

Background: TREX1 is a 3′–5′ DNA exonuclease, and mutations in human TREX1 are associated with autoimmune/inflammatory diseases.
Results: TREX1 interacts with poly(ADP-ribose) polymerase-1 (PARP1).
Conclusion: TREX1 contributes to maintenance of suitable PARP1 levels and its functions in the DNA damage response.
Significance: Identification of the molecular mechanism of TREX1 is necessary for understanding the development or progression of TREX1-associated diseases.
Keywords: Autoimmune Disease, DNA Damage Response, Immunology, Inflammation, Protein Translocation, PARP1, TREX1
Abstract
The main function of the 3′–5′ DNA exonuclease TREX1 is to digest cytosolic single-stranded DNA to prevent activation of cell-intrinsic responses to immunostimulatory DNA. TREX1 translocates to the nucleus following DNA damage with its nuclear activities being less well defined. Although mutations in human TREX1 have been linked to autoimmune/inflammatory diseases, the mechanisms contributing to the pathogenesis of these diseases remain incompletely understood. Here, using mass spectrometry and co-immunoprecipitation assays and in vivo overexpression models, we show that TREX1 interacts with poly(ADP-ribose) polymerase-1 (PARP1), a nuclear enzyme involved in the DNA damage response. Two zinc finger domains at the amino terminus of PARP1 were required for the interaction with TREX1 that occurs after nuclear translocation of TREX1 in response to DNA damage. Functional studies suggested that TREX1 may contribute to stabilization of PARP1 levels in the DNA damage response and its activity. These results provide new insights into the mechanisms of single-stranded DNA repair following DNA damage and alterations induced by gene mutations.
Introduction
Recognition of nucleic acids by innate immune receptors, exemplified by Toll-like receptors 3, 7/8, and 9, plays an important role in the clearance of viral infections. However, if self-nucleic acids are involved, recognition can also contribute to the pathogenesis of autoimmune diseases, such as systemic lupus erythematosus (SLE),4 by driving inappropriate activation of type I interferon (IFN1) through an innate immune signaling pathway (1, 2).
TREX1, the most abundant 3′–5′ DNA exonuclease in mammalian cells, specifically cleaves single-stranded DNA (ssDNA) or mispaired 3′ termini of DNA duplexes (3, 4). Mutations in the human TREX1 gene have been linked to four clinically overlapping autoimmune/inflammatory diseases: Aicardi-Goutières syndrome (AGS), familial chilblain lupus, SLE, and retinal vasculopathy with cerebral leukodystrophy (RVCL). These diseases exhibit both genetic and clinical overlaps but present as distinct clinical conditions (5). AGS, a lethal early onset disease characterized by an inflammatory encephalopathy and elevated levels of IFN1 in the cerebrospinal fluid, has been associated primarily with autosomal recessive mutations in TREX1 that impair its exonuclease activity (6). Patients with RVCL, caused by distinct dominant mutations in TREX1, present as adults with retinal and cerebral endotheliopathy together with cerebral leukodystrophy. A subset of these patients develops systemic vascular involvement manifested by Raynaud phenomenon or hepatic or renal dysfunction (7). All the TREX1 mutations described in RVCL cause C-terminal frameshifts that result in deletion of the predicted transmembrane domain responsible for its anchoring in the endoplasmic reticulum but leave enzymatic function intact. Familial chilblain lupus, an inherited cutaneous form of SLE caused by dominant mutations in TREX1, is not associated with neurological manifestations, but there is some overlap with AGS as some AGS patients develop chilblains and autoantibodies (8, 9). Dominant mutations in TREX1 have also been identified in 0.5–2% of SLE patients (10, 11).
Studies have shown that Trex1 knock-out mice die from a severe inflammatory cardiomyopathy with elevated expression of IFN1 and production of autoantibodies (12, 13). Cytosolic ssDNA fragments derived from endogenous retroelements (13) or aberrant replication intermediates (14) accumulate in TREX1-deficient cells of the knock-out mice, leading to the activation of autoimmunity through the IFN signaling pathway. Similar ssDNA fragments were identified in TREX1-deficient cells derived from a patient with AGS (14). Taken together, the studies of deficient mice provide a mechanistic link between accumulation of self-nucleic acids and induction of an IFN response and autoimmunity. However, because the mice do not exhibit pathologies that resemble the major manifestations characteristic of patients with AGS (encephalopathy) or RVCL (retinal and cerebral endotheliopathy), the mechanisms contributing to the pathogenesis of these human diseases remain incompletely understood.
Several roles for TREX1 have been identified in various cellular processes. First, TREX1 is a component of the SET complex that normally resides in the endoplasmic reticulum (15). This complex, including TREX1, translocates to the nucleus during granzyme A-mediated activation of apoptosis. In the nucleus, TREX1 removes ssDNA nicks linked by the SET complex endonuclease, NM23-H1, to inhibit rejoining of the nicked ends (15, 16). In fact, cells carrying the familial chilblain lupus-associated TREX1 mutation, D18N, are relatively resistant to granzyme A, suggesting the importance of TREX1 to granzyme A-activated DNA damage (17). Second, it was also found that following relocalization caused by DNA damage TREX1 is associated with replication foci and attenuates chronic activation of the ataxia telangiectasia mutated-dependent DNA damage response (14). These observations are indicative of roles for TREX1 in the nucleus, the details of which remain to be clearly defined. Interestingly, TREX1 and other genes found to be mutated among patients with AGS have been shown to affect the human immunodeficiency virus (HIV) in various stages of its intracellular replication cycle. TREX1 helps HIV evade innate immune recognition by degrading cytosolic ssDNA generated during reverse transcription (18). Thus, TREX1 has distinct function in both the nucleus and the cytoplasm.
The purpose of the current study was to clarify the underlying mechanisms by which TREX1 contributes to the development of autoimmune/inflammatory diseases. We hypothesized that as-yet-unknown proteins may interact with TREX1 and that these interactions may be affected by disease-causing mutations in the gene. Here, we identified a novel TREX1 binding partner, poly(ADP-ribose) polymerase-1 (PARP1), a nuclear enzyme that participates in the DNA damage response (19, 20). Although PARP1 is involved in many aspects of the cellular responses to DNA damage, it is essential for repair of ssDNA breaks predominantly through base excision repair (BER) mechanisms. We demonstrate that TREX1 translocates to the nucleus in response to DNA damage and interacts there with nuclear PARP1. Our findings suggest that TREX1 may have a previously unappreciated role in the DNA damage response and that mutation-induced alterations in TREX1 function could contribute to development or progression of human autoimmune/inflammatory diseases.
EXPERIMENTAL PROCEDURES
Cell Culture Conditions
Human embryonic kidney (HEK) 293, HeLa, and K562 cells were obtained from the American Type Culture Collection (Manassas, VA) and maintained in DMEM (Quality Biological, Gaithersburg, MD) supplemented with 10% fetal bovine serum (Quality Biological), 50 μm 2-mercaptoethanol (Invitrogen), and penicillin-streptomycin under 5% CO2 in air at 37 °C. To induce DNA damage, cells were treated with 2 mm sodium arsenite (Sigma-Aldrich) and 5 mm hydroxyurea (Sigma-Aldrich) for the indicated times.
