

Background: Hydroxylation of natural products is a key step for generating structural diversity.
Results: The revI gene disruptants accumulated reveromycin T, which showed stronger anticancer activity than reveromycin A.
Conclusion: Co-crystal structure analysis, site-directed mutagenesis, and biochemical characterization revealed how P450revI recognizes reveromycin T.
Significance: The co-crystal structure provides insight into the design of regio- and stereo-specific hydroxylation for novel spiroacetal compounds.
Keywords: Biosynthesis, Crystallography, Cytochrome P450, Enzyme Catalysis, Polyketide, Streptomyces, Reveromycin, Secondary Metabolite
Abstract
Numerous cytochrome P450s are involved in secondary metabolite biosynthesis. The biosynthetic gene cluster for reveromycin A (RM-A), which is a promising lead compound with anti-osteoclastic activity, also includes a P450 gene, revI. To understand the roles of P450revI, we comprehensively characterized the enzyme by genetic, kinetic, and structural studies. The revI gene disruptants (ΔrevI) resulted in accumulation of reveromycin T (RM-T), and revI gene complementation restored RM-A production, indicating that the physiological substrate of P450revI is RM-T. Indeed, the purified P450revI catalyzed the C18-hydroxylation of RM-T more efficiently than the other RM derivatives tested. Moreover, the 1.4 Å resolution co-crystal structure of P450revI with RM-T revealed that the substrate binds the enzyme with a folded compact conformation for C18-hydroxylation. To address the structure-enzyme activity relationship, site-directed mutagenesis was performed in P450revI. R190A and R81A mutations, which abolished salt bridge formation with C1 and C24 carboxyl groups of RM-T, respectively, resulted in significant loss of enzyme activity. The interaction between Arg190 and the C1 carboxyl group of RM-T elucidated why P450revI was unable to catalyze both RM-T 1-methyl ester and RM-T 1-ethyl ester. Moreover, the accumulation of RM-T in ΔrevI mutants enabled us to characterize its biological activity. Our results show that RM-T had stronger anticancer activity and isoleucyl-tRNA synthetase inhibition than RM-A. However, RM-T showed much less anti-osteoclastic activity than RM-A, indicating that hemisuccinate moiety is important for the activity. Structure-based P450revI engineering for novel hydroxylation and subsequent hemisuccinylation will help facilitate the development of RM derivatives with anti-osteoclast activity.
Introduction
Streptomyces produce a wide variety of natural products that are used for medicinal drugs (1) and bioprobes (2) to elucidate biological functions. Reveromycin A (RM-A)3 (3), which is a spiroacetal polyketide compound produced by Streptomyces sp. SN-593, inhibits bone resorption by specifically inducing apoptosis in osteoclasts (4). It has been demonstrated that RM-A inhibited bone metastasis of lung and prostate cancer cells through anti-osteoclastic activity (5–7). We recently reported that RM-A normalized bone metabolism and loss of alveolar bone during continuous tooth movement in osteoprotegerin-deficient (OPG−/−) mice (8). Despite extensive effort to optimize chemical synthesis of RM derivatives to increase biological activity, only limited success has been achieved (9, 10).
Understanding RM-A biosynthetic machinery and utilization of its unique enzymes are promising strategies for the creation of novel RM derivatives. Recently, we identified the RM-A biosynthetic gene cluster, which consists of 21 open reading frames spanning 91 kb (11). Gene disruption and complementation analyses revealed that polyketide synthase (PKS) genes (revA, revB, revC, and revD; Fig. 1A) are responsible for the generation of the polyketide skeleton (RM-A1a). Dihydroxy ketone synthase (revG) and spiroacetal synthase (revJ) genes that are essential for stereospecific spiroacetal formation have been identified (Fig. 1B).
FIGURE 1.

Gene organization (A) and RM-A biosynthetic pathway (B). A, the revI gene was organized in revJIH gene sets. B, formation of RM-T from post-PKS biosynthetic precursor RM-A1a into RM-T, C18-hydroxylation of RM-T, and subsequent hemisuccinylation.
We also identified the biosynthetic intermediate RM-T, which has been a promising target for the evaluation of biological activity because, in contrast to RM-A, it has a stable 6,6-spiroacetal structure due to the absence of hemisuccinate moiety. However, it was difficult to obtain enough RM-T from the wild-type strain because other biosynthetic reactions also occur.
Recently, we unexpectedly found novel RM-T derivatives (RM-T 1-methyl ester and RM-T 1-ethyl ester) in alcohol-added fermentation broth during feeding experiments (12). This finding prompted us to investigate the biological activity of RM-T derivatives. Although RM-A derivatives that were chemically modified at the C1 carboxyl group had reduced biological activity (9), the cytotoxic activities of the RM-T derivatives exhibited a 2–10-fold reduction of IC50 values against the HL-60 and K562 cell lines (12). Therefore, evaluating the biological activity of RM-T is also of interest.
The presence of only one P450 gene (revI) in the RM-A biosynthetic gene cluster indicates that the P450revI enzyme might be responsible for catalyzing C18-hydroxylation of RM-T. In this study, we disrupted the revI gene to evaluate its biosynthetic intermediate. Additionally, it remains unclear why no hydroxylated or hemisuccinylated metabolites derived from RM-T 1-esters were isolated from the culture broth, even in the presence of functional genes in the RM-A cluster. Kinetic analysis of P450revI is essential to obtain insight into the mechanism underlying this observation.
In this study, we comprehensively characterized RM-T C18-hydroxylase (P450revI) by gene disruption, co-crystal structure analysis, site-directed mutagenesis, and biochemical characterization. Taking advantage of the RM-T-accumulating strain, the biological activities of RM-T were also compared with those of other RM derivatives.
EXPERIMENTAL PROCEDURES
Chemicals
Ampicillin, kanamycin, and chloramphenicol were purchased from Nacalai Tesque, Inc. (Kyoto, Japan). Streptomycin, spectinomycin, thiostreptone, ribostamycin, NADPH, spinach ferredoxin (Fd), and spinach Fd-NADP+ reductase were purchased from Sigma-Aldrich. Carumonum was purchased from Takeda Pharmaceutical Co. Ltd. All other chemicals were of analytical grade. Spirofungin A (SF-A) and spirofungin B (SF-B) were chemically synthesized (13). RM-A1a, RM-A1b, RM-A1c, and RM-T were isolated from Streptomyces sp. SN-593, as described previously (3, 11). RM-T 1-methyl ester and RM-T 1-ethyl ester were isolated from alcohol-added fermentation of the wild-type strain (12). C18-hydroxy RM-T (RM-T1) was prepared as described previously, except that NaOH was used instead of LiOH (14).
Analytical Methods
The 1H and 13C NMR spectra were recorded on JEOL ECP-500 spectrometers in CD3OD. Chemical shifts were referenced to the residual solvent signal. UV spectra were measured using a JASCO V-630 BIO spectrophotometer. Optical rotations were recorded with a HORIBA SEPA-300 high-sensitivity polarimeter. The high-resolution mass spectrum was measured using a JEOL JMS-T100LC mass spectrometer. Thin layer chromatography was performed on a Merck 0.25-mm silica gel-precoated plate (60 F 254) with detection by UV light (254 nm) and/or 10% phosphomolybdic acid ethanol solution with heating.
