

Abstract
Pancreatitis is an increasingly common and sometimes severe disease that lacks a specific therapy. The pathogenesis of pancreatitis is still not well understood. Calcium (Ca2+) is a versatile carrier of signals regulating many aspects of cellular activity and plays a central role in controlling digestive enzyme secretion in pancreatic acinar cells. Ca2+ overload is a key early event and is crucial in the pathogenesis of many diseases. In pancreatic acinar cells, pathological Ca2+ signaling (stimulated by bile, alcohol metabolites and other causes) is a key contributor to the initiation of cell injury due to prolonged and global Ca2+ elevation that results in trypsin activation, vacuolization and necrosis, all of which are crucial in the development of pancreatitis. Increased release of Ca2+ from stores in the intracellular endoplasmic reticulum and/or increased Ca2+ entry through the plasma membrane are causes of such cell damage. Failed mitochondrial adenosine triphosphate (ATP) production reduces re-uptake and extrusion of Ca2+ by the sarco/endoplasmic reticulum Ca2+-activated ATPase and plasma membrane Ca2+-ATPase pumps, which contribute to Ca2+ overload. Current findings have provided further insight into the roles and mechanisms of abnormal pancreatic acinar Ca2+ signals in pancreatitis. The lack of available specific treatments is therefore an objective of ongoing research. Research is currently underway to establish the mechanisms and interactions of Ca2+ signals in the pathogenesis of pancreatitis.
Keywords: Pancreatitis, Calcium signaling, Pancreatic acinar cells, Overload, Cell injury
Core tip: Calcium (Ca2+) overload is crucial in the pathogenesis of pancreatitis, which results in trypsin activation, vacuolization and necrosis. Such cell injury results from increased Ca2+ released from intracellular endoplasmic reticulum Ca2+ stores, increased Ca2+ entry through the plasma membrane and Ca2+ pump defects. Current findings have provided further insight into the roles and mechanisms of Ca2+ overload in pancreatitis. The lack of specific treatments is a stimulus for ongoing research. This review summarizes recent advances in our understanding of Ca2+ signaling in the pathogenesis of pancreatitis, and discusses how research has guided our search for potential therapeutic targets.
INTRODUCTION
Pancreatitis remains a disease with significant morbidity and lethality, and is typically caused by alcohol abuse or complications arising from biliary disease[1]. The pathogenesis of pancreatitis is multi-factorial and has not yet been clarified[2-5]. In recent years, several pancreatic mechanisms have been proposed, such as trypsinogen activation[6], pancreatic microcirculation malfunction[7], calcium (Ca2+) overload[8-10] and inflammatory pathways[11-13]. Among these various theories, Ca2+ overload is receiving increasing attention and is being extensively investigated in the pathogenesis of pancreatitis[14]. Recent advances in our understanding of Ca2+ signaling of pancreatic acinar cells in the pathogenesis of pancreatitis are reviewed in this article, including a discussion on how research has guided our search for potential therapeutic targets.
PHYSIOLOGICAL AND PATHOLOGICAL CA2+ SIGNALS IN PANCREATIC ACINAR CELLS
As the most universal carrier of biological signals, intracellular Ca2+ is involved in the modulation of virtually all cellular functions, from its origin at fertilization to its end in the apoptotic process[15]. Intracellular Ca2+ acts both as a first and a second messenger to control cellular functions via regulating free-Ca2+ concentrations in the cytoplasm, for example, controlling the contraction and relaxation of muscles, and regulating secretion from exocrine glands[16]. Ca2+ signals elicited by physiological stimulation are transient and mostly localized in the granule-containing apical pole, whereas sustained global elevation of cytosolic Ca2+ concentrations can be fatal[17-19]. The digestive enzymes produced by pancreatic acinar cells are packaged in zymogen granules in the apical pole[20]. Physiological stimulation elicits proenzyme exocytosis exclusively through the apical membrane[21]. Ca2+ overload causes inappropriate intracellular trypsin activation, vacuolization and necrosis[20,22-26], which contribute to subsequent cell injury and are often fatal in human acute pancreatitis[27]. Pretreatment with pharmacological Ca2+ chelators or blockers was found to prevent premature digestive enzyme activation, vacuolization, skeletal disruption and acinar cell necrosis induced by Ca2+ overload[28].