Antibodies
Rabbit polyclonal anti-TREX1 antibody was kindly provided by Dr. Parul Kothari and Dr. John Atkinson (Washington University School of Medicine, St. Louis, MO). Other antibodies used in this study included antibodies to GFP and V5 (Invitrogen); those to PARP1, cleaved PARP1, GAPDH, and calreticulin; mouse monoclonal antibody to rabbit IgG (conformation-specific); and HRP-conjugated anti-mouse IgG antibody (Cell Signaling Technology, Danvers, MA); anti-lamin A/C antibody, HRP-conjugated anti-rabbit IgG antibody, and HRP-conjugated anti-goat IgG antibody (Santa Cruz Biotechnology, Santa Cruz, CA); those to FLAG, p53, and α-tubulin (Sigma-Aldrich); anti-histone H3 antibody (Millipore, Billerica, MA); and anti-poly(ADP-ribose) (PAR) antibody (Trevigen, Gaithersburg, MD).
DNA Constructs
To generate intensely fluorescent protein N-terminal portion (IFPN)-tagged TREX1 vectors (21), wild-type TREX1 coding regions were directionally subcloned into the IFPN protein complementation vector (VYF101 vector) kindly provided by Dr. Zhiyong Ding (University of Texas M. D. Anderson Cancer Center, Houston, TX). Briefly, the TREX1 coding sequence was amplified with forward primer (ATCGATATGGGCTCGCAGGCCCTG with ClaI site in italics) and reverse primer (GAATTCCTACTCCCCAGGTGTGGC with EcoRI site in italics), and the ClaI/EcoRI PCR fragment was subcloned into the ClaI/EcoRI-digested VYF101 vector. To generate FLAG-tagged TREX1 vectors, the coding region for TREX1 was subcloned into pCMV-3Tag-6 vector (Agilent Technologies, Santa Clara, CA). To generate V5-tagged PARP1 and its serial deletion mutant constructs (N1, N2, N3, N4, C1, C2, and C3), cDNA sequences corresponding to different regions were amplified by PCR and subcloned into pcDNA3.1/V5-His-TOPO vector (Invitrogen).
Stable Cell Lines
To generate stable cell lines expressing wild-type TREX1, HEK293 Tet-On cells (Clontech) were transfected with each construct in IFPN-tagged plasmids (in VYF101 vector). The cells were selected with 200 μg/ml Zeocin (Invitrogen) for at least 2 weeks, and the expression level of TREX1 was determined by Western blotting using anti-TREX1 or anti-GFP antibody.
Preparation of Cell Extracts and Co-immunoprecipitation
Whole cell lysates were prepared in Nonidet P-40 cell lysis buffer (50 mm Tris, pH 7.4, 250 mm NaCl, 5 mm EDTA, 50 mm NaF, 1 mm Na3VO4, 1% Nonidet P40, 0.02% NaN3) (Invitrogen) supplemented with protease inhibitor (Roche Applied Science), or nuclear and cytoplasmic extracts were prepared according to the manufacturer's protocol for the NE-PER nuclear and cytoplasmic extraction kit or for the subcellular protein fractionation kit (Thermo Scientific, Rockford, IL). For co-immunoprecipitation (co-IP) assays, protein G-coupled magnetic beads (Dynabeads Protein G, Invitrogen) were used according to the manufacturer's protocol. Briefly, 50 μl of magnetic beads were washed with PBS/Tween 20 and incubated with a primary antibody for 1 h at 4 °C. After washing the bead-antibody complex, cell lysates were cross-linked to the complex and incubated overnight at 4 °C. The bead-antibody-antigen complex was washed with PBS/Tween 20 three times and eluted in 20 μl of premixed NuPAGE lithium dodecyl sulfate sample buffer and NuPAGE sample reducing agent (Invitrogen) and incubated for 10 min at 70 °C. Samples were loaded onto a gel for SDS-PAGE followed by Western blotting.
Western Blotting
Protein expression levels were determined by SDS-PAGE and subsequent immunoblotting as described previously (22). Briefly, 20–30 μg of protein determined by the BCA protein assay method (Thermo Scientific) was separated on a NuPAGE Novex 4–12% Bis-Tris gel (Invitrogen) and transferred to a polyvinylidene difluoride membrane. After blocking with a 5% skim milk solution, the blot was incubated with a primary antibody according to the manufacturer's recommended dilution in the blocking solution containing 0.1% Tween 20. The antibody was detected with an HRP-conjugated secondary antibody, developed using an enhanced chemiluminescent substrate (SuperSignal West Pico, Thermo Scientific), exposed to film. When co-IP followed by Western blotting was performed, an anti-rabbit IgG conformation-specific antibody was used as a bridging antibody before incubation with an anti-mouse IgG-HRP antibody to minimize the denatured and reduced rabbit IgG light or heavy chains.
Mass Spectrometry and Protein Identification
Extracts from TREX1 stably transfected cell lines were immunoprecipitated with anti-GFP antibody, and the bead-antibody-antigen complexes were eluted and loaded onto a gel for SDS-PAGE. The gel was stained with Coomassie Brilliant Blue, and protein bands were dissected. Mass spectrometry for protein identification was performed by the Research Technologies Branch of National Institute of Allergy and Infectious Diseases, National Institutes of Health. Briefly, the gel slices containing protein bands were subjected to in-gel trypsin digestion. The released peptides and 50% acetonitrile extractions from in-gel digestion were dried by SpeedVac (Labconco, Kansas, MO). The recovered peptides were resuspended in 5 μl of solvent A (0.1% formic acid, 2% acetonitrile, 97.9% water). The bound peptides were separated at 500 nl/min using an AQ C18 reverse phase medium (3-μm particle size and 200-μm pore) packed in a pulled tip, nanochromatography column (0.100-mm inner diameter × 150-mm length) from Precision Capillary Columns (San Clemente, CA). Chromatography was performed in line with a LTQ-Velos Orbitrap mass spectrometer (Thermo Scientific), and the mobile phase consisted of a linear gradient prepared from solvent A and solvent B (0.1% formic acid, 2% water, 97.9% acetonitrile). Computer-controlled, data-dependent automated switching to MS/MS by Xcalibur 2.1 software was used for data acquisition and provided peptide sequence information. Data processing and data bank searching were performed with PD 1.2 and Mascot software (Matrix Science, Beachwood, OH). The data were searched against protein sequences deposited in the NCBI non-redundant protein database and a reverse sequence decoy database. Proteins were identified using a 1% false discovery rate cutoff and two peptides per protein minimums.
DNA Fragmentation Assay
Cells were lysed in 100 μl of lysis buffer (100 mm NaCl, 10 mm Tris-Cl, pH 8.0, 1 mm EDTA, pH 8.0, 0.1% SDS, 0.1 mg/ml Proteinase K) and incubated at 50 °C for 3 h followed by addition of 5 μl of SDS-OUT reagent (Thermo Scientific). The content was mixed and incubated on ice for 20 min followed by centrifugation at 10,000 × g for 10 min at 4 °C. Supernatants were collected in fresh centrifuge tubes and treated with RNase for 20 min on ice followed by quantification using a Nanodrop spectrophotometer (Thermo Scientific). Equal amounts of DNA (1.5 μg) were electrophoresed in a 2% agarose gel. Images of DNA agarose gels were captured using MultiImageTM Light Cabinet (Alpha Innotech Corp., San Leandro, CA). Densitometric analysis of the fragmented DNA was done using ImageJ software by subtracting the high molecular weight DNA density from total DNA density.