Bacterial Strains and Plasmids
In brief, Escherichia coli DH5α (Takara), E. coli GM2929 hsdS::Tn10 carrying pUB307-aph::Tn7 (15), and E. coli BL21 StarTM (DE3) (Invitrogen) were used for general DNA manipulation, for E. coli-Streptomyces sp. SN-593 conjugation, and for the preparation of recombinant protein, respectively. E. coli expression vector pET28b(+) (Novagen) was used for preparation of recombinant protein. A biosynthetic gene involved in RM-A biosynthesis was isolated from Streptomyces sp. SN-593.
Culture Conditions
E. coli strains were grown at 37 °C in LB broth (1% tryptone, 0.5% yeast extract, and 1% NaCl), LB agar Miller (Nacalai Tesque, Inc.), or Terrific broth (1.2% Bacto-tryptone, 2.4% yeast extract, 0.4% glycerol, 0.231% KH2PO4, and 1.254% K2HPO4). For the selection of plasmid-containing E. coli cells, ampicillin (100 μg ml−1), kanamycin (25 μg ml−1), chloramphenicol (15 μg ml−1), streptomycin (50 μg ml−1), or spectinomycin (100 μg ml−1) was added to the LB plate.
Streptomyces sp. SN-593 cultures were routinely grown at 28 °C in an MS (2% soy flour, 2% d-(−)-mannitol, 2% agar) plate. For analysis of metabolites, spores were inoculated into a 500-ml cylindrical flask containing 70 ml of SY medium (0.1% yeast extract, 0.1% NZ-amine, and 1% starch, pH 7.0) for 2 days at 28 °C on a rotary shaker at 150 rpm. One milliliter of the preculture was inoculated in a 500-ml cylindrical flask containing 70 ml of the RM-A-producing medium (2% potato dextrose (Difco), 1% malt extract (Difco), 1% dried yeast (Asahi beer), 5% tomato juice (TABLE LAND, Maruzen Foods), 0.1% K2HPO4, 0.1% NaCl, 0.03% MgSO4·7H2O, 0.01% NaNO3, 0.005% ZnSO4·7H2O, and 0.005% CuSO4·5H2O, pH 6.5, before autoclaving) for 5 days at 28 °C on the same rotary shaker at 150 rpm.
Gene Disruption and Complementation
The revI genes were inactivated by PCR-targeted gene replacement using plasmids (pKD46, pKD13, and pCP20) and E. coli BW25113, as described previously (16, 17). Ampicillin-resistant pKD46, containing the λ Red-mediated recombination functions, was used with pCC1FOS (EPICENTRE® Biotechnologies) fosmids that encode for chloramphenicol resistance. pCC1FOS-11A02 containing the revI gene was used for gene replacement. The pKD13 plasmid was used as the template for the FRT-flanked kanamycin resistance gene cassette (17, 18).
An ∼5-kb DNA fragment with the disrupted gene in the middle was amplified by PrimeSTAR®HS DNA polymerase (TaKaRa) under the following conditions: 98 °C for 10 s, 25 cycles of 98 °C for 10 s, 64 °C for 5 s, and 68 °C for 5 min. Then it was ligated into the HindIII site of the pIM (11) to construct a disruption vector (pIM-ΔrevI). Primers used for gene disruption and the hybridization probe are listed in Table 1. To construct a complementation vector, the revI gene was ligated into the BamHI and HindIII sites of pTYM19 (19) with the aphII promoter. The primers used for gene complementation are listed in Table 1. For intergeneric conjugation with Streptomyces sp. SN-593, E. coli GM2929 hsdS::Tn10 (pUB307::Tn7) (15) was used as the plasmid donor strain, as described previously (20).
TABLE 1.
The primers used in this study
Underlines (6-bp and 39-bp) show restriction enzyme sites and homologous sequences for the target DNA regions, respectively.

a Primers are also used to make Southern hybridization probe.
b The pET28b(+)-revI was used as template DNA.
c The codons changed to generate the desired mutation are shown in boldface.
Extraction and LC-MS Analysis
Wild-type and revI gene disruptants (ΔrevI) were grown in a 500-ml cylindrical flask containing SY medium (70 ml) for 2 days. One milliliter of the preculture was inoculated in a 500-ml cylindrical flask containing RM-A-producing medium (70 ml). Five days after inoculation, 4 ml of culture broth was extracted with an equal volume of acetone and then concentrated to remove acetone. The pH of the aqueous extract was adjusted to 4 by adding acetic acid and then extracted twice with an equal volume of ethyl acetate. The organic layer was concentrated in vacuo and dissolved in 1.2 ml of methanol. An electrospray ionization-MS analysis was performed using a Waters Alliance HPLC system equipped with a mass spectrometer (Q-TRAP, Applied Biosystems). The HPLC conditions were as follows: column, XTerra®MSC18, 5 μm (2.1 × 150 mm); flow rate, 0.2 ml min−1; solvent A, water containing 0.05% formic acid; solvent B, acetonitrile. After injection of 1 μl of extracted sample into a column equilibrated with 30% solvent B, the column was developed with a linear gradient from 30 to 100% solvent B over the course of 20 min and kept at 100% solvent B for another 20 min. Mass spectra were collected in the electrospray ionization-negative mode.
Plasmid Construction for Heterologous Gene Expression
A PCR fragment of 1,194 bp containing revI was amplified from pCC1FOS-11A02 by PrimeSTAR®HS DNA polymerase (TaKaRa) under the following conditions: 98 °C for 10 s, 25 cycles of 98 °C for 10 s, 64 °C for 5 s, and 68 °C for 75 s. The primers used for revI gene amplification are listed in Table 1. After restriction enzyme digestion, the insert was ligated to pET28b(+) vector (Novagen) to construct pET28b(+)-revI.
Site-directed Mutagenesis
Mutagenesis of the revI gene was performed by standard conditions given in QuikChange protocols (Stratagene). The revI gene inserted into the NdeI/XhoI restriction sites of the pET28b(+) vector were used in combination with Pfu Turbo DNA polymerase (Stratagene), and the primer pairs are listed in Table 1. Site-specific mutations were confirmed by DNA sequencing.
Purification of P450revI for Enzyme Kinetics and X-ray Crystal Study
After overnight preculture of E. coli BL21 StarTM (DE3) (Invitrogen) transformed with pET28b(+)-revI or mutated pET28b(+)-revI, cells were inoculated into 2 liters of Terrific broth medium containing kanamycin (50 μg ml−1). Preculture was added to reach an initial optical density of 0.1 at 600 nm (A600). Cells were then cultured at 30 °C. When the A600 reached a value of 0.6, 5-aminolevulinic acid and isopropyl-β-d-thiogalactopyranoside were added to a final concentration of 0.5 mm. After further growth for 6 h at 30 °C, cells were harvested by centrifugation and frozen at −80 °C. All subsequent steps were performed at 4 °C.
After thawing on ice, cells were suspended in 80 ml of buffer A (50 mm Tris-HCl (pH 7.5), 500 mm NaCl, 10 mm 2-mercaptoethanol, and 20% glycerol) including 0.5 mg of lysozyme ml−1 and 500 units of benzonase. The cell suspension was sonicated on ice for 10 × 10 s with a 1-min interval after each sonication treatment (TOMY UD-200). Cellular debris was removed by centrifugation (10,000 × g for 30 min) (TOMY SRX-201). The supernatant was applied to the nickel-nitrilotriacetic acid (Ni-NTA)-agarose column (2 × 4 cm) (Qiagen), equilibrated with buffer A, and washed with 100 ml of buffer A.
