

Background: Histone H3 methylation on lysines 4 and 36 stimulates interaction between histone deacetylase complexes and chromatin.
Results: The NuA4 lysine acetyltransferase complex also binds to methylated H3 and stimulates nucleosomal binding and H3 acetylation by SAGA.
Conclusion: Histone H3 methylation stimulates both nucleosomal acetylation and deacetylation.
Significance: H3 methylation is key in properly regulating the level of acetylation at a transcribed gene.
Keywords: Acetyltransferase, Chromatin Modification, Histone Acetylase, Histone Methylation, Transcription Elongation Factor, Esa1, NuA4, SAGA, Set1, Set2
Abstract
Cotranscriptional methylation of histone H3 lysines 4 and 36 by Set1 and Set2, respectively, stimulates interaction between nucleosomes and histone deacetylase complexes to block cryptic transcription in budding yeast. We previously showed that loss of all H3K4 and H3K36 methylation in a set1Δset2Δ mutant reduces interaction between native nucleosomes and the NuA4 lysine acetyltransferase (KAT) complex. We now provide evidence that NuA4 preferentially binds H3 tails mono- and dimethylated on H3K4 and di- and trimethylated on H3K36, an H3 methylation pattern distinct from that recognized by the RPD3C(S) and Hos2/Set3 histone deacetylase complexes (HDACs). Loss of H3K4 or H3K36 methylation in set1Δ or set2Δ mutants reduces NuA4 interaction with bulk nucleosomes in vitro and in vivo, and reduces NuA4 occupancy of transcribed coding sequences at particular genes. We also provide evidence that NuA4 acetylation of lysine residues in the histone H4 tail stimulates SAGA interaction with nucleosomes and its recruitment to coding sequences and attendant acetylation of histone H3 in vivo. Thus, H3 methylation exerts opposing effects of enhancing nucleosome acetylation by both NuA4 and SAGA as well as stimulating nucleosome deacetylation by multiple HDACs to maintain the proper level of histone acetylation in transcribed coding sequences.
Introduction
Chromatin regulates transcription by limiting the accessibility of the DNA to RNA polymerase II (Pol II).3 Transcription activators overcome the effects of chromatin by recruiting coactivator complexes that alter chromatin structure, allowing Pol II to use the DNA as a template to synthesize RNA.
The NuA4 coactivator complex contains the only essential lysine acetyltransferase (KAT) in Saccharomyces cerevisiae, Esa1 (1). Esa1 is a part of both NuA4 and a smaller subcomplex called Piccolo NuA4 (picNuA4) along with Epl1 and Yng2. Besides picNuA4, NuA4 contains other subcomplexes with subunits that are shared with the RPD3C(S) histone deacetylase complex and the SWR1 chromatin remodeling complex (2). Eaf1 is the only subunit unique to NuA4 and is important for complex integrity (3).
Although it was initially thought that the SAGA and NuA4 KAT complexes were restricted to promoters, we and others found that these complexes are also recruited to coding sequences (CDS) where they stimulate transcription elongation (4, 5). Nucleosome acetylation alters the contacts between DNA and histones as well as creating a binding platform for complexes containing bromodomains (6, 7). Gcn5 and Esa1, the catalytic subunits of SAGA and NuA4, respectively, stimulate transcription elongation, in part by stimulating the recruitment of bromodomain containing chromatin remodeling complexes RSC and SWI/SNF (4).
We have proposed a two-step mechanism for the recruitment of NuA4 to CDS. NuA4 gets into CDS through contact with the Pol II C-terminal domain (CTD) phosphorylated on serine 5 of the heptapeptide repeat (Ser-5p) and then acetylates methylated nucleosomes. NuA4 occupancy in CDS as well as interaction with histones is reduced in set1Δset2Δ cells (4).
Set1 and Set2 are histone methyltransferases (HMTs) responsible for cotranscriptional methylation of histone H3 lysines 4 (8) and 36 (9), respectively. Set1-mediated di- and trimethylation of H3K4 is enhanced by ubiquitination of histone H2B lysine 123 by the Rad6/Bre1 complex (10). H3K4 methylation is found in a gradient across genes, with trimethylation found predominantly at the 5′ end, dimethylation found in the middle, and monomethylation enriched toward the 3′ end of CDS (11). Set2-mediated H3K36 trimethylation is enriched toward the 3′ end of CDS (11).
Although H3K4 and H3K36 methylation are associated with actively transcribed genes, both have been shown to repress cryptic transcription by stimulating interaction between histone deacetylase complexes (HDACs) and nucleosomes. H3K4 dimethylation serves as a binding platform for the Hos2-containing Set3 complex (12). Although Set3C binds dimethylated H3K4, RPD3C(S) binds di- and trimethylated H3K36 (H3K36me2/3) (13). RPD3C(S) binds to H3K36me2/3 through the chromodomain (CHD) in its subunit Eaf3 and the plant homeobox domain (PHD) in its subunit Rco1 (13–16). There is also evidence that RPD3C(S) recruitment to the GAL1/GAL10 locus under repressive conditions is stimulated by H3K4 methylation (17). Set3C binds to dimethylated H3K4 through the Set3 PHD (12).
NuA4 also contains subunits harboring CHDs or PHDs that are likely important for its stable association with nucleosomes in CDS. Eaf3 is found in NuA4 as well as RPD3C(S). Esa1 also contains a CHD and Yng2 contains a PHD. The Esa1 CHD preferentially bound unmodified H3 tails versus H3 tails monomethylated on K4 or K4 and K9 in vitro (18). The isolated Yng2 PHD preferentially binds to trimethylated H3K4 in vitro (19).
SAGA subunits Gcn5 and Spt7 both contain bromodomains, which have been shown to recognize acetylated histones (20, 21). The Gcn5 bromodomain binds both acetylated H3 and tetra-acetylated H4 tails in vitro, whereas the Spt7 bromodomain only binds weakly to acetylated H3 tails (20). The role of these subunits in SAGA recruitment to CDS is unknown. Because of the two bromodomain-containing subunits, it is expected that SAGA interaction with nucleosomes would be stimulated by histone acetylation, which could involve NuA4.
Previous reports suggested that the primary function of histone H3 methylation by Set1 and Set2 is to stimulate the interaction of HDACs RPD3C(S) and Hos2/Set3 with nucleosomes and their recruitment to CDS. We have shown that complete loss of H3K4 and H3K36 methylation in a set1Δset2Δ double mutant also results in reduced NuA4-nucleosome interactions (4). These data have led us to the hypothesis that H3 methylation is a key regulator of nucleosome acetylation by NuA4 and SAGA, as well as deacetylation by HDACs. To test this hypothesis, we investigated how histone methylation of H3K4 and H3K36 stimulates NuA4 interaction with nucleosomes. We also examined how nucleosome acetylation by NuA4 stimulates SAGA interaction with nucleosomes and recruitment to CDS. Our results support a model in which NuA4 links H3 methylation to acetylation of both histones H3 and H4.
EXPERIMENTAL PROCEDURES
All strains used in this study are listed in Table 1. The wild-type strains BY4741, BY4743, and deletion derivatives described previously (22) were purchased from Research Genetics. All deletions were verified by PCR. The Myc-tagged strains were generated as described previously (23) and verified by PCR analysis of chromosomal DNA and Western blot analysis with anti-Myc antibodies. Strain DGY303 was created as described previously (24), amplifying the kanMX6 cassette of pFA6a-kanMX6 with the primers 5′-TAACCCACCTACCGTTAGTTGAAATAGAAACAAAGAAGAAGGCGGATCCCCGGGTTAATTAA-3′ and 5′-GGTATTTTTGTTCAGTTACGTTTTCTTTTCAGTTTGTTTTTTTCCATCTCGAATTCGAGCTCGTTTAAAC-3′. Loss of YNG2 was confirmed by PCR analysis of chromosomal DNA using the appropriate primers. DGY304 was created by sporulating strain DGY303, dissecting tetrads, and identifying spores with the same genotype as BY4741 except also containing yng2Δ::kanMX6. The absence of YNG2 in the resulting strain, DGY304, was confirmed by demonstrating a decrease in histone H4 acetylation by Western blotting of whole cell extracts (WCEs) using antibodies against tetra-acetylated H4. DGY353 was made as described previously (25), and replacement of kanMX by LEU2 was confirmed by PCR analysis of chromosomal DNA and demonstrating the ability to grow on SC-Leu but not medium containing kanamycin. SET2 was deleted from strains 3GS1-B-4 and YSB2156 to make DGY421 and DGY425, respectively, using homologous recombination with a LEU2 fragment generated by PCR amplification from chromosomal DNA of strain DGY353 using primers 5′-GAGAAGAAGCTGACTTCGACTATTG-3′ and 5′-AAAAATAAAGACACTTGAAACGCAC-3′. DGY443 was constructed as previously described (26), as were HHT2-TAP strains (27). Proper integration of the TAP tag was confirmed by PCR analysis of chromosomal DNA and Western blotting of WCEs with α-H3 and α-TAP antibodies. The H4 quadruple lysine mutant and isogenic WT strains, pJD62_H4_wild-type, Boeke-EMH-H4–171 K5,8,12,16R, and Boeke-EMH-H4–172 K5,8,12,16Q were purchased from Open Biosystems.
