Generation Sequencing[76] or DNA phylogenetic microarrays like the HuGChip[125], an in-vitro selection of the best strains should be possible, also based on functional analysis (e.g., transcriptomics and metabolomics), that would be then followed by rationale tests in animals/humanized gnotobiotic mice, before clinical research.
CONCLUSION
The diversity of the archaeal component of the human gut microbiota has been greatly renewed during these last few years and will likely continue to grow: difficulties to culture them in the laboratory has been counterpart by the generalization of Next Generation Sequencing technologies, used mainly in 16S diversity studies and metagenomics. Links with diseases/human health, if any, will therefore be established in the next years, considering the archaea as a diverse population, as it is for bacteria, with more discriminant methods than methane in breath for methanogens. The recently discovered 7th order of methanogens illustrates these points: the presence of different phylogenetic clusters may signify different, even opposite, incidence for the health, and therefore highly-discriminant tools are needed. This order was named Methanoplasmatales, and then Methanomassiliicoccales (according to the first described species)[28,29], the current official name[30]. Within, and besides Methanomassiliicoccaceae, we propose the family name of Methanomethylophilaceae for the clade encompassing the digestive species of the Candidatus genus Methanomethylophilus. This would highlight the unique dependence to methylated compounds among methanogens, for the whole order (methane-, methanogenic; methylophilus, that likes methyl compounds). Genomics has strongly helped in the determination of their putative roles in the GIT, and indicated their unique metabolic properties before experimental proofs. This shows the potent physiological importance of the still poorly known domain Archaea in the human gut, and the role of its members, beneficial or deleterious, remaining to be determined. It may also provide, as it is for the Methanomassiliicoccales, new directions in probiotics design.
Footnotes
Supported by PhD Scholarship from the French “Ministère de l’Enseignement Supérieur et de la Recherche” (To Nadia Gaci); Science Foundation Ireland through a postdoctoral grant of the Alimentary Pharmabiotic Centre (to Guillaume Borrel); PhD Scholarship of the European Union (UE) and the Auvergne Council (FEDER) (to William Tottey); Science Foundation Ireland through a Principal Investigator award and by an FHRI award to the ELDERMET project by the Department Agriculture, Fisheries and Marine of the Government of Ireland (to Paul W O’Toole)
P- Reviewer: Hold GL, Liu HK, Martinez-Zorzano VS S- Editor: Ma YJ L- Editor: A E- Editor: Liu XM
References
- 1.DeLong EF. Everything in moderation: archaea as ‘non-extremophiles’. Curr Opin Genet Dev. 1998;8:649–654. doi: 10.1016/s0959-437x(98)80032-4. [DOI] [PubMed] [Google Scholar]
- 2.Brochier-Armanet C, Boussau B, Gribaldo S, Forterre P. Mesophilic Crenarchaeota: proposal for a third archaeal phylum, the Thaumarchaeota. Nat Rev Microbiol. 2008;6:245–252. doi: 10.1038/nrmicro1852. [DOI] [PubMed] [Google Scholar]
- 3.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA. 1990;87:4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williams TA, Foster PG, Cox CJ, Embley TM. An archaeal origin of eukaryotes supports only two primary domains of life. Nature. 2013;504:231–236. doi: 10.1038/nature12779. [DOI] [PubMed] [Google Scholar]
- 5.Forterre P. The common ancestor of archaea and eukarya was not an archaeon. Archaea. 2013;2013:372396. doi: 10.1155/2013/372396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pereira SL, Reeve JN. Histones and nucleosomes in Archaea and Eukarya: a comparative analysis. Extremophiles. 1998;2:141–148. doi: 10.1007/s007920050053. [DOI] [PubMed] [Google Scholar]
- 7.Koga Y, Morii H. Biosynthesis of ether-type polar lipids in archaea and evolutionary considerations. Microbiol Mol Biol Rev. 2007;71:97–120. doi: 10.1128/MMBR.00033-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peretó J, López-García P, Moreira D. Ancestral lipid biosynthesis and early membrane evolution. Trends Biochem Sci. 2004;29:469–477. doi: 10.1016/j.tibs.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 9.Kandler O, König H. Cell wall polymers in Archaea (Archaebacteria) Cell Mol Life Sci. 1998;54:305–308. doi: 10.1007/s000180050156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borrel G, Jézéquel D, Biderre-Petit C, Morel-Desrosiers N, Morel JP, Peyret P, Fonty G, Lehours AC. Production and consumption of methane in freshwater lake ecosystems. Res Microbiol. 2011;162:832–847. doi: 10.1016/j.resmic.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 11.Reeburgh WS. Oceanic methane biogeochemistry. Chem Rev. 2007;107:486–513. doi: 10.1021/cr050362v. [DOI] [PubMed] [Google Scholar]
