

Background: The voltage-dependent anion channel 1 (VDAC1) is identified as a receptor of human plasminogen kringle 5 (K5), but the role and mechanisms of VDAC1 in K5-induced endothelial cell (EC) apoptosis remain elusive.
Results: K5 up-regulates VDAC1 through a AKT-GSK3β pathway.
Conclusion: A positive feedback loop of “VDAC1-AKT-GSK3β-VDAC1” mediates K5-induced EC apoptosis.
Significance: This finding provides new perspectives on the mechanisms of K5-induced apoptosis.
Keywords: Apoptosis, Bax, Cell Biology, Cell Signaling, Cell Surface Receptor, Cellular Regulation, Glycogen Synthase Kinase 3 (GSK-3), Molecular Cell Biology, Phosphorylation, PI 3-Kinase (PI3K)
Abstract
Human plasminogen kringle 5 (K5) is known to display its potent anti-angiogenesis effect through inducing endothelial cell (EC) apoptosis, and the voltage-dependent anion channel 1 (VDAC1) has been identified as a receptor of K5. However, the exact role and underlying mechanisms of VDAC1 in K5-induced EC apoptosis remain elusive. In the current study, we showed that K5 increased the protein level of VDAC1, which initiated the mitochondrial apoptosis pathway of ECs. Our findings also showed that K5 inhibited the ubiquitin-dependent degradation of VDAC1 by promoting the phosphorylation of VDAC1, possibly at Ser-12 and Thr-107. The phosphorylated VDAC1 was attenuated by the AKT agonist, glycogen synthase kinase (GSK) 3β inhibitor, and siRNA, suggesting that K5 increased VDAC1 phosphorylation via the AKT-GSK3β pathway. Furthermore, K5 promoted cell surface translocation of VDAC1, and binding between K5 and VDAC1 was observed on the plasma membrane. HKI protein blocked the impact of K5 on the AKT-GSK3β pathway by competitively inhibiting the interaction of K5 and cell surface VDAC1. Moreover, K5-induced EC apoptosis was suppressed by VDAC1 antibody. These data show for the first time that K5-induced EC apoptosis is mediated by the positive feedback loop of “VDAC1-AKT-GSK3β-VDAC1,” which may provide new perspectives on the mechanisms of K5-induced apoptosis.
Introduction
Angiogenesis, the process by which new blood vessels are formed, plays a critical role in the pathology of many angiogenesis-related diseases including cancer (1), diabetic retinopathy (2), and diabetic nephropathy (3). Given this, anti-angiogenic approaches have potential clinical applications. Over recent decades, much research has been focused on the relationship between angiogenic inhibitors and angiogenesis-related diseases.
Kringle 5 (K5),4 the fifth kringle domain of human plasminogen, has been shown to be the most active anti-angiogenic factor of all plasminogen proteolytic fragments (4). Previously, K5 has been found to display an inhibitory effect on alkali burn-induced corneal neovascularization and ischemia-induced retinal neovascularization (5, 6). Moreover, studies have shown K5 to act as an effective intervention against the growth of various solid tumors, such as glioma, colorectal, and hepatocellular carcinoma, as well as Lewis lung carcinoma via its anti-angiogenic activity (7–10). The inhibitory effect of K5 on neovascularization is based on the targeting of endothelial cells (ECs). It has been reported that K5 may cause cell cycle arrest and apoptosis of bFGF-stimulated endothelial cells (11).
The voltage-dependent anion channel (VDAC), also known as multifunctional mitochondrial porin, has three isoforms located on the outer mitochondrial membrane. Among them, VDAC1 is the most studied isoform and regulates a series of physiological and pathological events, such as the balance of cellular Ca2+, energy metabolism, and cell apoptosis (12). Up-regulation of VDAC1 has been shown to facilitate arsenic trioxide (As2O3)-induced human IM-9 multiple myeloma cell death (13). Down-regulation of VDAC1 by siRNA attenuated endostatin-induced human microvascular endothelial cell apoptosis (14). VDAC1-mediated apoptosis mainly occurs through interaction with pro-apoptotic protein Bax and causes the opening of the mitochondrial permeability transition pore (mPTP), which results in the release of cytochrome c into the cytoplasm and subsequent cleavage of caspase-9 (15). Thus, VDAC1 presents as a key factor in mitochondria-mediated apoptosis. Our previous investigation showed that the mitochondrial apoptosis pathway at least partly mediated K5-induced human umbilical vein endothelial cell (HUVEC) apoptosis by regulating the subcellular distribution of Bak and Bcl-xL (16). Therefore, we propose that VDAC1 may link the upstream signals to the mitochondrial apoptosis pathway in endothelial cells initiated by K5.
VDAC was originally considered to be located only on mitochondrial membrane. Later, increasing evidence suggested that VDAC may also be found on the plasma membrane (17). Furthermore, the plasma membrane VDAC1 was reported to be the receptor of K5 in HUVECs (18). However, the exact role and underlying mechanisms of VDAC1 in K5-induced EC apoptosis remain elusive. Here, we show for the first time that K5 induces VDAC1 expression both in plasma membrane and mitochondrial membrane through inhibiting its ubiquitin-dependent degradation via the AKT-GSK3β axis. The increased plasma membrane VDAC1, as a receptor, delivers the signal to modulate its own expression, and the increased mitochondrial membrane VDAC1 initiates the mitochondrial apoptosis pathway through binding with Bak. There is a positive feedback loop of “VDAC1-AKT-GSK3β-VDAC1” in K5-induced EC apoptosis. Our findings may provide new perspectives on the mechanisms of K5-induced apoptosis.
EXPERIMENTAL PROCEDURES
Cell Culture
This study conformed to the principles outlined in the Declaration of Helsinki and was approved by the Medical Ethics Committee of Sun Yat-sen University. Written informed consent was obtained from the donor. HUVECs were isolated from human umbilical cord with trypsin, as previously described (19). After dissociation, the cells were collected and cultured on gelatin-coated culture flasks in M199 medium with 15% FBS, 60 μg/ml of endothelial cell growth supplements, 100 μg/ml of penicillin, and 100 μg/ml of streptomycin. All cells were incubated in a humidified atmosphere containing 5% CO2 at 37 °C. Subcultures were performed with trypsin-EDTA. Medium were refreshed every 2 days. The cultured cells were serum-starved overnight and then carried out under 1% FBS and 20 ng/ml of bFGF for the indicated time periods. Studies were conducted on cells from passages 2–6.
Reagents and Antibodies
MG132 and AKT inhibitor IV were obtained from Merck Millipore (La Jolla, CA). The GSK3β inhibitor (CHIR99021) was purchased from Selleckchem (Sellekchem, TX). Insulin was obtained from Sigma. Hexokinase I (HKI) protein was from Abcam (Cambridge, UK). Antibodies of streptavidin-HRP, Na+/K+-ATPase, p-AKT (Ser-473), AKT, p-GSK3β (Ser-9), His tag, Myc tag, cytochrome c, and COXIV were purchased from Cell Signaling Technology. Antibodies of caspase-3, Bak, Bcl-xL, VDAC1 (20B12), GRP78 (N-20), GRP78 (H-129), and Ub (P4D1) were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-VDAC1/porin (20B12AF2), anti-pro-caspase-3, and anti-phosphoserine/threonine/tyrosine antibodies were from Abcam. The GSK3β antibody was obtained from BD Pharmingen. Antibodies of anti-β-actin and anti-GAPDH were obtained from Sigma.