RELEASE OF CA2+ FROM THE ENDOPLASMIC RETICULUM
There are two types of G protein-coupled receptors localized on the plasma membrane, namely, acetylcholine (ACh) and cholecystokinin (CCK) receptors[8]. ACh is a secretagogue that activates phospholipase C (PLC) through ACh receptor ligand binding, which in turn cleaves phosphatidylinositol 4,5-bisphosphate into the classic Ca2+-releasing messengers inositol 1,4,5-trisphosphate (IP3) and diacylglycerol to mobilize Ca2+ and activate protein kinase C respectively[29]. The other principal secretagogue in acinar cells is the hormone CCK, which exists in multiple molecular forms, such as CCK8 and CCK58. CCK interacts with its receptor and activates adenosine diphosphate-ribosyl cyclase to produce the novel Ca2+-releasing agent nicotinic acid adenine dinucleotide phosphate (NAADP) and cyclic adenosine diphosphate-ribose (cADPR).
There are two types of regulated Ca2+-release channels localized on the endoplasmic reticulum (ER) membrane, namely, the IP3 receptors (IP3R) and ryanodine receptors (RyR). IP3R are concentrated in the apical part of the acinar cell and binding of IP3 activates gated Ca2+ channels to release intracellular stored Ca2+ from the ER, which participates in the apical cytosolic Ca2+-spiking response to stimulation with physiological concentrations of ACh[10,19,30,31]. RyR in the basal region of acinar cells are activated by NAADP and cADPR, and oligomers form gated Ca2+ channels to release intracellular Ca2+ from ER stores[32] in response to stimulation with physiological concentrations of CCK[33-35]. Intriguingly, the Ca2+ response mediated by RyR was observed in the apical pole in mouse acinar cells and required functional IP3R, which could be interpreted as co-localization and coordination of RyR and IP3R[36].
Hyperstimulation with agents (in contrast to physiological stimulation) can induce acinar cell injury by IP3R-induced release of Ca2+ from the ER. The Ca2+ increase spreads from the apical pole to the basolateral part of the acinar cell, and a sustained global Ca2+ elevation causes pancreatitis-like cellular changes, such as abnormal intracellular enzyme activation, vacuolization and necrosis[20]. Treatment with IP3R inhibitors, such as caffeine and 2-aminoethoxydiphenyl borate, can reduce abnormal Ca2+ signals and the probability of ethanol-induced pancreatitis, but the low affinity and multiple actions restrict its therapeutic potential[37,38]. Hyperstimulation by CCK8 is specifically dependent on functional RyR, and induces toxic pancreatitis-like changes as a result of sustained global elevation of Ca2+ released from the ER. These aberrant Ca2+ signals and acinar cell injuries can be blocked in vitro and in vivo by pretreating with RyR inhibitors[8,39]. Hyperstimulation by CCK also activates PLC, which generates IP3 and elicits Ca2+ overload[20].
Although the ER is a large Ca2+ store in the basolateral part of pancreatic acinar cells, there are also extensive acidic Ca2+ stores present in the apical part, which similarly release Ca2+ into the cytoplasm through IP3, cADPR and NAADP signaling. Hyperstimulation from bile acids and alcohol metabolites can elicit pathological Ca2+ release from both the ER and acidic stores[40,41].
STORE-OPERATED CA2+ (SOC) INFLUX
Another abnormal Ca2+ signal in the pathogenesis of pancreatitis is extracellular Ca2+ entry, which is regulated at the plasma membrane of acinar cells by SOC channels[42]. Under physiological conditions, CCK and ACh induce the release of Ca2+ from the ER, followed by Ca2+ extrusion from the cell, suggesting that SOC entry is required to elevate intracellular Ca2+. The molecular mechanism underlying these pancreatic Ca2+-entry channels is ill-defined. Current research suggests that Ca2+-entry channels belong to the transient receptor potential family, including Ca2+ release-activated Ca2+ channel protein 1 (Orai1), transient receptor potential channel 1, and stromal interaction molecule (STIM) 1[43,44]. Recent studies indicate that STIM proteins serve as sensors, are concentrated in the ER membrane, and monitor the Ca2+ concentration in the ER lumen. When the luminal concentration is reduced in response to secretagogue stimulation, STIM proteins sense the changes, accumulate and translocate to the plasma membrane where they co-localize with and activate Orai1 channels[45-48].