- 12.Bridgham SD, Cadillo-Quiroz H, Keller JK, Zhuang Q. Methane emissions from wetlands: biogeochemical, microbial, and modeling perspectives from local to global scales. Glob Chang Biol. 2013;19:1325–1346. doi: 10.1111/gcb.12131. [DOI] [PubMed] [Google Scholar]
- 13.Angel R, Claus P, Conrad R. Methanogenic archaea are globally ubiquitous in aerated soils and become active under wet anoxic conditions. ISME J. 2012;6:847–862. doi: 10.1038/ismej.2011.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hackstein JH, van Alen TA. Fecal methanogens and vertebrate evolution. Evolution. 1996;50:559–572. doi: 10.1111/j.1558-5646.1996.tb03868.x. [DOI] [PubMed] [Google Scholar]
- 15.Ohkuma M, Noda S, Kudo T. Phylogenetic relationships of symbiotic methanogens in diverse termites. FEMS Microbiol Lett. 1999;171:147–153. doi: 10.1111/j.1574-6968.1999.tb13425.x. [DOI] [PubMed] [Google Scholar]
- 16.Bond JH, Engel RR, Levitt MD. Factors influencing pulmonary methane excretion in man. An indirect method of studying the in situ metabolism of the methane-producing colonic bacteria. J Exp Med. 1971;133:572–588. doi: 10.1084/jem.133.3.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakamura N, Lin HC, McSweeney CS, Mackie RI, Gaskins HR. Mechanisms of microbial hydrogen disposal in the human colon and implications for health and disease. Annu Rev Food Sci Technol. 2010;1:363–395. doi: 10.1146/annurev.food.102308.124101. [DOI] [PubMed] [Google Scholar]
- 18.Saengkerdsub S, Ricke SC. Ecology and characteristics of methanogenic archaea in animals and humans. Crit Rev Microbiol. 2014;40:97–116. doi: 10.3109/1040841X.2013.763220. [DOI] [PubMed] [Google Scholar]
- 19.Weiland P. Biogas production: current state and perspectives. Appl Microbiol Biotechnol. 2010;85:849–860. doi: 10.1007/s00253-009-2246-7. [DOI] [PubMed] [Google Scholar]
- 20.Conrad R. The global methane cycle: recent advances in understanding the microbial processes involved. Environ Microbiol Rep. 2009;1:285–292. doi: 10.1111/j.1758-2229.2009.00038.x. [DOI] [PubMed] [Google Scholar]
- 21.Moraes LE, Strathe AB, Fadel JG, Casper DP, Kebreab E. Prediction of enteric methane emissions from cattle. Glob Chang Biol. 2014;20:2140–2148. doi: 10.1111/gcb.12471. [DOI] [PubMed] [Google Scholar]
- 22.Eckburg PB, Lepp PW, Relman DA. Archaea and their potential role in human disease. Infect Immun. 2003;71:591–596. doi: 10.1128/IAI.71.2.591-596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levitt MD, Furne JK, Kuskowski M, Ruddy J. Stability of human methanogenic flora over 35 years and a review of insights obtained from breath methane measurements. Clin Gastroenterol Hepatol. 2006;4:123–129. doi: 10.1016/j.cgh.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 24.Lepp PW, Brinig MM, Ouverney CC, Palm K, Armitage GC, Relman DA. Methanogenic Archaea and human periodontal disease. Proc Natl Acad Sci USA. 2004;101:6176–6181. doi: 10.1073/pnas.0308766101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horz HP, Conrads G. Methanogenic Archaea and oral infections - ways to unravel the black box. J Oral Microbiol. 2011;3 doi: 10.3402/jom.v3i0.5940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mihajlovski A, Alric M, Brugère JF. A putative new order of methanogenic Archaea inhabiting the human gut, as revealed by molecular analyses of the mcrA gene. Res Microbiol. 2008;159:516–521. doi: 10.1016/j.resmic.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 27.Mihajlovski A, Doré J, Levenez F, Alric M, Brugère JF. Molecular evaluation of the human gut methanogenic archaeal microbiota reveals an age-associated increase of the diversity. Environ Microbiol Rep. 2010;2:272–280. doi: 10.1111/j.1758-2229.2009.00116.x. [DOI] [PubMed] [Google Scholar]
- 28.Paul K, Nonoh JO, Mikulski L, Brune A. “Methanoplasmatales,” Thermoplasmatales-related archaea in termite guts and other environments, are the seventh order of methanogens. Appl Environ Microbiol. 2012;78:8245–8253. doi: 10.1128/AEM.02193-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iino T, Tamaki H, Tamazawa S, Ueno Y, Ohkuma M, Suzuki K, Igarashi Y, Haruta S. Candidatus Methanogranum caenicola: a novel methanogen from the anaerobic digested sludge, and proposal of Methanomassiliicoccaceae fam. nov. and Methanomassiliicoccales ord. nov., for a methanogenic lineage of the class Thermoplasmata. Microbes Environ. 2013;28:244–250. doi: 10.1264/jsme2.ME12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol. 2006;56:1–6. doi: 10.1099/ijs.0.64188-0. [DOI] [PubMed] [Google Scholar]
- 31.Garcia JL, Patel BK, Ollivier B. Taxonomic, phylogenetic, and ecological diversity of methanogenic Archaea. Anaerobe. 2000;6:205–226. doi: 10.1006/anae.2000.0345. [DOI] [PubMed] [Google Scholar]