Plasmid Construction
A full-length cDNA fragment of K5 was amplified from human adjacent non-tumor tissues of hepatocellular carcinoma with a pigment epithelium-derived factor (PEDF, a secreted glycoprotein) signal peptide and an artificially synthesized signal peptide added as described in a previous study (20). A full-length cDNA fragment of VDAC1 was isolated from HUVECs. His-K5-artificial synthesized signal peptide-pcDNA 3.1(+), His-K5-PEDF signal peptide-pcDNA 3.1(+), and Myc-VDAC1-pcDNA 3.1(+) (wild type) plasmids were constructed by inserting amplified K5 and VDAC1 genes into pcDNA 3.1(+) between the restriction sites of BamHI and XhoI. Four site-directed mutant plasmids of VDAC1 (Myc-VDAC1-S12A/T51A/S103A/T107A-pcDNA3.1(+)) were constructed with the help of GENEray (Generay Biotech, Shanghai, China). The His tag and Myc tag were added in the C-terminal of the target genes. The primer sequences for extracting the target genes were as follows: K5 with a PEDF signal peptide, 5′-TTAGGATCCTGCTACTCCTCTGCATTGGAGCCCTCCTCGGGCACAGCAGCTGCGAAGAAGACTGTATGTTTGG-3′ (forward) and 5′-GGCGCTCGAGTTAATGGTGATGGTGATGGTGCGCACACTGAGGGACATC-3′ (reverse); K5 with an artificially synthesized signal peptide, 5′-TTAGGATCCATGTGGTGGCGCCTGTGGTGGCTGCTGCTGCTGCTGCTGCTGCTGTGGCCCATGGTGTGGGCCGAAGAAGACTGTATGTTTGG-3′ (forward) and 5′-GGCGCTCGAGTTAATGGTGATGGTGATGGTGCGCACACTGAGGGACATC-3′ (reverse); and VDAC1, 5′-TTAGGATCCATGGCTGTGCCACCCACGT-3′ (forward) and 5′-GGCGCTCGAGTTACAGATCCTCTTCAGAGATGAGTTTCTGCTCTGCTTGAAATTCCAGTCC-3′(reverse). Plasmids pBiFC-K5VN155 and pBIFC-VDAC1VC155 were constructed as follows. The oligonucleotides of K5 with a PEDF signal peptide sequence and VDAC1 were cloned into the SalI-XhoI restriction sites in bimolecular fluorescence complementation (BiFC) eukaryotic expression plasmids pBiFC-VN155 (I152L) and pBiFC-VC155 (pCMV-HA backbone), respectively. The positive control plasmids for the subsequent BiFC assay were pBiFC-bJunVN55 (I152L) and pBiFC-bFosVC155 (pCMV-HA backbone); and the negative control plasmids for the BiFC assay were pBiFC-bJunVN55 and pBiFC-bFos(ΔZIP)VC155. All of the BiFC plasmids were presented by Dr. Changdeng Hu (Purdue University). The oligonucleotide sequences of primers for the BiFC plasmids were as follows: K5, 5′-ATTGTCGACTGCTACTCCTCTGCATTGGAGCCCTCCTCGGGCACAGCAGCTGCGAAGAAGACTGTATGTTTGG-3′ (forward) and 5′-ATTCTCGAGTGCTACTCCTCTGCATTGGAGCCCTCCTCGGGCACAGCAGCTGCGAAGAAGACTGTATGTTTGG-3′(reverse); VDAC1, 5′-ATTGTCGACGATGGCTGTGCCACCCACGTAT-3′ (forward) and 5′-GGCGCTCGAGTGCTTGAAATTCCAGTCCTAG-3′ (reverse). All of the plasmids were purified with a NucleoBond Xtra Midi EF Kit (MACHEREY-NAGEL, Germany) following the manufacturer's protocol. Plasmids were delivered into HUVECs by electroporation using the NeonTM Transfection System (Invitrogen). The transfection of plasmids into HEK293 cells was carried out using Attractene reagent (Qiagen).
BiFC Assay
The BiFC assay was performed as previously described (21). Plasmids pBiFC-K5VN155 and pBIFC-VDAC1VC155 were constructed by fusing K5 and VDAC1 to non-fluorescent fragments derived from the N-terminal and C-terminal of fluorescent Venus, respectively. When K5 and VDAC1 bind, the two non-fluorescent fragments get close and an intact fluorescent Venus is reconstituted. Under a fluorescence microscope, yellow fluorescence can then be visualized. For the BiFC assay, HEK293 cells were seeded in 6-well plates and transfected with appropriate plasmids (0.25 μg each). Fluorescent images were similarly acquired 24 h post-transfection with YFP (EX515, EM528).
RNA Interference
The siRNAs for silencing VDAC1, glucose-regulated protein 78 (GRP78), and GSK3β genes, in addition to scrambled siRNA, which serves as a negative control, were purchased from RiboBio (Guangzhou, China). Transfection of synthetic siRNAs was performed using HiPerFect (Qiagen) according to the manufacturer's instructions. The sense sequences of the double-strand siRNA were as follows: siVDAC1, 5′-AGUGACGGGCAGUCUGGAATT-3′; siGRP78, 5′-CCAAGAUGCUGACAUUGAATT-3′; siGSK3β 001, 5′-AUAGUCCGAUUGCGUUAUU-3′; siGSK3β 002, 5′-AUCUUUGGAGCCACUGAUU-3′; and siGSK3β 003, 5′-GAUCUGUCUUGAAGGAGAA-3′.
Quantitative Real-time PCR (qPCR)
The qPCR was performed as previously described (22, 23). In brief, total RNA was extracted from HUVECs by TRIzol Reagent (Invitrogen). 500 ng of total RNA was used for reverse transcription and qPCR. Target mRNA was determined using the capillary-based Light Cycler 2.0 Systems (Roche Diagnostics Corporation). β-actin was used as an internal control. The cDNA was amplified with special primers for VDAC1 (forward, 5′-AGATCAGCTTGCACGTGGACTGAAG-3′; reverse, 5′-TCGAAATCCATGTCGCAGCCCA-3′) and β-actin (forward, 5′-GTTGGCGTACAGGTCTTTGC-3′; reverse, 5′-GCACTCTTCCAGCCTTCCTT-3′).
mPTP Opening Assay
The mPTP opening was measured with a MitoProbe Transition Pore Assay Kit according to the manufacturer's instructions (Molecular Probes). Briefly, cells were collected and incubated with 2 μm calcein AM containing CoCl2, which selectively labels mitochondria as a quencher of calcein fluorescence, and ionomycin at 37 °C for 15 min. Next, a flow cytometer was used to assess the fluorescence with excitation at 488 nm. Black (calcein AM only), green (calcein AM and CoCl2), and pink (calcein AM, CoCl2, and ionomycin) peak shape lines were employed to indicate different intensities of subgroup cells. The mPTP opening was displayed by a reduction of mitochondrial calcein signal (green subgroups).