TABLE 1.
Yeast strains used in this study
Coimmunoprecipitation experiments were carried out using WCEs as described previously (28) with the antibodies described below. Band intensity was quantified by laser densitometry using ImageJ software (29). Western blot analysis was conducted using WCEs made by trichloroacetic acid extraction as described previously (30) with the antibodies described below.
Peptide binding assays were conducted as described previously (31) with some modifications. Biotinylated H3K4 peptides were purchased from Millipore, and biotinylated H3K36 peptides were custom-made by Sigma Genosys. H3K4 binding buffer (25 mm Tris-HCl, pH 8.0, 500 mm NaCl, 1 mm dithiothreitol, 5% glycerol, 0.03% Nonidet P-40) or H3K36 binding buffer (25 mm Tris-HCl, pH 8.0, 400 mm NaCl, 1 mm dithiothreitol, 5% glycerol, 0.03% Nonidet P-40) was used instead of CTD-binding buffer. Peptide-binding assays were done with NuA4 containing CBD-tagged Eaf1 purified from yeast strain DGY443 as described previously (31). Western blots of input, bound, and supernatant fractions were done using α-TAP and α-Esa1 antibodies. Band intensity was quantified by laser densitometry using ImageJ software (29).
Nucleosome pull-downs were conducted as described previously (32) with some modifications. IgG Sepharose-bound chromatin from HHF2-TAP strains was incubated overnight with WCEs from EAF1-Myc strains in binding buffer (50 mm Tris-HCl, pH 7.5, 0.1% Nonidet P-40, 200 mm NaCl, 10% glycerol, and protease inhibitors). Binding reactions were washed four times with binding buffer, and binding was detected by Western blot analysis with α-Myc and α-H4 antibodies. ChIP assays were carried out as described previously (5) using PCR primers also described previously (4).
The following antibodies were used for ChIP, coimmunoprecipitation analysis, or Western blot analysis. Mouse monoclonal anti-Myc (11667149001; Roche Applied Science), rabbit monoclonal anti-Esa1 (ab4466; Abcam), mouse monoclonal anti-Rpb3 (W0012; Neoclone), anti-phospho-Ser-5 Rpb1 (H14; Covance), rabbit polyclonal anti-H3 (ab1791; Abcam), rabbit polyclonal anti-H3K4me (ab8895; Abcam), rabbit polyclonal anti-H3K4me2 (ab7766; Abcam), rabbit monoclonal anti-trimethyl (Lys-4) histone H3 (05-745: Upstate), rabbit polyclonal anti-H3K36me (ab9048; Abcam), rabbit polyclonal anti-H3K36me2 (07–369; Millipore), rabbit polyclonal anti-H3K36me3 (ab9050; Abcam), rabbit polyclonal anti-H3K79me2 (07–366; Millipore), rabbit polyclonal anti-H3Ac (06–0599; Millipore), mouse monoclonal anti-H4 (ab31827; Abcam), rabbit anti-H4Ac (06–866; Upstate), and rabbit polyclonal anti-TAP (CAB1001; Open Biosystems).
RESULTS
Based on prior knowledge of the binding specificities of NuA4 subunits Esa1, Eaf3, and Yng2 summarized above, we hypothesized that NuA4 binding to nucleosomes would be stimulated by methylation of H3K4 and H3K36. NuA4-mediated acetylation of methylated nucleosomes in CDS would help counteract the effects of increased HDAC recruitment. To test our model, we first sought to obtain direct evidence that H3 methylation enhances nucleosome binding by NuA4.
NuA4 Binds to Unmodified Nucleosomes and Nucleosomes Mono- or Dimethylated on H3K4 and Di- or Trimethylated on H3K36 in Vitro
To identify the nucleosome substrate for NuA4, we examined the binding of NuA4 to histone tail peptides in vitro. NuA4 was partially purified from strains containing EAF1-TAP. We used Eaf1 to purify NuA4, because it has been reported to be the only subunit unique to NuA4 and is important for complex integrity (3, 33). Biotinylated histone tail peptides were immobilized on streptavidin-coated magnetic beads and used to pull down the partially purified TAP-Eaf1 NuA4. Binding reactions were analyzed by Western blot with antibodies against the TAP tag to detect TAP-Eaf1 and antibodies against the NuA4 catalytic subunit Esa1. The fact that the amounts of TAP-Eaf1 and Esa1 in the bound fractions varied in parallel (Fig. 1, A and B, lanes labeled B) suggests that the NuA4 complex is intact in our binding assays.
FIGURE 1.
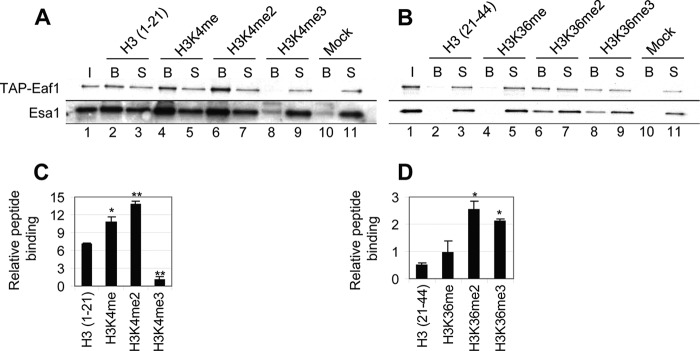
NuA4 preferentially binds to dimethylated H3K4 and H3K36 peptides. Histone tail peptide pull-downs of purified NuA4. A and B, 1.5 μg of biotinylated H3K4 (A) or H3K36 (B) peptide was immobilized on streptavidin-coated Dynal beads and incubated with TAP-Eaf1 NuA4 purified from strain DGY443. Binding reactions were washed three times and the bound (lanes labeled B), input (I), and supernatant (S) fractions were subjected to Western analysis using antibodies against the TAP tag or Esa1. C and D, the ratio of bead to supernatant signal intensity for each peptide was normalized to the same ratio for the Mock reaction. Relative peptide binding was then calculated as the average of those ratios for the two NuA4 subunits (n = 3). H3K4 methylation and H3K36 di- and trimethylation significantly increased NuA4 binding compared with the unmodified peptide as calculated by Student's t test (*, p < 0.01; **, p < 0.001).
We performed these binding assays in 500 mm NaCl for the H3K4 peptides and 400 mm NaCl for the H3K36 peptides, because at lower salt concentrations NuA4 bound equally to all peptides. Interestingly, we found that NuA4 interacted with unmodified as well as mono- or dimethylated H3K4 peptides (amino acids 1–21), with the highest level of NuA4 pulled down by the H3K4me2 peptide (Fig. 1A, lanes 2, 4, 6, 8, and 10). The fact that the amount of TAP-Eaf1 and Esa1 in the supernatant fractions did not change with an increasing amount in the bound fraction, is likely because we analyzed a much larger proportion of the bound fraction than the supernatant fraction and only a small proportion (<30%) of the input NuA4 was bound. We did not observe any interaction between NuA4 and the H3K4me3 peptide, or the streptavidin-coated beads without any peptide (Fig. 1A, lanes 8 and 10). This result was surprising, because the isolated Yng2 PHD preferentially bound to H3K4me3 peptides (19). Quantification of the results revealed that, even though NuA4 was able to bind to the unmodified H3(1–21) peptide (∼6-fold over background), binding was increased by ∼50% with monomethylation and 2-fold with dimethylation on H3K4 (Fig. 1C).
Similar to its interaction with H3K4 peptides, a greater amount of NuA4 was pulled down by di- and trimethylated H3K36 than by any of the other H3(21–44) peptides tested (Fig. 1, B and D). Combined with the H3(1–21) peptide binding results, these results suggest that, whereas NuA4 may bind unmodified H3 tails, its binding is stimulated by methylation of both H3K4 and H3K36. Interestingly, NuA4 binding to the H3K36me2 peptide is consistent with the previously established role of Eaf3, a subunit of NuA4 and RPD3C(S), in promoting the binding of RPD3C(S) to dimethylated H3K36 (13).