Orai1 channels are localized not only in the apical part of acinar cells, but also in the basal and lateral membranes, which cover about 95% of the pancreatic acinar cell surface[43]. Following ER Ca2+ store depletion, Orail1 interacts with STIM and activates SOC channels. The wide distribution of Orai1 channels enables sustained Ca2+ entry under physiological conditions, without the need for local Ca2+ concentrations, and refilling of the ER Ca2+ stores after agonist-elicited depletion[8]. However, Orai1 activity can result in an abnormal sustained global Ca2+ elevation following pathological stimulation, such as by high, toxic concentrations of CCK8, alcohol and bile acid, which all elicit intracellular Ca2+ overload that is mostly dependent on external Ca2+ influx[20,23,25]. Therefore, SOC entry may be crucial for the development of acute pancreatitis. Without external sustained global Ca2+ entry, cellular injury does not occur[20,22-25,49]. Removal of external Ca2+ or abrogation of elevated Ca2+ with a Ca2+ chelator can protect acinar cells against abnormal changes, such as trypsinogen activation and vacuolization[20,25,49]. SOC channel blockers might therefore be a possible therapeutic approach for the treatment of acute pancreatitis[7].
CA2+ PUMP DEFECTS
Sarco/endoplasmic reticulum Ca2+-activated adenosine triphosphate (ATP)ase (SERCA) is an ER Ca2+ pump which actively re-uptakes Ca2+ into the ER lumen to compensate for resting leakage into the cytosol[8,50]. Under normal physiological conditions, the elevation of intracellular Ca2+ can activate the SERCA pump[19,27,51], and Ca2+ release elicited by stimulation is followed by Ca2+ re-uptake. The rate of uptake decreases as luminal Ca2+ concentration increases until the uptake rate equals the resting leak rate[8]. Pathological stimulation by bile acids or fatty acids can elicit Ca2+ overload by inhibiting the SERCA pump and depolarizing the inner mitochondrial membrane, resulting in reduced ATP production, which in turn lessens the ability of the SERCA pump to restore ER Ca2+ stores[25]. Prolonged and uncompensated Ca2+ overload released from ER stores can cause thapsigargin activation and vacuolization in pancreatic acinar cells, which can be visualized directly[20].
All eukaryotic cells export Ca2+ through two pathways, the plasma membrane Ca2+-ATPase (PMCA; commonly called the Ca2+ pump) and the Na+-Ca2+ exchanger (NCE), to prevent Ca2+ overload and for the maintenance of intracellular Ca2+ at the appropriately low level[52,53]. The PMCA has high Ca2+ affinity but low transport capacity and is ATP-dependent. Any elevation of cytosolic Ca2+ can activate the PMCA to rapidly extrude Ca2+ in physiological conditions. Whereas ER-released Ca2+ is localized in the apical part and Ca2+ entry occurs across the basolateral surface, PMCA Ca2+ extrusion is confined to a small apical region only, which restricts the PMCA function as a fine-tuner of cell cytosolic Ca2+. Pathological stimulation can depolarize mitochondria and cause a deficiency in ATP production, which inhibits Ca2+ extrusion and aggravates the cytosolic Ca2+ overload[24,54].
The NCE has a low Ca2+ affinity and is a high-capacity transmembrane protein of the plasma membrane involved in Ca2+ homeostasis, and is especially important in excitable cells. Because of its high capacity, the NCE can extrude Ca2+ at a much higher rate than the PMCA, serving as the fast Ca2+ transporting system. For example, activation of the NCE prevents Ca2+ overload induced by pathological stimulation and cell death in neurons. Inactivation of the NCE can cause neuronal death, which can be visualized directly[55]. In pancreatic acinar cells, the NCE is of little quantitative importance, which explains why Ca2+ overloading is particularly dangerous in pancreatic acinar cells[19,27].
As another Ca2+ store, mitochondria also participate in maintaining cytosolic Ca2+ homeostasis in pancreatic acinar cells. Mitochondria surround the apical pole in a perigranular belt, separating zymogen granules from the basolateral part of the acinar cell, and are also positioned just beneath the plasma membrane and surrounding the nucleus[19,27,55-58]. The membrane potential across the inner mitochondrial membrane is the driving force behind mitochondrial uptake of Ca2+ into the matrix through the Ca2+ uniporter, a Ca2+-selective ion channel[59,60]. Mitochondria in pancreatic acinar cells play an important role in maintaining cytosolic Ca2+ homeostasis[56-58]. When cytosolic Ca2+ is elevated by physiological stimulation, mitochondria sense the Ca2+ in the environment and take up Ca2+ via the Ca2+ uniporter[60]. Ca2+ spikes released from the ER occurring in the apical region can cause immediate Ca2+ uptake into the mitochondrial matrix, preventing further spread of the Ca2+ signal into the basolateral part of the acinar cell, which contains the nucleus. Perigranular mitochondria function as a Ca2+ buffer barrier[8], causing Ca2+ uptake termination and Ca2+ removal via the mitochondrial NCE[61,62]. Mitochondrial Ca2+ uptake activates Krebs cycle enzymes and drives ATP production, supplying ATP for SERCA-mediated Ca2+ re-uptake into the ER and PMCA-mediated Ca2+ extrusion[27,63]. Pathological stimulation that can induce experimental pancreatitis, such as with bile salts, fatty acids and CCK or its analog, can depolarize the inner mitochondrial membrane, inducing further collapse of the mitochondrial membrane potential and impairment of ATP production[64]. This situation prevents perigranular mitochondrial Ca2+ re-uptake and mitochondria cannot buffer the apical Ca2+ elevation, causing the local Ca2+ signal to spread to the whole of the acinar cell[30]. Failure of ATP production reduces the ability of the SERCA and PMCA pumps to take Ca2+ back into the ER and for extrusion, which contributes to Ca2+ overload. This is the most likely explanation for why pretreatment with Ca2+ chelators can limit the global and sustained elevation of Ca2+.