Cell Apoptosis Detection
Double staining was performed using an Annexin V-FITC Apoptosis Detection Kit (KeyGEN, Nanjing, China) to measure apoptosis. Cells were cultured for 72 h and prepared as described elsewhere (24). Annexin V and propidium iodide were added for incubation in the dark for 15 min at 4 °C, and then cells were analyzed by a flow cytometer (EPICS XL-MCL or Gallios, Beckman). For determination of caspase-3/7 activity, a caspase-3/7 luminescent activity assay was performed using a Caspase-Glo-3/7 Assay kit (Promega).
Western Blotting Analysis
Cells were harvested and lysed with 1× SDS lysis buffer and the subsequent Western blot analysis was performed as described elsewhere (25).
Co-immunoprecipitation (Co-IP) Assay
The procedures for immunoprecipitation have been described elsewhere (26). Briefly, cells were washed with ice-cold PBS and lysed for 20 min on ice in RIPA lysis buffer (Applygen, Beijing, China) supplemented protease inhibitor mixture (Sigma) and PMSF (Sigma). Lysates were pre-cleared then incubated overnight at 4 °C with the indicated antibody and protein A-Sepharose CL-4B beads (500 μg of protein; 5 μg of IgG; 50 μl of beads; Invitrogen). Immunoprecipitates were washed twice with RIPA lysis buffer. Proteins were eluted from beads with SDS sample buffer and then subjected to Western blotting analysis.
Biotinylation of Cell Surface Proteins
The assay that involved analysis of the cell surface expression of VDAC1 was used as previously described (27). First, cell surface proteins were labeled with biotin using Sulfo-NHS-LC-Biotin (ThermoFisher Scientific) according to the manufacturer's instructions. Cells were extracted using RIPA lysis buffer as indicated previously and immunoprecipitation was used to detect the special protein on cell surface. Antibodies against VDAC1 and IgG were incubated with cell lysates overnight. Finally Western blotting was used to detect the cell surface VDAC1.
Cell Fractionation
Isolation of mitochondria and cytosol fractions was performed using a Mitochondria Isolation Kit (APPLYGEN, Beijing, China) according to the manufacturer's instructions. Cells were washed twice with chilled PBS. Next, cells were harvested by scraping in PBS and placed in a glass homogenizer for homogenizing. The resulting homogenates were centrifuged at 800 × g for 5 min at 4 °C. The participations, including cell nucleus, plasma membrane debris, and non-lysed cells, were discarded. Then the supernatants were again centrifuged at 800 × g for 5 min at 4 °C and the participations were discarded. Finally, the resulting supernatants were centrifuged at 1000 × g for 5 min at 4 °C. After centrifugation, the supernatants, which contained cytosol fractions, were collected and served as controls for subsequent experiments. The resulting participations were the mitochondria fractions and resuspended in Mito-Cyto Isolation Buffer supplemented with protease inhibitors. The resuspension solutions were homogenized 20 times on ice. Mitochondria and cytosol fractions were analyzed by Western blotting analysis.
Statistical Analysis
Data were expressed as mean ± S.D. Multiple comparisons were assessed by one-way analysis of variance using SPSS 13.0 software (SPSS, Chicago, IL) and differences with p < 0.05 were considered statistically significant. All experiments were performed at least three times.
RESULTS
VDAC1 Is Involved in K5-induced Activation of the Mitochondrial Apoptosis Pathway
As our previous study showed, K5-stimulated apoptosis of activated endothelial cells was mediated by the mitochondrial apoptosis pathway (16). To investigate whether VDAC1 plays a role in this process, mitochondrial and cytosol fractions were isolated after cells were treated with K5 for 24 h. As shown in Fig. 1A, K5 significantly augmented the expression of mitochondrial VDAC1. Furthermore, K5 promoted the interaction of Bak and VDAC1 while reducing the interaction of Bcl-xL and VDAC1 (Fig. 1B). Consequently, the mitochondrial calcein signal (green subgroups) was reduced to 55.53 ± 2.87% compared with that of control group, which indicated mitochondrial membrane damage and mPTP opening (Fig. 1C). In addition, we observed that K5 increased the cytosol cytochrome c level in a dose-dependent manner and activated caspase-3 (Fig. 1, D and E). Taken together, these results suggest that K5-induced endothelial cells apoptosis may be mediated by VDAC1.
FIGURE 1.

K5 induces apoptosis of HUVEC via mitochondrial pathway. A, cells were treated as described, and mitochondria and cytosol components of VDAC1 were detected. mito, mitochondria. COXIV (mitochondria), β-actin (cytoplasm), and Na+/K+-ATPase (plasma membrane) were analyzed to assess purity. B, after K5 treatment for 24 h, co-IP was used with anti-VDAC1 antibody or anti-Bax antibody to detect their interaction. C, HUVECs were starved overnight with M199 containing 0.2% FBS and then treated with K5 for 24 h. The mPTP opening assay was then employed to assess the direct damage of mitochondria integrity. Data shown are means of three independent experiments. *, p < 0.05. D, K5 dose-dependently increased cytoplasm cytochrome c after cells were starved overnight and treated with K5 for 24 h. E, cells were treated with 640 nmol/liter of K5 for 72 h. The protein levels of pro-caspase-3 and cleaved caspase-3 were tested. All results were repeated at least three times.
K5 Enhances the Expression of VDAC1 by Inhibiting the Ubiquitin-dependent Degradation of VDAC1 in HUVECs
Because the mitochondrial VDAC1 was up-regulated by K5, we next evaluated the effect of K5 on the expression of total VDAC1. We found that K5 dose dependently increased the expression of total VDAC1 at the protein level but not at the mRNA level (Fig. 2, A and B). Therefore, it is possible that K5 stabilizes the VDAC1 protein by inhibiting its ubiquitination. To test this hypothesis, we performed ubiquitination assays. The ubiquitination level of the VDAC1 protein was significantly decreased in the presence of K5 in HUVECs, demonstrating that K5 inhibited the ubiquitination of VDAC1 (Fig. 2C).
FIGURE 2.

K5 inhibits the ubiquitination degradation of VDAC1 in HUVECs. The VDAC1 level was measured after exposure to K5 for 24 h at the indicated concentrations. HUVECs were subjected to both Western blotting analysis (A) and qPCR analysis (B). Data are shown as mean ± S.D. *, p < 0.05; **, p < 0.01. C, cells were incubated with K5 for 4 h, and 1 μmol/liter of MG132 was added for another 20 h. Then co-IP analysis was used to detect the ubiquitination level of VDAC1. All results were repeated at least three times. DMSO, dimethyl sulfoxide; IB, immunoblot.