To confirm the results of the peptide pulldown assays, we examined NuA4 binding to nucleosomes in vitro. Nucleosomes were immobilized from whole cell extracts of HHF2-TAP strains with IgG beads, washed to remove chromatin-associated proteins, and then incubated with the same preparation of purified TAP-Eaf1 NuA4 used in the peptide pulldown experiments. The nucleosomes were taken from strains with deletions of the SET1 and SET2 HMTs. Nucleosomes from set1Δ cells should have no H3K4 methylation, nucleosomes from set2Δ cells should have no H3K36 methylation, and those from set1Δset2Δ cells should lack both methylation marks. It appeared that nucleosomes from set1Δ and set2Δ cells pulled down slightly less NuA4 than nucleosomes from WT cells (Fig. 2), whereas nucleosomes presumably devoid of any methylation on H3K4 or H3K36 purified from set1Δset2Δ cells pulled down ∼50% less NuA4 than did WT nucleosomes. This correlates with the peptide pulldown data, showing that NuA4-nucleosome interactions are stimulated by both H3K4 and H3K36 methylation. The decrease in NuA4 pulled down by chromatin from HMT mutant strains is not due to a lower level of nucleosomes immobilized on the beads, as equal amounts of TAP-H4 can be seen in each bead fraction (Fig. 2A). We observed no interaction between NuA4 and the IgG beads, because protein A is removed from the TAP tag during the purification process. Similar to the peptide pulldown results, we did not observe a reciprocal change in the amount of NuA4 in the supernatant fraction with changes in the bead fraction. This is likely due to the fact that we loaded the entire bead fraction and only a small portion of the supernatant fraction, and that only a small fraction of the input NuA4 bound to the nucleosomes.
FIGURE 2.
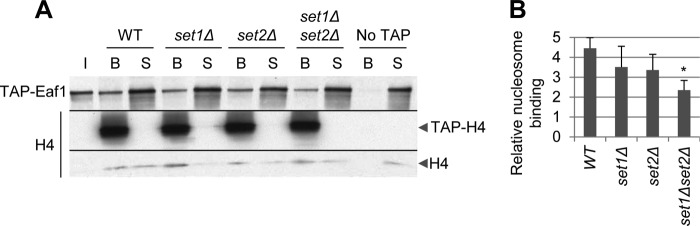
H3K4 and H3K36 methylation stimulates NuA4 binding to nucleosomes in vitro. Nucleosome pull-downs of purified NuA4. A, chromatin was extracted from mid-log phase YPD cultures of HHF2-TAP (3GS1-B-4); HHF2-TAP set1Δ (YSB2156), set2Δ (DGY421), and set1Δset2Δ (DGY425); and HHF2 (BY4741), immobilized on IgG-agarose beads, washed to remove chromatin-associated proteins, and incubated with TAP-Eaf1 NuA4 purified from DGY443. After washing, the entire bound and 10% of each supernatant fraction were subjected to Western analysis using antibodies against the TAP tag and histone H4. B, the ratio of bound to supernatant signal intensity was calculated for each strain and then normalized to the No TAP ratio (n = 3). Nucleosomes from set1Δset2Δ cells pulled down significantly less NuA4 than nucleosomes from WT cells as calculated by Student's t test (*, p < 0.01)
NuA4 Interaction with Nucleosomes in Vivo Is Stimulated by Both H3K4 and H3K36 Methylation
Our in vitro binding assays suggest that NuA4 can interact with hypomethylated nucleosomes, but that binding is stimulated by H3K4 and H3K36 methylation catalyzed by the Set1 complex and Set2, respectively. We extended those results by examining NuA4-nucleosome interactions in vivo using coimmunoprecipitation analysis of strains containing Myc-tagged Eaf1. Chromatin was immunoprecipitated from WCEs with anti-H3 antibodies. Input, pellet, and supernatant fractions were subjected to Western blotting with α-Myc and α-H3 antibodies. We compared NuA4-nucleosome interactions in a WT WCE to those lacking Set1, Set2, or both HMTs. To control for nonspecific Myc tag interactions, we also examined a strain containing Myc-tagged RLI1, as Rli1 is a ribosome-associated protein (34) with no known role in transcription and is not expected to interact with chromatin. Fig. 3A shows the results for the set1Δ, set2Δ, and set1Δset2Δ mutants. All three HMT mutants reduce NuA4 interaction with nucleosomes (Fig. 3A). Loss of H3K4 methylation in set1Δ cells reduces NuA4-nucleosome binding ∼50%, whereas binding is reduced ∼80% in set2Δ cells lacking H3K36 methylation, and in the set1Δset2Δ double mutant (Fig. 3B). The fact that both set1Δ and set2Δ reduce NuA4 interaction with nucleosomes suggests that H3K4 and H3K36 methylation both contribute to NuA4-nucleosome binding. The decreased signal in the pellet fractions cannot be attributed to inefficient immunoprecipitation, because the α-H3 blot shows equivalent pull down of TAP-H3 from each of the extracts.
FIGURE 3.

NuA4 interaction with nucleosomes in vivo is stimulated by H3K4 and H3K36 methylation. A and B, coimmunoprecipitation of Myc-Eaf1 with H3. WCEs of mid-log phase YPD cultures of HHT2-TAP EAF1-Myc (DGY498, DGY488, DGY490, and DGY492) or RLI1-Myc (DGY512) strains were immunoprecipitated (IP) with anti-H3 antibodies and subjected to Western (WB) analysis with anti-H3 and anti-Myc antibodies. All of the pellet and 10% of the input and supernatant fractions were loaded on the gel. In B, the ratio of pellet to supernatant Myc-Eaf1 signal was divided by the TAP-H3 pellet signal for each strain and then normalized to the WT ratio (n = 3). Loss of histone methylation significantly reduced the amount of NuA4 coimmunoprecipitated with histone H3 as calculated by Student's t test (*, p < 0.01; **, p < 0.001). C and D, ChIP analysis of Myc-Eaf1 (C) and Rpb3 (D) at GAL1. EAF1-Myc strains (DGY3, DGY191, DGY112, and DGY200) were grown to early log phase in SCRaf at 25 °C, treated with 2% galactose for 30 min, and ChIP was performed using anti-Myc (C) or anti-Rpb3 (D) antibodies and PCR primers to amplify the GAL1 UAS or 3′ ORF and chromosome V (Chr V) in the presence of SYBR Green. Occupancy was calculated, taking the ratio of signal intensities of PCR products for GAL1 versus the Chr V reference and dividing by the same ratio for input samples. Occupancy in galactose was then divided by the occupancy in raffinose. E–J, ChIP analysis of Myc-Eaf1 (E, G, and I) and Rpb3 (F, H, and J) at ADH1 (E and F), ARG1 (G and H), and PMA1 (I and J). EAF1-Myc strains (the same as in C and D with addition of DGY8) were grown in SC at 25 °C, treated with 0.6 μm SM for 30 min, and processed for ChIP as in C and D above with primers for the appropriate genes described previously (4). Strains in which occupancy was significantly different from WT as calculated by Student's t test are marked with an asterisk (*, p < 0.01).
We next asked whether the reductions observed in NuA4-nucleosome interactions in the set1Δ, set2Δ, and set1Δset2Δ mutants led to reductions in NuA4 occupancy at CDS of inducible and constitutively expressed genes. We previously reported a small reduction in NuA4 occupancy in the GAL1 coding sequence in the set1Δset2Δ double mutant under conditions of galactose induction (4). Here we found that the set1Δset2Δ double mutant and both set1Δ and set2Δ single mutants showed a similar ∼50% decrease in Myc-Eaf1 occupancy at the 3′ end of the GAL1 coding sequence (Fig. 3C, 3′ ORF), without affecting Myc-Eaf1 occupancy of the UAS. NuA4 occupancy had similar defects in both the set1Δ and set2Δ mutants as in the double mutant, suggesting that the NuA4 interaction with nucleosomes methylated by both HMTs is important for its recruitment to transcribed CDS. The loss of NuA4 occupancy at GAL1 is not due to impaired transcription, as Rpb3 occupancy is essentially the same in the promoter and 3′ CDS in all mutant and WT strains (Fig. 3D). Similar to what we observed at GAL1, NuA4 occupancy at the constitutively expressed ADH1 3′ ORF was decreased ∼50% in all three mutant strains (Fig. 3E), without any change in Pol II occupancy (Fig. 3F).