- 32.Liu Y, Whitman WB. Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. Ann N Y Acad Sci. 2008;1125:171–189. doi: 10.1196/annals.1419.019. [DOI] [PubMed] [Google Scholar]
- 33.Ferry JG. Enzymology of one-carbon metabolism in methanogenic pathways. FEMS Microbiol Rev. 1999;23:13–38. doi: 10.1111/j.1574-6976.1999.tb00390.x. [DOI] [PubMed] [Google Scholar]
- 34.Whiticar MJ, Faber E, Schoell M. Biogenic methane formation in marine and freshwater environments: CO2 reduction vs. acetate fermentation-Isotope evidence. Geochim Cosmochim Acta. 1986;50:693–709. [Google Scholar]
- 35.Friedrich MW. Methyl-coenzyme M reductase genes: unique functional markers for methanogenic and anaerobic methane-oxidizing Archaea. Methods Enzymol. 2005;397:428–442. doi: 10.1016/S0076-6879(05)97026-2. [DOI] [PubMed] [Google Scholar]
- 36.Luton PE, Wayne JM, Sharp RJ, Riley PW. The mcrA gene as an alternative to 16S rRNA in the phylogenetic analysis of methanogen populations in landfill. Microbiology. 2002;148:3521–3530. doi: 10.1099/00221287-148-11-3521. [DOI] [PubMed] [Google Scholar]
- 37.Scanlan PD, Shanahan F, Marchesi JR. Human methanogen diversity and incidence in healthy and diseased colonic groups using mcrA gene analysis. BMC Microbiol. 2008;8:79. doi: 10.1186/1471-2180-8-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Horz HP, Seyfarth I, Conrads G. McrA and 16S rRNA gene analysis suggests a novel lineage of Archaea phylogenetically affiliated with Thermoplasmatales in human subgingival plaque. Anaerobe. 2012;18:373–377. doi: 10.1016/j.anaerobe.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 39.Nava GM, Carbonero F, Croix JA, Greenberg E, Gaskins HR. Abundance and diversity of mucosa-associated hydrogenotrophic microbes in the healthy human colon. ISME J. 2012;6:57–70. doi: 10.1038/ismej.2011.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Janssen PH, Kirs M. Structure of the archaeal community of the rumen. Appl Environ Microbiol. 2008;74:3619–3625. doi: 10.1128/AEM.02812-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Angenent LT, Karim K, Al-Dahhan MH, Wrenn BA, Domíguez-Espinosa R. Production of bioenergy and biochemicals from industrial and agricultural wastewater. Trends Biotechnol. 2004;22:477–485. doi: 10.1016/j.tibtech.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 42.Bernalier-Donadille A. [Fermentative metabolism by the human gut microbiota] Gastroenterol Clin Biol. 2010;34 Suppl 1:S16–S22. doi: 10.1016/S0399-8320(10)70016-6. [DOI] [PubMed] [Google Scholar]
- 43.Rey FE, Gonzalez MD, Cheng J, Wu M, Ahern PP, Gordon JI. Metabolic niche of a prominent sulfate-reducing human gut bacterium. Proc Natl Acad Sci USA. 2013;110:13582–13587. doi: 10.1073/pnas.1312524110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stewart JA, Chadwick VS, Murray A. Carriage, quantification, and predominance of methanogens and sulfate-reducing bacteria in faecal samples. Lett Appl Microbiol. 2006;43:58–63. doi: 10.1111/j.1472-765X.2006.01906.x. [DOI] [PubMed] [Google Scholar]
- 45.Weaver GA, Krause JA, Miller TL, Wolin MJ. Incidence of methanogenic bacteria in a sigmoidoscopy population: an association of methanogenic bacteria and diverticulosis. Gut. 1986;27:698–704. doi: 10.1136/gut.27.6.698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O’Keefe SJ, O’Keefe EA, Burke E, Roberts P, Lavender R, Kemp T. Milk-induced malabsorption in malnourished African patients. Am J Clin Nutr. 1991;54:130–135. doi: 10.1093/ajcn/54.1.130. [DOI] [PubMed] [Google Scholar]
- 47.Hudson MJ, Tomkins AM, Wiggins HS, Drasar BS. Breath methane excretion and intestinal methanogenesis in children and adults in rural Nigeria. Scand J Gastroenterol. 1993;28:993–998. doi: 10.3109/00365529309098298. [DOI] [PubMed] [Google Scholar]
- 48.Segal I, Walker AR, Lord S, Cummings JH. Breath methane and large bowel cancer risk in contrasting African populations. Gut. 1988;29:608–613. doi: 10.1136/gut.29.5.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miller TL, Wolin MJ, Conway de Macario E, Macario AJ. Isolation of Methanobrevibacter smithii from human feces. Appl Environ Microbiol. 1982;43:227–232. doi: 10.1128/aem.43.1.227-232.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miller TL, Wolin MJ. Methanosphaera stadtmaniae gen. nov., sp. nov.: a species that forms methane by reducing methanol with hydrogen. Arch Microbiol. 1985;141:116–122. doi: 10.1007/BF00423270. [DOI] [PubMed] [Google Scholar]
- 51.Pochart P, Lémann F, Flourié B, Pellier P, Goderel I, Rambaud JC. Pyxigraphic sampling to enumerate methanogens and anaerobes in the right colon of healthy humans. Gastroenterology. 1993;105:1281–1285. doi: 10.1016/0016-5085(93)90129-z. [DOI] [PubMed] [Google Scholar]