TARGETS FOR POTENTIAL THERAPY
To date, there is no specific treatment for either acute or chronic pancreatitis[39,65-67]. The current therapy for pancreatitis is limited to the inhibition of proteolytic enzymes. Protease inhibitors have a modest preventative role in experimental animal models, however, they fail to show therapeutic value in clinical treatment[68,69]. An aberrant increase in cytosolic Ca2+ is a key molecular event in the pathogenesis of pancreatitis. Intracellular Ca2+ overload is a major reason for pancreatic acinar cell injury from toxin stimulation that induces pancreatitis[7,20,22-25,49]. Abnormal, prolonged, global Ca2+ signals lead to premature enzyme activation, vacuole formation and acinar cell damage. Thus, it is clinically relevant to identify the targets of the aberrant Ca2+ signals[70]. New avenues are required based on current findings in our understanding of Ca2+ signaling in the pathogenesis of pancreatitis (Figure 1). Possible interventions include: (1) inhibition of Ca2+ entry pathways; (2) enhancement of Ca2+ extrusion; and (3) inhibition of the primary Ca2+ release from the ER; and iv) protection of mitochondrial function, which can serve as potential therapeutic targets (Table 1). Recent progress in our understanding of Ca2+ signals of pancreatic acinar cells in the pathogenesis of pancreatitis now provides opportunities for the developments of better therapeutic approaches.
Figure 1.
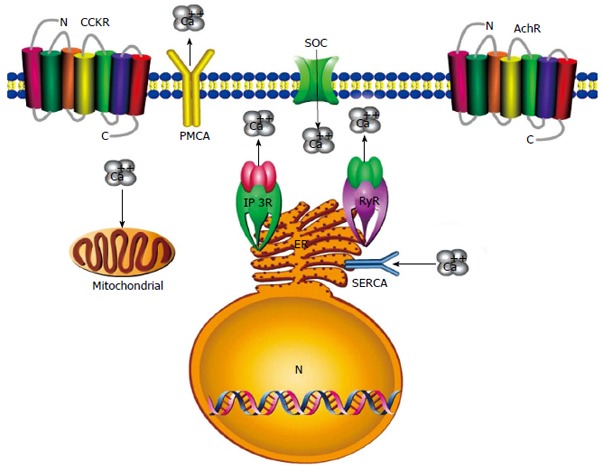
Diagram showing possible interventions for therapeutic targets of pancreatitis. Blockage of Ca2+ entry will probably depend on inhibition of the store-operated Ca2+ (SOC) channel in the pancreatic acinar cell. Activation of plasma membrane Ca2+-adenosine triphosphate (ATP)ase (PMCA) and sarco/endoplasmic reticulum Ca2+-activated ATPase (SERCA) would enhance Ca2+ extrusion from the cell and endoplasmic reticulum (ER). Intracellular Ca2+ release from the ER can be prevented through inhibition of the inositol 1,4,5-trisphosphate receptor (IP3R) and ryanodine receptor (RyR). Preconditioning strategies could protect mitochondrial function to ensure adequate ATP production extrusion by Ca2+ pumps and for pancreatic acinar cells to survive intact.
Table 1.
Potential therapeutic targets
| Potential therapeutic targets |
| Store-operated Ca2+ channel |
| IP3 receptor |
| Ryanodine receptor |
| SERCA |
| PMCA |
| Mitochondria |
IP3: 1,4,5-trisphosphate; PMCA: Plasma membrane Ca2+-adenosine triphosphate (ATP)ase; SERCA: Sarco/endoplasmic reticulum Ca2+-activated ATPase.