We next examined NuA4 occupancy at the inducible ARG1 gene. The activator for ARG1 is Gcn4, translation of which is up-regulated by amino acid starvation. To induce ARG1 expression, we treated cells with sulfometuron methyl, which causes Ile/Val starvation, and used a gcn4Δ strain as a negative control. ARG1 is not induced by sulfometuron methyl (SM) treatment in gcn4Δ cells, and we thus observed low levels of Eaf1 and Rpb3 occupancies in this strain (Fig. 3, G and H). In contrast to GAL1 and ADH1, NuA4 occupancy at ARG1 under conditions of induction by Gcn4 was reduced ∼40% in set1Δset2Δ cells but not in the single mutants (Fig. 3G). At PMA1, which is constitutively expressed, NuA4 occupancy was reduced ∼40% in set1Δ and set1Δset2Δ cells but not in the set2Δ single mutant (Fig. 3I). Pol II occupancy was unaffected by loss of histone methylation at each of these genes (Fig. 3, H and J). We have previously suggested a two-stage model for recruitment of NuA4 to CDS, wherein NuA4 first interacts with Pol II phosphorylated on Ser-5 of its CTD and then binds to nucleosomes in the coding sequence methylated on H3K4 and H3K36. The fact that histone methylation defects affected NuA4 occupancy in a gene-specific manner suggests that the relative contributions of Ser-5p and H3 methylation on K4 (by Set1) and K36 (by Set2) to NuA4 recruitment are also gene-specific.
H4 Acetylation by NuA4 Stimulates H3 Acetylation by SAGA
As described above, the SAGA complex is recruited to CDS, acetylates histone H3, and stimulates transcription elongation in a manner similar to NuA4. Although NuA4 contains subunits with domains that are thought to recognize methylated histones, SAGA subunits carry bromodomains that have been shown to recognize acetylated histones (20, 21). SAGA acetylation of H3 in vitro was shown to be stimulated by the presence of acetylated H4K16, presumably by enhancing the nucleosomal binding by the SAGA bromodomains (35). Thus, we hypothesized that loss of H4 acetylation in NuA4 mutants would lead to a decrease in H3 acetylation by SAGA as well.
We first examined H3 acetylation in bulk histones by Western blots on WCEs from NuA4 and SAGA mutant strains. We used mutants lacking subunits of NuA4 and SAGA important for KAT activity or complex integrity. Eaf1 contributes to NuA4 integrity (3), Yng2 stimulates nucleosome acetylation by Esa1 (36), and Esa1 is the KAT subunit of NuA4. Because Esa1 is essential, we used the esa1L254P temperature-sensitive allele, henceforth referred to simply as esa1 (37). Gcn5 is the KAT subunit of SAGA (38) and Ada1 is essential for SAGA integrity (39). To measure histone acetylation, we grew strains in YPD to mid-log phase at 30 °C. After incubation at 36 °C for 4 h, WCEs were prepared and subjected to Western blot analysis with antibodies against di-acetylated H3 (H3-ac), tetra-acetylated H4 (H4-ac), total H3 and total H4 (Fig. 4A). Acetylation per histone was then calculated as the ratio of acetylated signal to total histone signal (Fig. 4, B and C).
FIGURE 4.

NuA4 stimulates H3 acetylation by SAGA. A–C, effect of NuA4 and SAGA subunit mutations on H3 and H4 acetylation. Strains (BY4741, DGY326, DGY304, DGY150, 7285, 1038, and DGY154) were grown in YPD at 30 °C, transferred to 36 °C for 4 h, and WCEs were subjected to Western analysis with antibodies against diacetylated H3, the H3 C terminus, tetra-acetylated H4 and H4 C terminus. In B and C, acetylation per histone was calculated as the ratio of the acetylated histone signal to the total histone signal for each strain (n = 4). Strains in which H3 or H4 acetylation was determined to be significantly reduced compared with WT as calculated by Student's t test are indicated by asterisks (*, p < 0.01; **, p < 0.001). D and E, ChIP analysis of H3-ac (D) and Rpb3 (E) at ADH1. WT and KAT mutant strains (BY4741, DGY150, and DGY154) were grown in YPD at 30 °C, transferred to 36 °C for 1 h, and ChIP was performed using antibodies to diacetylated H3, H3 (D), and Rpb3 (E) and PCR primers to amplify the ADH1 promoter and 3′ ORF, a region of the chromosome VI telomere (Tel VI) (D), and chromosome V (Chr V) (E). H3-ac/H3 occupancy was calculated as the ratio of H3-ac occupancy to H3 occupancy. H3-ac/H3 in the KAT mutants was determined to be significantly less than WT by Student's t test (*, p < 0.01; **, p < 0.001). F and G, coimmunoprecipitation of Myc-Ada2 with H3. Myc-ADA2 or Myc-RLI1 strains (HQY392, DGY365, and YDH353) were grown in YPD at 30 °C and transferred to 36 °C for 1 h. WCEs were immunoprecipitated with anti-H3 antibodies and subjected to Western analysis with anti-H3 and anti-Myc antibodies. In G, relative binding was calculated as the ratio of Myc pellet to supernatant signal divided by the H3 pellet signal. Relative binding in esa1 cells was determined to be significantly less than in WT cells by Student's t test (*, p < 0.01). H–K, ChIP analysis of Myc-Ada2 (H and J) and Rpb3 (I and K) at ARG1 (H and I) and ADH1 (J and K). WT, esa1, and gcn4Δ strains (HQY392, DGY365, and HQY503) were grown in SC at 30 °C, transferred to 36 °C for 30 min, treated with 0.6 μm SM for 30 min, and ChIP was performed using anti-Myc (H and J) and anti-Rpb3 (I and K) antibodies and PCR primers to the ARG1 UAS and 3′ ORF (H and I), the ADH1 promoter and 3′ ORF (J and K), and Chr V. Myc-Ada2 3′ ORF occupancy in esa1 was determined to be significantly less than WT by Student's t test (*, p < 0.01).
As expected, H4-ac was reduced in yng2Δ and esa1 strains (Fig. 4, A and B). This is consistent with the fact that Yng2 and Esa1 are in both NuA4 and picNuA4 and both affect KAT activity, whereas Eaf1 occurs only in NuA4 and does not affect picNuA4. Surprisingly, H4-ac was also reduced ∼50% in gcn5Δ cells (Fig. 4, A and B), suggesting that Gcn5 stimulates NuA4 assembly, recruitment, or activity. Considering that ada1Δ destabilizes SAGA (39) but did not reduce H4 acetylation, this proposed role of Gcn5 might operate in the context of the ADA or SAGA complex. Interestingly, we observed a marked reduction in H3-ac in esa1 cells that, as just described, also showed the largest reduction in H4-ac (Fig. 4, A-C). This suggests that H4 acetylation by NuA4 stimulates H3 acetylation by SAGA. As expected, H3 acetylation was reduced by both gcn5Δ and ada1Δ, with the former having the greater effect (Fig. 4, A and C). These reductions were relatively more severe than those elicited by the NuA4 mutants (Fig. 4C).
To confirm the results seen in bulk histones, we investigated the effect of esa1 on H3 acetylation at ADH1 by ChIP. Cultures were grown in YPD to early log phase at 30 °C and then transferred to 36 °C for 4 h before cross-linking. ChIP was performed using α-H3 and α-H3-ac antibodies and PCR primers to amplify the ADH1 core promoter (Pro) and 3′ ORF and a region in the telomere of chromosome VI (Tel VI). Acetylation and histone H3 occupancy were calculated as the ratio of ADH1 to Tel VI for the immunoprecipitated samples divided by the same ratio for the input samples. To correct for the effects of histone occupancy changes on acetylation, we calculated acetylation per nucleosome (H3-ac/H3), dividing the H3-ac occupancy by the H3 occupancy. Remarkably, H3-ac/H3 was reduced by ∼60% in the esa1 mutant at both the promoter and 3′ ORF of ADH1. Relative to the esa1 mutant, H3-ac/H3 was reduced slightly more at the promoter and to the same extent at the 3′ ORF in the gcn5Δ strain (Fig. 4D). As expected, the gcn5Δesa1 double mutant displayed the greatest reduction in both promoter and 3′ ORF H3-ac/H3 ratios (Fig. 4D). The decrease in H3-ac cannot be attributed to decreased transcription as Rpb3 (Pol II) levels in the KAT mutants were even higher than in WT (Fig. 4E). Thus, H4 acetylation by NuA4 appears to exert a strong stimulatory effect on H3 acetylation by SAGA in vivo.