- 52.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Samuel BS, Gordon JI. A humanized gnotobiotic mouse model of host-archaeal-bacterial mutualism. Proc Natl Acad Sci USA. 2006;103:10011–10016. doi: 10.1073/pnas.0602187103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Samuel BS, Hansen EE, Manchester JK, Coutinho PM, Henrissat B, Fulton R, Latreille P, Kim K, Wilson RK, Gordon JI. Genomic and metabolic adaptations of Methanobrevibacter smithii to the human gut. Proc Natl Acad Sci USA. 2007;104:10643–10648. doi: 10.1073/pnas.0704189104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hansen EE, Lozupone CA, Rey FE, Wu M, Guruge JL, Narra A, Goodfellow J, Zaneveld JR, McDonald DT, Goodrich JA, et al. Pan-genome of the dominant human gut-associated archaeon, Methanobrevibacter smithii, studied in twins. Proc Natl Acad Sci USA. 2011;108 Suppl 1:4599–4606. doi: 10.1073/pnas.1000071108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lurie-Weinberger MN, Peeri M, Gophna U. Contribution of lateral gene transfer to the gene repertoire of a gut-adapted methanogen. Genomics. 2012;99:52–58. doi: 10.1016/j.ygeno.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 57.Lurie-Weinberger MN, Peeri M, Tuller T, Gophna U. Extensive Inter-Domain Lateral Gene Transfer in the Evolution of the Human Commensal Methanosphaera stadtmanae. Front Genet. 2012;3:182. doi: 10.3389/fgene.2012.00182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fricke WF, Seedorf H, Henne A, Krüer M, Liesegang H, Hedderich R, Gottschalk G, Thauer RK. The genome sequence of Methanosphaera stadtmanae reveals why this human intestinal archaeon is restricted to methanol and H2 for methane formation and ATP synthesis. J Bacteriol. 2006;188:642–658. doi: 10.1128/JB.188.2.642-658.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jones BV, Begley M, Hill C, Gahan CG, Marchesi JR. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc Natl Acad Sci USA. 2008;105:13580–13585. doi: 10.1073/pnas.0804437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kulik EM, Sandmeier H, Hinni K, Meyer J. Identification of archaeal rDNA from subgingival dental plaque by PCR amplification and sequence analysis. FEMS Microbiol Lett. 2001;196:129–133. doi: 10.1111/j.1574-6968.2001.tb10553.x. [DOI] [PubMed] [Google Scholar]
- 61.Belay N, Mukhopadhyay B, Conway de Macario E, Galask R, Daniels L. Methanogenic bacteria in human vaginal samples. J Clin Microbiol. 1990;28:1666–1668. doi: 10.1128/jcm.28.7.1666-1668.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ferrari A, Brusa T, Rutili A, Canzi E, Biavati B. Isolation and characterization of Methanobrevibacter oralis sp. nov. Curr Microbiol. 1994;29:7–12. [Google Scholar]
- 63.Khelaifia S, Garibal M, Robert C, Raoult D, Drancourt M. Draft Genome Sequencing of Methanobrevibacter oralis Strain JMR01, Isolated from the Human Intestinal Microbiota. Genome Announc. 2014;2:e00073–14. doi: 10.1128/genomeA.00073-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dridi B, Henry M, El Khéchine A, Raoult D, Drancourt M. High prevalence of Methanobrevibacter smithii and Methanosphaera stadtmanae detected in the human gut using an improved DNA detection protocol. PLoS One. 2009;4:e7063. doi: 10.1371/journal.pone.0007063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto JM, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Claesson MJ, Jeffery IB, Conde S, Power SE, O’Connor EM, Cusack S, Harris HM, Coakley M, Lakshminarayanan B, O’Sullivan O, et al. Gut microbiota composition correlates with diet and health in the elderly. Nature. 2012;488:178–184. doi: 10.1038/nature11319. [DOI] [PubMed] [Google Scholar]
- 67.Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Liggett CM, Nelson KE. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tyakht AV, Kostryukova ES, Popenko AS, Belenikin MS, Pavlenko AV, Larin AK, Karpova IY, Selezneva OV, Semashko TA, Ospanova EA, et al. Human gut microbiota community structures in urban and rural populations in Russia. Nat Commun. 2013;4:2469. doi: 10.1038/ncomms3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fernandes J, Wang A, Su W, Rozenbloom SR, Taibi A, Comelli EM, Wolever TM. Age, dietary fiber, breath methane, and fecal short chain fatty acids are interrelated in Archaea-positive humans. J Nutr. 2013;143:1269–1275. doi: 10.3945/jn.112.170894. [DOI] [PubMed] [Google Scholar]
- 71.O’Keefe SJ, Chung D, Mahmoud N, Sepulveda AR, Manafe M, Arch J, Adada H, van der Merwe T. Why do African Americans get more colon cancer than Native Africans? J Nutr. 2007;137:175S–182S. doi: 10.1093/jn/137.1.175S. [DOI] [PubMed] [Google Scholar]