Footnotes
Supported by grants from the National Natural Science Foundation of China No. 30171167, No. 30901945 and the Specialized Research Fund for the Doctoral Program of Higher Education No. 20130201130009
P- Reviewer: Seicean A, Smith RC S- Editor: Ding Y L- Editor: AmEditor E- Editor: Liu XM
References
- 1.Gerasimenko JV, Lur G, Ferdek P, Sherwood MW, Ebisui E, Tepikin AV, Mikoshiba K, Petersen OH, Gerasimenko OV. Calmodulin protects against alcohol-induced pancreatic trypsinogen activation elicited via Ca2+ release through IP3 receptors. Proc Natl Acad Sci USA. 2011;108:5873–5878. doi: 10.1073/pnas.1016534108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garip G, Sarandöl E, Kaya E. Effects of disease severity and necrosis on pancreatic dysfunction after acute pancreatitis. World J Gastroenterol. 2013;19:8065–8070. doi: 10.3748/wjg.v19.i44.8065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mayerle J, Hlouschek V, Lerch MM. Current management of acute pancreatitis. Nat Clin Pract Gastroenterol Hepatol. 2005;2:473–483. doi: 10.1038/ncpgasthep0293. [DOI] [PubMed] [Google Scholar]
- 4.Dambrauskas Z, Giese N, Gulbinas A, Giese T, Berberat PO, Pundzius J, Barauskas G, Friess H. Different profiles of cytokine expression during mild and severe acute pancreatitis. World J Gastroenterol. 2010;16:1845–1853. doi: 10.3748/wjg.v16.i15.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang J, Rouse RL. Histopathology and pathogenesis of caerulein-, duct ligation-, and arginine-induced acute pancreatitis in Sprague-Dawley rats and C57BL6 mice. Histol Histopathol. 2014;29:1135–1152. doi: 10.14670/HH-29.1135. [DOI] [PubMed] [Google Scholar]
- 6.Chiari H. Über die Selbstverdauung des menschlichen Pankreas. Zeitschrift für Heilkunde. 1896;17:69–96. [Google Scholar]
- 7.Feng JY, Li YY. Alteration and role of heat shock proteins in acute pancreatitis. J Dig Dis. 2010;11:277–283. doi: 10.1111/j.1751-2980.2010.00450.x. [DOI] [PubMed] [Google Scholar]
- 8.Petersen OH, Sutton R. Ca2+ signalling and pancreatitis: effects of alcohol, bile and coffee. Trends Pharmacol Sci. 2006;27:113–120. doi: 10.1016/j.tips.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 9.Booth DM, Mukherjee R, Sutton R, Criddle DN. Calcium and reactive oxygen species in acute pancreatitis: friend or foe? Antioxid Redox Signal. 2011;15:2683–2698. doi: 10.1089/ars.2011.3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Orabi AI, Luo Y, Ahmad MU, Shah AU, Mannan Z, Wang D, Sarwar S, Muili KA, Shugrue C, Kolodecik TR, et al. IP3 receptor type 2 deficiency is associated with a secretory defect in the pancreatic acinar cell and an accumulation of zymogen granules. PLoS One. 2012;7:e48465. doi: 10.1371/journal.pone.0048465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gu H, Werner J, Bergmann F, Whitcomb DC, Büchler MW, Fortunato F. Necro-inflammatory response of pancreatic acinar cells in the pathogenesis of acute alcoholic pancreatitis. Cell Death Dis. 2013;4:e816. doi: 10.1038/cddis.2013.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shrivastava P, Bhatia M. Essential role of monocytes and macrophages in the progression of acute pancreatitis. World J Gastroenterol. 2010;16:3995–4002. doi: 10.3748/wjg.v16.i32.3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sah RP, Dawra RK, Saluja AK. New insights into the pathogenesis of pancreatitis. Curr Opin Gastroenterol. 2013;29:523–530. doi: 10.1097/MOG.0b013e328363e399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frick TW. The role of calcium in acute pancreatitis. Surgery. 2012;152:S157–S163. doi: 10.1016/j.surg.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 15.Whitaker M. Calcium at fertilization and in early development. Physiol Rev. 2006;86:25–88. doi: 10.1152/physrev.00023.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Low JT, Shukla A, Thorn P. Pancreatic acinar cell: new insights into the control of secretion. Int J Biochem Cell Biol. 2010;42:1586–1589. doi: 10.1016/j.biocel.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 17.Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 18.Montell C. The latest waves in calcium signaling. Cell. 2005;122:157–163. doi: 10.1016/j.cell.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 19.Ashby MC, Tepikin AV. Polarized calcium and calmodulin signaling in secretory epithelia. Physiol Rev. 2002;82:701–734. doi: 10.1152/physrev.00006.2002. [DOI] [PubMed] [Google Scholar]