Nonspecifically bound proteins were washed with 50 ml of buffer B (50 mm Tris-HCl (pH 7.5), 500 mm NaCl, 10 mm 2-mercaptoethanol, and 10% glycerol) containing 5 mm imidazole and 0.2% Tween 20. After washing with 200 ml of buffer B containing 5 mm imidazole, the column was further washed with 50 ml of buffer B containing 40 mm imidazole. His tag fusion protein was eluted with 35 ml of buffer B containing 250 mm imidazole. The purity of P450revI (CYP107E6) was confirmed by SDS-PAGE (Fig. 2A). For the enzyme assay, the purified proteins were dialyzed against 50 mm Tris-HCl (pH 7.5) containing 20% glycerol and frozen at −80 °C.
FIGURE 2.
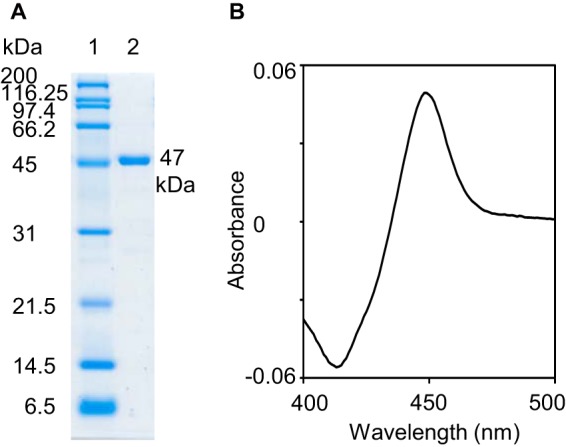
12.5% SDS-PAGE analysis of the His8 tag purified P450revI (A) and CO difference spectrum (B). A, lane 1, molecular mass markers; lane 2, 1 μg of wild-type P450revI was applied.
For x-ray analysis of P450revI, 60 units of thrombin was added to the Ni-NTA fraction (20 mg of P450revI) and dialyzed against 1 liter of buffer B containing 5 mm imidazole. After three buffer changes, the sample was passed through an Ni-NTA column (2 × 7 cm) to remove the His tag fragment. Then the passing fraction (50 ml) was applied to a benzamidine-Sepharose column to remove thrombin. His tag-free P450revI was dialyzed against buffer C (50 mm Tris-HCl (pH 7.5) and 10% glycerol) and applied to a Mono QTM 5/50 GL column (GE Healthcare) that was previously equilibrated with buffer C at a flow rate of 1 ml min−1. After washing for 5 min, a linear NaCl concentration gradient, from 0 to 500 mm, was established over 55 min. The P450revI fraction was concentrated with Amicon Ultracel-30K (Millipore Corp.), and the buffer was replaced with 10 mm Tris-HCl (pH 7.5). Finally, 10 mg of His tag-free P450revI was concentrated to 43 mg ml−1 for crystal analysis.
To investigate multimeric status, His tag-free P450revI was dialyzed against gel filtration buffer (50 mm Tris-HCl (pH 7.5), 200 mm NaCl, and 10% glycerol) and applied to a Superdex200 column (GE Healthcare), which was previously equilibrated with the same buffer, at a flow rate of 0.8 ml min−1. The column was calibrated with ferritin (440 kDa), aldolase (158 kDa), conalbumin (75 kDa), ovalbumin (43 kDa), carbonic anhydrase (29 kDa), ribonuclease A (13.7 kDa), and aprotinin (6.5 kDa). The elution was established over 80 min. The P450revI elution peak corresponded to a molecular mass of 42 kDa, suggesting that P450revI was in the monomeric form.
CO Difference Spectra
CO difference spectroscopy was performed to quantify and normalize the amount of correctly folded P450 enzyme used in enzyme activity assays. The amount of functional P450 was calculated using the extinction coefficient of 91 mm−1 cm−1 at 450 nm (Fig. 2B) (21).
Measurement of P450revI Activity
Kinetic assays were performed in 1.5-ml tubes with a final reaction volume of 0.2 ml containing 50 mm Tris-HCl (pH 7.5), 1 mm NADPH, 0.1 mg ml−1 spinach Fd, 1 unit ml−1 spinach Fd-NADP+ reductase, 0.5–15 μm RM-T, and 1 pmol of P450revI (measured by CO difference spectroscopy).
Substrate concentrations of RM-A1c varied from 5 to 30 μm in the presence of 100 pmol of P450revI, and those of SF-A varied from 34 to 224 μm in the presence of 200 pmol of P450revI. After preincubation at 30 °C for 5 min, the reactions were initiated by the addition of NADPH. After incubation for 1 min (RM-T), 0.5 min (RM-A1c), and 10 min (SF-A), the reaction was terminated by the rapid addition of 4 μl of acetic acid. After extraction with ethyl acetate (0.4 × 2 ml), the supernatant was dried and dissolved in 0.2 ml of methanol.
The sample (20 μl) was subjected to LC-MS in the conditions described above. The product peak was calculated from the standard curve, which was obtained from the maximum UV wavelength (238 nm) of the RM-T standard. The enzyme-specific activity (pmol of product formed min−1 pmol of enzyme−1) was calculated by time-dependent product formation. The kinetic constants were calculated by a nonlinear regression fit to the Michaelis-Menten equation using SigmaPlot11. Kinetic analyses of mutant enzymes were performed in the presence of 100 pmol of R81A, 10 pmol of F91G, 1 pmol of L287F, and 50 pmol of A292V. The concentrations of RM-T varied from 7.5 to 150 μm. After incubation for 0.5 min (F91G and A292V) or 1 min (R81A and L287F), mutant enzyme reactions were terminated and analyzed, as described above.
Crystallization
Crystallization of P450revI was performed using sitting drop vapor diffusion. The reservoir solution contained 0.2 m sodium tartrate and 22% polyethylene glycol 3350. The substrate-saturated solution was added to the enzyme solution (15 mg/ml) at a ratio of 1:3, just before crystallization. The substrate-bound form of the enzyme solution (0.5 μl) was mixed with an equal volume of reservoir solution and incubated at 20 °C.
Single crystals appeared within 2–3 days. The crystals were cryoprotected by a solution containing 0.2 m sodium tartrate, 25% polyethylene glycol 3350, and 30% glycerol. The crystals were flash-frozen and stored in liquid nitrogen until data collection.
Data Collection and Structure Determination
All data were collected at SPring-8, beamline BL44B2, with a quantum 210 detector (ADSC). The data were processed with HKL2000 (22). The crystal was in the space group P21 with the cell dimensions a = 52.78, b = 73.43, and c = 58.31 Å and β = 112.11°. The asymmetric unit contained one molecule of the enzyme. The experimental phases and the initial structural model for P450revI were obtained using the multiple-wavelength anomalous diffraction method and the program SOLVE/RESOLVE (23). Further model building was conducted using ARP/wARP. Manual model building was done with COOT (24), and structural refinement was performed with REFMAC5 from CCP4 (25). A simulated annealing composite omit map was calculated using PHENIX (26). All figures were drawn with PyMOL (version 1.7.2.1, Schrödinger, LLC).
Growth-inhibiting Activity against Animal Cells
The HL-60, K562, and tsFT210 cell lines were cultured in RPMI 1640 (Invitrogen), supplemented with fetal calf serum (10% for HL-60 and K562) and calf serum (5% for tsFT210) (27), 50 units/ml penicillin, and 50 μg/ml streptomycin at 37 °C for HL-60 and K562 and at 32 °C for tsFT210 in a humidified atmosphere containing 5% CO2.