- 72.Million M, Maraninchi M, Henry M, Armougom F, Richet H, Carrieri P, Valero R, Raccah D, Vialettes B, Raoult D. Obesity-associated gut microbiota is enriched in Lactobacillus reuteri and depleted in Bifidobacterium animalis and Methanobrevibacter smithii. Int J Obes (Lond) 2012;36:817–825. doi: 10.1038/ijo.2011.153. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 73.Million M, Angelakis E, Maraninchi M, Henry M, Giorgi R, Valero R, Vialettes B, Raoult D. Correlation between body mass index and gut concentrations of Lactobacillus reuteri, Bifidobacterium animalis, Methanobrevibacter smithii and Escherichia coli. Int J Obes (Lond) 2013;37:1460–1466. doi: 10.1038/ijo.2013.20. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 74.Dridi B, Henry M, Richet H, Raoult D, Drancourt M. Age-related prevalence of Methanomassiliicoccus luminyensis in the human gut microbiome. APMIS. 2012;120:773–777. doi: 10.1111/j.1600-0463.2012.02899.x. [DOI] [PubMed] [Google Scholar]
- 75.Brugère JF, Mihajlovski A, Missaoui M, Peyret P. Tools for stools: the challenge of assessing human intestinal microbiota using molecular diagnostics. Expert Rev Mol Diagn. 2009;9:353–365. doi: 10.1586/erm.09.16. [DOI] [PubMed] [Google Scholar]
- 76.Claesson MJ, Wang Q, O’Sullivan O, Greene-Diniz R, Cole JR, Ross RP, O’Toole PW. Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucleic Acids Res. 2010;38:e200. doi: 10.1093/nar/gkq873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rieu-Lesme F, Delbès C, Sollelis L. Recovery of partial 16S rDNA sequences suggests the presence of Crenarchaeota in the human digestive ecosystem. Curr Microbiol. 2005;51:317–321. doi: 10.1007/s00284-005-0036-8. [DOI] [PubMed] [Google Scholar]
- 78.Hatzenpichler R, Lebedeva EV, Spieck E, Stoecker K, Richter A, Daims H, Wagner M. A moderately thermophilic ammonia-oxidizing crenarchaeote from a hot spring. Proc Natl Acad Sci USA. 2008;105:2134–2139. doi: 10.1073/pnas.0708857105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hoffmann C, Dollive S, Grunberg S, Chen J, Li H, Wu GD, Lewis JD, Bushman FD. Archaea and fungi of the human gut microbiome: correlations with diet and bacterial residents. PLoS One. 2013;8:e66019. doi: 10.1371/journal.pone.0066019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hatzenpichler R. Diversity, physiology, and niche differentiation of ammonia-oxidizing archaea. Appl Environ Microbiol. 2012;78:7501–7510. doi: 10.1128/AEM.01960-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nam YD, Chang HW, Kim KH, Roh SW, Kim MS, Jung MJ, Lee SW, Kim JY, Yoon JH, Bae JW. Bacterial, archaeal, and eukaryal diversity in the intestines of Korean people. J Microbiol. 2008;46:491–501. doi: 10.1007/s12275-008-0199-7. [DOI] [PubMed] [Google Scholar]
- 82.Oxley AP, Lanfranconi MP, Würdemann D, Ott S, Schreiber S, McGenity TJ, Timmis KN, Nogales B. Halophilic archaea in the human intestinal mucosa. Environ Microbiol. 2010;12:2398–2410. doi: 10.1111/j.1462-2920.2010.02212.x. [DOI] [PubMed] [Google Scholar]
- 83.Khelaifia S, Garibal M, Robert C, Raoult D, Drancourt M. Draft Genome Sequence of a Human-Associated Isolate of Methanobrevibacter arboriphilicus, the Lowest-G+C-Content Archaeon. Genome Announc. 2014;2 doi: 10.1128/genomeA.01181-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nguyen-Hieu T, Khelaifia S, Aboudharam G, Drancourt M. Methanogenic archaea in subgingival sites: a review. APMIS. 2013;121:467–477. doi: 10.1111/apm.12015. [DOI] [PubMed] [Google Scholar]
- 85.Dridi B, Fardeau ML, Ollivier B, Raoult D, Drancourt M. Methanomassiliicoccus luminyensis gen. nov., sp. nov., a methanogenic archaeon isolated from human faeces. Int J Syst Evol Microbiol. 2012;62:1902–1907. doi: 10.1099/ijs.0.033712-0. [DOI] [PubMed] [Google Scholar]
- 86.Borrel G, Harris HM, Tottey W, Mihajlovski A, Parisot N, Peyretaillade E, Peyret P, Gribaldo S, O’Toole PW, Brugère JF. Genome sequence of “Candidatus Methanomethylophilus alvus” Mx1201, a methanogenic archaeon from the human gut belonging to a seventh order of methanogens. J Bacteriol. 2012;194:6944–6945. doi: 10.1128/JB.01867-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Borrel G, Harris HM, Parisot N, Gaci N, Tottey W, Mihajlovski A, Deane J, Gribaldo S, Bardot O, Peyretaillade E, et al. Genome Sequence of “Candidatus Methanomassiliicoccus intestinalis” Issoire-Mx1, a Third Thermoplasmatales-Related Methanogenic Archaeon from Human Feces. Genome Announc. 2013;1:e00453–13. doi: 10.1128/genomeA.00453-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gorlas A, Robert C, Gimenez G, Drancourt M, Raoult D. Complete genome sequence of Methanomassiliicoccus luminyensis, the largest genome of a human-associated Archaea species. J Bacteriol. 2012;194:4745. doi: 10.1128/JB.00956-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Borrel G, O’Toole PW, Harris HM, Peyret P, Brugère JF, Gribaldo S. Phylogenomic data support a seventh order of Methylotrophic methanogens and provide insights into the evolution of Methanogenesis. Genome Biol Evol. 2013;5:1769–1780. doi: 10.1093/gbe/evt128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lapage SP, Sneath PHA, Lessel EF, Skerman VBD, Seeliger HPR, Clark WA, editors . International Code of Nomenclature of Bacteria: Bacteriological Code, 1990 Revision. Washington (DC): ASM Press; 1992. [PubMed] [Google Scholar]
