

Background: To elucidate the rate-determining steps of cellobiohydrolase Cel7A from T. reesei, variants with lower substrate affinity were designed.
Results: Mutant (W38A) had reduced substrate affinity but a 2-fold increase in the maximum quasi-steady-state rate.
Conclusion: Dissociation of stalled TrCel7A is the rate-limiting step in the initial phase of hydrolysis.
Significance: This work offers a new perspective for the design of faster cellulases.
Keywords: Biodegradation, Biofuel, Cellobiohydrolase, Cellulose, Enzyme Kinetics, Trichoderma reesei, Crystalline Cellulose Degradation
Abstract
Cellobiohydrolases are exo-active glycosyl hydrolases that processively convert cellulose to soluble sugars, typically cellobiose. They effectively break down crystalline cellulose and make up a major component in industrial enzyme mixtures used for deconstruction of lignocellulosic biomass. Identification of the rate-limiting step for cellobiohydrolases remains controversial, and recent reports have alternately suggested either association (on-rate) or dissociation (off-rate) as the overall bottleneck. Obviously, this uncertainty hampers both fundamental mechanistic understanding and rational design of enzymes with improved industrial applicability. To elucidate the role of on- and off-rates, respectively, on the overall kinetics, we have expressed a variant in which a tryptophan residue (Trp-38) in the middle of the active tunnel has been replaced with an alanine. This mutation weakens complex formation, and the population of substrate-bound W38A was only about half of the wild type. Nevertheless, the maximal, steady-state rate was twice as high for the variant enzyme. It is argued that these opposite effects on binding and activity can be reconciled if the rate-limiting step is after the catalysis (i.e. in the dissociation process).
Introduction
Enzymatic breakdown of crystalline cellulose has proven challenging to describe in molecular detail. This is probably the result of a complex enzyme architecture with different domains and moieties that interacts with the heterogeneous surface of the insoluble substrate (1–4). Moreover, the linear β-1,4-glucan chains in the highly condensed substrate are connected by strong intermolecular forces, which compete with enzyme-substrate interactions during the hydrolysis and hence add to the complexity of the process (5, 6). The most extensively studied cellulase is the cellobiohydrolase Cel7A from Trichoderma reesei (an anamorph of the fungus Hypocrea jecorina). TrCel7A is a multidomain enzyme comprising a catalytic domain (core), which is connected by a highly glycosylated linker peptide to a family 1 carbohydrate-binding module (CBM1)3 (7). The three-dimensional crystal structure of the catalytic domain in complex with different cello-oligosaccharides has been solved, revealing a 50-Å-long active site tunnel with 10 glucosyl-binding sites, numbered from −7 at the entrance of the tunnel to +3 at the exit (8). The enzyme hydrolyzes cellulose processively from the reducing end with the scissile bond between subsite −1 and +1, hence leaving cellobiose in the +1,+2 expulsion site. Analysis of processive hydrolysis requires consideration of a number of steps, including adsorption, surface diffusion, recognition, “threading,” complex formation, catalysis, product expulsion, processive sliding, and dissociation (4, 9). Many of these steps remain poorly understood partly because their isolation has proven difficult in experiments (4) and partly as a result of the heterogeneous environment in which these enzyme work (2, 10).
It is of particular importance to identify the slowest and hence rate-determining step in the catalytic cycle. Numerous earlier studies have addressed this question, and interestingly, it appears as if the conclusions fall mainly into two mutually conflicting groups. Thus, a number of works have used either direct experimental evidence or re-examination of broader, previously published material to conclude that the overall rate is governed by the association of enzyme and substrate, particularly the placement of a cellulose strand in the long active tunnel (11–18). In contrast to this, other studies, including work by this group on the pre-steady state kinetics of wild type Cel7A, have suggested that the formation of the activated complex is quite rapid and that dissociation of enzyme, which is in a position where further processive movement is hindered, is the rate-limiting step (19–25). To elucidate this question, we have designed, expressed, and purified variants of TrCel7A with lower affinity for the substrate. The idea was that the kinetic behavior of an enzyme with a less stable substrate complex could reveal whether the bottleneck was located before or after the activated complex (i.e. whether rate limitation was one of the associating or dissociating steps). Specifically, we replaced Trp-38 located in subsite −4 (Fig. 1) in the active tunnel with Ala. Previous work has suggested that this tryptophan residue interacts with the substrate (8, 26), and its replacement with a non-aromatic residue was expected to weaken the complex. Here, we report a comparative analysis of steady-state kinetics, adsorption, and processivity for wild type TrCel7A and the Trp-38 → Ala variant, henceforth termed WT and W38A, respectively. To assess possible effects of the CBM, we have made this comparison both for the intact enzyme and the catalytic domain alone.
FIGURE 1.

Structure of TrCel7A. Shown is a surface representation of TrCel7A in complex with a cellulose chain and highlighted tryptophan residues in the catalytic tunnel. The image was made in PyMOL (Schrödinger, LLC, New York) using the crystal structure of the catalytic domain (Protein Data Bank code 8CEL) and carbon binding module (Protein Data Bank code 1CBH).
EXPERIMENTAL PROCEDURES
All experiments were conducted in a standard 50 mm acetate buffer, 2 mm CaCl2, pH 5.0.
Mutagenesis, Expression, and Purification
All enzymes were expressed heterologously in Aspergillus oryzae and purified as described elsewhere (27). Core variants (i.e. the catalytic domain alone) were made by introducing a BamHI restriction-site in the TrCel7A gene at the end of the core domain, making it possible to splice out the core domain from the TrCel7A plasmid or W38A plasmid. All purified enzyme stocks showed a single band in SDS NuPAGE 4–12% BisTris gel (GE Healthcare), and the absence of specific contamination by β-glucosidase was confirmed by the lack of activity against p-nitrophenyl β-d-glucopyranoside. Henceforth, we will use subscript “intact” for enzymes with and “core” for enzymes without the CBM. Enzyme concentrations were determined by absorbance at 280 nm using molar extinction coefficients of 84,810 m−1 cm−1, 79,120 m−1 cm−1, 79,210 m−1 cm−1, and 73,520 m−1 cm−1 for WTintact, W38Aintact, WTcore, and W38Acore, respectively.