- 20.Raraty M, Ward J, Erdemli G, Vaillant C, Neoptolemos JP, Sutton R, Petersen OH. Calcium-dependent enzyme activation and vacuole formation in the apical granular region of pancreatic acinar cells. Proc Natl Acad Sci USA. 2000;97:13126–13131. doi: 10.1073/pnas.97.24.13126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Palade G. Intracellular aspects of the process of protein synthesis. Science. 1975;189:347–358. doi: 10.1126/science.1096303. [DOI] [PubMed] [Google Scholar]
- 22.Krüger B, Albrecht E, Lerch MM. The role of intracellular calcium signaling in premature protease activation and the onset of pancreatitis. Am J Pathol. 2000;157:43–50. doi: 10.1016/S0002-9440(10)64515-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Voronina S, Longbottom R, Sutton R, Petersen OH, Tepikin A. Bile acids induce calcium signals in mouse pancreatic acinar cells: implications for bile-induced pancreatic pathology. J Physiol. 2002;540:49–55. doi: 10.1113/jphysiol.2002.017525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim JY, Kim KH, Lee JA, Namkung W, Sun AQ, Ananthanarayanan M, Suchy FJ, Shin DM, Muallem S, Lee MG. Transporter-mediated bile acid uptake causes Ca2+-dependent cell death in rat pancreatic acinar cells. Gastroenterology. 2002;122:1941–1953. doi: 10.1053/gast.2002.33617. [DOI] [PubMed] [Google Scholar]
- 25.Criddle DN, Raraty MG, Neoptolemos JP, Tepikin AV, Petersen OH, Sutton R. Ethanol toxicity in pancreatic acinar cells: mediation by nonoxidative fatty acid metabolites. Proc Natl Acad Sci USA. 2004;101:10738–10743. doi: 10.1073/pnas.0403431101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Voronina S, Sherwood M, Barrow S, Dolman N, Conant A, Tepikin A. Downstream from calcium signalling: mitochondria, vacuoles and pancreatic acinar cell damage. Acta Physiol (Oxf) 2009;195:161–169. doi: 10.1111/j.1748-1716.2008.01931.x. [DOI] [PubMed] [Google Scholar]
- 27.Reed AM, Husain SZ, Thrower E, Alexandre M, Shah A, Gorelick FS, Nathanson MH. Low extracellular pH induces damage in the pancreatic acinar cell by enhancing calcium signaling. J Biol Chem. 2011;286:1919–1926. doi: 10.1074/jbc.M110.158329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petersen O. Can specific calcium channel blockade be the basis for a drug-based treatment of acute pancreatitis? Expert Rev Gastroenterol Hepatol. 2014;8:339–341. doi: 10.1586/17474124.2014.896192. [DOI] [PubMed] [Google Scholar]
- 29.Mikoshiba K, Hisatsune C. Encyclopedia of Biological Chemistry 2nd ed. Lennarz WJ, Lane MD, editor. New York: Academic Press; 2013. pp. 653–656. [Google Scholar]
- 30.Petersen OH. Ca2+ signalling and Ca2+-activated ion channels in exocrine acinar cells. Cell Calcium. 2005;38:171–200. doi: 10.1016/j.ceca.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 31.Williams JA, Yule DI. Physiology of the Gastrointestinal Tract. 5th ed. New York: Academic Press; 2012. pp. 1361–1398. [Google Scholar]
- 32.Orabi AI, Muili KA, Javed TA, Jin S, Jayaraman T, Lund FE, Husain SZ. Cluster of differentiation 38 (CD38) mediates bile acid-induced acinar cell injury and pancreatitis through cyclic ADP-ribose and intracellular calcium release. J Biol Chem. 2013;288:27128–27137. doi: 10.1074/jbc.M113.494534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leite MF, Burgstahler AD, Nathanson MH. Ca2+ waves require sequential activation of inositol trisphosphate receptors and ryanodine receptors in pancreatic acini. Gastroenterology. 2002;122:415–427. doi: 10.1053/gast.2002.30982. [DOI] [PubMed] [Google Scholar]