- 91.Li CL, Liu DL, Jiang YT, Zhou YB, Zhang MZ, Jiang W, Liu B, Liang JP. Prevalence and molecular diversity of Archaea in subgingival pockets of periodontitis patients. Oral Microbiol Immunol. 2009;24:343–346. doi: 10.1111/j.1399-302X.2009.00514.x. [DOI] [PubMed] [Google Scholar]
- 92.Horz HP, Conrads G. The discussion goes on: What is the role of Euryarchaeota in humans? Archaea. 2010;2010:967271. doi: 10.1155/2010/967271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Paul L, Ferguson DJ, Krzycki JA. The trimethylamine methyltransferase gene and multiple dimethylamine methyltransferase genes of Methanosarcina barkeri contain in-frame and read-through amber codons. J Bacteriol. 2000;182:2520–2529. doi: 10.1128/jb.182.9.2520-2529.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Srinivasan G, James CM, Krzycki JA. Pyrrolysine encoded by UAG in Archaea: charging of a UAG-decoding specialized tRNA. Science. 2002;296:1459–1462. doi: 10.1126/science.1069588. [DOI] [PubMed] [Google Scholar]
- 95.Borrel G, Gaci N, Peyret P, O’Toole PW, Gribaldo S, Brugère JF. Unique characteristics of the pyrrolysine system in the 7th order of methanogens: implications for the evolution of a genetic code expansion cassette. Archaea. 2014;2014:374146. doi: 10.1155/2014/374146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brugère JF, Borrel G, Gaci N, Tottey W, O’Toole PW, Malpuech-Brugère C. Archaebiotics: proposed therapeutic use of archaea to prevent trimethylaminuria and cardiovascular disease. Gut Microbes. 2014;5:5–10. doi: 10.4161/gmic.26749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Poulsen M, Schwab C, Jensen BB, Engberg RM, Spang A, Canibe N, Højberg O, Milinovich G, Fragner L, Schleper C, et al. Methylotrophic methanogenic Thermoplasmata implicated in reduced methane emissions from bovine rumen. Nat Commun. 2013;4:1428. doi: 10.1038/ncomms2432. [DOI] [PubMed] [Google Scholar]
- 98.Panigrahi P, Sule M, Sharma R, Ramasamy S, Suresh CG. An improved method for specificity annotation shows a distinct evolutionary divergence among the microbial enzymes of the cholylglycine hydrolase family. Microbiology. 2014;160:1162–1174. doi: 10.1099/mic.0.077586-0. [DOI] [PubMed] [Google Scholar]
- 99.Borrel G, Parisot N, Harris HM, Peyretaillade E, Gaci N, Tottey W, Bardot O, Raymann K, Gribaldo S, Peyret P, et al. Comparative genomics highlights the unique biology of Methanomassiliicoccales, a Thermoplasmatales-related seventh order of methanogenic archaea that encodes pyrrolysine. BMC Genomics. 2014;15:679. doi: 10.1186/1471-2164-15-679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Blais Lecours P, Marsolais D, Cormier Y, Berberi M, Haché C, Bourdages R, Duchaine C. Increased prevalence of Methanosphaera stadtmanae in inflammatory bowel diseases. PLoS One. 2014;9:e87734. doi: 10.1371/journal.pone.0087734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Leung DT, Robertshaw AM, Tadesse K. Breath methane excretion in Hong Kong Chinese children. J Pediatr Gastroenterol Nutr. 1992;14:275–278. doi: 10.1097/00005176-199204000-00007. [DOI] [PubMed] [Google Scholar]
- 102.Peled Y, Gilat T, Liberman E, Bujanover Y. The development of methane production in childhood and adolescence. J Pediatr Gastroenterol Nutr. 1985;4:575–579. doi: 10.1097/00005176-198508000-00013. [DOI] [PubMed] [Google Scholar]
- 103.McKay LF, Eastwood MA, Brydon WG. Methane excretion in man--a study of breath, flatus, and faeces. Gut. 1985;26:69–74. doi: 10.1136/gut.26.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Peled Y, Weinberg D, Hallak A, Gilat T. Factors affecting methane production in humans. Gastrointestinal diseases and alterations of colonic flora. Dig Dis Sci. 1987;32:267–271. doi: 10.1007/BF01297052. [DOI] [PubMed] [Google Scholar]
- 105.Polag D, Leiß O, Keppler F. Age dependent breath methane in the German population. Sci Total Environ. 2014;481:582–587. doi: 10.1016/j.scitotenv.2014.02.086. [DOI] [PubMed] [Google Scholar]
- 106.Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO. Development of the human infant intestinal microbiota. PLoS Biol. 2007;5:e177. doi: 10.1371/journal.pbio.0050177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Abell GC, Conlon MA, McOrist AL. Methanogenic archaea in adult human faecal samples are inversely related to butyrate concentration. Microb Ecol Health Dis. 2006;18:154–160. [Google Scholar]