Recombinant Protein Expression and Purification
Escherichia coli strain BL21 harboring a pET-15b construct expressing human PARP1 proteins was grown in LB broth supplemented with ampicillin at 37 °C and induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside. Bacterial cells expressing His-tagged proteins were suspended with lysis buffer and lysed by sonication. Soluble His6-tagged proteins were separated from insoluble cell debris by centrifugation at 14,000 rpm and purified by affinity chromatography on nickel-nitrilotriacetic acid-agarose beads (Thermo Scientific). For expression of GST fusion proteins, cDNAs for human TREX1 were subcloned into pGEX-4T-3 (GE Healthcare). GST-TREX1 was expressed in E. coli strain BL21. Cells were lysed in lysis buffer supplemented with a mixture of protease inhibitors, and the proteins were purified with glutathione-Sepharose 4B (GE Healthcare) according to the standard protocol.
GST Pulldown Assay
Pulldown experiments were performed with purified recombinant proteins. Briefly, GST-tagged recombinant proteins were incubated with glutathione-Sepharose 4B beads for 4 h at 4 °C. Beads were then washed thoroughly with PBS five times and added to the His-tagged protein followed by incubation overnight at 4 °C. After washing, bound proteins were analyzed by SDS-PAGE and immunoblotting using anti-PARP antibody. GST-TREX1 was analyzed by SDS-PAGE and Coomassie Brilliant Blue staining.
Transfections with Plasmids and siRNAs
Cells were transfected with the plasmids or TREX1 siRNAs using Lipofectamine LTX or RNAiMAX (Invitrogen), respectively. The TREX1 siRNA was designed by Santa Cruz Biotechnology as a pool of three target-specific siRNAs of 20–25 nucleotides. A scrambled non-targeting siRNA (Santa Cruz Biotechnology) was used as a negative control.
Cell Proliferation Assay
siRNA-treated HeLa cells were incubated in 96-well plates for 48 h and assayed by 4-[3-(2-methoxy-4-nitrophenyl)-2-[4-nitrophenyl]-2H-5-tetrazolio]-1,3-benzenedisulfonate sodium salt (WST-8) using Cell Counting Kit-8 (Sigma-Aldrich). Absorbance at 450 nm was measured for each culture using an ELISA plate reader (SpectraMax, Molecular Devices, Sunnyvale, CA).
Measurement of PARP1 Activity
PARP1 activity in cell extracts was analyzed using the HT colorimetric PARP/apoptosis assay (Trevigen) based on the ELISA method, which semiquantitatively detects PAR deposited onto histone proteins in a 96-well format. Briefly, 100-ng protein lysates prepared from siRNA-transfected cells in triplicate were incubated with 2 mm nicotine adenine dinucleotide (NAD) and activated DNA in the histone-coated plate and developed with an anti-PAR monoclonal antibody, an anti-mouse IgG-HRP antibody, and a TACS-Sapphire colorimetric substrate. The reaction was read at 450 nm using an ELISA plate reader.
Statistical Analysis
Data are reported as the mean ± S.E. The paired t test was used to test for differences. p values <0.05 were considered to be statistically significant.
RESULTS
Identification of PARP1 as a Novel Protein Partner for TREX1
To identify TREX1-interacting proteins, we first generated stable cell lines expressing human TREX1 protein in HEK293 cells. We confirmed that the expression levels of IFPN-tagged TREX1 were higher than that of endogenous TREX1 and that the IFPN-tagged TREX1 protein could be detected by Western blotting with anti-GFP antibody (Fig. 1A). IFPN-tagged TREX1 was purified from the cell lines with anti-GFP antibody, and a single protein band was detected on the Coomassie Brilliant Blue-stained protein gel (Fig. 1B). The results of peptide sequences defined by mass spectrometry identified PARP1 as a candidate partner protein for TREX1 (Fig. 1B). To confirm the mass spectrometry result, purified cell extracts precipitated with anti-GFP antibody were analyzed by Western blotting with antibodies to PARP1 and TREX1 (Fig. 1C). This approach showed that the anti-GFP antibody co-immunoprecipitated PARP1, confirming a specific interaction of TREX1 with PARP1. We further examined the in vitro interaction of TREX1 and PARP1 using a GST pulldown assay. PARP1 protein was pulled down by immobilized GST-TREX1 but not GST alone, indicating direct interactions of these proteins (Fig. 1D).
FIGURE 1.

Identification of PARP1 as a novel protein partner for TREX1. A, expression levels of both IFPN-tagged TREX1 and endogenous TREX1 proteins from HEK293 stable cell lines expressing TREX1 were determined by Western blotting. α-Tubulin served as an endogenous loading control. B, IFPN-tagged TREX1 was purified with anti-GFP antibody from the cell extracts, eluted in SDS buffer, and subjected to SDS-PAGE. The proteins were visualized by Coomassie Brilliant Blue (CBB) staining. Peptide sequences of the PARP1 protein were obtained by mass spectrometry. Two of seven peptide sequences that identified PARP1 are shown. Note that the IFPN-TREX1 target protein of size 50 kDa is almost overlapped by the heavy chain IgG band. HC, heavy chain; LC, light chain. C, the whole-cell extracts from the stable cell lines were immunoprecipitated with anti-GFP antibody and immunoblotted with anti-PARP1 or anti-TREX1 antibody. Protein input was assessed by Western blotting with the indicated antibodies. D, purified PARP1 was incubated with GST or GST-TREX1 coupled to glutathione-Sepharose 4B. Proteins retained on Sepharose were eluted and then blotted with the indicated antibodies.
Identification of PARP1 Domains Required for Interaction with TREX1
PARP1 is a highly conserved multidomain protein containing an N-terminal DNA-binding domain with zinc finger (ZF) motifs 1 and 2 (ZF1 and ZF2), an automodification domain with a breast cancer 1 protein (BRCA1) C terminus motif, and a C-terminal catalytic domain (19). A third ZF domain (ZF3), which has recently been characterized, is distinct in structure and function from ZF1 and ZF2 (23). To identify the domains of PARP1 that interact with TREX1, we generated a V5-tagged full-length PARP1 and a panel of deletion mutants (Fig. 2A). These constructs were co-transfected with FLAG-tagged TREX1 followed by co-IP with anti-V5 antibody and Western blotting with anti-FLAG antibody and vice versa (Fig. 2B). We confirmed that full-length PARP1 interacted with TREX1 (Fig. 2B, lane Wild type) but not with empty vector (lane Mock). Furthermore, we found that TREX1 interacted with PARP1 domains included in N1, N2, N3, and N4 (lanes N1–N4) but not with domains included in C1, C2, and C3 (lanes C1–C3, gray rectangles). These results indicate that only ZF1 and ZF2 are required for binding. A possible contribution from the recently described ZF3 was not examined and thus cannot be ruled out.
FIGURE 2.