- 34.Gerasimenko JV, Maruyama Y, Yano K, Dolman NJ, Tepikin AV, Petersen OH, Gerasimenko OV. NAADP mobilizes Ca2+ from a thapsigargin-sensitive store in the nuclear envelope by activating ryanodine receptors. J Cell Biol. 2003;163:271–282. doi: 10.1083/jcb.200306134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamasaki M, Thomas JM, Churchill GC, Garnham C, Lewis AM, Cancela JM, Patel S, Galione A. Role of NAADP and cADPR in the induction and maintenance of agonist-evoked Ca2+ spiking in mouse pancreatic acinar cells. Curr Biol. 2005;15:874–878. doi: 10.1016/j.cub.2005.04.033. [DOI] [PubMed] [Google Scholar]
- 36.Ashby MC, Petersen OH, Tepikin AV. Spatial characterisation of ryanodine-induced calcium release in mouse pancreatic acinar cells. Biochem J. 2003;369:441–445. doi: 10.1042/BJ20021039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petersen OH, Gerasimenko OV, Gerasimenko JV. Pathobiology of acute pancreatitis: focus on intracellular calcium and calmodulin. F1000 Med Rep. 2011;3:15. doi: 10.3410/M3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Choi KJ, Kim KS, Kim SH, Kim DK, Park HS. Caffeine and 2-Aminoethoxydiphenyl Borate (2-APB) Have Different Ability to Inhibit Intracellular Calcium Mobilization in Pancreatic Acinar Cell. Korean J Physiol Pharmacol. 2010;14:105–111. doi: 10.4196/kjpp.2010.14.2.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Husain SZ, Orabi AI, Muili KA, Luo Y, Sarwar S, Mahmood SM, Wang D, Choo-Wing R, Singh VP, Parness J, et al. Ryanodine receptors contribute to bile acid-induced pathological calcium signaling and pancreatitis in mice. Am J Physiol Gastrointest Liver Physiol. 2012;302:G1423–G1433. doi: 10.1152/ajpgi.00546.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parekh AB, Putney JW. Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 41.Gerasimenko JV, Sherwood M, Tepikin AV, Petersen OH, Gerasimenko OV. NAADP, cADPR and IP3 all release Ca2+ from the endoplasmic reticulum and an acidic store in the secretory granule area. J Cell Sci. 2006;119:226–238. doi: 10.1242/jcs.02721. [DOI] [PubMed] [Google Scholar]
- 42.Petersen OH, Gerasimenko OV, Tepikin AV, Gerasimenko JV. Aberrant Ca(2+) signalling through acidic calcium stores in pancreatic acinar cells. Cell Calcium. 2011;50:193–199. doi: 10.1016/j.ceca.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 43.Lur G, Sherwood MW, Ebisui E, Haynes L, Feske S, Sutton R, Burgoyne RD, Mikoshiba K, Petersen OH, Tepikin AV. InsP3receptors and Orai channels in pancreatic acinar cells: co-localization and its consequences. Biochem J. 2011;436:231–239. doi: 10.1042/BJ20110083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ambudkar IS. Ca2+ signaling and regulation of fluid secretion in salivary gland acinar cells. Cell Calcium. 2014;55:297–305. doi: 10.1016/j.ceca.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 46.Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luik RM, Wu MM, Buchanan J, Lewis RS. The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J Cell Biol. 2006;174:815–825. doi: 10.1083/jcb.200604015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Criddle DN, Murphy J, Fistetto G, Barrow S, Tepikin AV, Neoptolemos JP, Sutton R, Petersen OH. Fatty acid ethyl esters cause pancreatic calcium toxicity via inositol trisphosphate receptors and loss of ATP synthesis. Gastroenterology. 2006;130:781–793. doi: 10.1053/j.gastro.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 50.Garside VC, Kowalik AS, Johnson CL, DiRenzo D, Konieczny SF, Pin CL. MIST1 regulates the pancreatic acinar cell expression of Atp2c2, the gene encoding secretory pathway calcium ATPase 2. Exp Cell Res. 2010;316:2859–2870. doi: 10.1016/j.yexcr.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burdakov D, Petersen OH, Verkhratsky A. Intraluminal calcium as a primary regulator of endoplasmic reticulum function. Cell Calcium. 2005;38:303–310. doi: 10.1016/j.ceca.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 52.Guerini D, Coletto L, Carafoli E. Exporting calcium from cells. Cell Calcium. 2005;38:281–289. doi: 10.1016/j.ceca.2005.06.032. [DOI] [PubMed] [Google Scholar]