Each cell line was used to seed a 96-well culture plate (1.5 × 104 cells/well/100 μl for HL-60 and K562 and 1.6 × 104 cells/well/100 μl for tsFT210) (IWAKI) and exposed to test compounds for 48 h. Cell growth was measured using Cell Count Reagent SF (Nacalai Tesque), per the manufacturer's instructions. Briefly, one-tenth volume of the WST-8 solution was added to each well, and the plates were incubated at 37 °C (HL-60 and K562) or 32 °C (tsFT210) for 1 h. Subsequently, cell growth was measured at 450 nm on a microplate reader (PerkinElmer Life Sciences).
Antibacterial Test
E. coli (HO141 strain) was precultured in a medium containing 0.5% polypeptone, 0.5% meat extract, 0.3% NaCl, and 0.001% SDS at 37 °C. E. coli solution was prepared such that the absorbance at 600 nm was 0.005, and 100 μl of the solution was seeded in a 96-well plate. Each chemical was added to the medium at a final concentration of 0.5% (v/v), prior to incubation at 37 °C for 6 h. A600 was measured using a microplate reader.
Growth-inhibiting Activity against Yeast
Budding yeast (MLC30M strain) was precultured in a medium containing 2% polypeptone, 1% yeast extract, 2% glucose, 0.02% adenine, and 0.001% SDS at 30 °C. Yeast solution was prepared such that the absorbance was 0.05, and 100 μl of the solution was seeded in a 96-well plate. Each chemical was added at a final concentration of 0.5% (v/v), prior to incubation at 30 °C. Eighteen hours after the addition of the agent, A600 was measured using a microplate reader.
Cytotoxic Activity against Osteoclasts
Bone marrow cells were harvested from thigh and shin bones of 5-week-old male ddY mice (Japan SLC, Inc.) and seeded in a type I collagen-coated plate (IWAKI) in α-minimum Eagle's medium (Sigma-Aldrich) with 10% fetal bovine serum, 0.5% penicillin/streptomycin solution, 50 ng ml−1 human M-CSF (Leukoprol, Kyowa Hakko), and 1 ng ml−1 human TGF-β1 (R&D Systems). Bone marrow cells were then maintained at 37 °C in a humidified atmosphere containing 5% CO2 for 3 days. Cells were washed twice with PBS, and cells adhered onto the plate were then used as bone marrow macrophage cells.
Bone marrow macrophage cells were further maintained in a culture medium in which α-minimum Eagle's medium was added with 10% fetal bovine serum, 0.5% penicillin/streptomycin solution, 50 ng ml−1 human M-CSF, and 50 ng ml−1 human soluble RANKL (PeproTech) at 37 °C in a humidified atmosphere containing 5% CO2 for 3 days to differentiate into osteoclasts. Each chemical was added to osteoclasts at 0.5% (v/v), and the culture was maintained at 37 °C in a humidified atmosphere containing 5% CO2 for 24 h. Subsequently, cells were treated with PBS solution containing 3.7% formalin at room temperature for 30 min. After the solution was removed, the cells were further treated with acetone/ethanol solution (1:1, v/v) at room temperature for 1 min, and the solution was removed to dry the cells. Immobilized cells were subjected to a reaction in TRAP solution (50 mm sodium tartrate, 90 mm sodium acetate, 0.01% naphthol AS-MX phosphate (Sigma), 0.05% fast red violet LB salt (Sigma), pH 5.0) at room temperature for 30 min and then washed with distilled water. The number of TRAP-positive multinucleated osteoclasts was counted, and the rate of survival was determined.
Inhibitory Activity against Isoleucyl tRNA Synthetase
To a solution for enzymatic reaction (20 mm imidazole (pH 7.5), 75 mm MgCl2, 0.5 mm DTT, 1 unit ml−1 tRNA (E. coli origin, Sigma), 3 mm ATP, 1 μm isoleucine, 10 μCi ml−1 [3H]isoleucine (GE Healthcare), and 10 μg of protein (HT1080 cell lysate)), each chemical was added at a final concentration of 1% (v/v) to attain a final volume of 100 μl, and the reaction was carried out at 25 °C for 20 min. Subsequently, 1 mg ml−1 BSA solution (400 μl) and 10% TCA solution (500 μl) were then added to terminate the reaction, following which the reaction mixtures were left to stand at 4 °C overnight. A precipitate obtained by centrifugation was transferred onto a GF-C filter (Whatman) and washed three times with 5% TCA solution, and the filter was dried. Two milliliters of Aquasol-2 (PerkinElmer Life Sciences) and the filter were placed in a vial and vigorously stirred. The amount of [3H]isoleucine was measured by a liquid scintillation counter (Beckman) to determine the rate of enzyme activity.
RESULTS
Accumulation of RM-T in the revI Gene Disruptants
Modification of hydroxyl groups by tailoring enzymes is often observed in the biosynthesis of secondary metabolites. We expected that the P450 gene (revI) found in the RM-A gene cluster might catalyze the key hydroxylation step for subsequent hemisuccinylation (Fig. 1B). Therefore, to identify the role of P450revI, we performed revI gene disruption by homologous recombination (Fig. 3, A and B). The metabolite analysis of the ΔrevI mutant strain revealed accumulation of the biosynthetic intermediate RM-T (Fig. 3, C–E). Restoration of RM-A production by the reintroduction of the revI gene into ΔrevI (Fig. 3F) strongly indicated that RM-T is consumed by P450revI.
FIGURE 3.

Gene disruption of revI and metabolite analysis. A, scheme of revI disruption and restriction map of the wild-type and ΔrevI mutant strains. The bar shows the expected fragment size (in bp) of BamHI digestion. An ∼5-kb DNA fragment was amplified from pIM-ΔrevI and used as a Southern hybridization probe. B, Southern blot analysis of the wild-type (lane 2) and mutant strains (lanes 3–6; individual isolations). Genomic DNA digested with BamHI was resolved on a 0.9% agarose gel and stained by EtBr. The arrows indicate the expected size of the DNA fragments from the wild-type (solid) and mutant (open) strains. C–F, LC-MS analysis of RM standards (C) and RMs from the culture extract of the wild-type strain (D), ΔrevI mutant strain (E), and the ΔrevI mutant strain complemented by pTYM19-Paph-revI (F).
P450revI Catalyzes the C18 Hydroxylation of RM-T
In order to characterize the function of P450revI, the enzyme was heterologously expressed in E. coli and purified by an Ni-NTA column (Fig. 2A). Proper protein folding was confirmed by CO difference spectroscopy (Fig. 2B). Because of the accumulation of RM-T in the ΔrevI mutant strain, P450revI reaction was examined using RM-T. As expected, P450revI efficiently converted RM-T into RM-T1 (Fig. 4, A and B). The LC-MS analysis clearly demonstrated the P450revI reaction product (m/z 559 [M-H]−). The retention time (12.82 min), UV, and mass spectrum of the reaction product were identical to that of the standard of RM-T1, prepared from RM-A.
FIGURE 4.
