

Abstract
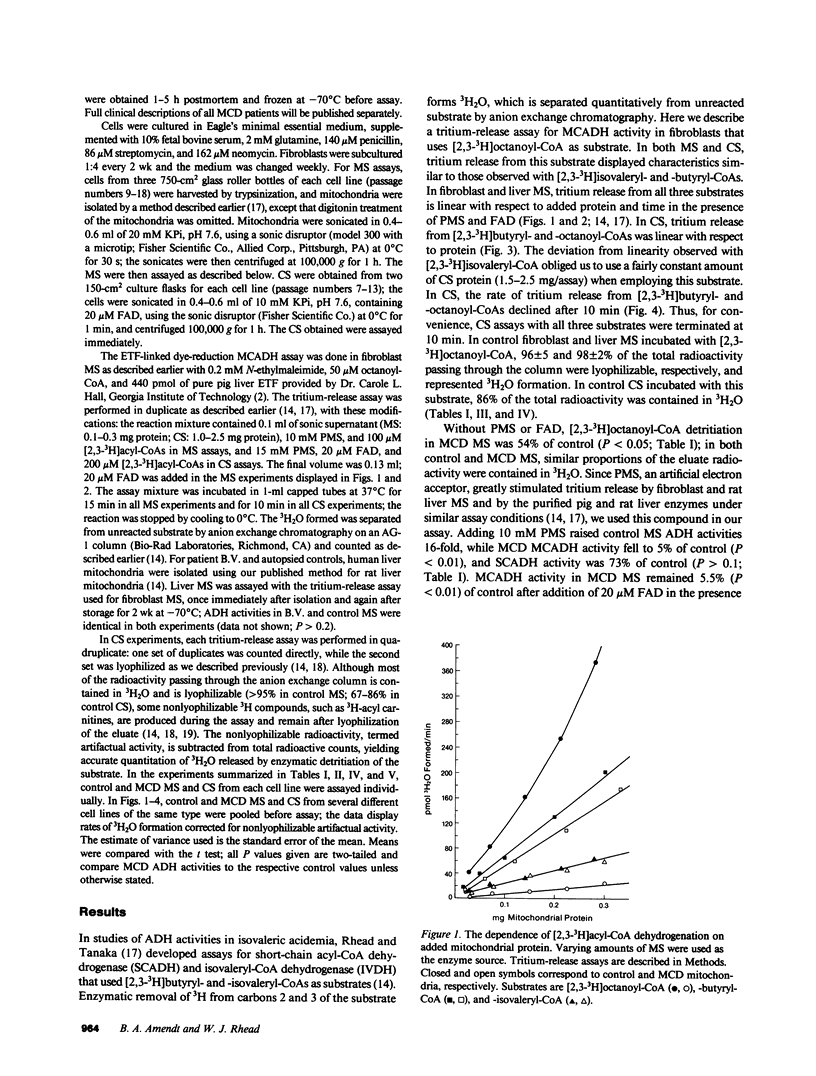
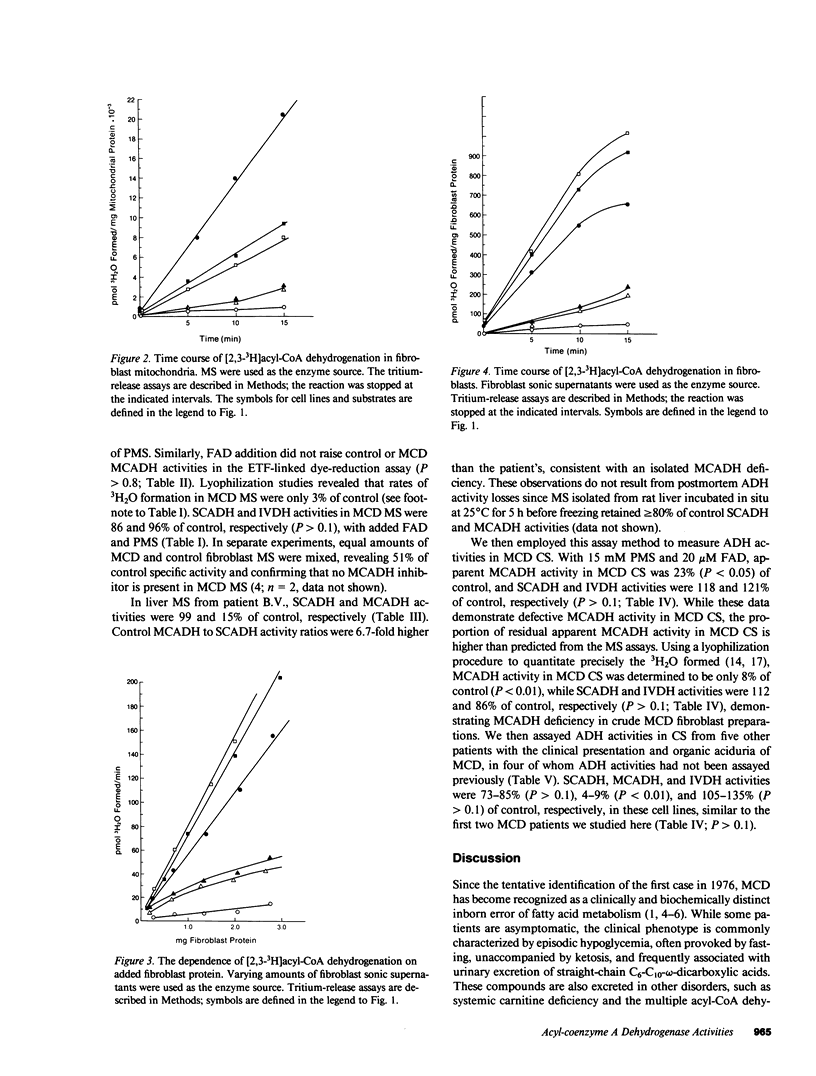
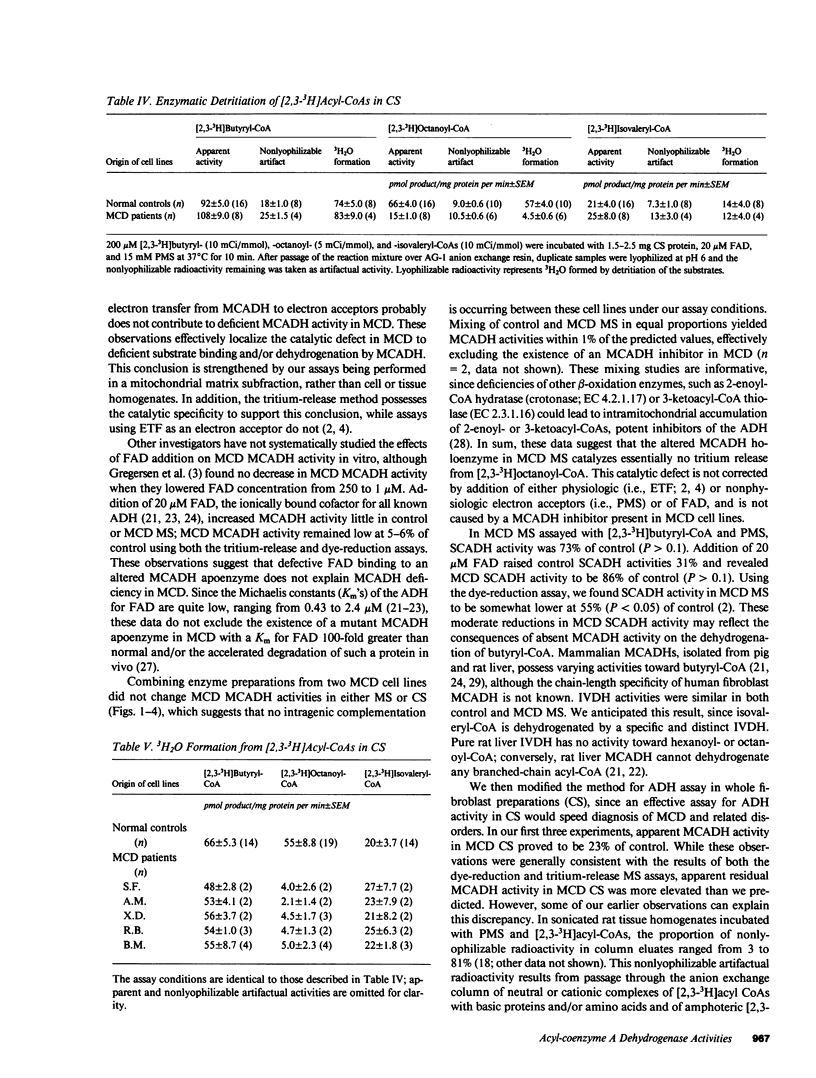
Medium-chain acyl-coenzyme A (CoA) dehydrogenase (MCADH; EC 1.3.99.3) deficiency (MCD) is an inborn error of beta-oxidation. We measured 3H2O formed by the dehydrogenation of [2,3-3H]acyl-CoAs in a 3H-release assay. Short-chain acyl-CoA dehydrogenase (SCADH; EC 1.3.99.2), MCADH, and isovaleryl-CoA dehydrogenase (IVDH; EC 1.3.99.10) activities were assayed with 100 microM [2,3-3H]butyryl-, -octanoyl-, and -isovaleryl-CoAs, respectively, in fibroblasts cultured from normal controls and MCD patients. Without the artificial electron acceptor phenazine methosulfate (PMS), MCADH activity in fibroblast mitochondrial sonic supernatants (MS) was 54% of control in two MCD cell lines (P less than 0.05). Addition of 10 mM PMS raised control acyl-CoA dehydrogenase activities 16-fold and revealed MCADH and SCADH activities to be 5 (P less than 0.01) and 73% (P greater than 0.1) of control, respectively. Thus, the catalytic defect in MCD involves substrate binding and/or dehydrogenation by MCADH and not the subsequent reoxidation of reduced MCADH by electron acceptors. 20 microM flavin adenine dinucleotide (FAD) did not stimulate MCD MCADH activity in either the 3H-release or electron-transfer(ring) flavoprotein-linked dye-reduction assays. Mixing experiments revealed no MCADH inhibitor in MCD MS; IVDH activities were identical in both control and MCD MS. In postmortem liver MS from another MCD patient, 3H2O formation from [2,3-3H]octanoyl-CoA was 15% of control. When 3H2O formation was assayed with 200 microM [2,3-3H]acyl-CoAs, 15 mM PMS, and 20 microM FAD in fibroblast sonic supernatants from seven MCD cell lines, SCADH, MCADH, and IVDH activities were 72-112% (P greater than 0.1), 4-9% (P less than 0.01), and 86-135% (P greater than 0.1) of control, respectively, revealing no significant biochemical heterogeneity among these patients.
Full text
PDF




Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Berge R. K. Purification and characterization of a long-chain acyl-CoA hydrolase from rat liver microsomes. Biochim Biophys Acta. 1979 Aug 30;574(2):321–333. doi: 10.1016/0005-2760(79)90013-4. [DOI] [PubMed] [Google Scholar]
- Borg L., Lindstedt S., Steen G., Hjalmarson O. Aliphatic C 6 -C 14 dicarboxylic acids in urine from an infant with fatal congenital lactic acidosis. Clin Chim Acta. 1972 Oct;41:363–366. doi: 10.1016/0009-8981(72)90532-3. [DOI] [PubMed] [Google Scholar]
- Coates P. M., Hale D. E., Stanley C. A., Glasgow A. M. Systemic carnitine deficiency simulating Reye syndrome. J Pediatr. 1984 Oct;105(4):679–679. doi: 10.1016/s0022-3476(84)80460-6. [DOI] [PubMed] [Google Scholar]
- Davidson B., Schulz H. Separation, properties, and regulation of acyl coenzyme A dehydrogenases from bovine heat and liver. Arch Biochem Biophys. 1982 Jan;213(1):155–162. doi: 10.1016/0003-9861(82)90450-7. [DOI] [PubMed] [Google Scholar]
- Divry P., David M., Gregersen N., Kølvraa S., Christensen E., Collet J. P., Dellamonica C., Cotte J. Dicarboxylic aciduria due to medium chain acyl CoA dehydrogenase defect. A cause of hypoglycemia in childhood. Acta Paediatr Scand. 1983 Nov;72(6):943–949. doi: 10.1111/j.1651-2227.1983.tb09849.x. [DOI] [PubMed] [Google Scholar]
- Dosman J., Crawhall J. C., Klassen G. A., Mamer O. A., Neumann P. Urinary excretion of C6-C10 dicarboxylic acids in glycogen storage disease types I and 3. Clin Chim Acta. 1974 Feb 28;51(1):93–101. doi: 10.1016/0009-8981(74)90065-5. [DOI] [PubMed] [Google Scholar]
- Furuta S., Miyazawa S., Hashimoto T. Induction of acyl-CoA dehydrogenases and electron transfer flavoprotein and their roles in fatty acid oxidation in rat liver mitochondria. J Biochem. 1981 Dec;90(6):1751–1756. doi: 10.1093/oxfordjournals.jbchem.a133652. [DOI] [PubMed] [Google Scholar]
- Furuta S., Miyazawa S., Hashimoto T. Purification and properties of rat liver acyl-CoA dehydrogenases and electron transfer flavoprotein. J Biochem. 1981 Dec;90(6):1739–1750. doi: 10.1093/oxfordjournals.jbchem.a133651. [DOI] [PubMed] [Google Scholar]