Because SAGA subunits Gcn5 and Spt7 contain bromodomains thought to recognize acetylated histones, we thought it likely that the decrease in H3 acetylation in the NuA4 mutants would result from decreased interaction between SAGA and nucleosomes. To test this prediction, we measured SAGA-nucleosome association by coimmunoprecipitation analysis of ADA2-Myc and RLI1-Myc strains, as Ada2 is a subunit of SAGA. We grew the strains in YPD to early log phase at 30 °C, transferred them to 36 °C for 4 h, and immunoprecipitated histone H3 from whole cell extracts. We analyzed the immune complexes by Western blotting with α-Myc and α-H3 antibodies. Consistent with the hypothesis that H4 acetylation by NuA4 stimulates SAGA interaction with nucleosomes, we found that ∼75% less Myc-Ada2 coimmunoprecipitates with H3 from an esa1 mutant than from WT cells (Fig. 4, F, cf. lanes 1–3 and 4–6, and G). The decrease in Myc-Ada2:H3 association is not attributable to a decrease in the efficiency of coimmunoprecipitation, because we observed equal amounts of H3 in the pellet fractions. These results suggest that SAGA preferentially binds to nucleosomes acetylated on histone H4.
We next asked whether the reduction in nucleosome binding we observed in the esa1 mutant would lead to reduced SAGA occupancy of transcribed CDS. To this end, we measured the occupancy of SAGA subunit Myc-Ada2 by ChIP at ARG1 and ADH1. Strains were grown in SC medium to early log phase at 30 °C, transferred to 36 °C for 3.5 h, and treated with 0.6 μm SM for 30 min at 36 °C before cross-linking. Consistent with our coimmunoprecipitation results, we observed significant reductions in Myc-Ada2 occupancy at the ARG1 and ADH1 CDS, but not UAS (Fig. 4, H and J), without a decrease in Rpb3 occupancy in esa1 cells (Fig. 4, I and K). These results suggest that H4 acetylation stimulates SAGA interaction with nucleosomes and that this interaction is important for recruitment of SAGA to transcribed CDS.
NuA4 has been reported to acetylate ∼95 non-histone targets (40). It is possible that the effects we observed on H3 acetylation and SAGA interaction with nucleosomes in esa1 cells are not due to loss of H4 acetylation, but rather decreased acetylation of another NuA4 substrate. To determine whether H4 acetylation is truly important for SAGA interaction with nucleosomes, we examined H3 acetylation and SAGA occupancy in strains expressing H4 variants with Arg or Gln substitutions of the four residues acetylated by NuA4, namely lysines 5, 8, 12, and 16 designated hhfsK8,8,12,16R and hhfsK5,8,12,16Q, respectively. We first examined H3 acetylation in bulk histones from WCEs of WT, hhfsK5,8,12,16Q, and hhfsK5,8,12,16R strains. Similar to NuA4 mutants, we observed an ∼50% reduction in H3 acetylation in the hhfsK5,8,12,16R cells, but not the hhfsK5,8,12,16Q cells (Fig. 5, A and B). As arginine cannot be acetylated by NuA4 and maintains the positive charge of H4, whereas glutamine may serve as an acetyl-lysine mimic, these findings support our hypothesis that acetylation of H3 by SAGA is enhanced by H4 acetylation by NuA4. Consistent with the Western blot results, ChIP analysis revealed that H3 acetylation was significantly decreased in the coding sequence of ADH1 specifically in the hhfsK5,8,12,16R mutant (Fig. 5C).
FIGURE 5.
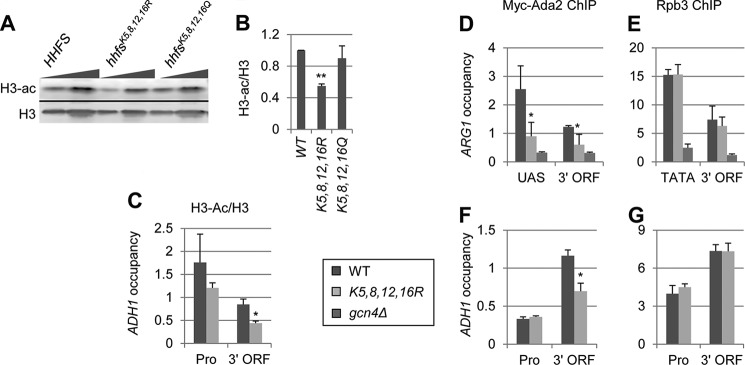
H4 acetylation stimulates H3 acetylation by SAGA. A and B, the effects of H4 tail Lys substitutions on H4 and H3 acetylation. WT and H4 mutant strains harboring the hhfsK5,8,12,16R or hhfsK5,8,12,16Q alleles (pJD62_H4_WT, Boeke-EMH-H4–172, and Boeke-EMH-H4–171, respectively) were grown to stationary phase in YPD and WCEs were subjected to Western analysis with antibodies against diacetylated H3 and the H3 C terminus. B, acetylation per histone was calculated as the ratio of H3-ac signal to the total H3 signal (n = 4). C–G, ChIP analysis of H3 and H3-ac (C), Myc-Ada2 (D and F), and Rpb3 (E and G) at ARG1 (D and E), and ADH1 (C, F, and G). WT, hhfsK5,8,12,16R, and gcn4Δ Myc-ADA2 strains (DGY655, DGY658, and DGY654) were grown to early log phase in SC at 30 °C, treated with 0.6 μm SM for 30 min, and ChIP was performed using anti-H3-ac and anti-H3 (C), anti-Myc (D and F), and anti-Rpb3 (E and G) antibodies and primers for the promoter and coding sequence of ADH1 (C, F, and G) or the enhancer and coding sequence of ARG1 (D and E). H3-ac occupancy was normalized to H3 occupancy. H3-ac/H3 and Myc-Ada2 occupancies in hhfsK5,8,12,16R cells were determined to be significantly less than in WT by Student's t test (*, p < 0.01).
We also asked whether the hhfsK5,8,12,16R mutant would affect SAGA recruitment in a manner similar to the NuA4 mutants, by analyzing SAGA occupancy using ChIP of Myc-Ada2. Although SAGA occupancy at ARG1 decreased at the 3′ ORF by ∼25% in esa1 cells (Fig. 4H), SAGA occupancy was reduced ∼60% at the promoter and 50% at the coding sequence in the H4 mutant (Fig. 5D). The greater defect in the histone mutant compared with esa1 may be due to a greater loss of H4 acetylation. Surprisingly, these last results suggest that H4 acetylation stimulates SAGA occupancy at promoters, as well as CDS. SAGA occupancy at promoters has been thought to occur through direct interaction between its Tra1 subunit and activators bound to the UAS elements (41).
Similar to esa1 cells, SAGA occupancy in hhfsK5,8,12,16R mutant cells was reduced ∼50% in the coding sequence of ADH1 with no decrease at the promoter (Fig. 5F). The fact that Pol II occupancy was unchanged at ARG1 and ADH1 in the H4 mutant cells suggests that the SAGA recruitment defects at these genes do not result from reduced transcription (Fig. 5, E and G). The greater SAGA occupancy defects at induced ARG1 may be due to the fact that this gene is expressed at higher levels than ADH1 (42). Together, our results suggest that acetylation of lysine residues in the N-terminal tail of H4 by NuA4 stimulates SAGA recruitment and H3 acetylation.
Loss of Histone Methylation Does Not Lead to an Increase in Histone Acetylation
If H3K4 and H3K36 methylation functions solely to stimulate HDAC interaction with nucleosomes and attendant nucleosome deacetylation, we would expect global histone acetylation levels to increase in set1Δ and set2Δ mutant cells. In contrast, if methylation is important for nucleosome binding by both HDACs and NuA4, then set1Δ and set2Δ cells might show no change in acetylation of bulk histones compared with WT cells. To test this hypothesis, we analyzed H4 tetra-acetylation in bulk histones in whole cell extracts of set1Δ, set2Δ, set1Δset2Δ, dot1Δ, and bre1Δ cells. Dot1 is responsible for H3K79 methylation and is associated with transcription elongation and maintenance of heterochromatin boundaries (43). Bre1 is the E3 ubiquitin ligase responsible for H2BK123 monoubiquitination (44), which is required for Set1-mediated H3K4 di- and trimethylation (Fig. 6C) and Dot1-mediated H3K79 methylation (10).
FIGURE 6.