Kinetic Measurements
The cellobiose production was monitored by an amperometric enzyme biosensor based on cellobiose dehydrogenase from Phanerochaete chrysosporium adsorbed onto the surface of a benzoquinone-modified carbon paste electrode as described in detail previously (28). Measurements were carried out at 25 °C in a 5-ml water-jacketed glass cell connected to a water bath. Avicel PH 101 (Sigma-Aldrich) suspensions were stirred at 600 rpm, and enzyme was injected to a final concentration of 100 nm from a Chemyx Fusion 100 syringe pump with an injection time of 1.0 s. All measurements were made in duplicate with calibration before and after each run, using the average sensitivity as the calibration factor (28).
Adsorption
The adsorption of enzyme on Avicel was measured in 96-well microtiter plates by adding substrate and enzyme to a final volume of 300 μl in each well. The substrate load was 10 g/liter for enzymes with CBM and 30 g/liter for the core variants, and the enzyme concentration was varied from 0 to 5 μm. The plate was sealed and incubated for 1 h at 25 °C and 1200 rpm orbital mixing. After incubation, the plate was centrifuged at 3500 rpm for 5 min, and 100 μl of supernatant was transferred to black 96-well microtiter plates. Intrinsic protein fluorescence was determined by measuring the emission at 340 nm at an excitation wavelength of 280 nm. In all experiments, an enzyme standard curve was made using the enzyme in question as reference.
Product Profile
The production of glucose, cellobiose, and cellotriose was measured at different time points by taking 500-μl subsets of a 50-ml reaction mixture, containing 10 g/liter Avicel and 0.50 μm enzyme at 25.0 °C. Subsets were quenched by mixing with the same volume 0.1 m NaOH, and following centrifugation, the concentrations of glucose, cellobiose, and cellotriose in the supernatants were measured on a Dionex ICS-5000 ion chromatograph (Thermo Fisher Scientific, Waltham, MA) as described elsewhere (27, 29).
RESULTS
Kinetics
Fig. 2 shows real-time data obtained by the P. chrysosporium cellobiose dehydrogenase biosensor. The two top panels illustrate the cellobiose production for the enzymes with CBM, WTintact, and W38Aintact. Analogously, the bottom panels present data for the two core variants, WTcore and W38Acore. The enzyme concentration was 100 nm in all trials, whereas the load of Avicel was varied over 2 orders of magnitude (0.5–60 g/liter). The results show that enzymes with the W38A mutation produce more cellobiose than the corresponding wild type except at very low Avicel loads.
FIGURE 2.

Real-time progress curves. Shown is the initial hydrolysis (0–300 s) of the four investigated TrCel7A variants, WTintact, W38Aintact, WTcore, and W38Acore. The enzyme concentration was 100 nm, and the Avicel load ranged from 0.5 to 60 g/liter.
We have previously suggested (23, 30) that experimental data for processive enzymes may be analyzed by a reaction scheme (Scheme 1), according to which the free enzyme, E, combines with a cellulose strand, Cm, to form an activated complex, ECm. The enzyme subsequently moves along the strand, which is sequentially shortened by one cellobiose, moiety C, for each step (so the strand is converted to Cm-1, Cm-2, etc.). In this process, the activated complexes (ECm-1, ECm-2 …) are all allowed two reaction pathways. They may either go through another catalytic cycle to produce a cellobiose and a correspondingly shortened cellulose strand, or they may dissociate. This reaction mechanism can be kinetically described by four parameters. These are the rate constants for association (kon), catalysis (kcat), and dissociation (koff) and the processivity number, n (the average number of sequential steps made following the attack of one cellulose strand).
SCHEME 1.
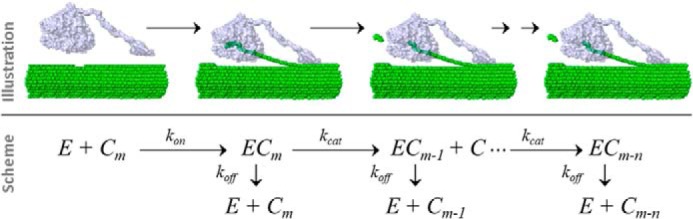
Simplified reaction scheme and its structural interpretation. The scheme defines the three rate constants (kon, kcat, and koff) used and the processivity number (n).
At quasi-steady state, the rate of cellobiose production according to the processive reaction in Scheme 1, pVSS, attains the usual hyperbolic form (23),
 |
where S is the load of substrate in g/liter, and the processive kinetic parameters, pKm and pVmax, are defined by the processivity, n, and the rate constants from Scheme 1.
 |
and
Equations 1–3 rely on the assumption that the substrate is in excess (i.e. that the concentration of attack sites for enzyme on the substrate surface is much larger than the enzyme concentration). This has previously been shown to be a reasonable assumption for conditions similar to those used here (22, 23). We note that the main difference between the parameters for processive enzyme activity given in Equations 2 and 3 and the analogous parameters in the conventional Michaelis-Menten equation is the constant, β. This so-called kinetic processivity coefficient is defined, β = 1 − (kcat/(kcat + koff))n and may be interpreted as the probability that the enzyme will dissociate from the cellulose strand before completing the average number (n) of processive steps (23).
The experimental data in Fig. 2 are typical for cellulases in the sense that a truly constant reaction rate (i.e. a fully linear progress curve) is never observed. As argued previously (23), this requires some compromise in the definition of the steady-state rate, and it was recommended to assign pVSS as the slope of the concentration trace after the initial burst, which represents a non-steady-state condition (23, 31) but before the rate has been significantly reduced by other (as of yet partially unknown) factors. In accordance with this, we fitted a line to the data from 100 to 150 s in Fig. 2 and used the slope as a measure of pVSS. Results are plotted as a function of the Avicel load in Fig. 3. Comparisons of data for the two enzymes with CBM (WTintact and W38Aintact) and the two catalytic domains (WTcore and W38Acore), respectively, again show that the W38A mutation slightly decreases activity at very low substrate load but significantly promotes pVSS when the Avicel load approaches saturating levels. Fig. 3 also shows that Equation 1 accounts well for the experimental data for all four enzymes, and the kinetic parameters derived from the regression analysis based on Equation 1 are listed in Table 1. These parameters reveal that the two W38A variants have 1.6–2 times higher pVmax compared with the corresponding enzymes without this mutation. Conversely, the affinity for the substrate is lowered by the W38A mutation (pKm is increased 4 times for the intact enzyme and 2 times for the core variant).
FIGURE 3.