K5 Promotes VDAC1 Phosphorylation via the AKT-GSK3β Axis
The phosphorylation of VDAC1 contributes to the stability of VDAC1 (14). Moreover, the AKT-GSK3β signaling pathway was reported to potentiate VDAC1 phosphorylation (28). We therefore tried to demonstrate the role of AKT-GSK3β axis on modulation of VDAC1 in our study. As shown in Fig. 3A, K5 may inhibit the activity of AKT by decreasing p-AKT (Ser-473), which represents an active form of AKT, and subsequently activating GSK3β by reducing p-GSK3β (Ser-9), which stands for a non-active form of GSK3β. On activation of AKT with insulin, K5-induced up-regulation of VDAC1 was blocked (Fig. 3B). Similarly, GSK3β knockdown also reduced the VDAC1 protein level and diminished the effect of K5 on VDAC1 (Fig. 3C). Using co-IP analysis, we found that phosphorylated VDAC1 was increased by K5, and this effect was enhanced by AKT inhibitor, whereas attenuated by GSK3β inhibitor (Fig. 3D). In this experiment, β-catenin was employed as a validity indicator of AKT and GSK3β inhibitors. As shown in Fig. 3D, β-catenin was significantly reduced by the AKT inhibitor, whereas markedly up-regulated by the GSK3β inhibitor, suggesting that these inhibitors were indeed effective. In addition, we constructed four site-directed mutant plasmids (S12A/T51A/S103A/T107A) to investigate the phosphorylation sites responsible for VDAC1 stability by transfecting HUVECs and HEK293 cells. Two VDAC1 mutants (S12A and T107A) were shown to reduce the VDAC1 protein level (Fig. 3E). To further test the correlation between phosphorylation and degradation of VDAC1, two other VDAC1 mutants (S12D and T107D), which simulate constant phosphorylation of VDAC1, were constructed. As shown in Fig. 3A, S12D and T107D were both observed to increase the protein level of VDAC1 compared with that of the wild type. These results indicate that increasing VDAC1 phosphorylation through the AKT-GSK3β axis probably disturbs its degradation and results in VDAC1 accumulation under K5 treatment. On the other hand, VDAC1 per se may mediate the modulation of K5 on the AKT-GSK3β axis.
FIGURE 3.

K5 increases the phosphorylation of VDAC1 via the AKT-GSK3β pathway. A, AKT and GSK3β activity were determined by Western blotting analysis after treatment with K5 for 9 h. p-AKT (Ser-473) represents an active form of AKT, and p-GSK3β (Ser-9) is a non-inactivated form of GSK3β. B, 200 nmol/liter of insulin was used as an activator of AKT and added to cells for 30 min before K5 treatment for 24 h. The protein levels of VDAC1, p-AKT (Ser-473), AKT, p-GSK3β (Ser-9), and GSK3β were detected by Western blotting analysis. C, GSK3β siRNAs (siGSK3β 001, 002, and 003) and nonspecific siRNA were transfected into HUVECs for 24 h. Then cells were exposed to K5 for another 24 h. The protein levels of p-GSK3β (Ser-9), GSK3β, and VDAC1 were measured by Western blotting analysis. D, 1 μmol/liter of AKT or GSK3β inhibitor was added to cells for 30 min. DMSO served as a solvent control. The cells were then treated by K5 for another 24 h. HUVECs lysates were prepared for co-IP analysis with a monoclonal antibody against VDAC1. The phosphorylated VDAC1 was probed with an anti-phosphoserine/threonine/tyrosine-reactive antibody. The quantitation analysis was performed and presented as mean ± S.D. *, p < 0.05. E, Myc-tagged wild-type VDAC1 (WT) or four VDAC1 mutated at Ser-12, Thr-51, Ser-103, and Thr-107 to alanine (S12A, T51A, S103A, and T107A) were constructed into pcDNA3.1(+) vector. HUVECs and HEK293 cells were transfected with the aforementioned plasmids. After 24 h post-transfection, the cells were lysed to detect the extrinsic expression of VDAC1 with anti-Myc tag antibody. F, after HUVECs transfection with mutated VDAC1 plasmids (Ser-12 and Thr-107 to alanine and aspartic acid) for 24 h, the protein level of extrinsic VDAC1 was detected by anti-Myc tag antibody. All results were repeated at least three times. DMSO, dimethyl sulfoxide.
Cell Surface VDAC1 Is Up-regulated and Acts as a Receptor in Charge of AKT-GSK3β-VDAC1 Signal Transduction with K5 Treatment
In addition to occurrence on mitochondrial, VDAC1 was also noted on the cell membrane and was identified as a receptor of K5 (18). To verify the binding between K5 and VDAC1, plasmids of K5 and VDAC1 were constructed. The plasmids of K5 were added with a PEDF signal peptide (His-K5–1) and an artificially synthesized signal peptide (His-K5–2), which may favor secretion of the K5 protein to mimic the action of the extracellular recombinant K5 protein. The His-K5–1 plasmid was identified as being a more efficient secreting K5 protein into the supernatant than the His-K5-2 plasmid (Fig. 4A). Hence, we chose the His-K5-1 plasmid for the co-IP assay. We showed that K5 could bind with VDAC1 after treatment of HUVECs with K5 containing conditioned medium for 10 min (Fig. 4B). This K5/VDAC1 direct interaction was also confirmed using the BiFC assay by co-transfecting cells with pBiFC-K5VN155 containing a PEDF signal peptide and pBIFC-VDAC1VC155 plasmids. It was observed that there was a number of Venus fluorescence (yellow color) dispersed in the cytoplasm and cell surface (Fig. 4C). To further determine the receptor action of VDAC1, HKI protein, an important agonist of cell surface VDAC1 (29), and siRNAs of VDAC1 were employed. Our results showed that the HKI protein and siRNAs of VDAC1 diminished the inhibitory effect of K5 on p-AKT and p-GSK3β (Fig. 4, D and E). Moreover, using the biotinylation of cell surface protein assay, we observed that both K5 and overexpression of VDAC1 increased the protein levels of total and plasma membrane VDAC1 (Fig. 4, F and G). The impact of K5 on the expression of total and plasma membrane VDAC1 was attenuated by treatment with HKI protein (Fig. 4F). Collectively, these results suggest that K5 can interact with VDAC1 on the plasma membrane. VDAC1, as a receptor of K5, mediates the AKT-GSK3β-VDAC1 signaling pathway.
FIGURE 4.