H3K4 and K36 methylation stimulates both H4 acetylation and deacetylation. A–C, among HMT mutants, only bre1Δ alters bulk H4 acetylation. Strains BY4741, DGY183, 1257, DGY184, 4276, and 3771 were grown to stationary phase in YPD. In A, WCEs were analyzed by Western blot with anti-tetra-acetylated H4 and anti-H4 C terminus antibodies. In B, acetylation per histone was calculated as the ratio of H4-ac signal to H4 signal for each strain (n = 4). H4 acetylation in bre1Δ cells was determined to be significantly greater than WT by Student's t test (**, p < 0.001). C, Bre1 stimulates di- and trimethylation of H3K4. Strains DGY3, DGY191, and DGY427 were grown to stationary phase in YPD and WCEs were analyzed by Western blot with anti-H3K4me3, anti-H3K4me2, anti-H3K4me, and anti-H3 antibodies. D and E, coimmunoprecipitation of Myc-Eaf1 with H3 is unaltered by bre1Δ and dot1Δ. In D, strains DGY498, DGY506, DGY509, and DGY512 were grown to mid-log phase in YPD medium and WCEs were immunoprecipitated (IP) with anti-H3 antibodies and subjected to Western (WB) analysis with anti-H3 and anti-Myc antibodies. In E, relative binding was calculated as the ratio of Myc pellet to supernatant signal divided by the H3 pellet signal (n = 3). F, ChIP analysis of Myc-Eaf1 at GAL1. WT, bre1Δ, dot1Δ, and gcn4Δ strains (DGY3, DGY427, DGY56, and DGY8) were grown in SCRaf at 30 °C, treated with 2% galactose for 30 min, and ChIP was performed using anti-Myc antibodies and PCR primers to amplify the GAL1 UAS or 3′ ORF and chromosome V in the presence of [α-33P]dATP. Occupancy was calculated, taking the ratio of radioactivities of PCR products for GAL1 versus the chromosome V reference and dividing by the same ratio for input samples. Occupancy in galactose was divided by the occupancy in raffinose.
When we examined H4 acetylation in bulk histones in mutants defective for H3K4, H3K36, or H3K79 methylation, we found no significant changes in any of the four HMT deletion mutants, whereas H4-ac levels increased ∼2-fold in bre1Δ cells (Fig. 6, A and B). As previously shown, di- and trimethylation of H3K4 are dramatically reduced in the bre1Δ mutant (10) (Fig. 6C), and it was shown previously that binding of the Set3/Hos2 HDAC to nucleosomes is stimulated by these H3 modifications (12). We found that NuA4, on the other hand, can bind monomethylated H3K4 (Fig. 1, A and C), and this H3K4 isoform is relatively unaffected by the bre1Δ mutation (Fig. 6C). Thus, the increase in H4 acetylation in the bre1Δ mutant is likely due to the loss of Set3/Hos2 interaction with nucleosomes and attendant reduction in H4-deacetylation without any loss of NuA4-nucleosome association and H4-acetylation. Consistent with this explanation, we found that NuA4 interaction with nucleosomes was unaffected in bre1Δ and dot1Δ cells (Fig. 6, D and E), and that NuA4 recruitment to the GAL1 coding sequence was unaffected by the bre1Δ mutation (Fig. 6F). The fact that NuA4 occupancy at the GAL1 coding sequence was reduced in set1Δ cells (Fig. 4C) but not bre1Δ cells (Fig. 6F) supports the hypothesis that H3K4 monomethylation stimulates NuA4, but not Set3C, interaction with nucleosomes. The fact that H4 acetylation did not increase in set1Δ and set2Δ cells is consistent with the model that H3K4 and H3K36 methylation stimulates both H4 acetylation by NuA4 and deacetylation by Set3/Hos2 and RPD3C(S).
DISCUSSION
The work presented herein provides evidence for a mechanism by which histone H3 methylation can regulate the level of both H3 and H4 acetylation in the transcribed CDS of budding yeast genes. Previous work focused on how histone H3 methylation stimulated interaction of histone deacetylase complexes Set3/Hos2 and RDP3C(S) with nucleosomes and thus promoted nucleosome deacetylation and repressed cryptic transcription (12–15, 45, 46). Here we provide evidence that H3 methylation stimulates NuA4 interaction with nucleosomes, and that H4 acetylation by NuA4 stimulates H3 acetylation by SAGA. Thus, it appears that H3 methylation can control the relative recruitment of KATs and HDACs to regulate the level of nucleosome acetylation in transcribed coding regions.
We have presented four lines of evidence indicating that NuA4 preferentially binds to H3 tails methylated on Lys-4 by the Set1 complex and on Lys-36 by Set2. First, by assaying the binding of purified NuA4 to immobilized peptides comprising distinct segments of the H3 tail (residues 1–20 or 21–44), we observed greater NuA4 binding to peptides that were mono- or dimethylated on Lys-4, or di- or trimethylated on Lys-36 (Fig. 1). NuA4 also bound to the unmodified H3K4 peptide, which is consistent with the previous finding that Esa1 CHD preferentially binds unmodified H3 tails (18). The high salt concentrations needed to see differences in NuA4 binding to the different peptides (500 mm for the H3K4 peptides and 400 mm for the H3K36 peptides) suggests that NuA4 binds strongly to the isolated H3 tail. The different salt concentrations needed for the H3K4 versus the H3K36 peptides might indicate that NuA4 binds more strongly to the N-terminal 21 amino acids than to the next 23 amino acids of histone H3.
Second, by assaying the binding of purified NuA4 to nucleosomes isolated from strains lacking Set1, Set2, or both HMTs, we found that nucleosomes lacking both Set1- and Set2-dependent modifications bound NuA4 at substantially reduced levels compared with nucleosomes lacking only Set1- or Set2-dependent modifications. The nucleosomes lacking only Set1- or Set2-dependent modifications supported NuA4 binding that was not significantly lower than that of nucleosomes purified from WT cells (Fig. 2). Thus, even though NuA4 binding to H3 tail peptides is stimulated by methylation of both Lys-4 and Lys-36 (Fig. 1), the presence of either modification appears to be sufficient for robust NuA4 binding to purified nucleosomes. We also observed significant binding to unmodified nucleosomes in these last experiments, which is consistent with the peptide pulldown assays where NuA4 bound to the unmodified H3(1–21) peptide. Thus, we conclude that NuA4 can bind hypomethylated nucleosomes, but that H3K4 and H3K36 methylation by the Set1 complex and Set2, respectively, make overlapping stimulatory contributions to bulk nucleosome association by NuA4 in vitro.
Third, we assayed association of NuA4 with native bulk nucleosomes by coimmunoprecipitation of Myc-tagged Eaf1 (a subunit unique to NuA4 (3)) with histone H3 from cell extracts. Here we observed that the absence of either Set1 or Set2 in cells evoked a strong reduction in NuA4-nucleosome association, with the set2Δ mutant displaying essentially the same strong reduction observed for the set1Δset2Δ double mutant (Fig. 3, A and B). These findings suggest that both H3K4 methylation by Set1 and H3K36 methylation by Set2 are required for robust NuA4 association with nucleosomes in vivo, with H3K36/Set2 making the more critical contribution. The greater binding defects observed for the HMT mutants in these last experiments compared with those where NuA4 binding to purified nucleosomes was assayed in vitro (Fig. 2) might be the result of other proteins that compete with NuA4 for interaction with hypomethylated nucleosomes in vivo. This competition would intensify the reductions in NuA4 association with native nucleosomes evoked by the loss of one or both methylation reactions in the HMT mutants. That both H3K4 methylation by Set1 and H3K36 methylation by Set2 were found to stimulate NuA4 interaction with native nucleosomes (Fig. 3, A and B) is consistent with the fact that NuA4 contains subunits with domains (CHD and PHD) thought to recognize one or the other methylated lysines.
Fourth, we found that NuA4 occupancy of the CDS of GAL1, ADH1, ARG1, and PMA1 was reduced to some degree in at least one of the HMT mutants (Fig. 3, C, E, G, and I). The greatest defects were seen at GAL1 and ADH1, where NuA4 occupancy decreased ∼50% in all of the mutants (Fig. 3, C and E). In contrast, NuA4 occupancy was reduced only in the set1Δset2Δ mutant at ARG1 and in set1Δ cells at PMA1 (Fig. 3, G and I). The different effect on NuA4 recruitment at various genes suggests that the contributions of histone methylation to NuA4 occupancy may be gene-specific. Overall, the effects of HMT mutants on native NuA4-nucleosome interaction (Fig. 3, A and B) were greater than their effects on coding sequence occupancies (Fig. 3, C–J). This is likely to be due to the two-stage recruitment mechanism of NuA4 to CDS, wherein NuA4 occupancy of CDS is stimulated by its interaction with the Pol II CTD (4). Thus, NuA4 can likely remain associated with CDS through its interaction with Pol II, even when it cannot bind directly to methylated nucleosomes. The gene-specific effects on NuA4 occupancy may be attributable to varying contributions of CTD and nucleosome interaction on recruiting NuA4 and maintaining its association with coding sequence nucleosomes at different genes. The variable effects of histone methylation on NuA4 occupancy at different CDS might also reflect differences in the rate or efficiency of transcription at those genes.