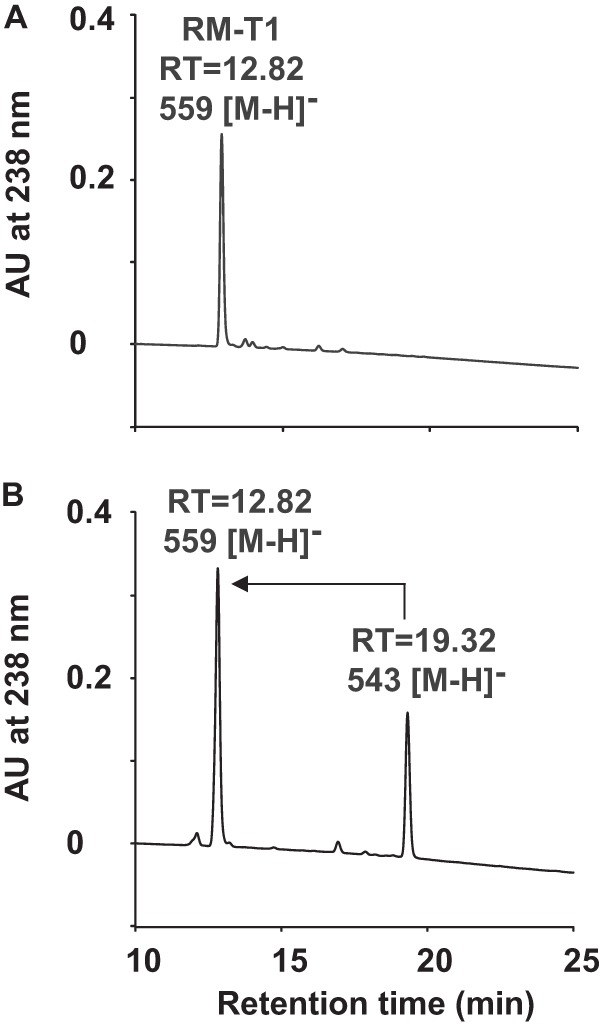
HPLC chromatogram of P450revI assay. A, HPLC chromatogram of the synthetic standard of RM-T1. B, HPLC profile of the P450revI reaction with RM-T. The enzyme (50 pmol) was incubated with 40 μm of RM-T for 1 min, and the reaction products were analyzed by LC-MS as described under “Experimental Procedures.”
To address substrate specificity, we tested the P450revI reaction with RM derivatives obtained from both biosynthesis and chemical synthesis research (11–13). P450revI was unable to catalyze the compounds containing 15R spiroacetal stereochemistry, such as SF-B; acyclic biosynthetic precursor RM-A1a; biosynthetic shunt product RM-A1b, which lost spiroacetal integrity; and the RM-T 1-methyl ester and RM-T 1-ethyl ester, which are modified at the C1 carboxyl group of RM-T. In contrast, the enzyme produced monooxygenated products from SF-A and RM-A1c containing 15S spiroacetal stereochemistry (Fig. 5).
FIGURE 5.

Structure-function analyses of substrates catalyzed by P450revI.
To characterize enzymatic properties, the kinetic parameters were also investigated for RM-T, SF-A, and RM-A1c. We found that P450revI showed the highest catalytic efficiency for RM-T (Table 2). A comparative study using SF-A and SF-B suggested the importance of 15S spiroacetal stereochemistry for substrate recognition by P450revI. Comparison of RM-A1c with RM-A1a and RM-A1b indicates that the integrity of the spiroacetal core structure was required for substrate recognition.
TABLE 2.
Comparison of the P450revI kinetic constants for RM-T and its derivatives
| Substrate | Km | kcat | kcat/Km |
|---|---|---|---|
| μm | min−1 | min−1μm−1 | |
| RM-T | 0.89 ± 0.05 | 331 ± 5 | 372 |
| RM-A1c | 11.7 ± 1.7 | 27.6 ± 1.7 | 2.36 |
| SF-A | 39.0 ± 2.6 | 8.04 ± 0.15 | 0.206 |
Loss of the catalytic activity for RM-T 1-methyl ester and RM-T 1-ethyl ester strongly indicated the importance of the C1 carboxyl group of RM-T for substrate recognition by P450revI (Fig. 5). Comparison of kcat/Km values between RM-A1c and RM-T suggested an involvement of the C24 carboxyl group in substrate recognition by P450revI. Additionally, comparison of kcat/Km values between SF-A and RM-T suggested that the C18 butyl group attached to the spiroacetal structure has a significant role for substrate recognition by P450revI. Based on gene disruption and biochemical analysis, we concluded that RM-T is a physiological substrate of P450revI.
Co-Crystal Structure of P450revI with Physiological Substrate RM-T
In order to understand the RM-T recognition by P450revI and the substrate structure-reactivity relationship, we determined the crystal structure of P450revI complexed with RM-T. Crystallographic and refinement statistics are summarized in Table 3. P450revI adopts the typical triangular P450 fold with a long I helix, which has two highly conserved residues, Glu244 and Thr245, which are important for oxygen activation (Fig. 6, A and B).
TABLE 3.
Data collection and refinement statistics
Numbers in parentheses show the values for the highest resolution shell.

a Rsym = ∑hkl|I(hkl) − 〈I(hkl)〉|/∑hkl|I(hkl)|, where 〈I(hkl)〉 is the average of I(hkl).
b Figure of merit.
c Rfactor = ∑hkl|Fobs(hkl) − Fcalc(hkl) /∑hklFobs(hkl), where Fobs and Fcalc are observed and calculated structure factor amplitudes, respectively. Free R-factor is the R-factor calculated using 5% of the selected reflections not included for refinement.
FIGURE 6.

Overall structure of P450revI (A), close-up view of the substrate binding site (B), and substrate binding pocket of P450revI-RM-T complex (C). Glu244 and Thr245 in the I helix, heme axial ligand Cys353, and the substrate are shown as stick models. BC- and FG-loops and a loop following the K helix are indicated by arrows. Heme and RM-T are shown by gray and green stick models, respectively.
The I helix forms the wall of the large substrate binding cavity, which has a volume of 2,446 Å3. The cavity is covered by a BC and FG loop. A loop region following the K helix also lines the cavity (Fig. 6C). The substrate side chain with the C1 carboxyl group aligns in parallel with the butyl chain at C18 to fit into the cavity, and the substrate thus has a compact conformation (Fig. 7A). The side chain with the C24 carboxyl group is roughly perpendicular to these two substrate side chains. One of the spiroacetal rings is above the heme D ring and is parallel to the heme.
FIGURE 7.

Substrate conformation and P450revI-substrate interactions. Simulated annealing-composite omit maps for the substrate (A) as well as side chains and a water molecule that interact with the substrate (B) are shown as mesh at 1.0 σ level. C, amino acid side chains, which make hydrogen bonding or hydrophobic interactions with the substrate.
The other spiroacetal ring is almost perpendicular to the heme. Arg190 makes a salt bridge with the C1 carboxyl group at a distance of 2.8 Å. Arg81, which is at the top of the substrate binding cavity, forms a bifurcated hydrogen bond with the C5 hydroxyl and C24 carboxyl groups. The oxygen atom of the spiroacetal ring is hydrogen-bonded with a water molecule (Wat246), which also makes a hydrogen bond with the heme 7-propionate group. Leu176, Ile240, Leu287, and Val391 come in contact with the hydrophobic moiety of the substrate, which includes C4–C13 atoms.
The phenyl ring of Phe91 is parallel to the butyl side chain of RM-T (Fig. 7B). Although P450revI hydroxylates the C18 atom, and no other hydroxylation product was detected, the nearest carbon atom to the heme iron is C16 (4.5 Å), and C18 is the third nearest one (5.2 Å). Ala292 is located in the loop that adopts two conformations (Gly290–Ala293), and this side chain in conformer A faces the substrate (Fig. 8). Very similar conformational ambiguity was also found at the Thr284–Phe286 region in P450 MycG, which catalyzes sequential hydroxylation and epoxidation in mycinamicin biosynthesis. Carbon atoms that are monooxygenated are relatively distant from the heme iron (9–10 Å), and reorientation of the substrate allows for the required monooxygenation. Such relocation was supported by NMR relaxation experiments in solution, and the attainment of the productive orientation of the substrate is probably coupled with conformational fluctuation of the Thr284–Phe286 (28). Therefore, for P450revI, the flexible loop region, Gly290–Ala293 also probably contributes to translocation of the substrate to allow C18-hydroxylation.