Steady-state kinetics. The steady-state hydrolytic rate, pVSS, for the four investigated enzymes is plotted as a function of the substrate load. Symbols represent experimental values derived from the slopes of linear fits between 100 and 150 s in Fig. 2. The lines are best fits of Equation 1. The kinetic parameters pKm and pVmax (Equation 1) are given in Table 1.
TABLE 1.
Steady-state kinetic and adsorption parameters for the four investigated TrCel7A variants, WTintact, W38Aintact, WTcore, and W38Acore
| Enzyme | Kinetic parameters |
Adsorption parameters |
||||
|---|---|---|---|---|---|---|
| pVmax | pKm | pVmax/pKm | Γmax | Kd | Γmax/Kd | |
| nm/s | g/liter | nmol/s/g | nmol/g | μm | liter/g | |
| WTintact | 19.5 ±0.6 | 1.54 ± 0.23 | 12.7 ± 1.93 | 195 ± 7 | 0.145 ± 0.019 | 1.345 ± 0.183 |
| W38Aintact | 38.4 ± 0.7 | 5.95 ± 0.34 | 6.5 ± 0.39 | 114 ± 4 | 0.544 ± 0.061 | 0.210 ± 0.025 |
| WTcore | 34.4 ± 1.8 | 12.6 ± 1.82 | 2.7 ± 0.42 | 30 ± 2 | 1.43 ± 0.195 | 0.021 ± 0.003 |
| W38Acore | 53.2 ± 2.9 | 24.3 ± 2.87 | 2.2 ± 0.28 | 22 ± 3 | 2.97 ± 0.620 | 0.008 ± 0.002 |
Adsorption
The effect of the W38A mutation on substrate affinity was further investigated by adsorption measurements. Results in Fig. 4 show the characteristic adsorption saturation and a strong effect of the CBM, which have been discussed in detail earlier both for TrCel7A and numerous related cellulases (32–34). In the current context, it is more important to note that the W38A mutation significantly reduces binding both in the intact enzyme and in the core variant. To quantify this, we analyzed the data with respect to a standard Langmuir isotherm, Γ, where the variables E and Γ represent free concentration and adsorbed amount of enzyme, respectively, and Γmax and Kd are the usual Langmuir parameters (saturation coverage and dissociation constant). The Langmuir isotherm was found to account well for the experimental data, and the parameters are listed in Table 1. It appears that the mutation weakens the interaction significantly because Kd increased 3.7 times in the intact enzyme and 2.1 times in the core variant. The saturation coverage, Γmax, was also reduced (30–40%) by the mutation, indicating a reduced substrate accessibility for the mutant.
FIGURE 4.

Adsorption isotherms for the four investigated enzymes. The coverage of enzyme on Avicel, Γ, is plotted as a function of the free enzyme concentration, E. Points represent the experimental data, and the lines are best fits of the Langmuir isotherm.
Product Profile
Progress curves for glucose, cellobiose, and cellotriose produced by WTintact and W38Aintact are shown in Fig. 5. These measurements were made to assess the influence of the W38A mutation on the processivity. It has previously been suggested that processivity can be estimated from the cellobiose/cellotriose ratio of either concentrations or steady-state production rates (31, 35, 36). Applying these principles, we found processivities of 23–24 for WTintact and 15–16 for W38Aintact. Product profiles were only measured for one enzyme concentration, and the following discussion of processivity hence neglects a possible dependence of n on the enzyme/substrate ratio. We note, however, that the enzyme/substrate ratio in the product profile measurements (Fig. 5) was comparable with the ratio in the kinetic measurements (Fig. 2) for substrate loads around pKm and hence that the two types of experiments appear compatible. The processivity of the wild type TrCel7A on Avicel has been measured before (15, 22, 31), and the current result is in line with these earlier reports. We note that relationships between product profiles and processivity have been extensively discussed (4, 35, 37), and somewhat deviant principles for the calculation of n have been put forward (22, 31, 36, 38). A discussion of the validity and weaknesses of these strategies is beyond the current scope, but using different approaches, we consistently found that the processivity of W38A was about 25% lower than the wild type. Hence, we conclude that the product profile suggests a moderately decreased processivity in the W38A variant compared with the wild type enzyme.
FIGURE 5.

Product profile. Shown are concentrations of glucose, cellobiose, and cellotriose produced by 0.5 μm WTintact and W38Aintact, respectively, during hydrolysis of Avicel at 25 °C and an initial load of 10 g/liter.
DISCUSSION
Tryptophan residues in the active site of glycoside hydrolases are important for processive hydrolysis (8, 39–42). Many mutational studies in which Trp has been replaced with non-aromatic residues have shown negative effects on activity and processivity of, for example, cellulases (16, 36, 38, 43–46) and chitinases (47–49). One suggested role of Trp is that “stacking” interactions of π-electrons in its aromatic ring and the α-face of the carbohydrate (50) enable the necessary sliding of the bound chain (4, 51), possibly through stabilization of the transition state between adjacent positions in the processive movement (52, 53). Tryptophan residues near the entrance of the active tunnel are also important for the initial “threading” and recognition of the cellulose strand, particularly on crystalline substrates (16, 26, 38, 44, 54). In the current work, we used the W38A mutation in T. reesei Cel7A to obtain an enzyme with weakened substrate affinity. The purpose was to study the kinetics of a low affinity variant over a broad range of substrate loads and use this information to elucidate whether the rate-limiting step was before or after the activated complex, ECm-1 in Scheme 1. Detailed molecular analysis of Trp-carbohydrate interactions, on the other hand, was beyond the current scope. In the simplified view of Scheme 1, the question we are addressing is whether kon or koff limits the overall reaction. As outlined in the Introduction, this question has recently attracted widespread interest, and its elucidation appears to be central for the fundamental understanding of processive cellulases. We chose Trp-38 as target for a low affinity mutant because this residue has previously been shown to interact with the cellulose chain (8, 26). Moreover, it is located in the middle of the tunnel quite far from the scissile bond unlike, for example, Trp-367 and Trp-376, which are directly involved in the twist of the cellulose strand that is important for the actual catalysis (55).