We have provided the first in vivo evidence that H3 acetylation by SAGA is stimulated by NuA4-mediated acetylation of H4. Previous data had shown that retention of SAGA on nucleosome arrays could be stimulated by either H3 or H4 acetylation, and that SAGA acetylation of H3 was stimulated by acetylation of H4K16 in vitro (21, 35). Here we showed that loss of H4 acetylation in esa1 cells led to decreased H3 acetylation in both bulk histones (Fig. 4, A–C) and at ADH1 (Fig. 4, D and E), decreased nucleosomal binding by SAGA (Fig. 4, F and G), and decreased SAGA occupancy at the CDS of ARG1 and ADH1 (Fig. 4, H–K). Similar to what we observed with NuA4 and loss of methylation, the Ada2 recruitment defect measured by ChIP was of lesser magnitude than the reduction in Ada2 coimmunoprecipitation with native nucleosomes in the esa1 mutant. This too can be explained by the fact that SAGA, similar to NuA4, is recruited to CDS through interaction with the CTD (5). SAGA has also been shown to bind to trimethylated H3K4 through its Sgf29 subunit, which could also contribute to occupancy in CDS (47, 48). Thus, our findings suggest that SAGA interaction with nucleosomes and its recruitment to transcribed CDS are stimulated by NuA4 acetylation of H4.
It was possible that the defects we observed in SAGA interaction with nucleosomes are not due to loss of H4 acetylation, but to reduced acetylation of another non-histone NuA4 target (40). Our finding that an H4 mutant in which the four lysines acetylated by NuA4 are substituted with arginines exhibits reductions in H3 acetylation and Ada2 occupancy of CDS similar to the defects seen in the esa1 mutant suggests that SAGA interaction with nucleosomes is indeed stimulated by acetylation of these H4 lysines by NuA4. This conclusion is consistent with the fact that SAGA subunits Gcn5 and Spt7 both contain bromodomains. Surprisingly, we observed a dramatic reduction in SAGA occupancy at the ARG1 UAS, as well as in the coding sequence in H4 mutant cells. This result suggests that, at least at some genes, H4 acetylation stimulates SAGA recruitment to promoters in addition to interaction of the Tra1 subunit of SAGA with activators. Although H3 acetylation decreased at both the promoter and coding sequence of ADH1 in esa1 cells, we only observed a significant decrease at the coding sequence in hhfsK5,8,12,16R cells. This might be explained by proposing that H4 mutant cells have adapted to the mutation in a manner that did not occur during the 30 min at the non-permissive temperature in the esa1 mutant.
We have shown previously that H3 acetylation decreases, whereas H4 acetylation increases, at GAL1 in a gcn5Δ mutant (4). Our finding here that esa1 reduces both H3 and H4 acetylation can explain why the esa1 mutant displayed larger reductions in the transcription elongation rate and SWI/SNF recruitment than did a gcn5Δ mutant. Our finding of even larger defects in the gcn5Δesa1 double mutant (4) is to be expected from our finding here that H3 acetylation is decreased more in the double mutant than in the esa1 single mutant.
Together, our findings suggest a mechanism by which histone methylation controls the level of acetylation of transcribed CDS through NuA4 and multiple HDACs (Fig. 7). We propose that hypomethylated nucleosomes containing H3K4me or H3K36me are recognized by NuA4, which acetylates them on H4. These H4-acetylated nucleosomes then become a substrate for SAGA, which catalyzes H3 acetylation. Nucleosomes carrying dimethylated H3K4 and K36 can still be recognized and acetylated by NuA4, but they are also recognized by the Set3/Hos2 (12) and RPD3C(S) (14, 15, 45, 46) complexes, thus evoking competition between acetylation by NuA4 and deacetylation by Hos2 and Rpd3.
FIGURE 7.

H3K4 and H3K36 methylation regulates H3 and H4 acetylation and deacetylation during transcription elongation. A, nucleosomes monomethylated on H3K4 by Set1 or H3K36 by Set2 (depicted by dotted arrows) are bound by NuA4, which acetylates the H4 tail (H4-ac). Nucleosomes containing H4-ac are bound by SAGA, which acetylates the H3 tail (H3-ac). NuA4 interaction with nucleosomes and attendant H4-ac formation, SAGA recruitment, and H3-ac depends primarily on Set1 and H3K4me in the 5′ end of CDS and Set2/H3K36me further downstream. B, on dimethylated nucleosomes, HDACs Set3/Hos2 and RPD3C(S) compete with NuA4 for binding to H3K4me2 and H3K36me2, respectively. As HDACs can now interact with these nucleosomes, acetylation is reduced. C, neither NuA4 nor Set3/Hos2 interacts with H3K4me3 in the 5′ ORF, whereas NuA4 and RPD3C(S) continue to oppose each other by interacting competitively with H3K36me3 at the 3′ ORF.
Genome-wide studies have shown that H3K4 trimethylation peaks at the 5′ ends of CDS, dimethylation peaks in the middle of CDS, and monomethylation peaks at the 3′ end of CDS (49). Similar to H3K4 monomethylation, H3K36 trimethylation peaks at the 3′ end of CDS with dimethylation showing a slightly broader range (49, 50). Because NuA4 can recognize both methylated H3K4 and H3K36, it can bind and acetylate nucleosomes over most of the coding sequence, whereas Set3/Hos2 is likely to be restricted to the 5′ half of an ORF and RPD3C(S) is enriched toward the 3′ end of the ORF (12, 46). We propose that H4 acetylation by NuA4 is counteracted by HDACs through two mechanisms. In the 5′ half of CDS, both NuA4 and Set3C can bind nucleosomes dimethylated on H3K4. Thus, not only can Hos2 deacetylate these nucleosomes, but the Set3 complex may also prevent NuA4 from binding. In the latter half of CDS, both NuA4 and RPD3C(S) recognize H3K36me2 and likely compete for binding nucleosomes containing that modification. More evidence for competition between NuA4 and RPD3C(S) is that NuA4 recruitment to the MAT double strand break is increased in an rco1 mutant (51). In addition to competing for nucleosome binding, RPD3C(S) can directly deacetylate nucleosomes that have been acetylated by SAGA and NuA4. Thus histone methylation by both Set1 and Set2 regulates the level of acetylation in transcribed CDS through a dynamic interplay among NuA4, SAGA, Set3C, and RPD3C(S).
Histone acetylation by NuA4 and SAGA is required to stimulate nucleosome eviction and transcription elongation (4, 5). On the other hand, excessive nucleosomal acetylation can activate cryptic promoters, interfering with the production of protein-coding transcripts (52). Hence, it is critical to maintain the proper balance of KAT and HDAC activities in CDS. Our results show that HMTs are key players in achieving this goal.
Acknowledgments
We thank Stephen Buratowski for strains. We thank Hongfang Qiu, Chhabi Govind, and Tae Soo Kim for advice and help with experiments.
This work was supported, in whole or in part, by the National Institutes of Health Intramural Research Program and the LIU Post Campus Faculty Research Committee.
- Pol II
- RNA polymerase II
- KAT
- lysine acetyltransferase
- CDS
- coding sequences
- HMT
- histone methyltransferase
- CTD
- Pol II C-terminal domain
- HDAC
- histone deacetylase complex
- CHD
- chromodomain
- PHD
- plant homeobox domain
- WCE
- whole cell extract
- SM
- sulfometuron methyl.