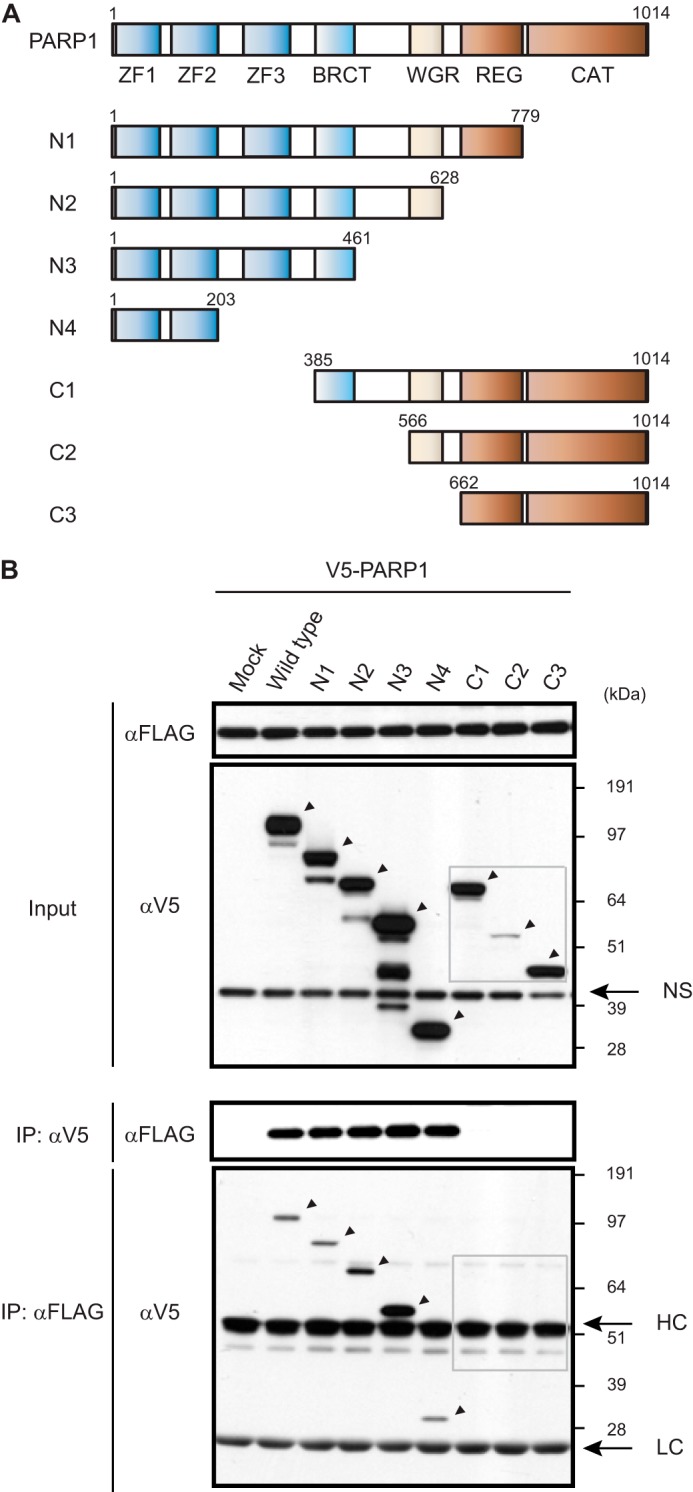
Identification of PARP1 domains required for interaction with TREX1. A, V5-tagged full-length PARP1 and deletion mutants used in this study are schematically represented. Numbers denote amino acid residues. BRCT, breast cancer 1 protein (BRCA1) C terminus domain; WGR, tryptophan-glycine-arginine domain; REG, regulatory domain; CAT, catalytic domain. B, cell extracts from HEK293 cells expressing V5-tagged full-length or deletion mutants of PARP1 with FLAG-TREX1 were immunoprecipitated with anti-V5 antibody followed by Western blotting with anti-FLAG antibody or vice versa. Arrows show specific protein identification. NS, nonspecific band; HC, heavy chain; LC, light chain.
DNA Damage Induces Nuclear Translocation of TREX1
TREX1 is localized to endoplasmic reticulum through its C-terminal transmembrane region (24), and previous reports have shown that TREX1 translocates to the nucleus following DNA damage induced by UV or γ-irradiation, hydroxyurea, or granzyme A (14, 15, 25). We hypothesized that nuclear translocation of TREX1 is required for interaction with nuclear PARP1 following DNA damage. To test this hypothesis, we first examined subcellular localization of TREX1 following induction of DNA damage. K562 cells were treated with sodium arsenite and hydroxyurea, which rapidly induce DNA breaks (26), and cell extracts recovered at various times after treatment were used to generate cytoplasmic and nuclear fractions. Western blotting showed that TREX1 was normally localized to cytoplasm and that the expression of nuclear TREX1 increased following treatment with arsenite (Fig. 3A) and hydroxyurea (Fig. 3B) in a time-dependent manner. The lower panel in Fig. 3A shows that nuclear TREX1 increased within 15 min and reached maximum levels at 60 min, suggesting that this translocation is rapid, but when compared with cytoplasmic levels, only a small amount of TREX1 translocated to the nucleus. TREX1 expression in cells treated with hydroxyurea was also increased about 3-fold at 240 min compared with the control level (Fig. 3B). We then proceeded to fractionate cell extracts into different cellular compartments, including cytoplasmic, cytoplasmic membrane, soluble nuclear, and chromatin-bound fractions using HeLa cells either untreated or treated with arsenite for 60 min. Western blotting and the analyses of band intensities showed that the levels of TREX1 clearly increased in both the soluble nuclear and chromatin-bound fractions following arsenite treatment and that the levels of PARP1 increased in chromatin-bound fractions but not in nuclear fractions (Fig. 3C). These results demonstrate that following DNA damage TREX1 translocated to the nucleus, especially to chromatin, in concert with PARP1, suggesting that TREX1 is associated with the DNA damage response by PARP1.
FIGURE 3.

DNA damage induces nuclear translocation of TREX1. A, subcellular localization of TREX1 was determined by Western blotting using cytoplasmic and nuclear fractions of K562 cells treated with sodium arsenite (SA) (2 mm) for the indicated periods of time (n = 3). GAPDH and lamin A/C served as endogenous loading controls for cytoplasmic and nuclear proteins, respectively. Densitometric analysis of the blots is shown in the lower panel, and values are given as the mean ± S.E. (error bars) of nuclear TREX1 levels relative to lamin A/C levels from three independent experiments. *, p < 0.05; **, p < 0.005 by paired t test. B, subcellular localization of TREX1 was determined by Western blotting using cytoplasmic and nuclear fractions of K562 cells treated with hydroxyurea (HU) (5 mm) for the indicated periods of time. Tubulin and lamin A/C served as endogenous loading controls for cytoplasmic and nuclear proteins, respectively. Densitometric analysis of the blots is shown in the lower panel. C, subcellular localizations of TREX1 and PARP1 were determined by Western blotting using cytoplasmic and nuclear fractions (CE, cytoplasmic extract; ME, membrane extract; NE, nuclear extract; CB, chromatin-bound extract) of HeLa cells treated with or without sodium arsenite (2 mm) for 1 h. Equal amounts of protein were loaded in each lane. Calreticulin, lamin A/C, and histone H3 served as loading controls for the membrane, soluble nuclear, and chromatin fractions, respectively. Densitometric analyses of the blots for TREX1 and PARP1 levels are shown in the lower panels. The values in soluble nuclear and chromatin-bound fractions are normalized to lamin A/C and histone H3 levels, respectively.