Substrate Affinity
Results in Table 1 confirmed that the affinity for Avicel was reduced by the W38A mutation. This was seen both for the enzymes with CBM (WTintact versus W38Aintact) and when comparing the two core variants, WTcore and W38Acore. Moreover, lower affinity was reflected in both kinetic and adsorption measurements (both pKm and Kd were increased by the W38A mutation). The absolute values of pKm and Kd cannot be directly compared because the insoluble nature of the substrate prevents conversion of pKm to molar units. However, the contribution of Trp-38 to the affinity may be assessed from the ratio of parameters, Ki(W38A)/Ki(WT), where Ki represents either pKm or Kd. For the intact enzyme, this ratio was 3.8 for Kd and 3.9 for pKm (Table 1). Thus, affinity changes derived independently by either kinetic or adsorption measurements were in perfect accord, and we may estimate the contribution to the standard free energy of binding brought about by Trp-38 as follows.
Insertion of the above values for the intact enzyme yields ΔΔGW380 = −3.3 kJ/mol. For the core variants (WTcore and W38Acore), the ratios were 2.1 (Kd) and 1.9 (pKm), and this translates into ΔΔGW380 = 1.7 kJ/mol. Provided that alanine in position 38 interacts negligibly with the cellulose strand (50) and that the overall conformation of the mutant is mainly unchanged, this suggests that Trp-38 contributes −2 to −3 kJ/mol to the standard free energy of substrate binding. This value perfectly matches general assessments of the interaction free energy for carbohydrates and aromatic side chains (50, 56), but it is lower than the contribution from Trp-38 to ligand binding (−15.9 kJ/mol) derived from molecular dynamics simulations (57). We note that a similar analysis of the contribution of the CBM (again using the appropriate ratios of parameters from Table 1) suggested ΔΔGCBM0 values of −5.4 kJ/mol for the wild type (WTintact versus WTcore), and −3.9 kJ/mol for the W38A mutant (W38Aintact versus W38Acore). This result for the wild type is in accord with values calculated from the adsorption isotherms reported by Stahlberg et al. (32). Comparing this and the results for the Trp mutant suggests that the CBM contributes about twice as much to the net affinity compared with Trp-38. Because the CBM has three subsites, a 1:3 ratio might have been expected, but the somewhat weaker subsite contribution in the CBM may reflect differences between interactions of a planar interface (CBM) and a tunnel (core). Moreover, the CBM has Tyr in all three subsites, and this residue has previously been shown to bind less tightly than Trp (58).
We note that contributions from either Trp-38 or the CBM were not simply additive. Thus, the W38A mutation was more detrimental to binding of the intact enzyme compared with the core, and the favorable effect of the CBM was larger in the wild type than in the W38A mutant. Origins of this non-additivity cannot be assessed on the basis of the current data, but we note that a related behavior has previously been observed by Palonen et al. (59). These workers found that the affinity of Cel7A core was much lower than the intact enzyme, thus suggesting a major role of the CBM for adsorption. However, when purified CBM was tested in separate experiments, it did not show high affinity, and this clearly suggests a synergy (non-additivity) in the adsorption process.
Kinetics
The remainder of this discussion will address the respective influence of association and dissociation on the overall rate. As outlined in the Introduction, this focus was motivated partly by recent discussions of rate limitation, which have highlighted these two steps, and partly by the deduction that the hydrolytic step (governed by kcat in Scheme 1) is an unlikely candidate for rate determination. The latter relies on numerous measurements of kcat for TrCel7A, which have shown that at room temperature this parameter is in the range of 2–10 s−1 (19, 21, 22, 24, 43, 55, 60, 61), and this is much faster than typically measured specific rates. Because the overall rate is obviously dictated by the slowest step, this discrepancy strongly suggests that catalysis is not the bottleneck and that rate determination must be some much slower process. To illustrate this for the current system, we note that the maximal specific rate at steady-state, pVmax/E0, for wild type TrCel7A on Avicel was about 0.2 s−1 (Table 1; E0 was 100 nm). This is over an order of magnitude less than kcat for the same system (4.8 s−1) (24), and we will therefore not consider this parameter further in the discussion of rate limitation. We note, however, that the above arguments pertain to hydrolysis by monocomponent TrCel7A, and different conclusions regarding kcat may be reached for mixtures of synergistically acting cellulases (21, 55). Hence, Jalak et al. (21) found that hydrolysis of bacterial cellulose by an optimally composed mixture of Cel7A and the endo-active enzyme Cel5A could reach a specific hydrolysis rate at steady state (about 2 s−1), which was close to the rate constant for the inner catalytic cycle (including hydrolysis, product expulsion, and processive movement). This implies that, under these conditions, kcat was indeed important for rate limitation. More recently, Knott et al. (55) have used molecular dynamics simulation and free energy calculation to suggest that the rate-limiting step in the hydrolytic chain of reactions is glycosylation of the nucleophile (Glu-212), which had a rate constant of about 11 s−1. This theoretical result is in remarkable accordance with kcat values derived from either high speed atomic force microscopy (60) or pre-steady-state kinetic measurements (24). Knott et al. (55) also used computational methods to analyze the kinetics of other steps, including processive motion, deglycosylation, catalytic activation, and “threading” the cellulose strand into the active tunnel, but these latter steps were found to be faster than glycosylation, which was therefore suggested to be potentially rate-limiting. The off-rate was not assessed by Knott et al. A combined interpretation of these results suggests that glycosylation is likely to be rate-limiting for the conditions studied by Jalak et al. However, specific rates reported for both monocomponent cellobiohydrolases (15, 16, 19, 30, 62) and enzyme mixtures (63, 64) are well below 1 s−1 and hence suggest a rate-limiting step outside the catalytic cycle.
The results we present here show that the W38A mutation in TrCel7A decreases substrate affinity and processivity but increases the specific hydrolytic rate for both enzymes with and without CBM. Although this decoupling appears puzzling at first, it may form the basis for a qualitative interpretation of the results. This is illustrated, at least to a first approximation, in the simplified energy diagram in Fig. 6. Thus, a variant with lowered substrate affinity (green line) compared with the wild type (blue line) will obviously show less binding (the ECm complex for the variant has higher free energy). Moreover, the variant also has a lower activation barrier, ΔΔGD≠, for dissociation (c.f. Fig. 6), and if ΔΔGD≠ > ΔΔGA≠ so that dissociation is rate-limiting, the low affinity variant will show a faster overall reaction. It follows that decoupling of affinity and activity, as seen in Table 1, may be rationalized along the lines of Fig. 6, provided that dissociation is rate-limiting. One corollary of a dissociation-controlled rate is that not all adsorbed enzyme can be catalytically active at the same time (20, 23), and this is congruent with suggestions of a bound but inactive population of processive cellulases. This may rely on irregularities in the cellulose structure, which obstruct processive movement (20, 30, 65) or “traffic jams” in which enzymes collide and stall (60). In any case, slow release will be associated with an adsorbed but catalytically inactive population of enzyme. It has been argued that the kinetic processivity factor, β (Equation 3), provides a measure of the catalytically active population and that it may be as low as 3–8% for TrCel7A (23). If so, a dissociation-controlled mechanism can be understood as a situation where dissociation of predominantly inactive ECm-1 complexes (and hence recruitment for attacks on new cellulose strands) is rate-limiting.