FIGURE 8.

Alternate conformations at Gly290–Ala293. The orange and green loop models show the alternate conformation (A). Shown is a simulated annealing-composite omit map for side chains and a water molecule, which interact with the substrate (B).
P450revI shows significant structural similarity to CYP105A1 (P450 SU-1), which is isolated from Streptomyces griseolus and can catalyze vitamin D3 hydroxylation. Fig. 9 illustrates the superposed structures of a vitamin D3-bound mutant CYP105A1 and RM-T-bound P450revI. CYP105A1 also has a large substrate binding cavity that can accommodate vitamin D3 (29). The bound substrate makes a salt bridge between Arg193 and the 3β-OH of the substrate, and a very similar interaction is found in the P450revI and RM-T complex, where Arg190 makes a salt bride with the C1 carboxyl group (Fig. 7B).
FIGURE 9.

Overlay structures of P450revI and CYP105A1. P450revI complexed with RM-T and CYP105A1 are shown as green and magenta loop models, respectively. Hemes and RM-T are shown as stick models (A). B, hydrogen bond between Arg193 in CYP105A1 and dihydroxyvitamin D3.
Site-directed Mutagenesis of P450revI
To gain further insight into the mode of substrate recognition by P450revI, site-directed mutagenesis was introduced at the amino acid positions of Arg81, Phe91, Arg190, Leu287, and Ala292, which can interact with RM-T. After purification of mutant P450revIs, CO difference spectra were determined. Like wild-type P450revI, the mutant enzymes showed clear absorption maxima at 450 nm (Fig. 10), which is consistent with that seen for properly folded enzymes that possess the heme cofactor oriented in the correct electron spin state. After estimation of the amount of properly folded enzyme, kinetic properties of the mutant enzymes were characterized. Arg190, which constructed a salt bridge with the C1 carboxyl group of RM-T (Fig. 7B), was converted to Ala190. The abolishment of the salt bridge in the R190A mutant completely eliminated the RM-T hydroxylase activity (Table 4).
FIGURE 10.
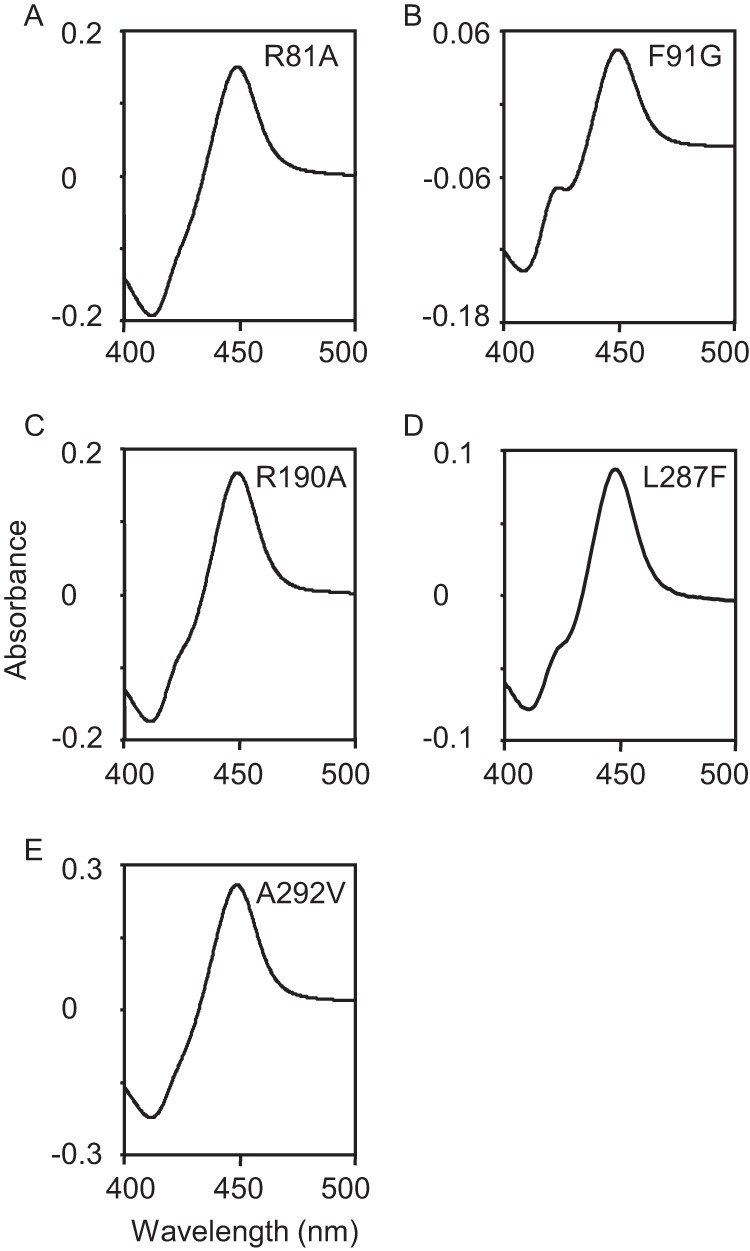
CO difference spectra of mutant P450revIs.
TABLE 4.
Comparison of the kinetic constants of wild type P450revI and several site-specific mutants for the substrate RM-T
The wild type kcat/Km value of 372 min−1 μm−1 was considered 100%.
| Enzyme | Km | kcat | kcat/Km | Relative activity |
|---|---|---|---|---|
| μm | min−1 | min−1 μm−1 | % | |
| Wild typea | 0.89 ± 0.05 | 331 ± 5 | 372 | 100 |
| L287F | 5.58 ± 0.82 | 89.5 ± 3.5 | 16.0 | 4.3 |
| F91G | 12.5 ± 2.0 | 197 ± 11 | 15.8 | 4.2 |
| A292V | 13.1 ± 1.1 | 143 ± 3 | 10.9 | 2.9 |
| R81A | 75.2 ± 9.4 | 84.4 ± 5.2 | 1.12 | 0.3 |
| R190A | NAb | NA | NA |
a Data are from Table 2.
b NA, no activity.
Consistently, wild-type P450revI was unable to catalyze RM-T 1-methyl ester and RM-T 1-ethyl ester (Fig. 5). Similarly, Arg81, which constructed a salt bridge with the C24 carboxyl group of RM-T (Fig. 7B), was converted to Ala81. The elimination of the salt bridge in the R81A mutant revealed 330-fold lower catalytic efficiency than that of the wild-type P450revI (Table 4). Similarly, wild-type P450revI showed 160-fold lower catalytic efficiency for RM-A1c, which is absent from the C24 carboxyl group (Fig. 5). Phe91 in the BC-loop constructed hydrophobic pocket accommodated the butyl residue at C18 of the spiroacetal core structure (Fig. 7B).
The F91G mutant was constructed to evaluate RM-T substrate recognition. As expected, the mutant showed 24-fold lower catalytic efficiency for RM-T than that of the wild-type (Table 4). The recognition of C18 alkyl residue of spiroacetal core structure by wild-type P450revI was also evaluated using SF-A, which has a methyl group instead of a butyl group, which is found in RM-T. Wild-type P450revI showed significantly low catalytic efficiency for SF-A (Table 2 and Fig. 5). Comparative studies support the recognition of C18 butyl residues by Phe91. Moreover, based on co-crystal structure (Figs. 7 and 8), the roles of Leu287 and Ala292, which interact with the substrate, on catalytic efficiency of C18-hydroxylation of RM-T were evaluated. A larger hydrophobic residue was introduced by site-directed mutagenesis to produce L287F and A292V. In this experiment, we expected a different regio-specific hydroxylation product from the mutants. However, the mutants dominantly produced RM-T1. Additionally, the mutants showed 23 and 34 times lower catalytic efficiency than that of the wild-type P450revI, primarily because of the higher Km values of these mutants (Table 4). These results suggest that P450revI does not have extra room to facilitate relocation of the substrate and that the substrate binding site of this enzyme is highly optimized for C18-hydroxylation of RM-T.