Endogenous TREX1 Interacts with PARP1 in Response to DNA Damage
To examine the physiological interaction between TREX1 and PARP1, we performed co-IP using endogenous proteins of HeLa cells treated with arsenite to induce DNA damage. PARP1 was detected in TREX1 immunoprecipitates of whole-cell extracts from arsenite-treated cells but not in the TREX1 immunoprecipitates of cells cultured under normal conditions or in immunoprecipitates with a control IgG (Fig. 4A). The interaction was not affected by pretreatment with DNase I (Fig. 4B), demonstrating that TREX1 interacts with PARP1 independently of interactions with DNA. We also confirmed this interaction using nuclear extracts (Fig. 4C). Next, we examined the effect of arsenite treatment on the binding capacity of TREX1 to PARP1 over time (Fig. 4D) because nuclear translocation of TREX1 would likely be associated with PARP1 interactions as predicted from the results in Fig. 3. We found that interactions of the proteins increased significantly in the nucleus following DNA damage (Fig. 4D), suggesting that TREX1 may act to modulate the function of PARP1 in the nucleus. These data suggest that the DNA damage is necessary for TREX1 and PARP1 to interact under physiologic conditions. It is worth noting that direct binding was seen in cells without intentional DNA damage in the TREX1 overexpression models shown in Figs. 1 and 2. It may be that the high levels of TREX1 expressed in these cells enabled detection of the interaction even in cells with minimal DNA damage.
FIGURE 4.

Endogenous TREX1 interacts with PARP1 in response to DNA damage. A, whole-cell extracts from HeLa cells treated with or without sodium arsenite (2 mm) for 1 h were immunoprecipitated with anti-TREX1 or anti-rabbit IgG antibody and immunoblotted with anti-PARP1 or anti-TREX1 antibody. Input of the proteins was assessed by Western blotting with the indicated antibodies. B, DNase I was added to the extracts at 200 units for 1 h at 32 °C prior to co-IP of TREX1. C, nuclear extracts from HeLa cells were immunoprecipitated with anti-TREX1 antibody and immunoblotted with anti-PARP1 or anti-TREX1 antibody. Lamin A/C served as an endogenous loading control for nuclear proteins. D, nuclear extracts from HeLa cells treated with or without sodium arsenite (2 mm) for the indicated times were immunoprecipitated with anti-TREX1 antibody and immunoblotted with anti-PARP1 or anti-TREX1 antibody.
Knockdown of TREX1 Promotes PARP1 Cleavage and Loss of PARP1 Enzymatic Activity
To understand the physiological effects of TREX1 on PARP1 function, we examined cells treated with a TREX1-specific siRNA. We found that the knockdown effect of TREX1 by siRNA was specific for the target molecule and was greater at 48 than 24 h (Fig. 5A). Studies of cell proliferation using a WST-8 assay showed that the TREX1 knockdown resulted in significantly reduced cell expansion (Fig. 5B). This result is consistent with data from an earlier study that showed that cells from Trex1 knock-out mice were retarded in G1/S transition and proliferated more slowly, resulting in a prolonged cell doubling time (14). This difference in cell numbers in our study could be due to an inhibition of cell cycle progression or cell death, raising the possibility that TREX1 may normally be associated with cell cycle control or inhibition of cell death. To probe these possibilities, we examined the effect of TREX1 knockdown on levels of p53, PARP1, and cleaved PARP1 proteins by Western blotting (Fig. 5C). The results showed that levels of p53, a classic indicator of DNA damage (27), were significantly increased and that the knockdown of TREX1 was associated with reduced levels of PARP1 and increased levels of cleaved PARP1. Previous studies showed that PARP1 is cleaved at the same site by caspase-3 or granzyme B to yield an N-terminal DNA-binding domain and a C-terminal catalytic domain, resulting in PARP1 inactivation (28). We confirmed this by using a DNA fragmentation assay. Total genomic DNA was isolated from HeLa cells after 48 h of transfection with TREX1-specific siRNA or control siRNA. As shown in Fig. 5D, DNA fragmentation was only induced in cells transfected with TREX1 siRNA as compared with control siRNA. These data suggest that reduction of TREX1 levels was associated with induction of apoptosis through decreased PARP1 activity. To test this concept, we determined PARP1 enzymatic activity using an ELISA-based assay. Fig. 5E compares the PARP1 activity in TREX1 siRNA-transfected cells with that of untreated cells or cells treated with a control siRNA. The results showed that PAPR1 activity was clearly reduced in TREX1 siRNA-treated cells. Taken together, our findings suggest that TREX1 is involved in preservation of PARP1 levels and function.
FIGURE 5.

Knockdown of TREX1 promotes PARP1 cleavage and loss of PARP1 enzymatic activity. A, HeLa cells were transfected with TREX1-specific or control siRNAs at the indicated concentrations (10–100 nm) and time course (24 or 48 h). Expression levels of endogenous TREX1 proteins were determined by Western blotting. α-Tubulin served as an endogenous loading control. (−), no siRNA treatment. B, cell proliferation assessed by the tetrazolium method was used with TREX1-specific or control siRNA-treated cells (50 nm). The cells were incubated for the indicated times (24–72 h), and the absorbance was measured for each culture using an ELISA plate reader (n = 3). Data are given as the mean ± S.E. (error bars). *, p < 0.01 by paired t test. C, HeLa cells were transfected with TREX1-specific or control siRNA (50 nm) for 48 h, and the expression levels of the indicated proteins were determined by Western blotting (n = 4). Densitometric analyses of the blots are shown in the lower panel, and values are given as the mean ± S.E. (error bars) for TREX1, p53, PARP1, and cleaved PARP1 levels relative to GAPDH levels from the four independent experiments. *, p < 0.05; **, p < 0.005; ***, p < 0.0005 by paired t test. (−), no siRNA treatment. D, agarose gel electrophoresis of genomic DNA isolated from cells transfected with TREX1 and control siRNA for 48 h. The intensity of fragmented DNA in the lower panel was calculated, and mean with S.D. (error bars) was plotted. The p value was calculated by comparing with the control sample using paired t test. AU, arbitrary units. E, HeLa cells were transfected with TREX1-specific or control siRNA (50 nm) for 48 h, and PARP1 activity in cell extracts (100 ng of protein) was measured by quantification of PAR from histones attached to strip wells in a 96-well format using the HT colorimetric PARP/apoptosis assay kit (n = 3). Data are given as the mean ± S.E. (error bars). *, p < 0.05; **, p < 0.005 by paired t test. (−), no siRNA treatment. n.s., not significant.