FIGURE 6.

Simplified energy diagram for TrCel7A catalysis with emphasis on association and dissociation. The blue curve represents a possible course for the wild type, and the green curve illustrates differences for a variant with lower substrate affinity (such as W38A). The reaction has been condensed into the same steps as in Scheme 1, and this is illustrated by the drawings at the top. The plot emphasizes how lower substrate affinity will tend to decrease the energy barrier for dissociation (ΔGD‡) rather than the barrier for association (ΔGA‡).
None of the rate constants in Scheme 1 can be calculated directly from the parameters in Table 1, but some of them may be assessed on the basis of the relationship,
 |
provided that kcat ≫ koff (23). This latter assumption has previously proven valid for TrCel7A hydrolyzing both Avicel (24) and other substrates (19, 22). If we insert the processivities for WTintact (n = 23) and W38Aintact (n = 16) found in Fig. 5 and pVmax values from Table 1 into Equation 5, we find off-rate constants of 0.0085 s−1 for WTintact and 0.024 s−1 for the W38Aintact mutant. Expressed as half-times for the dissociation of enzyme-substrate complexes (t½ = ln2/koff), this translates into 82 and 29 s, respectively. When off-rate constants and pKm values are known, the (second order) on-rate constants can be readily calculated from Equation 2. We found kon values of 0.0055 (g/liter)−1 s−1 and 0.0040 (g/liter)−1 s−1 for WTintact and W38Aintact, respectively. In conclusion, this analysis suggests that the lowered net affinity of the W38A mutant reflected a moderate (∼27%) reduction in kon and significant (∼2.8-fold) increase in koff. Because kon and koff are of different kinetic orders, their values cannot be directly compared. However, an apparent on-rate constant at a fixed substrate load, konS (in units of s−1) can be compared with koff to assess the importance of association and dissociation, respectively. If, for example, we consider a very dilute substrate, such as S = 1 g/liter, together with the rate constants given above, we find that koff > konS. This implies an association controlled reaction. However, the apparent on-rate constant grows proportionally with S, whereas koff is independent of the substrate load, and this means that at, for example, 10 g/liter, the situation is reversed, and konS > koff. To put this in more general terms, we note that the processive Michaelis constant was defined (Equation 5) as pKm = koff/kon. It follows that the apparent on-rate constant, konS, is equal to koff when S = pKm and that konS > koff (i.e. off-rate controlled hydrolysis) when S > pKm. This analysis allows a more detailed interpretation of the rate limitation. Thus, off-rate limitation, as suggested above, only occurs for S > pKm, because at lower loads, the scarcity of attack sites inevitably delays association to the extent where it becomes rate-limiting. This distinction is obviously important in discussions of rate limitation, and conclusions based on experiments at a single load will only provide an incomplete picture of the limiting step. As seen from Table 1, the changeover from on- to off-rate limitation occurs at low loads for the wild type enzyme (pKm = 1.5 g/liter), but it increases significantly for the low affinity variants and reaches 24 g/liter for W38Acore. This shows that the potential of fast hydrolysis of this latter enzyme only materializes at quite high substrate loads. This may be interesting from an applied perspective because plans for industrial conversion of biomass usually include very high solid loads of 100–400 g/liter (66, 67). Although typically only around half of this dry mass is cellulose, it appears most likely that these conditions are in the regime, where the initial rate is governed by koff.
Some studies on processive cellulases have found that replacement of Trp residues near the entrance of the catalytic tunnel leads to enhanced activity against soluble and/or amorphous substrates, whereas the hydrolysis of highly crystalline substrate is reduced in comparison with the wild type (16, 38, 44). In these cases, the activity was only tested at one (low) substrate load (0.7–2.5 g/liter). Because the activity curves in Fig. 3 showed a crossover at intermediate substrate loads, it appears that a broader range of loads may be necessary to reach a conclusion on the effect of the mutation on activity. We emphasize, however, that the W38A mutants studied here for their activity against Avicel (with an intermediate crystallinity) could behave differently on other substrates and hence that further experimental work would be required to elucidate possible kinetic differences between Trp mutations near the opening (16, 38, 44) and (as Trp-38) in the middle of the catalytic tunnel, respectively.
Conclusion
The W38A mutation in Cel7A moderately reduced the strength of binding to cellulose. Specifically, the standard free energy of association was about 3 kJ/mol less negative compared with the wild type enzyme. Kinetic analysis suggested that the reduced net affinity can be broken into a small reduction of kon and a larger increase of koff. At substrate loads approaching saturating levels, these changes led to a 2-fold reduction in adsorption but a 2-fold increase in activity. Conversely, at very low substrate loads, both activity and adsorption were lessened by the mutation. These observations strongly suggest that except in very dilute Avicel suspensions, where infrequent enzyme-substrate collisions predictably become limiting, the rate-determining step must be in the dissociation process. The results cannot be reconciled with an association-limited interpretation because the on-rate constant for the (more active) mutant was lower than the on-rate constant for the (less active) wild type. Scheme 1 represents a simplification, where several potential steps (see the Introduction) have been pooled into one association and one dissociation reaction. It follows that kon and koff are composite rate constants reflecting the kinetics of passing through a sequence of steps. The dissociation process, which we have found to be the bottleneck, may, for example, be partitioned into the separate release of core and CBM domains. We cannot unambiguously separate these steps on the basis of the current data, but we note that the W38A mutation brought about similar kinetic changes in both the intact enzyme and the separate core domain. In light of this, we speculate that the release of substrate from the active tunnel is rate-limiting under the conditions studied here. As discussed in the Introduction, other studies have reached quite different conclusions regarding the rate-limiting step for cellobiohydrolases, and this raises the question of whether rate determination may be different for different conditions and particularly whether it may shift as the substrate is gradually modified during hydrolysis. This cannot be addressed from the current measurements of initial rates, but it would be relevant for future work.