Biological Activity of RM-T
RM-T accumulation in the ΔrevI mutant strain (Fig. 3) enabled us to evaluate its biological activity (Table 5). RM-T efficiently inhibited the activity of isoleucyl tRNA synthetase, which is the molecular target of RM-A (30). Consistently, RM-T showed stronger cytotoxic activity against cancer cell lines than any other RM derivatives tested. RM-T also demonstrated growth-inhibitory activity against yeast cells, suggesting its application as an antifungal agent. On the other hand, RM-T showed much less anti-osteoclastic activity than RM-A, suggesting that the hemisuccinate moiety in addition to the C1 and C24 carboxyl groups was essential to exhibit the strong activity (10).
TABLE 5.
Biological activity of RM derivatives
The assay conditions are described under “Experimental Procedures.”

DISCUSSION
In natural product biosynthesis, introduction of a hydroxyl group by P450 is a key step for subsequent modifications, such as glycosylation, acetylation, and methylation. Understanding a variety of substrate recognitions and reactions by P450s will expand the chemical diversity for drug leads (31). Here, we comprehensively characterized P450revI involved in RM-A biosynthesis. We demonstrated that the C18-hydroxylation of RM-T by P450revI is essential for subsequent hemisuccinate formation (Figs. 3 and 4). The 1.4 Å resolution co-crystal structure of P450revI with RM-T demonstrated the mode of substrate recognition (Figs. 6–8). The biosynthesis of RM-A is unique because C18-hydroxylation to afford the tertiary alcohol was followed by ester formation, which chemically required high pressure (1.5 gigapascals) (32, 33).
Structure-function analyses demonstrated that P450revI efficiently catalyzed substrates with 15S spiroacetal structure whose stereochemistry was produced by RevG and RevJ in the RM-A biosynthetic pathway (11). Moreover, the interaction between Arg190 of P450revI and the C1 carboxyl group of RM-T (Fig. 7B) clearly explained the reason why alcohol-added fermentation resulted in the accumulation of RM-T 1-methyl ester and RM-T 1-ethyl ester, the latter of which does not contain hemisuccinylated moiety. Additionally, this finding led us to speculate that RM-T 1-methyl and 1-ethyl ester intermediates might be released as a post-PKS biosynthetic precursor when either methanol or ethanol was added to the culture medium.
Interestingly, RM-T showed stronger anticancer and isoleucyl tRNA synthetase inhibition activity than RM-A (Table 5). Because the presence of hemisuccinate moiety at the C18 position causes the conversion of bioactive 6,6-spiroacetal (RM-A) into inactive 5,6-spiroacetal structure (RM-B) (Fig. 1B) (11), the stability of 6,6-spiroacetal structure of RM-T might be one of the reasons for the strong biological activities observed. Unfortunately, RM-T showed weak anti-osteoclastic activity, suggesting that hemisuccinate moiety is important for efficient incorporation into osteoclasts in an acidic environment (4).
To minimize the conversion of 5,6-spiroacetal formation, mutagenesis in P450revI for novel regio- and stereo-specific hydroxylation in 6,6-spiroaceal structure and subsequent hemisuccinylation might be an approach to enhance the stability of RM derivatives that retain strong anti-osteoclast activity. Therefore, future research should focus on the engineering of P450revI to catalyze novel regio- and stereo-specific hydroxylation.
Reintroduction of the engineered P450revI into the ΔrevI mutant is promising for the production of novel RM derivatives. Based on co-crystal structure (Fig. 7B), C16 of RM-T was located closer to the heme-iron reaction center (4.5 Å) than C18 (5.2 Å). Therefore, changing the hydroxylation position from C18 to C16 will be the first step. The co-crystal structure of P450revI-RM-T provided insight into the rational design of novel hydroxylation products. Phe91, Leu287, and flexible loop Gly290–Ala293 found in P450revI (Figs. 7B and 8) will be the target amino acid residues for research on combinatorial mutagenesis.
Moreover, because of the biological activity of SF-A, which is structurally similar to RM-A (Table 5) (10, 34, 35), creating its hemisuccinylated product is an intriguing approach to evaluate anti-osteoclastic activity. Gene organization similar to the RM-A biosynthetic gene cluster has been found in the genome sequence of Streptomyces violaceusniger Tü4113 (NC_015957), which is a strain that produces both SF-A and SF-B (34). Consistent with the structure of SF-A (Fig. 5), cytochrome P450 and possible modification genes were not found in the putative cluster.
For the creation of novel hemisuccinylated compounds, one approach is the introduction of modification genes (revI, revK, revL, and revM) found in the RM-A gene cluster into S. violaceusniger Tü4113. The other approach is to perform domain exchange of butylmalonyl-CoA-specific acyltransferase (AT4) from RM-PKS into methylmalonyl-CoA-specific AT from the putative SF-A gene cluster. In the latter combinatorial biosynthesis approach, the utilization of the ΔrevI strain and reintroduction of P450revI mutants catalyzing regio- and stereo-specific hydroxylation will further expand chemical diversity for drug leads.
Acknowledgments
We thank E. Oowada, H. Takagi, and H. Aono for technical assistance.
This work was partially supported by Japan Society for the Promotion of Science (JSPS) KAKENHI Grants 24248022 (to H. O.) and 24380052 (to S. T.) by the science and technology research promotion program for agriculture, forestry, fisheries, and the food industry and by a grant for the “project focused on developing key technology of discovering and manufacturing drugs for next-generation treatment and diagnosis” from the Ministry of Economy, Trade, and Industry (METI), Japan.
The atomic coordinates and structure factors (code 3WVS) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- RM
- reveromycin
- CYP or P450
- cytochrome P450
- PKS
- polyketide synthase
- SF
- spirofungin
- Ni-NTA
- nickel-nitrilotriacetic acid
- IRS
- isoleucyl tRNA synthetase.