VDAC1 acting as a receptor is responsible for AKT-GSK3β signals transduction with K5 treatment. A, after transfection for 24 h, cell lysates and 100 μg of supernatant protein of HEK293 cells were extracted for Western blotting analysis. The protein level of K5 was detected with anti-His tag antibody. Vector, pcDNA3.1(+); His-K5–1, His-K5-artificially synthesized signal peptide-pcDNA 3.1(+); His-K5–2, His-K5-PEDF signal peptide-pcDNA 3.1(+). B, HUVECs were treated with the conditioned medium from K5-overexpressed HEK293 cells for 10 min, then immunoprecipitated with VDAC1 antibody. The K5 level was determined using anti-His tag antibody. C, fluorescence microscopy of K5-VDAC1 binding using BiFC assay. HEK293 cells were co-transfected with pCMV-K5VN155 and pCMV-VDAC1VC155 plasmids. Fluorescence images were acquired 24 h post-transfection. Yellow color (Venus) representing K5-VDAC1 interaction was observed under the microscope. Scale bar, 20 μm. The activity of AKT and GSK3β was examined to confirm the receptor role of VDAC1. VDAC1 blockade assay by HKI (D) or VDAC1 siRNAs (E) was used. 100 nmol/liter of HKI was added to the cells for 30 min prior to the addition of K5. HUVECs were transfected with VDAC1 siRNAs for 24 h before K5 addition. Data are shown as mean ± S.D. **, p < 0.01. F, for biotinylation of cell surface proteins, cells were incubated with biotin for 30 min and then quenched with glycine. After that, cell lysates were subjected to co-IP and Western blotting assay. G, HEK293 cells were transfected with Myc-VDAC1-pcDNA3.1(+) for 24 h. Then total and plasma membrane VDAC1 were detected with antibodies of VDAC1 and streptavidin-HRP. All results were repeated at least three times.
VDAC1 Exhibits Its Receptor Function in K5-induced Endothelial Cell Apoptosis
To functionally validate that VDAC1 acts as a receptor, we thus applied an antibody blockade assay. HUVECs were treated with antibody at a concentration of 2 μg/ml. Two hours later, K5 was added for another 48 h. Then cells were incubated with Annexin V and propidium iodide dye and detected by a flow cytometer. Our results showed that the apoptosis of HUVECs induced by K5 was eliminated when VDAC1 antibody was used (Fig. 5A). Because GRP78 is also reported to be a receptor of K5, we explored the effect of GRP78 antibody on cell apoptosis (27, 30). In contrast, GRP78 antibody showed no inhibitory effect on K5-induced HUVEC apoptosis (Fig. 5A). Consistently, under K5 treatment, the protein level of pro-caspase-3 was restored when exposed to VDAC1 antibody, whereas GRP78 antibody did not show this effect (Fig. 5B). Similarly, after VDAC1 knockdown using siRNA, K5 lost its impact with declining pro-caspase-3, whereas K5 could still decrease the pro-caspase-3 protein level even when GRP78 was knocked down (Fig. 5C). Accordingly, K5-stimulated caspase-3/7 activity was inhibited by VDAC1 knockdown but not by GRP78 knockdown (Fig. 5D). Taken together, our results suggest that VDAC1 is the receptor involved in K5-induced apoptosis rather than GRP78.
FIGURE 5.

VDAC1 is the receptor for K5-induced apoptosis rather than GRP78. A, to test the protein by which K5-induced endothelial cells apoptosis, antibody blockage assay was used. Cells were harvested after 72 h for FITC-Annexin V/propidium iodide staining and quantitative analysis of apoptotic cells was performed using flow cytometry. Data for FITC-Annexin V/propidium iodide flow cytometry in a representative experiment are presented as graphs. Quantification of apoptotic cells are shown as mean ± S.D. of triplicates. *, p < 0.05 versus control group; #, p < 0.05 versus GRP78 antibody group. n.s., non-significant. B, the protein level of pro-caspase-3 was tested by Western blotting analysis. Data are shown as mean ± S.D. *, p < 0.05. In parallel, siRNAs of VDAC1 and GRP78 were applied to knockdown the expression of target genes. The protein level of pro-caspase-3 and the activity of caspase-3/7 were measured by Western blotting analysis (C) and a caspase-3/7 activity assay kit (D), respectively. Data are shown as mean ± S.D. *, p < 0.05 versus NC siRNA group; #, p < 0.05 versus siGRP78 group. All results were repeated at least three times.
DISCUSSION
In apoptosis, VDAC1 has been proposed as a crucial regulator in the mitochondrial apoptosis pathway by gating the release of cytochrome c. This critical step generally needs assistance of the Bcl-2 family, such as pro-apoptotic proteins Bak and Bax. By interaction with Bak and Bax, VDAC1 allows the mitochondrial outer membrane permeabilization to occur, and thus cytochrome c diffuses into the cytoplasm (31). This process has been reported in various studies. For example, As2O3-, ethanol-, endostatin-, and cisplain-induced cell apoptosis were abolished by preventing the VDAC1-Bax interaction using antibodies or siRNA of VDAC1 (13, 14, 32, 33). In our study, we showed that K5 increased the VDAC1-Bak interaction, which led to mPTP opening, an increase of the cytoplasm cytochrome c level and ultimate activation of caspase-3. This is the first study to identify the involvement of mitochondrial VDAC1 in K5-induced HUVEC apoptosis, which provides evidence that VDAC1 may be a potential therapeutic target for angiogenesis-related diseases.
The enhanced interaction of VDAC1 and Bak was due to up-regulation of mitochondrial-expressed VDAC1. Our study further revealed that K5 induced the expression of total VDAC1, and that overexpressing VDAC1 may result in increased mitochondrial subcellular VDAC1 (data not shown). Hence, K5-induced total VDAC1 expression accounted for up-regulated distribution of VDAC1 on mitochondria. Of note, K5 did not affect the mRNA level of VDAC1. Subsequently, we observed that K5 inhibited the ubiquitination of VDAC1 and promoted its stability and accumulation. Therefore, the next area of investigation is the regulatory mechanism concerning VDAC1 stability. Post-translational modifications are associated with protein degradation. Among many post-translational modifications, phosphorylation and acetylation are most frequently studied. A number of studies focus on the phosphorylation of VDAC1. Numerous phosphorylation sites have been documented by means of mass spectrometry or site-directed mutagenesis (34). Phosphorylated VDAC1 on serines 12 and 103 yielded stable VDAC1 and cells sensitized to apoptosis (14). In addition, VDAC1 phosphorylated on threonine 51 disrupted its binding with hexokinase II (HKII) and potentiated cell apoptosis (28). In the present report, we observed that the VDAC1 protein level was reduced by the mutation of Ser-12 and Thr-107 to alanine, but not by the sites of Thr-51 and Ser-103 in HUVECs and HEK293 cells. The different result between our study and previous report on Ser-103 may be attributed to the fact that we used different cell types (14). Furthermore, VDAC1 mutants at sites of Ser-12 and Thr-107 to aspartic acid decreased its degradation. Herein, our study was the first to correlate Thr-107 with modulation of the VDAC1 protein level.