Footnotes
- CBM
- carbohydrate-binding module
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1. Teeri T. T. (1997) Crystalline cellulose degradation: new insight into the function of cellobiohydrolases. Trends Biotechnol. 15, 160–167 [Google Scholar]
- 2. Zhang Y. H., Lynd L. R. (2004) Toward an aggregated understanding of enzymatic hydrolysis of cellulose: noncomplexed cellulase systems. Biotechnol. Bioeng. 88, 797–824 [DOI] [PubMed] [Google Scholar]
- 3. Bansal P., Hall M., Realff M. J., Lee J. H., Bommarius A. S. (2009) Modeling cellulase kinetics on lignocellulosic substrates. Biotechnol. Adv. 27, 833–848 [DOI] [PubMed] [Google Scholar]
- 4. Beckham G. T., Ståhlberg J., Knott B. C., Himmel M. E., Crowley M. F., Sandgren M., Sørlie M., Payne C. M. (2014) Towards a molecular-level theory of carbohydrate processivity in glycoside hydrolases. Curr. Opin. Biotechnol. 27, 96–106 [DOI] [PubMed] [Google Scholar]
- 5. Beckham G. T., Matthews J. F., Peters B., Bomble Y. J., Himmel M. E., Crowley M. F. (2011) Molecular-level origins of biomass recalcitrance: decrystallization free energies for four common cellulose polymorphs. J. Phys. Chem. B 115, 4118–4127 [DOI] [PubMed] [Google Scholar]
- 6. Payne C. M., Jiang W., Shirts M. R., Himmel M. E., Crowley M. F., Beckham G. T. (2013) Glycoside hydrolase processivity is directly related to oligosaccharide binding free energy. J. Am. Chem. Soc. 135, 18831–18839 [DOI] [PubMed] [Google Scholar]
- 7. Abuja P. M., Schmuck M., Pilz I., Tomme P., Claeyssens M., Esterbauer H. (1988) Structural and functional domains of cellobiohydrolase I from Trichoderma reesei. Eur. Biophys. J. 15, 339–342 [Google Scholar]
- 8. Divne C., Ståhlberg J., Teeri T. T., Jones T. A. (1998) High-resolution crystal structures reveal how a cellulose chain is bound in the 50 Å long tunnel of cellobiohydrolase I from Trichoderma reesei. J. Mol. Biol. 275, 309–325 [DOI] [PubMed] [Google Scholar]
- 9. Chundawat S. P. S., Beckham G. T., Himmel M. E., Dale B. E. (2011) Deconstruction of lignocellulosic biomass to fuels and chemicals. Annu. Rev. Chem. Biomol. Eng. 2, 121–145 [DOI] [PubMed] [Google Scholar]
- 10. Himmel M. E., Ding S. Y., Johnson D. K., Adney W. S., Nimlos M. R., Brady J. W., Foust T. D. (2007) Biomass recalcitrance: engineering plants and enzymes for biofuels production. Science 315, 804–807 [DOI] [PubMed] [Google Scholar]
- 11. Shang B. Z., Chang R., Chu J. W. (2013) Systems-level modeling with molecular resolution elucidates the rate-limiting mechanisms of cellulose decomposition by cellobiohydrolases. J. Biol. Chem. 288, 29081–29089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fox J. M., Jess P., Jambusaria R. B., Moo G. M., Liphardt J., Clark D. S., Blanch H. W. (2013) A single-molecule analysis reveals morphological targets for cellulase synergy. Nat. Chem. Biol. 9, 356–361 [DOI] [PubMed] [Google Scholar]
- 13. von Ossowski I., Ståhlberg J., Koivula A., Piens K., Becker D., Boer H., Harle R., Harris M., Divne C., Mahdi S., Zhao Y., Driguez H., Claeyssens M., Sinnott M. L., Teeri T. T. (2003) Engineering the exo-loop of Trichoderma reesei cellobiohydrolase, Cel7A. A comparison with Phanerochaete chrysosporium Cel7D. J. Mol. Biol. 333, 817–829 [DOI] [PubMed] [Google Scholar]
- 14. Wilson D. B. (2009) Cellulases and biofuels. Curr. Opin. Biotechnol. 20, 295–299 [DOI] [PubMed] [Google Scholar]
- 15. Nakamura A., Watanabe H., Ishida T., Uchihashi T., Wada M., Ando T., Igarashi K., Samejima M. (2014) Trade-off between processivity and hydrolytic velocity of cellobiohydrolases at the surface of crystalline cellulose. J. Am. Chem. Soc. 136, 4584–4592 [DOI] [PubMed] [Google Scholar]
- 16. Nakamura A., Tsukada T., Auer S., Furuta T., Wada M., Koivula A., Igarashi K., Samejima M. (2013) The tryptophan residue at the active site tunnel entrance of Trichoderma reesei cellobiohydrolase Cel7A is important for initiation of degradation of crystalline cellulose. J. Biol. Chem. 288, 13503–13510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fox J. M., Levine S. E., Clark D. S., Blanch H. W. (2012) Initial- and processive-cut products reveal cellobiohydrolase rate limitations and the role of companion enzymes. Biochemistry 51, 442–452 [DOI] [PubMed] [Google Scholar]
- 18. Maurer S. A., Bedbrook C. N., Radke C. J. (2012) Cellulase adsorption and reactivity on a cellulose surface from flow ellipsometry. Ind. Eng. Chem. Res. 51, 11389–11400 [Google Scholar]
- 19. Kurasin M., Väljamäe P. (2011) Processivity of cellobiohydrolases is limited by the substrate. J. Biol. Chem. 286, 169–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jalak J., Väljamäe P. (2010) Mechanism of initial rapid rate retardation in cellobiohydrolase catalyzed cellulose hydrolysis. Biotechnol. Bioeng. 106, 871–883 [DOI] [PubMed] [Google Scholar]
- 21. Jalak J., Kurašin M., Teugjas H., Väljamäe P. (2012) Endo-exo synergism in cellulose hydrolysis revisited. J. Biol. Chem. 287, 28802–28815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cruys-Bagger N., Elmerdahl J., Praestgaard E., Tatsumi H., Spodsberg N., Borch K., Westh P. (2012) Pre-steady-state kinetics for hydrolysis of insoluble cellulose by cellobiohydrolase Cel7A. J. Biol. Chem. 287, 18451–18458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cruys-Bagger N., Elmerdahl J., Praestgaard E., Borch K., Westh P. (2013) A steady-state theory for processive cellulases. FEBS J. 280, 3952–3961 [DOI] [PubMed] [Google Scholar]
- 24. Cruys-Bagger N., Tatsumi H., Ren G. R., Borch K., Westh P. (2013) Transient kinetics and rate-limiting steps for the processive cellobiohydrolase Cel7A: effects of substrate structure and carbohydrate binding domain. Biochemistry 52, 8938–8948 [DOI] [PubMed] [Google Scholar]
- 25. Cruys-Bagger N., Tatsumi H., Borch K., Westh P. (2014) A graphene screen-printed carbon electrode for real-time measurements of unoccupied active sites in a cellulase. Anal. Biochem. 447, 162–168 [DOI] [PubMed] [Google Scholar]
- 26. Taylor C. B., Payne C. M., Himmel M. E., Crowley M. F., McCabe C., Beckham G. T. (2013) Binding site dynamics and aromatic-carbohydrate interactions in processive and non-processive family 7 glycoside hydrolases. J. Phys. Chem. B 117, 4924–4933 [DOI] [PubMed] [Google Scholar]
- 27. Westh P., Kari J., Olsen J., Borch K., Jensen K., Krogh K. B. R. M. (May 1, 2014) PCT International Patent Application WO/2014/064115, C12N 9/00 Ed.