- 108.Miller TL, Wolin MJ. Stability of Methanobrevibacter smithii populations in the microbial flora excreted from the human large bowel. Appl Environ Microbiol. 1983;45:317–318. doi: 10.1128/aem.45.1.317-318.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Woodmansey EJ, McMurdo ME, Macfarlane GT, Macfarlane S. Comparison of compositions and metabolic activities of fecal microbiotas in young adults and in antibiotic-treated and non-antibiotic-treated elderly subjects. Appl Environ Microbiol. 2004;70:6113–6122. doi: 10.1128/AEM.70.10.6113-6122.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Claesson MJ, Cusack S, O’Sullivan O, Greene-Diniz R, de Weerd H, Flannery E, Marchesi JR, Falush D, Dinan T, Fitzgerald G, et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci USA. 2011;108 Suppl 1:4586–4591. doi: 10.1073/pnas.1000097107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dridi B, Fardeau ML, Ollivier B, Raoult D, Drancourt M. The antimicrobial resistance pattern of cultured human methanogens reflects the unique phylogenetic position of archaea. J Antimicrob Chemother. 2011;66:2038–2044. doi: 10.1093/jac/dkr251. [DOI] [PubMed] [Google Scholar]
- 112.Lewis S, Cochrane S. Alteration of sulfate and hydrogen metabolism in the human colon by changing intestinal transit rate. Am J Gastroenterol. 2007;102:624–633. doi: 10.1111/j.1572-0241.2006.01020.x. [DOI] [PubMed] [Google Scholar]
- 113.Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, Wu Y, Hazen SL. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013;368:1575–1584. doi: 10.1056/NEJMoa1109400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zeisel SH, daCosta KA, Youssef M, Hensey S. Conversion of dietary choline to trimethylamine and dimethylamine in rats: dose-response relationship. J Nutr. 1989;119:800–804. doi: 10.1093/jn/119.5.800. [DOI] [PubMed] [Google Scholar]
- 115.Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19:576–585. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhang AQ, Mitchell SC, Smith RL. Dietary precursors of trimethylamine in man: a pilot study. Food Chem Toxicol. 1999;37:515–520. doi: 10.1016/s0278-6915(99)00028-9. [DOI] [PubMed] [Google Scholar]
- 117.Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wang Z, Tang WH, Buffa JA, Fu X, Britt EB, Koeth RA, Levison BS, Fan Y, Wu Y, Hazen SL. Prognostic value of choline and betaine depends on intestinal microbiota-generated metabolite trimethylamine-N-oxide. Eur Heart J. 2014;35:904–910. doi: 10.1093/eurheartj/ehu002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mitchell SC, Smith RL. Trimethylaminuria: the fish malodor syndrome. Drug Metab Dispos. 2001;29:517–521. [PubMed] [Google Scholar]
- 120.Mackay RJ, McEntyre CJ, Henderson C, Lever M, George PM. Trimethylaminuria: causes and diagnosis of a socially distressing condition. Clin Biochem Rev. 2011;32:33–43. [PMC free article] [PubMed] [Google Scholar]
- 121.Spellacy E, Watts RW, Goolamali SK. Trimethylaminuria. J Inherit Metab Dis. 1980;2:85–88. doi: 10.1007/BF01805663. [DOI] [PubMed] [Google Scholar]
- 122.Motika MS, Zhang J, Cashman JR. Flavin-containing monooxygenase 3 and human disease. Expert Opin Drug Metab Toxicol. 2007;3:831–845. doi: 10.1517/17425255.3.6.831. [DOI] [PubMed] [Google Scholar]
- 123.Feria-Gervasio D, Denis S, Alric M, Brugère JF. In vitro maintenance of a human proximal colon microbiota using the continuous fermentation system P-ECSIM. Appl Microbiol Biotechnol. 2011;91:1425–1433. doi: 10.1007/s00253-011-3462-5. [DOI] [PubMed] [Google Scholar]
- 124.Feria-Gervasio D, Tottey W, Gaci N, Alric M, Cardot JM, Peyret P, Martin JF, Pujos E, Sébédio JL, Brugère JF. Three-stage continuous culture system with a self-generated anaerobia to study the regionalized metabolism of the human gut microbiota. J Microbiol Methods. 2014;96:111–118. doi: 10.1016/j.mimet.2013.11.015. [DOI] [PubMed] [Google Scholar]
- 125.Tottey W, Denonfoux J, Jaziri F, Parisot N, Missaoui M, Hill D, Borrel G, Peyretaillade E, Alric M, Harris HM, et al. The human gut chip “HuGChip”, an explorative phylogenetic microarray for determining gut microbiome diversity at family level. PLoS One. 2013;8:e62544. doi: 10.1371/journal.pone.0062544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Human Microbiome Project Consortium. A framework for human microbiome research. Nature. 2012;486:215–221. doi: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Haines A, Metz G, Dilawari J, Blendis L, Wiggins H. Breath-methane in patients with cancer of the large bowel. Lancet. 1977;2:481–483. doi: 10.1016/s0140-6736(77)91605-1. [DOI] [PubMed] [Google Scholar]
- 128.Piqué JM, Pallarés M, Cusó E, Vilar-Bonet J, Gassull MA. Methane production and colon cancer. Gastroenterology. 1984;87:601–605. [PubMed] [Google Scholar]
- 129.Karlin DA, Jones RD, Stroehlein JR, Mastromarino AJ, Potter GD. Breath methane excretion in patients with unresected colorectal cancer. J Natl Cancer Inst. 1982;69:573–576. [PubMed] [Google Scholar]
- 130.Hoff G, Bjørneklett A, Moen IE, Jenssen E. Epidemiology of polyps in the rectum and sigmoid colon. Evaluation of breath methane and predisposition for colorectal neoplasia. Scand J Gastroenterol. 1986;21:193–198. doi: 10.3109/00365528609034646. [DOI] [PubMed] [Google Scholar]