TREX1 Influences PARP1 Cleavage and Poly(ADP-ribosyl)ation of PARP1 during DNA Damage Response
To further validate TREX1 as a regulator of PARP1 protein expression, we assessed PARP1 cleavage in TREX1-overexpressing cells in response to apoptotic induction. As shown in Fig. 6A, overexpression of TREX1, but not a control FLAG vector, was associated with ∼3-fold increased full-length PARP1 protein levels in HeLa cells treated with staurosporine. Cleaved PARP1, however, was decreased approximately by half, suggesting that TREX1 represses PARP1 cleavage. PARP1 is activated by DNA strand breaks and catalyzes poly(ADP-ribosyl)ation of acceptor proteins, including itself (19, 20). To assess this, we determined the effect of TREX1 on poly(ADP-ribosyl)ation of PARP1 in cells undergoing DNA damage and performed co-IP experiments using stably TREX1 overexpressed cell lines treated with arsenite (Fig. 6B). In TREX1 stable cell lines, poly(ADP-ribosyl)ation of PARP1 was clearly higher than in control cell lines under normal conditions, whereas poly(ADP-ribosyl)ation was enhanced by arsenite treatment peaking at 10 min. These data suggest that TREX1 can stabilize PARP1 levels and enhance arsenite-induced poly(ADP-ribosyl)ation of PARP1, supporting the findings presented in Fig. 5. It is also possible that these findings could be explained by TREX1 itself having antiapoptotic functions quite separate from its interactions with PARP1.
FIGURE 6.

TREX1 influences PARP1 cleavage and poly(ADP-ribosyl)ation of PARP1 during DNA damage response. A, HeLa cells transfected with FLAG-TREX1 or a control FLAG vector were cultured with or without staurosporine (1 μm) for 3 h before harvest. Whole protein extracts prepared with Nonidet P-40 cell lysis buffer were subjected to SDS-PAGE and then immunoblotted with antibodies against PARP1, FLAG, or tubulin. Signal intensities of endogenous full-length and cleaved PARP1 were quantified and normalized to tubulin. Data are given as the mean ± S.E. (error bars). *, p < 0.05; **, p < 0.005 by paired t test. B, TREX1 stably overexpressing or control HEK293 cells were treated with or without sodium arsenite (2 mm) for 10 or 30 min. Whole-cell extracts were immunoprecipitated with anti-PARP1 antibody and immunoblotted with anti-PAR or anti-PARP1 antibody. Densitometric analyses of the blots for PAR levels normalized to PARP1 levels are shown in the lower panels. PARlated, poly(ADP-ribosyl)ated.
DISCUSSION
Previous studies have identified both nuclear and cytoplasmic activities of TREX1. As an interacting component of the endoplasmic reticulum-resident SET complex, TREX1 translocates to the nucleus of cells to degrade nicked DNA during granzyme A-mediated cell death (15) or following exposure to genotoxic stress. Normal nuclear functions may include excision of mispaired DNA 3′ termini (29) and BER. Cytoplasmic activities, such as digestion of cytosolic HIV DNA (18) and enhancement of lysosomal biogenesis (30), have only recently been identified. Proteins that partner with TREX1 to mediate these functions are reasonably well known for some activities (15) but not at all for others. Identification of partnering proteins involved in both nuclear and cytoplasmic activities of TREX1 is likely to be of importance for understanding the pathogenesis of differing human diseases caused by distinct mutations in TREX1: inflammatory/autoimmune disorders, including AGS (6), SLE (10, 11), and familial chilblain lupus (8, 9), and the endotheliopathy RVCL (7). This study, undertaken to detect potential additional interacting proteins, identified PARP1 as a previously unappreciated partner for TREX1. The results of this study demonstrated that TREX1 interacts with PARP1 after nuclear translocation of the TREX1 in response to DNA damage and that nuclear TREX1 may contribute to the regulation of PARP1 activity in the DNA damage response. Previous studies showed that these proteins play important roles in response to DNA damage, including ssDNA break repair. We confirmed the interaction using mass spectrometry and co-IP methods using in vivo overexpression models and in vitro; however, the physiological interaction was identified only under conditions involving DNA damage.
We found that the two N-terminal ZF domains of PARP1, ZF1 and ZF2, were critical for its interaction with TREX1. The fact that this interaction was not dependent on the presence of DNA is indicative of a direct interaction between the proteins rather than one based on the ability of both to bind DNA. An earlier study showed that PARP1 recognizes nicked DNA by utilizing the ZF motifs in its DNA-binding domain. However, these regions can also mediate protein-protein interactions as they contain sequences responsible for interactions between ZF1 and ZF2 (31). Furthermore, the ZFs can also act as an interface with various protein partners, including the BER pathway protein XRCC1 (32, 33). A number of other PARP1 partner proteins have been identified, including BIN1 (34), SIRT6 (35), and IκB kinase, protein inhibitor of activated STAT protein γ, and ataxia telangiectasia mutated (36), although the PARP1 domains required for these interactions have not been carefully mapped. It is possible that the ZF-dependent interaction between TREX1 and PARP1 could interfere with or augment the ability of PARP1 to engage in these interactions.
The main function of TREX1 is to digest ssDNA derived from endogenous retroelements or from processing of aberrant replication intermediates to prevent their accumulation and activation of cell-intrinsic responses to immunostimulatory DNA (13, 14). However, the physiological function of nuclear TREX1 remained unclear. It was shown previously that TREX1 translocates to the nucleus following DNA damage induced by UV or γ-irradiation, hydroxyurea (14, 25), granzyme A (15), the environmental carcinogen benzo(a)pyrene (25), or the DNA topoisomerase I inhibitor camptothecin (37). Chowdhury et al. (15) showed that TREX1 works as a proofreading exonuclease for BER because TREX1 degrades DNA in concert with endonuclease NM23-H1 during granzyme A-mediated cell death, indicating that TREX1 acts in the nucleus and may function in DNA repair. Our results show that TREX1 translocated to the nucleus of K562 and HeLa cells after treatment with arsenite. The new finding from the current study is the association of TREX1 with PARP1 following nuclear translocation. Although PARP1 is involved in many aspects of the cellular response to DNA damage, it plays a key role in BER of ssDNA break (19, 20). In this way, TREX1 and PARP1 proteins have similar functions, suggesting that our findings may provide new insights into the mechanisms of ssDNA repair following DNA damage. Furthermore, we found that translocated TREX1 became chromatin-associated in concert with an increase in chromatin-bound PARP1. In this regard, it is noteworthy that SET, which translocates to the nucleus with TREX1 during granzyme A-mediated DNA damage, gains access to chromatin to promote transcription with PARP1 (38). These reports support the hypothesis that TREX1 may be a component of a chromatin-bound protein complex that also includes SET and PARP1.
We also examined the functional role of TREX1 in the regulation of PARP1 stability and function and found that knockdown of TREX1 reduced cell expansion in association with a loss of PARP1 enzymatic activity. This finding was dependent on the induction of PARP1 cleavage and an increase in p53 levels, directly linking TREX1-PARP1 interactions to the DNA damage response (39). Furthermore, we found that cells with increasing levels of transfected TREX1 exhibited progressively increased PARP1 stability and that TREX1 stimulated increased autopoly(ADP-ribosyl)ation of PARP1 following DNA damage. Although nuclear translocation of TREX1 seems to be essential for PARP1 stabilization in the DNA damage response, the exact molecular mechanisms of its regulation of PARP1 are only partially understood. It remains to be determined how TREX1 is associated with stabilization of PARP1 or how the TREX1-PARP1 complex is targeted to DNA nicks. It will also be important to determine whether the TREX1-PARP1 interaction is affected by mutations responsible for the various TREX1-related diseases.