- 28. Cruys-Bagger N., Ren G., Tatsumi H., Baumann M. J., Spodsberg N., Andersen H. D., Gorton L., Borch K., Westh P. (2012) An amperometric enzyme biosensor for real-time measurements of cellobiohydrolase activity on insoluble cellulose. Biotechnol. Bioeng. 109, 3199–3204 [DOI] [PubMed] [Google Scholar]
- 29. Alasepp K., Borch K., Cruys-Bagger N., Badino S., Jensen K., Sørensen T. H., Windahl M. S., Westh P. (2014) In situ stability of substrate associated cellulases studied by scanning calorimetry. Langmuir 30, 7134–7142 [DOI] [PubMed] [Google Scholar]
- 30. Praestgaard E., Elmerdahl J., Murphy L., Nymand S., McFarland K. C., Borch K., Westh P. (2011) A kinetic model for the burst phase of processive cellulases. FEBS J. 278, 1547–1560 [DOI] [PubMed] [Google Scholar]
- 31. Kipper K., Väljamäe P., Johansson G. (2005) Processive action of cellobiohydrolase Cel7A from Trichoderma reesei is revealed as “burst” kinetics on fluorescent polymeric model substrates. Biochem. J. 385, 527–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ståhlberg J., Johansson G., Pettersson G. (1991) A new model for enzymatic hydrolysis of cellulose based on the two-domain structure of cellobiohydrolase I. Nat. Biotechnol. 9, 286–290 [Google Scholar]
- 33. Wahlström R., Rahikainen J., Kruus K. (2014) Cellulose hydrolysis and binding with Trichoderma reesei Cel5A and Cel7A and their core domains in ionic liquid solutions. Biotechnol. Bioeng. 111, 726–733 [DOI] [PubMed] [Google Scholar]
- 34. Igarashi K., Wada M., Hori R., Samejima M. (2006) Surface density of cellobiohydrolase on crystalline celluloses. FEBS J. 273, 2869–2878 [DOI] [PubMed] [Google Scholar]
- 35. Horn S. J., Sørlie M., Vårum K. M., Väljamäe P., Eijsink V. G. (2012) Measuring processivity. Methods Enzymol. 510, 69–95 [DOI] [PubMed] [Google Scholar]
- 36. Vuong T. V., Wilson D. B. (2009) Processivity, synergism, and substrate specificity of Thermobifida fusca Cel6B. Appl. Environ. Microbiol. 75, 6655–6661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wilson D. B., Kostylev M. (2012) Cellulase processivity. Methods Mol. Biol. 908, 93–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kostylev M., Alahuhta M., Chen M., Brunecky R., Himmel M. E., Lunin V. V., Brady J., Wilson D. B. (2014) Cel48A from Thermobifida fusca: structure and site directed mutagenesis of key residues. Biotechnol. Bioeng. 111, 664–673 [DOI] [PubMed] [Google Scholar]
- 39. Rouvinen J., Bergfors T., Teeri T., Knowles J. K., Jones T. A. (1990) Three-dimensional structure of cellobiohydrolase II from Trichoderma reesei. Science 249, 380–386 [DOI] [PubMed] [Google Scholar]
- 40. Divne C., Ståhlberg J., Reinikainen T., Ruohonen L., Pettersson G., Knowles J. K. C., Teeri T. T., Jones T. A. (1994) The three-dimensional crystal structure of the catalytic core of cellobiohydrolase I from Trichoderma reesei. Science 265, 524–528 [DOI] [PubMed] [Google Scholar]
- 41. van Aalten D. M. F., Synstad B., Brurberg M. B., Hough E., Riise B. W., Eijsink V. G. H., Wierenga R. K. (2000) Structure of a two-domain chitotriosidase from Serratia marcescens at 1.9-angstrom resolution. Proc. Natl. Acad. Sci. U.S.A. 97, 5842–5847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Breyer W. A., Matthews B. W. (2001) A structural basis for processivity. Protein Sci. 10, 1699–1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Igarashi K., Koivula A., Wada M., Kimura S., Penttilä M., Samejima M. (2009) High speed atomic force microscopy visualizes processive movement of Trichoderma reesei cellobiohydrolase I on crystalline cellulose. J. Biol. Chem. 284, 36186–36190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Koivula A., Kinnari T., Harjunpää V., Ruohonen L., Teleman A., Drakenberg T., Rouvinen J., Jones T. A., Teeri T. T. (1998) Tryptophan 272: an essential determinant of crystalline cellulose degradation by Trichoderma reesei cellobiohydrolase Cel6A. FEBS Lett. 429, 341–346 [DOI] [PubMed] [Google Scholar]
- 45. Zhang S., Irwin D. C., Wilson D. B. (2000) Site-directed mutation of noncatalytic residues of Thermobifida fusca exocellulase Cel6B. Eur. J. Biochem. 267, 3101–3115 [DOI] [PubMed] [Google Scholar]
- 46. Kostylev M., Wilson D. (2013) Two-parameter kinetic model based on a time-dependent activity coefficient accurately describes enzymatic cellulose digestion. Biochemistry 52, 5656–5664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Horn S. J., Sikorski P., Cederkvist J. B., Vaaje-Kolstad G., Sørlie M., Synstad B., Vriend G., Vårum K. M., Eijsink V. G. H. (2006) Costs and benefits of processivity in enzymatic degradation of recalcitrant polysaccharides. Proc. Natl. Acad. Sci. U.S.A. 103, 18089–18094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zakariassen H., Aam B. B., Horn S. J., Vårum K. M., Sørlie M., Eijsink V. G. H. (2009) Aromatic residues in the catalytic center of chitinase A from Serratia marcescens affect processivity, enzyme activity, and biomass converting