- 53.Ferreira-Gomes MS, González-Lebrero RM, de la Fuente MC, Strehler EE, Rossi RC, Rossi JP. Calcium occlusion in plasma membrane Ca2+-ATPase. J Biol Chem. 2011;286:32018–32025. doi: 10.1074/jbc.M111.266650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Voronina SG, Barrow SL, Gerasimenko OV, Petersen OH, Tepikin AV. Effects of secretagogues and bile acids on mitochondrial membrane potential of pancreatic acinar cells: comparison of different modes of evaluating DeltaPsim. J Biol Chem. 2004;279:27327–27338. doi: 10.1074/jbc.M311698200. [DOI] [PubMed] [Google Scholar]
- 55.Bano D, Young KW, Guerin CJ, Lefeuvre R, Rothwell NJ, Naldini L, Rizzuto R, Carafoli E, Nicotera P. Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell. 2005;120:275–285. doi: 10.1016/j.cell.2004.11.049. [DOI] [PubMed] [Google Scholar]
- 56.Tinel H, Cancela JM, Mogami H, Gerasimenko JV, Gerasimenko OV, Tepikin AV, Petersen OH. Active mitochondria surrounding the pancreatic acinar granule region prevent spreading of inositol trisphosphate-evoked local cytosolic Ca(2+) signals. EMBO J. 1999;18:4999–5008. doi: 10.1093/emboj/18.18.4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Park MK, Ashby MC, Erdemli G, Petersen OH, Tepikin AV. Perinuclear, perigranular and sub-plasmalemmal mitochondria have distinct functions in the regulation of cellular calcium transport. EMBO J. 2001;20:1863–1874. doi: 10.1093/emboj/20.8.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Voronina S, Sukhomlin T, Johnson PR, Erdemli G, Petersen OH, Tepikin A. Correlation of NADH and Ca2+ signals in mouse pancreatic acinar cells. J Physiol. 2002;539:41–52. doi: 10.1113/jphysiol.2001.013134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360–364. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- 60.Leo S, Bianchi K, Brini M, Rizzuto R. Mitochondrial calcium signalling in cell death. FEBS J. 2005;272:4013–4022. doi: 10.1111/j.1742-4658.2005.04855.x. [DOI] [PubMed] [Google Scholar]
- 61.Nicholls DG. Mitochondria and calcium signaling. Cell Calcium. 2005;38:311–317. doi: 10.1016/j.ceca.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 62.Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev. 2006;86:369–408. doi: 10.1152/physrev.00004.2005. [DOI] [PubMed] [Google Scholar]
- 63.Mukherjee R, Criddle DN, Gukovskaya A, Pandol S, Petersen OH, Sutton R. Mitochondrial injury in pancreatitis. Cell Calcium. 2008;44:14–23. doi: 10.1016/j.ceca.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gukovskaya AS, Gukovsky I. Which way to die: the regulation of acinar cell death in pancreatitis by mitochondria, calcium, and reactive oxygen species. Gastroenterology. 2011;140:1876–1880. doi: 10.1053/j.gastro.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Frossard JL, Steer ML, Pastor CM. Acute pancreatitis. Lancet. 2008;371:143–152. doi: 10.1016/S0140-6736(08)60107-5. [DOI] [PubMed] [Google Scholar]
- 66.Brock C, Nielsen LM, Lelic D, Drewes AM. Pathophysiology of chronic pancreatitis. World J Gastroenterol. 2013;19:7231–7240. doi: 10.3748/wjg.v19.i42.7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jung J, Lee MG. Role of calcium signaling in epithelial bicarbonate secretion. Cell Calcium. 2014;55:376–384. doi: 10.1016/j.ceca.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 68.Seifert G, Kurzinger RP, Hopt UT, Wittel UA. Systemic differential gene regulation of the inter-α-trypsin inhibitor family in acute necrotizing pancreatitis in mice. J Surg Res. 2013;180:e83–e90. doi: 10.1016/j.jss.2012.03.061. [DOI] [PubMed] [Google Scholar]
- 69.Seta T, Noguchi Y, Shikata S, Nakayama T. Treatment of acute pancreatitis with protease inhibitors administered through intravenous infusion: an updated systematic review and meta-analysis. BMC Gastroenterol. 2014;14:102. doi: 10.1186/1471-230X-14-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Voronina S, Tepikin A. Mitochondrial calcium in the life and death of exocrine secretory cells. Cell Calcium. 2012;52:86–92. doi: 10.1016/j.ceca.2012.03.007. [DOI] [PubMed] [Google Scholar]