In conclusion, this study has identified and characterized an interaction between TREX1 and PARP1 in response to DNA damage in vivo and in vitro. Functional studies suggested that TREX1 is associated with stabilization of PARP1 levels and function. At present, we are not able to distinguish between the possibility that the effects we see on apoptosis associated with depletion and overexpression of TREX1 might be due to antiapoptotic effects of TREX1 itself or that the binding of TREX1 to PARP1 may be important to the regulation of PARP1 in the DNA damage response. Further studies are necessary to determine whether this partnering is important to the development or progression of TREX1-associated diseases.
Acknowledgments
We thank Parul Kothari and John Atkinson (Washington University School of Medicine) for providing specific antibodies and plasmids and detailed responses to our queries, Farokh Dotiwala and Judy Lieberman (Harvard Medical School) for helpful advice related to this work, Robert Crouch (National Institute of Child Health and Human Development) for critical reading of the manuscript, and Ming Zhao (Research Technologies Branch, National Institute of Allergy and Infectious Diseases) for assistance in mass spectrometry analyses. We also thank our colleagues Shweta Jain and Alexander Kovalchuk for useful discussions.
This work was supported, in whole or in part, by the National Institutes of Health Intramural Research Program of the NIAID.
- SLE
- systemic lupus erythematosus
- AGS
- Aicardi-Goutières syndrome
- BER
- base excision repair
- IFN1
- type I interferon
- PARP1
- poly(ADP-ribose) polymerase-1
- RVCL
- retinal vasculopathy with cerebral leukodystrophy
- TREX1
- 3′ repair exonuclease 1
- ZF
- zinc finger
- ssDNA
- single-stranded DNA
- PAR
- poly(ADP-ribose)
- IFPN
- intensely fluorescent protein N-terminal portion
- co-IP
- co-immunoprecipitation
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1. Barbalat R., Ewald S. E., Mouchess M. L., Barton G. M. (2011) Nucleic acid recognition by the innate immune system. Annu. Rev. Immunol. 29, 185–214 [DOI] [PubMed] [Google Scholar]
- 2. Barber G. N. (2011) Innate immune DNA sensing pathways: STING, AIMII and the regulation of interferon production and inflammatory responses. Curr. Opin. Immunol. 23, 10–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Höss M., Robins P., Naven T. J., Pappin D. J., Sgouros J., Lindahl T. (1999) A human DNA editing enzyme homologous to the Escherichia coli DnaQ/MutD protein. EMBO J. 18, 3868–3875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mazur D. J., Perrino F. W. (1999) Identification and expression of the TREX1 and TREX2 cDNA sequences encoding mammalian 3′→5′ exonucleases. J. Biol. Chem. 274, 19655–19660 [DOI] [PubMed] [Google Scholar]
- 5. Kavanagh D., Spitzer D., Kothari P. H., Shaikh A., Liszewski M. K., Richards A., Atkinson J. P. (2008) New roles for the major human 3′–5′ exonuclease TREX1 in human disease. Cell Cycle 7, 1718–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Crow Y. J., Hayward B. E., Parmar R., Robins P., Leitch A., Ali M., Black D. N., van Bokhoven H., Brunner H. G., Hamel B. C., Corry P. C., Cowan F. M., Frints S. G., Klepper J., Livingston J. H., Lynch S. A., Massey R. F., Meritet J. F., Michaud J. L., Ponsot G., Voit T., Lebon P., Bonthron D. T., Jackson A. P., Barnes D. E., Lindahl T. (2006) Mutations in the gene encoding the 3′–5′ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat. Genet. 38, 917–920 [DOI] [PubMed] [Google Scholar]
- 7. Richards A., van den Maagdenberg A. M., Jen J. C., Kavanagh D., Bertram P., Spitzer D., Liszewski M. K., Barilla-Labarca M. L., Terwindt G. M., Kasai Y., McLellan M., Grand M. G., Vanmolkot K. R., de Vries B., Wan J., Kane M. J., Mamsa H., Schäfer R., Stam A. H., Haan J., de Jong P. T., Storimans C. W., van Schooneveld M. J., Oosterhuis J. A., Gschwendter A., Dichgans M., Kotschet K. E., Hodgkinson S., Hardy T. A., Delatycki M. B., Hajj-Ali R. A., Kothari P. H., Nelson S. F., Frants R. R., Baloh R. W., Ferrari M. D., Atkinson J. P. (2007) C-terminal truncations in human 3′–5′ DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat. Genet. 39, 1068–1070 [DOI] [PubMed] [Google Scholar]
- 8. Lee-Kirsch M. A., Gong M., Schulz H., Rüschendorf F., Stein A., Pfeiffer C., Ballarini A., Gahr M., Hubner N., Linné M. (2006) Familial chilblain lupus, a monogenic form of cutaneous lupus erythematosus, maps to chromosome 3p. Am. J. Hum. Genet. 79, 731–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rice G., Newman W. G., Dean J., Patrick T., Parmar R., Flintoff K., Robins P., Harvey S., Hollis T., O'Hara A., Herrick A. L., Bowden A. P., Perrino F. W., Lindahl T., Barnes D. E., Crow Y. J. (2007) Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am. J. Hum. Genet. 80, 811–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee-Kirsch M. A., Gong M., Chowdhury D., Senenko L., Engel K., Lee Y. A., de Silva U., Bailey S. L., Witte T., Vyse T. J., Kere J., Pfeiffer C., Harvey S., Wong A., Koskenmies S., Hummel O., Rohde K., Schmidt R. E., Dominiczak A. F., Gahr M., Hollis T., Perrino F. W., Lieberman J., Hübner N. (2007) Mutations in the gene encoding the 3′–5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat. Genet. 39, 1065–1067 [DOI] [PubMed] [Google Scholar]
- 11. Namjou B., Kothari P. H., Kelly J. A., Glenn S. B., Ojwang J. O., Adler A., Alarcón-Riquelme M. E., Gallant C. J., Boackle S. A., Criswell L. A., Kimberly R. P., Brown E., Edberg J., Stevens A. M., Jacob C. O., Tsao B. P., Gilkeson G. S., Kamen D. L., Merrill J. T., Petri M., Goldman R. R., Vila L. M., Anaya J. M., Niewold T. B., Martin J., Pons-Estel B. A., Sabio J. M., Callejas J. L., Vyse T. J., Bae S. C., Perrino F. W., Freedman B. I., Scofield R. H., Moser K. L., Gaffney P. M., James J. A., Langefeld C. D., Kaufman K. M., Harley J. B., Atkinson J. P. (2011) Evaluation of the TREX1 gene in a large multi-ancestral lupus cohort. Genes Immun. 12, 270–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Morita M., Stamp G., Robins P., Dulic A., Rosewell I., Hrivnak G., Daly G., Lindahl T., Barnes D. E. (2004) Gene-targeted mice lacking the Trex1 (DNase III) 3′→5′ DNA exonuclease develop inflammatory myocarditis. Mol. Cell. Biol. 24, 6719–6727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stetson D. B., Ko J. S., Heidmann T., Medzhitov R. (2008) Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134, 587–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang Y. G., Lindahl T., Barnes D. E. (2007) Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell 131, 873–886 [DOI] [PubMed] [Google Scholar]