Some candidate protein kinases catalyzing the phosphorylation of VDAC1 have been identified, such as GSK3β, CaMII, CKI, and PKC as well as p38 MAPK. Among these, GSK3β has been shown to be involved in almost all VDAC1 phosphorylation sites. GSK3β was reported to be responsible for VDAC1 phosphorylation at Ser-12/102/103/104/136 and Thr-51/107 (34). Consistent with previous studies, we found that GSK3β knockdown decreased the total protein level of VDAC1 and blocked the effect of K5 on VDAC1. Accordingly, GSK3β inhibitors reduced phosphorylated VDAC1 and abrogated the effect of K5. GSK3β has been reported to be inactivated through the phosphorylation of Ser-9, and AKT has been shown to inhibit the GSK3β activation (35, 36). So, insulin, an agonist of AKT, had the same effect as the GSK3β inhibitor on obliterating VDAC1 protein, even under treatment with K5. In contrast, both total and phosphorylated VDAC1 were increased by the AKT inhibitor. These results established an important role of the AKT-GSK3β signaling pathway in VDAC1 phosphorylation in the presence of K5.
However, as an extracellular protein, it was important to understand the mechanism whereby K5 sends signals into the cell to regulate the AKT-GSK3β signaling pathway. In other words, what is the receptor that mediates the effects of K5? In the present study, the HKI protein that could naturally bind with VDAC1 was found to block the effects of K5 on the AKT-GSK3β signaling pathway. Furthermore, VDAC1 knockdown confirmed that K5-induced GSK3β activation is VDAC1-dependent. These data suggest that VDAC1 enables the transmission of K5-activated signals into cells.
Several publications have reported extra-mitochondrial localizations of VDAC1. VDAC1 has been detected in the plasma membrane of human B lymphocytes (37). Within plasma, VDAC1 has been found to be located in caveolae or caveolae-related domains of established T lymphoid-like cell lines (38). The VDAC1 N-terminal was reputed to assume its trafficking to cell membrane (39, 40). Moreover, VDAC1 has been recently identified as the receptor for K5 on HUVEC membrane (18). These findings have been corroborated by our study using co-IP and BiFC assays. The interaction between K5 and VDAC1 was detected in the K5-treated group. Controversially, GRP78 has also been regarded as a receptor for K5 in some other investigations (27, 30). However, in our study GRP78 was not involved in K5-induced apoptosis. Collectively, our results show that VDAC1 is the receptor for K5 that mediates the AKT-GSK3β signaling pathway and cell apoptosis. Ca2+, as a classic second messenger, may bridge the VDAC1 and AKT-GSK3β signaling pathways. K5 can induce an influx of Ca2+ from the extracellular environment toward the cytosol (18, 29). The influx of Ca2+ was the mediator of cell apoptosis (41, 42). Therefore, we deduced that, through binding with cell surface VDAC1, K5 leads to an increase of intracellular Ca2+, which initials the AKT-GSK3β signaling pathway. The connection between Ca2+ and the AKT-GSK3β signaling pathway awaits further elucidation.
VDAC1 serves as a necessary plasma membrane receptor for K5 to exert its biological function. The up-regulation of the total VDAC1 protein level by K5 not only led to an increase in mitochondrial membrane VDAC1, but also cell membrane VDAC1. The elevated cell surface VDAC1 could in turn amplify the apoptotic-inducing effect of K5. This regulatory model is termed a “positive feed-back loop,” which is universal in the process of cell growth controlled by growth factors such as PDGF/PDGFβ, EGF/EGFR, and VEGF/VEGFR (43–45). The positive feedback loop is an economically efficient way for ligands to orchestrate cell behavior with important physiological significance. Taken together, our study established the presence of a positive feedback loop in K5-induced HUVEC apoptosis.
In summary, this study elucidated the function of VDAC1 in K5-induced HUVEC apoptosis: on the one hand, as a channel protein on mitochondrial membrane, VDAC1 mediates cell apoptosis through releasing cytochrome c to cytoplasm; on the other hand, as a receptor for K5 on the cell surface, VDAC1 transmits K5-triggering signals that regulate its own protein level and cell apoptosis (Fig. 6). Our study is the first to propose a positive feedback loop of VDAC1-AKT-GSK3β-VDAC1, which provides new perspectives on the mechanisms of K5-induced apoptosis.
FIGURE 6.
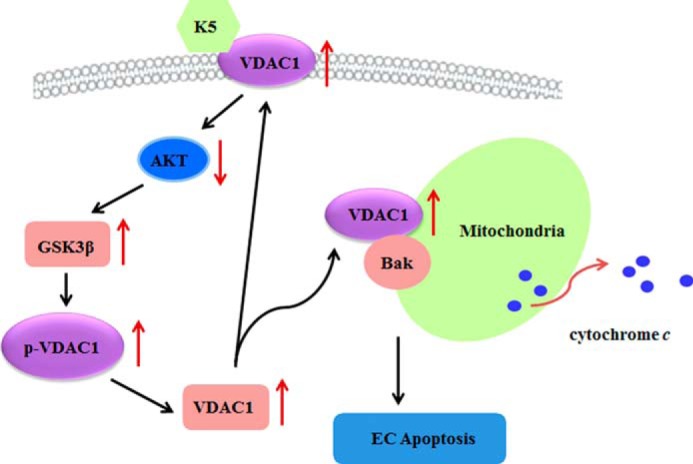
Schematic overview of the underlying mechanism of K5-induced EC apoptosis. With binding to VDAC1 in cell membrane, K5 inactivates AKT and thus stimulates the activity of GSK3β, which promotes the phosphorylation of VDAC1 (p-VDAC1). p-VDAC1 blocks the ubiquitin-dependent degradation of VDAC1 and facilitates its accumulation. Subsequently, the up-regulated VDAC1 partially translocates onto mitochondrial membrane, combines with Bak, and initiates cytochrome c release and cell apoptosis. In addition, the remainder of the VDAC1 transports onto cell surface, and in turn as a receptor amplifies the apoptotic-inducing effect of K5. Collectively, a positive feedback loop of VDAC1-AKT-GSK3β-VDAC1 mediates K5-induced EC apoptosis.
Acknowledgment
We thank Dr. Changdeng Hu (Purdue University) for BiFC plasmids.
This work was supported by National Nature Science Foundation of China Grants 30973449, 81172163, 81272515, 81272338, 81200706, 81471033, and 81400639, National Key Sci-Tech Special Project of China Grant 2009ZX09103-642, 2013ZX09102053, Program for Doctoral Station in University Grants 20120171110053 and 20130171110053, Key Project of Nature Science Foundation of Guangdong Province, Grant 10251008901000009, Key Sci-tech Research Project of Guangdong Province, Grant 2011B031200006, Guandong Natural Science Fund Grants S2012010009250 and S2012040006986, Key Sci-tech Research Project of Guangzhou Municipality Grants 2011Y1–00017–8 and 12A52061519, and the Innovative talents project of doctoral candidates of Sun Yat-sen University.
- K5
- kringle 5
- BiFC
- bimolecular fluorescence complementation
- co-IP
- co-immunoprecipitation
- EC
- endothelial cell
- GRP78
- glucose-regulated protein 78
- HUVECs
- human vein endothelial cells
- mPTP
- mitochondrial permeability transition pore
- PEDF
- pigment epithelium-derived factor
- VDAC
- voltage-dependent anion channel
- qPCR
- qualitative PCR
- HKI
- hexokinase I.