- Goodman S. I., Frerman F. E. Glutaric acidaemia type II (multiple acyl-CoA dehydrogenation deficiency). J Inherit Metab Dis. 1984;7 (Suppl 1):33–37. doi: 10.1007/BF03047371. [DOI] [PubMed] [Google Scholar]
- Gregersen N., Kølvraa S. Medium chain acyl-CoA dehydrogenase deficiency: apparent Km and Vmax values for fibroblast acyl-CoA dehydrogenase towards octanoyl CoA in patient and control cell lines. J Inherit Metab Dis. 1984;7 (Suppl 2):105–106. doi: 10.1007/978-94-009-5612-4_27. [DOI] [PubMed] [Google Scholar]
- Gregersen N., Lauritzen R., Rasmussen K. Suberylglycine excretion in the urine from a patient with dicarboxylic aciduria. Clin Chim Acta. 1976 Aug 2;70(3):417–425. doi: 10.1016/0009-8981(76)90355-7. [DOI] [PubMed] [Google Scholar]
- Hall C. L. Acyl-CoA dehydrogenases and electron-transferring flavoprotein. Methods Enzymol. 1978;53:502–518. doi: 10.1016/s0076-6879(78)53053-x. [DOI] [PubMed] [Google Scholar]
- Hall C. L., Kamin H. The purification and some properties of electron transfer flavoprotein and general fatty acyl coenzyme A dehydrogenase from pig liver mitochondria. J Biol Chem. 1975 May 10;250(9):3476–3486. [PubMed] [Google Scholar]
- Howat A. J., Bennett M. J., Variend S., Shaw L. Deficiency of medium chain fatty acylcoenzyme A dehydrogenase presenting as the sudden infant death syndrome. Br Med J (Clin Res Ed) 1984 Mar 31;288(6422):976–976. doi: 10.1136/bmj.288.6422.976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda Y., Dabrowski C., Tanaka K. Separation and properties of five distinct acyl-CoA dehydrogenases from rat liver mitochondria. Identification of a new 2-methyl branched chain acyl-CoA dehydrogenase. J Biol Chem. 1983 Jan 25;258(2):1066–1076. [PubMed] [Google Scholar]
- Ikeda Y., Tanaka K. Purification and characterization of isovaleryl coenzyme A dehydrogenase from rat liver mitochondria. J Biol Chem. 1983 Jan 25;258(2):1077–1085. [PubMed] [Google Scholar]