- 131.Kashtan H, Rabau M, Peled Y, Milstein A, Wiznitzer T. Methane production in patients with colorectal carcinoma. Isr J Med Sci. 1989;25:614–616. [PubMed] [Google Scholar]
- 132.Zhang H, DiBaise JK, Zuccolo A, Kudrna D, Braidotti M, Yu Y, Parameswaran P, Crowell MD, Wing R, Rittmann BE, et al. Human gut microbiota in obesity and after gastric bypass. Proc Natl Acad Sci USA. 2009;106:2365–2370. doi: 10.1073/pnas.0812600106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Million M, Thuny F, Angelakis E, Casalta JP, Giorgi R, Habib G, Raoult D. Lactobacillus reuteri and Escherichia coli in the human gut microbiota may predict weight gain associated with vancomycin treatment. Nutr Diabetes. 2013;3:e87. doi: 10.1038/nutd.2013.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Armougom F, Henry M, Vialettes B, Raccah D, Raoult D. Monitoring bacterial community of human gut microbiota reveals an increase in Lactobacillus in obese patients and Methanogens in anorexic patients. PLoS One. 2009;4:e7125. doi: 10.1371/journal.pone.0007125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pimentel M, Mayer AG, Park S, Chow EJ, Hasan A, Kong Y. Methane production during lactulose breath test is associated with gastrointestinal disease presentation. Dig Dis Sci. 2003;48:86–92. doi: 10.1023/a:1021738515885. [DOI] [PubMed] [Google Scholar]
- 136.Pimentel M, Chow EJ, Lin HC. Normalization of lactulose breath testing correlates with symptom improvement in irritable bowel syndrome. a double-blind, randomized, placebo-controlled study. Am J Gastroenterol. 2003;98:412–419. doi: 10.1111/j.1572-0241.2003.07234.x. [DOI] [PubMed] [Google Scholar]
- 137.Bratten JR, Spanier J, Jones MP. Lactulose breath testing does not discriminate patients with irritable bowel syndrome from healthy controls. Am J Gastroenterol. 2008;103:958–963. doi: 10.1111/j.1572-0241.2008.01785.x. [DOI] [PubMed] [Google Scholar]
- 138.Attaluri A, Jackson M, Valestin J, Rao SS. Methanogenic flora is associated with altered colonic transit but not stool characteristics in constipation without IBS. Am J Gastroenterol. 2010;105:1407–1411. doi: 10.1038/ajg.2009.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lee KM, Paik CN, Chung WC, Yang JM, Choi MG. Breath methane positivity is more common and higher in patients with objectively proven delayed transit constipation. Eur J Gastroenterol Hepatol. 2013;25:726–732. doi: 10.1097/MEG.0b013e32835eb916. [DOI] [PubMed] [Google Scholar]
- 140.Chatterjee S, Park S, Low K, Kong Y, Pimentel M. The degree of breath methane production in IBS correlates with the severity of constipation. Am J Gastroenterol. 2007;102:837–841. doi: 10.1111/j.1572-0241.2007.01072.x. [DOI] [PubMed] [Google Scholar]
- 141.Pimentel M, Chatterjee S, Chow EJ, Park S, Kong Y. Neomycin improves constipation-predominant irritable bowel syndrome in a fashion that is dependent on the presence of methane gas: subanalysis of a double-blind randomized controlled study. Dig Dis Sci. 2006;51:1297–1301. doi: 10.1007/s10620-006-9104-6. [DOI] [PubMed] [Google Scholar]
- 142.Majewski M, McCallum RW. Results of small intestinal bacterial overgrowth testing in irritable bowel syndrome patients: clinical profiles and effects of antibiotic trial. Adv Med Sci. 2007;52:139–142. [PubMed] [Google Scholar]
- 143.Hwang L, Low K, Khoshini R, Melmed G, Sahakian A, Makhani M, Pokkunuri V, Pimentel M. Evaluating breath methane as a diagnostic test for constipation-predominant IBS. Dig Dis Sci. 2010;55:398–403. doi: 10.1007/s10620-009-0778-4. [DOI] [PubMed] [Google Scholar]
- 144.Fiedorek SC, Pumphrey CL, Casteel HB. Breath methane production in children with constipation and encopresis. J Pediatr Gastroenterol Nutr. 1990;10:473–477. doi: 10.1097/00005176-199005000-00010. [DOI] [PubMed] [Google Scholar]
- 145.Vianna ME, Conrads G, Gomes BP, Horz HP. Identification and quantification of archaea involved in primary endodontic infections. J Clin Microbiol. 2006;44:1274–1282. doi: 10.1128/JCM.44.4.1274-1282.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Vianna ME, Holtgraewe S, Seyfarth I, Conrads G, Horz HP. Quantitative analysis of three hydrogenotrophic microbial groups, methanogenic archaea, sulfate-reducing bacteria, and acetogenic bacteria, within plaque biofilms associated with human periodontal disease. J Bacteriol. 2008;190:3779–3785. doi: 10.1128/JB.01861-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lira EA, Ramiro FS, Chiarelli FM, Dias RR, Feres M, Figueiredo LC, Faveri M. Reduction in prevalence of Archaea after periodontal therapy in subjects with generalized aggressive periodontitis. Aust Dent J. 2013;58:442–447. doi: 10.1111/adj.12123. [DOI] [PubMed] [Google Scholar]
- 148.Brochier-Armanet C, Forterre P, Gribaldo S. Phylogeny and evolution of the Archaea: one hundred genomes later. Curr Opin Microbiol. 2011;14:274–281. doi: 10.1016/j.mib.2011.04.015. [DOI] [PubMed] [Google Scholar]