REFERENCES
- 1. Folkman J. (1971) Tumor angiogenesis: therapeutic implications. N. Engl. J. Med. 285, 1182–1186 [DOI] [PubMed] [Google Scholar]
- 2. Campochiaro P. A. (2004) Ocular neovascularisation and excessive vascular permeability. Expert Opin. Biol. Ther. 4, 1395–1402 [DOI] [PubMed] [Google Scholar]
- 3. Yamamoto Y., Maeshima Y., Kitayama H., Kitamura S., Takazawa Y., Sugiyama H., Yamasaki Y., Makino H. (2004) Tumstatin peptide, an inhibitor of angiogenesis, prevents glomerular hypertrophy in the early stage of diabetic nephropathy. Diabetes 53, 1831–1840 [DOI] [PubMed] [Google Scholar]
- 4. Cao Y., Chen A., An S. S., Ji R. W., Davidson D., Llinás M. (1997) Kringle 5 of plasminogen is a novel inhibitor of endothelial cell growth. J. Biol. Chem. 272, 22924–22928 [DOI] [PubMed] [Google Scholar]
- 5. Cai W., Ma J., Li C., Yang Z., Yang X., Liu W., Liu Z., Li M., Gao G. (2005) Enhanced anti-angiogenic effect of a deletion mutant of plasminogen kringle 5 on neovascularization. J. Cell. Biochem. 96, 1254–1261 [DOI] [PubMed] [Google Scholar]
- 6. Zhang Z., Ma J. X., Gao G., Li C., Luo L., Zhang M., Yang W., Jiang A., Kuang W., Xu L., Chen J., Liu Z. (2005) Plasminogen kringle 5 inhibits alkali-burn-induced corneal neovascularization. Invest. Ophthalmol. Vis. Sci. 46, 4062–4071 [DOI] [PubMed] [Google Scholar]
- 7. Perri S. R., Nalbantoglu J., Annabi B., Koty Z., Lejeune L., François M., Di Falco M. R., Béliveau R., Galipeau J. (2005) Plasminogen kringle 5-engineered glioma cells block migration of tumor-associated macrophages and suppress tumor vascularization and progression. Cancer Res. 65, 8359–8365 [DOI] [PubMed] [Google Scholar]
- 8. Liu X. Y., Qiu S. B., Zou W. G., Pei Z. F., Gu J. F., Luo C. X., Ruan H. M., Chen Y., Qi Y. P., Qian C. (2005) Effective gene-virotherapy for complete eradication of tumor mediated by the interaction of hTRAIL (TNFSF10) and plasminogen K5. Mol. Ther. 11, 531–541 [DOI] [PubMed] [Google Scholar]
- 9. Yang X., Cheng R., Li C., Cai W., Ma J. X., Liu Q., Yang Z., Song Z., Liu Z., Gao G. (2006) Kringle 5 of human plasminogen suppresses hepatocellular carcinoma growth both in grafted and xenografted mice by anti-angiogenic activity. Cancer Biol. Ther. 5, 399–405 [DOI] [PubMed] [Google Scholar]
- 10. Cai W. B., Zhang Y., Cheng R., Wang Z., Fang S. H., Xu Z. M., Yang X., Yang Z. H., Ma J. X., Shao C. K., Gao G. Q. (2012) Dual inhibition of plasminogen kringle 5 on angiogenesis and chemotaxis suppresses tumor metastasis by targeting HIF-1α pathway. PLoS One 7, e53152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lu H., Dhanabal M., Volk R., Waterman M. J., Ramchandran R., Knebelmann B., Segal M., Sukhatme V. P. (1999) Kringle 5 causes cell cycle arrest and apoptosis of endothelial cells. Biochem. Biophys. Res. Commun. 258, 668–673 [DOI] [PubMed] [Google Scholar]
- 12. Shoshan-Barmatz V., Ben-Hail D. (2012) VDAC, a multi-functional mitochondrial protein as a pharmacological target. Mitochondrion 12, 24–34 [DOI] [PubMed] [Google Scholar]
- 13. Zheng Y., Shi Y., Tian C., Jiang C., Jin H., Chen J., Almasan A., Tang H., Chen Q. (2004) Essential role of the voltage-dependent anion channel (VDAC) in mitochondrial permeability transition pore opening and cytochrome c release induced by arsenic trioxide. Oncogene 23, 1239–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yuan S., Fu Y., Wang X., Shi H., Huang Y., Song X., Li L., Song N., Luo Y. (2008) Voltage-dependent anion channel 1 is involved in endostatin-induced endothelial cell apoptosis. FASEB J. 22, 2809–2820 [DOI] [PubMed] [Google Scholar]
- 15. Shimizu S., Narita M., Tsujimoto Y. (1999) Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 399, 483–487 [DOI] [PubMed] [Google Scholar]
- 16. Gu X., Yao Y., Cheng R., Zhang Y., Dai Z., Wan G., Yang Z., Cai W., Gao G., Yang X. (2011) Plasminogen K5 activates mitochondrial apoptosis pathway in endothelial cells by regulating Bak and Bcl-x(L) subcellular distribution. Apoptosis 16, 846–855 [DOI] [PubMed] [Google Scholar]
- 17. De Pinto V., Messina A., Accardi R., Aiello R., Guarino F., Tomasello M. F., Tommasino M., Tasco G., Casadio R., Benz R., De Giorgi F., Ichas F., Baker M., Lawen A. (2003) New functions of an old protein: the eukaryotic porin or voltage dependent anion selective channel (VDAC). Ital. J. Biochem. 52, 17–24 [PubMed] [Google Scholar]
- 18. Gonzalez-Gronow M., Kalfa T., Johnson C. E., Gawdi G., Pizzo S. V. (2003) The voltage-dependent anion channel is a receptor for plasminogen kringle 5 on human endothelial cells. J. Biol. Chem. 278, 27312–27318 [DOI] [PubMed] [Google Scholar]
- 19. Jaffe E. A., Nachman R. L., Becker C. G., Minick C. R. (1973) Culture of human endothelial cells derived from umbilical veins: identification by morphologic and immunologic criteria. J. Clin. Invest. 52, 2745–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Barash S., Wang W., Shi Y. (2002) Human secretory signal peptide description by hidden Markov model and generation of a strong artificial signal peptide for secreted protein expression. Biochem. Biophys. Res. Commun. 294, 835–842 [DOI] [PubMed] [Google Scholar]
- 21. Kodama Y., Hu C. D. (2010) An improved bimolecular fluorescence complementation assay with a high signal-to-noise ratio. BioTechniques 49, 793–805 [DOI] [PubMed] [Google Scholar]
- 22. Wang R., Xie H., Huang Z., Ma J., Fang X., Ding Y., Sun Z. (2011) T cell factor 1 regulates thymocyte survival via a RORγt-dependent pathway. J. Immunol. 187, 5964–5973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu G., Zhang L., Wang D. Y., Xu R., Liu Z., Han D. M., Wang X. D., Zuo K. J., Li H. B. (2010) Opposing roles of IL-17A and IL-25 in the regulation of TSLP production in human nasal epithelial cells. Allergy 65, 581–589 [DOI] [PubMed] [Google Scholar]