- Inestrosa N. C., Bronfman M., Leighton F. Purification of the peroxisomal fatty acyl-CoA oxidase from rat liver. Biochem Biophys Res Commun. 1980 Jul 16;95(1):7–12. doi: 10.1016/0006-291x(80)90696-8. [DOI] [PubMed] [Google Scholar]
- Karpati G., Carpenter S., Engel A. G., Watters G., Allen J., Rothman S., Klassen G., Mamer O. A. The syndrome of systemic carnitine deficiency. Clinical, morphologic, biochemical, and pathophysiologic features. Neurology. 1975 Jan;25(1):16–24. doi: 10.1212/wnl.25.1.16. [DOI] [PubMed] [Google Scholar]
- Kølvraa S., Gregersen N., Christensen E., Hobolth N. In vitro fibroblast studies in a patient with C6-C10-dicarboxylic aciduria: evidence for a defect in general acyl-CoA dehydrogenase. Clin Chim Acta. 1982 Nov 24;126(1):53–67. doi: 10.1016/0009-8981(82)90361-8. [DOI] [PubMed] [Google Scholar]
- Mamer O. A., Montgomery J. A., Colle E. Profiles in altered metabolism. II--(omega -- 1)-hydroxyacid excretion in a case of episodic hypoglycemia. Biomed Mass Spectrom. 1980 Feb;7(2):53–57. doi: 10.1002/bms.1200070203. [DOI] [PubMed] [Google Scholar]
- McKean M. C., Beckmann J. D., Frerman F. E. Subunit structure of electron transfer flavoprotein. J Biol Chem. 1983 Feb 10;258(3):1866–1870. [PubMed] [Google Scholar]
- Mortensen P. B., Gregersen N. The biological origin of ketotic dicarboxylic aciduria. II. In vivo and in vitro investigations of the beta-oxidation of C8-C16-dicarboxylic acids in unstarved, starved and diabetic rats. Biochim Biophys Acta. 1982 Mar 12;710(3):477–484. doi: 10.1016/0005-2760(82)90132-1. [DOI] [PubMed] [Google Scholar]
- Mortensen P. B., Gregersen N. The biological origin of ketotic dicarboxylic aciduria. In vivo and in vitro investigations of the omega-oxidation of C6-C16-monocarboxylic acids in unstarved, starved and diabetic rats. Biochim Biophys Acta. 1981 Dec 23;666(3):394–404. doi: 10.1016/0005-2760(81)90298-8. [DOI] [PubMed] [Google Scholar]
- Osumi T., Hashimoto T. Acyl-CoA oxidase of rat liver: a new enzyme for fatty acid oxidation. Biochem Biophys Res Commun. 1978 Jul 28;83(2):479–485. doi: 10.1016/0006-291x(78)91015-x. [DOI] [PubMed] [Google Scholar]