efficiency. J. Biol. Chem. 284, 10610–10617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Watanabe T., Ariga Y., Sato U., Toratani T., Hashimoto M., Nikaidou N., Kezuka Y., Nonaka T., Sugiyama J. (2003) Aromatic residues within the substrate-binding cleft of Bacillus circulans chitinase A1 are essential for hydrolysis of crystalline chitin. Biochem. J. 376, 237–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Laughrey Z. R., Kiehna S. E., Riemen A. J., Waters M. L. (2008) Carbohydrate-π interactions: what are they worth? J. Am. Chem. Soc. 130, 14625–14633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sørlie M., Zakariassen H., Norberg A. L., Eijsink V. G. H. (2012) Processivity and substrate-binding in family 18 chitinases. Biocatal. Biotransform. 30, 353–365 [Google Scholar]
- 52. Parsiegla G., Reverbel C., Tardif C., Driguez H., Haser R. (2008) Structures of mutants of cellulase Cel48F of Clostridium cellulolyticum in complex with long hemithiocellooligosaccharides give rise to a new view of the substrate pathway during processive action. J. Mol. Biol. 375, 499–510 [DOI] [PubMed] [Google Scholar]
- 53. Parsiegla G., Reverbel-Leroy C., Tardif C., Belaich J. P., Driguez H., Haser R. (2000) Crystal structures of the cellulase Cel48F in complex with inhibitors and substrates give insights into its processive action. Biochemistry 39, 11238–11246 [DOI] [PubMed] [Google Scholar]
- 54. Ghattyvenkatakrishna P. K., Alekozai E. M., Beckham G. T., Schulz R., Crowley M. F., Uberbacher E. C., Cheng X. (2013) Initial recognition of a cellodextrin chain in the cellulose-binding tunnel may affect cellobiohydrolase directional specificity. Biophys. J. 104, 904–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Knott B. C., Haddad Momeni M., Crowley M. F., Mackenzie L. F., Götz A. W., Sandgren M., Withers S. G., Ståhlberg J., Beckham G. T. (2014) The mechanism of cellulose hydrolysis by a two-step, retaining cellobiohydrolase elucidated by structural and transition path sampling studies. J. Am. Chem. Soc. 136, 321–329 [DOI] [PubMed] [Google Scholar]
- 56. Kiehna S. E., Laughrey Z. R., Waters M. L. (2007) Evaluation of a carbohydrate-π interaction in a peptide model system. Chem. Commun. 4026–4028 [DOI] [PubMed] [Google Scholar]
- 57. Taylor C. B., Talib M. F., McCabe C., Bu L., Adney W. S., Himmel M. E., Crowley M. F., Beckham G. T. (2012) Computational investigation of glycosylation effects on a family 1 carbohydrate-binding module. J. Biol. Chem. 287, 3147–3155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lehtiö J., Sugiyama J., Gustavsson M., Fransson L., Linder M., Teeri T. T. (2003) The binding specificity and affinity determinants of family 1 and family 3 cellulose binding modules. Proc. Natl. Acad. Sci. U.S.A. 100, 484–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Palonen H., Tenkanen M., Linder M. (1999) Dynamic interaction of Trichoderma reesei cellobiohydrolases Cel6A and Cel7A and cellulose at equilibrium and during hydrolysis. Appl. Environ. Microbiol. 65, 5229–5233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Igarashi K., Uchihashi T., Koivula A., Wada M., Kimura S., Okamoto T., Penttilä M., Ando T., Samejima M. (2011) Traffic jams reduce hydrolytic efficiency of cellulase on cellulose surface. Science 333, 1279–1282 [DOI] [PubMed] [Google Scholar]
- 61. Gruno M., Väljamae P., Pettersson G., Johansson G. (2004) Inhibition of the Trichoderma reesei cellulases by cellobiose is strongly dependent on the nature of the substrate. Biotechnol. Bioeng. 86, 503–511 [DOI] [PubMed] [Google Scholar]
- 62. Murphy L., Baumann M. J., Borch K., Sweeney M., Westh P. (2010) An enzymatic signal amplification system for calorimetric studies of cellobiohydrolases. Anal. Biochem. 404, 140–148 [DOI] [PubMed] [Google Scholar]
- 63. Monschein M., Reisinger C., Nidetzky B. (2013) Enzymatic hydrolysis of microcrystalline cellulose and pretreated wheat straw: a detailed comparison using convenient kinetic analysis. Bioresour. Technol. 128, 679–687 [DOI] [PubMed] [Google Scholar]
- 64. Olsen S. N., Lumby E., McFarland K., Borch K., Westh P. (2011) Kinetics of enzymatic high-solid hydrolysis of lignocellulosic biomass studied by calorimetry. Appl. Biochem. Biotechnol. 163, 626–635 [DOI] [PubMed] [Google Scholar]
- 65. Eriksson T., Karlsson J., Tjerneld F. (2002) A model explaining declining rate in hydrolysis of lignocellulose substrates with cellobiohydrolase I (Cel7A) and endoglucanase I (Cel7B) of Trichoderma reesei. Appl. Biochem. Biotechnol. 101, 41–60 [DOI] [PubMed] [Google Scholar]
- 66. Jørgensen H., Kristensen J. B., Felby C. (2007) Enzymatic conversion of lignocellulose into fermentable sugars: challenges and opportunities. Biofuels Bioprod. Biorefin. 1, 119–134 [Google Scholar]
- 67. Larsen J., Østergaard Petersen M., Thirup L., Wen Li H., Krogh Iversen F. (2008) The IBUS process: lignocellulosic bioethanol close to a commercial reality. Chem. Eng. Technol. 31, 765–772 [Google Scholar]