- 24. Lin C. S., Xie S. B., Liu J., Zhao Z. X., Chong Y. T., Gao Z. L. (2010) Effect of revaccination using different schemes among adults with low or undetectable anti-HBs titers after hepatitis B virus vaccination. Clin. Vaccine Immunol. 17, 1548–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xu G., Cheng L., Wen W., Oh Y., Mou Z., Shi J., Xu R., Li H. (2007) Inverse association between T-cell immunoglobulin and mucin domain-1 and T-bet in a mouse model of allergic rhinitis. Laryngoscope 117, 960–964 [DOI] [PubMed] [Google Scholar]
- 26. Blomgran R., Zheng L., Stendahl O. (2007) Cathepsin-cleaved Bid promotes apoptosis in human neutrophils via oxidative stress-induced lysosomal membrane permeabilization. J. Leukoc. Biol. 81, 1213–1223 [DOI] [PubMed] [Google Scholar]
- 27. McFarland B. C., Stewart J., Jr., Hamza A., Nordal R., Davidson D. J., Henkin J., Gladson C. L. (2009) Plasminogen kringle 5 induces apoptosis of brain microvessel endothelial cells: sensitization by radiation and requirement for GRP78 and LRP1. Cancer Res. 69, 5537–5545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pastorino J. G., Hoek J. B., Shulga N. (2005) Activation of glycogen synthase kinase 3β disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 65, 10545–10554 [DOI] [PubMed] [Google Scholar]
- 29. Gonzalez-Gronow M., Kaczowka S. J., Payne S., Wang F., Gawdi G., Pizzo S. V. (2007) Plasminogen structural domains exhibit different functions when associated with cell surface GRP78 or the voltage-dependent anion channel. J. Biol. Chem. 282, 32811–32820 [DOI] [PubMed] [Google Scholar]
- 30. Davidson D. J., Haskell C., Majest S., Kherzai A., Egan D. A., Walter K. A., Schneider A., Gubbins E. F., Solomon L., Chen Z., Lesniewski R., Henkin J. (2005) Kringle 5 of human plasminogen induces apoptosis of endothelial and tumor cells through surface-expressed glucose-regulated protein 78. Cancer Res. 65, 4663–4672 [DOI] [PubMed] [Google Scholar]
- 31. Green D. R. (2006) At the gates of death. Cancer Cell 9, 328–330 [DOI] [PubMed] [Google Scholar]
- 32. Adachi M., Higuchi H., Miura S., Azuma T., Inokuchi S., Saito H., Kato S., Ishii H. (2004) Bax interacts with the voltage-dependent anion channel and mediates ethanol-induced apoptosis in rat hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 287, G695–G705 [DOI] [PubMed] [Google Scholar]
- 33. Tajeddine N., Galluzzi L., Kepp O., Hangen E., Morselli E., Senovilla L., Araujo N., Pinna G., Larochette N., Zamzami N., Modjtahedi N., Harel-Bellan A., Kroemer G. (2008) Hierarchical involvement of Bak, VDAC1 and Bax in cisplatin-induced cell death. Oncogene 27, 4221–4232 [DOI] [PubMed] [Google Scholar]
- 34. Kerner J., Lee K., Tandler B., Hoppel C. L. (2012) VDAC proteomics: post-translation modifications. Biochim. Biophys. Acta 1818, 1520–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Frame S., Cohen P. (2001) GSK3 takes centre stage more than 20 years after its discovery. Biochem. J. 359, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cross D. A., Alessi D. R., Cohen P., Andjelkovich M., Hemmings B. A. (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378, 785–789 [DOI] [PubMed] [Google Scholar]
- 37. Thinnes F. P., Götz H., Kayser H., Benz R., Schmidt W. E., Kratzin H. D., Hilschmann N. (1989) Identification of human porins. I. Purification of a porin from human B-lymphocytes (Porin 31HL) and the topochemical proof of its expression on the plasmalemma of the progenitor cell. Biol. Chem. Hoppe Seyler 370, 1253–1264 [PubMed] [Google Scholar]
- 38. Bàthori G., Parolini I., Tombola F., Szabò I., Messina A., Oliva M., De Pinto V., Lisanti M., Sargiacomo M., Zoratti M. (1999) Porin is present in the plasma membrane where it is concentrated in caveolae and caveolae-related domains. J. Biol. Chem. 274, 29607–29612 [DOI] [PubMed] [Google Scholar]
- 39. Buettner R., Papoutsoglou G., Scemes E., Spray D. C., Dermietzel R. (2000) Evidence for secretory pathway localization of a voltage-dependent anion channel isoform. Proc. Natl. Acad. Sci. U.S.A. 97, 3201–3206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schwarzer C., Becker S., Awni L. A., Cole T., Merker R., Barnikol-Watanabe S., Thinnes F. P., Hilschmann N. (2000) Human voltage-dependent anion-selective channel expressed in the plasmalemma of Xenopus laevis oocytes. Int. J. Biochem. Cell Biol. 32, 1075–1084 [DOI] [PubMed] [Google Scholar]
- 41. Yanamandra N., Buzzeo R. W., Gabriel M., Hazlehurst L. A., Mari Y., Beaupre D. M., Cuevas J. (2011) Tipifarnib-induced apoptosis in acute myeloid leukemia and multiple myeloma cells depends on Ca2+ influx through plasma membrane Ca2+ channels. J. Pharmacol. Exp. Ther. 337, 636–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gao L. J., Gu P. Q., Fan W. M., Liu Z., Qiu F., Peng Y. Z., Guo X. R. (2011) The role of gC1qR in regulating survival of human papillomavirus 16 oncogene-transfected cervical cancer cells. Int. J. Oncol. 39, 1265–1272 [DOI] [PubMed] [Google Scholar]
- 43. Eriksson A., Nistér M., Leveen P., Westermark B., Heldin C. H., Claesson-Welsh L. (1991) Induction of platelet-derived growth factor α- and β-receptor mRNA and protein by platelet-derived growth factor BB. J. Biol. Chem. 266, 21138–21144 [PubMed] [Google Scholar]
- 44. Earp H. S., Austin K. S., Blaisdell J., Rubin R. A., Nelson K. G., Lee L. W., Grisham J. W. (1986) Epidermal growth factor (EGF) stimulates EGF receptor synthesis. J. Biol. Chem. 261, 4777–4780 [PubMed] [Google Scholar]
- 45. Adham S. A., Coomber B. L. (2009) Glucose is a key regulator of VEGFR2/KDR in human epithelial ovarian carcinoma cells. Biochem. Biophys. Res. Commun. 390, 130–135 [DOI] [PubMed] [Google Scholar]