REFERENCES
- 1. Clardy J., Walsh C. (2004) Lessons from natural molecules. Nature 432, 829–837 [DOI] [PubMed] [Google Scholar]
- 2. Usui T., Osada H. (2000) Biochemical Tools for Investigating Cell Function. in Bioprobes (Osada H., ed) pp. 125–305, Springer, Tokyo [Google Scholar]
- 3. Osada H., Koshino H., Isono K., Takahashi H., Kawanishi G. (1991) Reveromycin A, a new antibiotic which inhibits the mitogenic activity of epidermal growth factor. J. Antibiot. 44, 259–261 [DOI] [PubMed] [Google Scholar]
- 4. Woo J. T., Kawatani M., Kato M., Shinki T., Yonezawa T., Kanoh N., Nakagawa H., Takami M., Lee K. H., Stern P. H., Nagai K., Osada H. (2006) Reveromycin A, an agent for osteoporosis, inhibits bone resorption by inducing apoptosis specifically in osteoclasts. Proc. Natl. Acad. Sci. U.S.A. 103, 4729–4734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Muguruma H., Yano S., Kakiuchi S., Uehara H., Kawatani M., Osada H., Sone S. (2005) Reveromycin A inhibits osteolytic bone metastasis of small-cell lung cancer cells, SBC-5, through an antiosteoclastic activity. Clin. Cancer Res. 11, 8822–8828 [DOI] [PubMed] [Google Scholar]
- 6. Yano A., Tsutsumi S., Soga S., Lee M. J., Trepel J., Osada H., Neckers L. (2008) Inhibition of Hsp90 activates osteoclast c-Src signaling and promotes growth of prostate carcinoma cells in bone. Proc. Natl. Acad. Sci. U.S.A. 105, 15541–15546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hiraoka K., Zenmyo M., Watari K., Iguchi H., Fotovati A., Kimura Y. N., Hosoi F., Shoda T., Nagata K., Osada H., Ono M., Kuwano M. (2008) Inhibition of bone and muscle metastases of lung cancer cells by a decrease in the number of monocytes/macrophages. Cancer Sci. 99, 1595–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tanaka M., Miyazawa K., Tabuchi M., Yabumoto T., Kadota M., Yoshizako M., Yamane C., Kawatani M., Osada H., Maeda H., Goto S. (2012) Effect of Reveromycin A on experimental tooth movement in OPG−/− mice. J. Dent. Res. 91, 771–776 [DOI] [PubMed] [Google Scholar]
- 9. Shimizu T., Usui T., Machida K., Furuya K., Osada H., Nakata T. (2002) Chemical modification of reveromycin A and its biological activities. Bioorg. Med. Chem. Lett. 12, 3363–3366 [DOI] [PubMed] [Google Scholar]
- 10. Shimizu T., Usui T., Fujikura M., Kawatani M., Satoh T., Machida K., Kanoh N., Woo J. T., Osada H., Sodeoka M. (2008) Synthesis and biological activities of reveromycin A and spirofungin A derivatives. Bioorg. Med. Chem. Lett. 18, 3756–3760 [DOI] [PubMed] [Google Scholar]
- 11. Takahashi S., Toyoda A., Sekiyama Y., Takagi H., Nogawa T., Uramoto M., Suzuki R., Koshino H., Kumano T., Panthee S., Dairi T., Ishikawa J., Ikeda H., Sakaki Y., Osada H. (2011) Reveromycin A biosynthesis uses RevG and RevJ for stereospecific spiroacetal formation. Nat. Chem. Biol. 7, 461–468 [DOI] [PubMed] [Google Scholar]
- 12. Nogawa T., Takahashi S., Sekiyama Y., Takagi H., Uramoto M., Koshino H., Kawatani M., Shimizu T., Osada H. (2013) Creation of novel reveromycin derivatives by alcohol-added fermentation. J. Antibiot. 66, 247–250 [DOI] [PubMed] [Google Scholar]
- 13. Shimizu T., Satoh T., Murakoshi K., Sodeoka M. (2005) Asymmetric total synthesis of (−)-spirofungin A and (+)-spirofungin B. Org. Lett. 7, 5573–5576 [DOI] [PubMed] [Google Scholar]
- 14. Koshino H., Takahashi H., Osada H., Isono K. (1992) Reveromycins, new inhibitors of eukaryotic cell growth. III. Structures of reveromycins A, B, C and D. J. Antibiot. 45, 1420–1427 [DOI] [PubMed] [Google Scholar]
- 15. Jiang J., Tetzlaff C. N., Takamatsu S., Iwatsuki M., Komatsu M., Ikeda H., Cane D. E. (2009) Genome mining in Streptomyces avermitilis: a biochemical Baeyer-Villiger reaction and discovery of a new branch of the pentalenolactone family tree. Biochemistry 48, 6431–6440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gust B., Challis G. L., Fowler K., Kieser T., Chater K. F. (2003) PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc. Natl. Acad. Sci. U.S.A. 100, 1541–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Datsenko K. A., Wanner B. L. (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cherepanov P. P., Wackernagel W. (1995) Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158, 9–14 [DOI] [PubMed] [Google Scholar]
- 19. Onaka H., Taniguchi S., Ikeda H., Igarashi Y., Furumai T. (2003) pTOYAMAcos, pTYM18, and pTYM19, actinomycete-Escherichia coli integrating vectors for heterologous gene expression. J. Antibiot. 56, 950–956 [DOI] [PubMed] [Google Scholar]
- 20. Takahashi S., Takagi H., Toyoda A., Uramoto M., Nogawa T., Ueki M., Sakaki Y., Osada H. (2010) Biochemical characterization of a novel indole prenyltransferase from Streptomyces sp. SN-593. J. Bacteriol. 192, 2839–2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Omura T., Sato R. (1964) The carbon monoxide-binding pigment of liver microsomes. I. Evidence for its hemoprotein nature. J. Biol. Chem. 239, 2370–2378 [PubMed] [Google Scholar]
- 22. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 23. Terwilliger T. C., Berendzen J. (1999) Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr. 55, 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Collaborative Computational Project, Number 4 (1994) The Ccp4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 26. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Osada H., Cui C. B., Onose R., Hanaoka F. (1997) Screening of cell cycle inhibitors from microbial metabolites by a bioassay using a mouse cdc2 mutant cell line, tsFT210. Bioorg. Med. Chem. 5, 193–203 [DOI] [PubMed] [Google Scholar]
- 28. Li S., Tietz D. R., Rutaganira F. U., Kells P. M., Anzai Y., Kato F., Pochapsky T. C., Sherman D. H., Podust L. M. (2012) Substrate recognition by the multifunctional cytochrome P450 MycG in mycinamicin hydroxylation and epoxidation reactions. J. Biol. Chem. 287, 37880–37890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sugimoto H., Shinkyo R., Hayashi K., Yoneda S., Yamada M., Kamakura M., Ikushiro S., Shiro Y., Sakaki T. (2008) Crystal structure of CYP105A1 (P450SU-1) in complex with 1α,25-dihydroxyvitamin D3. Biochemistry 47, 4017–4027 [DOI] [PubMed] [Google Scholar]
- 30. Miyamoto Y., Machida K., Mizunuma M., Emoto Y., Sato N., Miyahara K., Hirata D., Usui T., Takahashi H., Osada H., Miyakawa T. (2002) Identification of Saccharomyces cerevisiae isoleucyl-tRNA synthetase as a target of the G1-specific inhibitor reveromycin A. J. Biol. Chem. 277, 28810–28814 [DOI] [PubMed] [Google Scholar]
- 31. Guengerich F. P. (2002) Cytochrome P450 enzymes in the generation of commercial products. Nat. Rev. Drug Discov. 1, 359–366 [DOI] [PubMed] [Google Scholar]
- 32. Shimizu T., Masuda T., Hiramoto K., Nakata T. (2000) Total synthesis of reveromycin A. Org. Lett. 2, 2153–2156 [DOI] [PubMed] [Google Scholar]
- 33. Shimizu T., Hiramoto K., Nakata T. (2001) Succinylation of tertiary alcohols under high pressure. Synthesis 10.1055/s-2001-14564 [DOI] [Google Scholar]
- 34. Höltzel A., Kempter C., Metzger J. W., Jung G., Groth I., Fritz T., Fiedler H. P. (1998) Spirofungin, a new antifungal antibiotic from Streptomyces violaceusniger Tu 4113. J. Antibiot. 51, 699–707 [DOI] [PubMed] [Google Scholar]
- 35. Marjanovic J., Kozmin S. A. (2007) Spirofungin A: stereoselective synthesis and inhibition of isoleucyl-tRNA synthetase. Angew. Chem. Int. Ed. Engl. 46, 8854–8857 [DOI] [PubMed] [Google Scholar]