- Pettersen J. E., Jellum E., Eldjarn L. The occurrence of adipic and suberic acid in urine from ketotic patients. Clin Chim Acta. 1972 Apr;38(1):17–24. doi: 10.1016/0009-8981(72)90202-1. [DOI] [PubMed] [Google Scholar]
- Rhead W. J., Amendt B. A., Fritchman K. S., Felts S. J. Dicarboxylic aciduria: deficient [1-14C]octanoate oxidation and medium-chain acyl-CoA dehydrogenase in fibroblasts. Science. 1983 Jul 1;221(4605):73–75. doi: 10.1126/science.6857268. [DOI] [PubMed] [Google Scholar]
- Rhead W. J., Hall C. L., Tanaka K. Novel tritium release assays for isovaleryl-CoA and butyryl-CoA dehydrogenases. J Biol Chem. 1981 Feb 25;256(4):1616–1624. [PubMed] [Google Scholar]
- Rhead W. J., Tanaka K. Demonstration of a specific mitochondrial isovaleryl-CoA dehydrogenase deficiency in fibroblasts from patients with isovaleric acidemia. Proc Natl Acad Sci U S A. 1980 Jan;77(1):580–583. doi: 10.1073/pnas.77.1.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg L. E. Vitamin-responsive inherited metabolic disorders. Adv Hum Genet. 1976;6:1–74. doi: 10.1007/978-1-4615-8264-9_1. [DOI] [PubMed] [Google Scholar]
- Saudubray J. M., Coudé F. X., Demaugre F., Johnson C., Gibson K. M., Nyhan W. L. Oxidation of fatty acids in cultured fibroblasts: a model system for the detection and study of defects in oxidation. Pediatr Res. 1982 Oct;16(10):877–881. doi: 10.1203/00006450-198210000-00015. [DOI] [PubMed] [Google Scholar]
- Stanley C. A., Hale D. E., Coates P. M., Hall C. L., Corkey B. E., Yang W., Kelley R. I., Gonzales E. L., Williamson J. R., Baker L. Medium-chain acyl-CoA dehydrogenase deficiency in children with non-ketotic hypoglycemia and low carnitine levels. Pediatr Res. 1983 Nov;17(11):877–884. doi: 10.1203/00006450-198311000-00008. [DOI] [PubMed] [Google Scholar]
- Truscott R. J., Hick L., Pullin C., Halpern B., Wilcken B., Griffiths H., Silink M., Kilham H., Grunseit F. Dicarboxylic aciduria: the response to fasting. Clin Chim Acta. 1979 May 16;94(1):31–39. doi: 10.1016/0009-8981(79)90183-9. [DOI] [PubMed] [Google Scholar]