- 15. Chowdhury D., Beresford P. J., Zhu P., Zhang D., Sung J. S., Demple B., Perrino F. W., Lieberman J. (2006) The exonuclease TREX1 is in the SET complex and acts in concert with NM23-H1 to degrade DNA during granzyme A-mediated cell death. Mol. Cell 23, 133–142 [DOI] [PubMed] [Google Scholar]
- 16. Fan Z., Beresford P. J., Oh D. Y., Zhang D., Lieberman J. (2003) Tumor suppressor NM23-H1 is a granzyme A-activated DNase during CTL-mediated apoptosis, and the nucleosome assembly protein SET is its inhibitor. Cell 112, 659–672 [DOI] [PubMed] [Google Scholar]
- 17. Lee-Kirsch M. A., Chowdhury D., Harvey S., Gong M., Senenko L., Engel K., Pfeiffer C., Hollis T., Gahr M., Perrino F. W., Lieberman J., Hubner N. (2007) A mutation in TREX1 that impairs susceptibility to granzyme A-mediated cell death underlies familial chilblain lupus. J. Mol. Med. 85, 531–537 [DOI] [PubMed] [Google Scholar]
- 18. Yan N., Regalado-Magdos A. D., Stiggelbout B., Lee-Kirsch M. A., Lieberman J. (2010) The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat. Immunol. 11, 1005–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rouleau M., Patel A., Hendzel M. J., Kaufmann S. H., Poirier G. G. (2010) PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 10, 293–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gibson B. A., Kraus W. L. (2012) New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 13, 411–424 [DOI] [PubMed] [Google Scholar]
- 21. Ding Z., Liang J., Lu Y., Yu Q., Songyang Z., Lin S. Y., Mills G. B. (2006) A retrovirus-based protein complementation assay screen reveals functional AKT1-binding partners. Proc. Natl. Acad. Sci. U.S.A. 103, 15014–15019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee C. H., Melchers M., Wang H., Torrey T. A., Slota R., Qi C. F., Kim J. Y., Lugar P., Kong H. J., Farrington L., van der Zouwen B., Zhou J. X., Lougaris V., Lipsky P. E., Grammer A. C., Morse H. C., 3rd. (2006) Regulation of the germinal center gene program by interferon (IFN) regulatory factor 8/IFN consensus sequence-binding protein. J. Exp. Med. 203, 63–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Langelier M. F., Planck J. L., Roy S., Pascal J. M. (2012) Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science 336, 728–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brucet M., Querol-Audí J., Serra M., Ramirez-Espain X., Bertlik K., Ruiz L., Lloberas J., Macias M. J., Fita I., Celada A. (2007) Structure of the dimeric exonuclease TREX1 in complex with DNA displays a proline-rich binding site for WW Domains. J. Biol. Chem. 282, 14547–14557 [DOI] [PubMed] [Google Scholar]
- 25. Christmann M., Tomicic M. T., Aasland D., Berdelle N., Kaina B. (2010) Three prime exonuclease I (TREX1) is Fos/AP-1 regulated by genotoxic stress and protects against ultraviolet light and benzo(a)pyrene-induced DNA damage. Nucleic Acids Res. 38, 6418–6432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ying S., Myers K., Bottomley S., Helleday T., Bryant H. E. (2009) BRCA2-dependent homologous recombination is required for repair of arsenite-induced replication lesions in mammalian cells. Nucleic Acids Res. 37, 5105–5113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang J., Duerksen-Hughes P. (1998) A new approach to identifying genotoxic carcinogens: p53 induction as an indicator of genotoxic damage. Carcinogenesis 19, 1117–1125 [DOI] [PubMed] [Google Scholar]
- 28. Zhu P., Martinvalet D., Chowdhury D., Zhang D., Schlesinger A., Lieberman J. (2009) The cytotoxic T lymphocyte protease granzyme A cleaves and inactivates poly(adenosine 5′-diphosphate-ribose) polymerase-1. Blood 114, 1205–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mazur D. J., Perrino F. W. (2001) Excision of 3′ termini by the Trex1 and TREX2 3′→5′ exonucleases. Characterization of the recombinant proteins. J. Biol. Chem. 276, 17022–17029 [DOI] [PubMed] [Google Scholar]
- 30. Hasan M., Koch J., Rakheja D., Pattnaik A. K., Brugarolas J., Dozmorov I., Levine B., Wakeland E. K., Lee-Kirsch M. A., Yan N. (2013) Trex1 regulates lysosomal biogenesis and interferon-independent activation of antiviral genes. Nat. Immunol. 14, 61–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ali A. A., Timinszky G., Arribas-Bosacoma R., Kozlowski M., Hassa P. O., Hassler M., Ladurner A. G., Pearl L. H., Oliver A. W. (2012) The zinc-finger domains of PARP1 cooperate to recognize DNA strand breaks. Nat. Struct. Mol. Biol. 19, 685–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Masson M., Niedergang C., Schreiber V., Muller S., Menissier-de Murcia J., de Murcia G. (1998) XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol. Cell. Biol. 18, 3563–3571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shall S., de Murcia G. (2000) Poly(ADP-ribose) polymerase-1: what have we learned from the deficient mouse model? Mutat. Res. 460, 1–15 [DOI] [PubMed] [Google Scholar]
- 34. Pyndiah S., Tanida S., Ahmed K. M., Cassimere E. K., Choe C., Sakamuro D. (2011) c-MYC suppresses BIN1 to release poly(ADP-ribose) polymerase 1: a mechanism by which cancer cells acquire cisplatin resistance. Sci. Signal. 4, ra19. [DOI] [PubMed] [Google Scholar]
- 35. Mao Z., Hine C., Tian X., Van Meter M., Au M., Vaidya A., Seluanov A., Gorbunova V. (2011) SIRT6 promotes DNA repair under stress by activating PARP1. Science 332, 1443–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stilmann M., Hinz M., Arslan S. C., Zimmer A., Schreiber V., Scheidereit C. (2009) A nuclear poly(ADP-ribose)-dependent signalosome confers DNA damage-induced IκB kinase activation. Mol. Cell 36, 365–378 [DOI] [PubMed] [Google Scholar]
- 37. Wang C. J., Lam W., Bussom S., Chang H. M., Cheng Y. C. (2009) TREX1 acts in degrading damaged DNA from drug-treated tumor cells. DNA Repair 8, 1179–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gamble M. J., Fisher R. P. (2007) SET and PARP1 remove DEK from chromatin to permit access by the transcription machinery. Nat. Struct. Mol. Biol. 14, 548–555 [DOI] [PubMed] [Google Scholar]
- 39. Kanai M., Hanashiro K., Kim S. H., Hanai S., Boulares A. H., Miwa M., Fukasawa K. (2007) Inhibition of Crm1-p53 interaction and nuclear export of p53 by poly(ADP-ribosyl)ation. Nat. Cell Biol. 9, 1175–1183 [DOI] [PubMed] [Google Scholar]