REFERENCES
- 1. Allard S., Utley R. T., Savard J., Clarke A., Grant P., Brandl C. J., Pillus L., Workman J. L., Côté J. (1999) NuA4, an essential transcription adaptor/histone H4 acetyltransferase comple containing Esa1p and the ATM-related cofactor Tra1p. EMBO J. 18, 5108–5119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Doyon Y., Côté J. (2004) The highly conserved and multifunctional NuA4 HAT complex. Curr. Opin. Genet. Dev. 14, 147–154 [DOI] [PubMed] [Google Scholar]
- 3. Auger A., Galarneau L., Altaf M., Nourani A., Doyon Y., Utley R. T., Cronier D., Allard S., Côté J. (2008) Eaf1 is the platform for NuA4 molecular assembly that evolutionarily links chromatin acetylation to ATP-dependent exchange of histone H2A variants. Mol. Cell. Biol. 28, 2257–2270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ginsburg D. S., Govind C. K., Hinnebusch A. G. (2009) NuA4 lysine acetyltransferase Esa1 is targeted to coding regions and stimulates transcription elongation with Gcn5. Mol. Cell. Biol. 29, 6473–6487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Govind C. K., Zhang F., Qiu H., Hofmeyer K., Hinnebusch A. G. (2007) Gcn5 promotes acetylation, eviction, and methylation of nucleosomes in transcribed coding regions. Mol. Cell 25, 31–42 [DOI] [PubMed] [Google Scholar]
- 6. Brower-Toland B., Wacker D. A., Fulbright R. M., Lis J. T., Kraus W. L., Wang M. D. (2005) Specific contributions of histone tails and their acetylation to the mechanical stability of nucleosomes. J. Mol. Biol. 346, 135–146 [DOI] [PubMed] [Google Scholar]
- 7. Mujtaba S., Zeng L., Zhou M. M. (2007) Structure and acetyl-lysine recognition of the bromodomain. Oncogene 26, 5521–5527 [DOI] [PubMed] [Google Scholar]
- 8. Briggs S. D., Bryk M., Strahl B. D., Cheung W. L., Davie J. K., Dent S. Y., Winston F., Allis C. D. (2001) Histone H3 lysine 4 methylation is mediated by Set1 and required for cell growth and rDNA silencing in Saccharomyces cerevisiae. Genes Dev. 15, 3286–3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Strahl B. D., Grant P. A., Briggs S. D., Sun Z. W., Bone J. R., Caldwell J. A., Mollah S., Cook R. G., Shabanowitz J., Hunt D. F., Allis C. D. (2002) Set2 is a nucleosomal histone H3-selective methyltransferase that mediates transcriptional repression. Mol. Cell. Biol. 22, 1298–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee J. S., Shukla A., Schneider J., Swanson S. K., Washburn M. P., Florens L., Bhaumik S. R., Shilatifard A. (2007) Histone crosstalk between H2B monoubiquitination and H3 methylation mediated by COMPASS. Cell 131, 1084–1096 [DOI] [PubMed] [Google Scholar]
- 11. Li B., Carey M., Workman J. L. (2007) The role of chromatin during transcription. Cell 128, 707–719 [DOI] [PubMed] [Google Scholar]
- 12. Kim T., Buratowski S. (2009) Dimethylation of H3K4 by Set1 recruits the Set3 histone deacetylase complex to 5′ transcribed regions. Cell 137, 259–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li B., Jackson J., Simon M. D., Fleharty B., Gogol M., Seidel C., Workman J. L., Shilatifard A. (2009) Histone H3 lysine 36 dimethylation (H3K36me2) is sufficient to recruit the Rpd3s histone deacetylase complex and to repress spurious transcription. J. Biol. Chem. 284, 7970–7976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Joshi A. A., Struhl K. (2005) Eaf3 chromodomain interaction with methylated H3-K36 links histone deacetylation to Pol II elongation. Mol. Cell 20, 971–978 [DOI] [PubMed] [Google Scholar]
- 15. Keogh M. C., Kurdistani S. K., Morris S. A., Ahn S. H., Podolny V., Collins S. R., Schuldiner M., Chin K., Punna T., Thompson N. J., Boone C., Emili A., Weissman J. S., Hughes T. R., Strahl B. D., Grunstein M., Greenblatt J. F., Buratowski S., Krogan N. J. (2005) Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell 123, 593–605 [DOI] [PubMed] [Google Scholar]
- 16. Li B., Gogol M., Carey M., Lee D., Seidel C., Workman J. L. (2007) Combined action of PHD and chromo domains directs the Rpd3S HDAC to transcribed chromatin. Science 316, 1050–1054 [DOI] [PubMed] [Google Scholar]
- 17. Pinskaya M., Gourvennec S., Morillon A. (2009) H3 lysine 4 di- and tri-methylation deposited by cryptic transcription attenuates promoter activation. EMBO J. 28, 1697–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jacobs S. A., Taverna S. D., Zhang Y., Briggs S. D., Li J., Eissenberg J. C., Allis C. D., Khorasanizadeh S. (2001) Specificity of the HP1 chromo domain for the methylated N-terminus of histone H3. EMBO J. 20, 5232–5241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shi X., Hong T., Walter K. L., Ewalt M., Michishita E., Hung T., Carney D., Peña P., Lan F., Kaadige M. R., Lacoste N., Cayrou C., Davrazou F., Saha A., Cairns B. R., Ayer D. E., Kutateladze T. G., Shi Y., Côté J., Chua K. F., Gozani O. (2006) ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 442, 96–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hassan A. H., Awad S., Al-Natour Z., Othman S., Mustafa F., Rizvi T. A. (2007) Selective recognition of acetylated histones by bromodomains in transcriptional co-activators. Biochem. J. 402, 125–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hassan A. H., Prochasson P., Neely K. E., Galasinski S. C., Chandy M., Carrozza M. J., Workman J. L. (2002) Function and selectivity of bromodomains in anchoring chromatin-modifying complexes to promoter nucleosomes. Cell 111, 369–379 [DOI] [PubMed] [Google Scholar]
- 22. Winzeler E. A., Shoemaker D. D., Astromoff A., Liang H., Anderson K., Andre B., Bangham R., Benito R., Boeke J. D., Bussey H., Chu A. M., Connelly C., Davis K., Dietrich F., Dow S. W., El Bakkoury M., Foury F., Friend S. H., Gentalen E., Giaever G., Hegemann J. H., Jones T., Laub M., Liao H., Liebundguth N., Lockhart D. J., Lucau-Danila A., Lussier M., M'Rabet N., Menard P., Mittmann M., Pai C., Rebischung C., Revuelta J. L., Riles L., Roberts C. J., Ross-MacDonald P., Scherens B., Snyder M., Sookhai-Mahadeo S., Storms R. K., Véronneau S., Voet M., Volckaert G., Ward T. R., Wysocki R., Yen G. S., Yu K., Zimmermann K., Philippsen P., Johnston M., Davis R. W. (1999) Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285, 901–906 [DOI] [PubMed] [Google Scholar]
- 23. Swanson M. J., Qiu H., Sumibcay L., Krueger A., Kim S.-J., Natarajan K., Yoon S., Hinnebusch A. G. (2003) A Multiplicity of coactivators is required by Gcn4p at individual promoters in vivo. Mol. Cell. Biol. 23, 2800–2820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Longtine M. S., McKenzie A., 3rd, Demarini D. J., Shah N. G., Wach A., Brachat A., Philippsen P., Pringle J. R. (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961 [DOI] [PubMed] [Google Scholar]
- 25. Voth W. P., Jiang Y. W., Stillman D. J. (2003) New “marker swap” plasmids for converting selectable markers on budding yeast gene disruptions and plasmids. Yeast 20, 985–993 [DOI] [PubMed] [Google Scholar]
- 26. Ghaemmaghami S., Huh W. K., Bower K., Howson R. W., Belle A., Dephoure N., O'Shea E. K., Weissman J. S. (2003) Global analysis of protein expression in yeast. Nature 425, 737–741 [DOI] [PubMed] [Google Scholar]
- 27. Puig O., Caspary F., Rigaut G., Rutz B., Bouveret E., Bragado-Nilsson E., Wilm M., Séraphin B. (2001) The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods 24, 218–229 [DOI] [PubMed] [Google Scholar]
- 28. Zhang F., Sumibcay L., Hinnebusch A. G., Swanson M. J. (2004) A triad of subunits from the Gal11/tail domain of Srb mediator is an in vivo target of transcriptional activator Gcn4p. Mol. Cell. Biol. 24, 6871–6886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rasband W. S. (1997–2014) ImageJ, U. S. National Institutes of Health, Bethesda, MD [Google Scholar]
- 30. Reid G. A., Schatz G. (1982) Import of proteins into mitochondria: yeast cells grown in the presence of carbonyl cyanide m-chlorophenylhydrazone accumulate massive amounts of some mitochondrial precursor polypeptides. J. Biol. Chem. 257, 13056–13061 [PubMed] [Google Scholar]
- 31. Qiu H., Hu C., Hinnebusch A. G. (2009) Phosphorylation of the Pol II CTD by KIN28 enhances BUR1/BUR2 recruitment and Ser2 CTD phosphorylation near promoters. Mol. Cell 33, 752–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kim T., Buratowski S. (2007) Two Saccharomyces cerevisiae JmjC domain proteins demethylate histone H3 Lys-36 in transcribed regions to promote elongation. J. Biol. Chem. 282, 20827–20835 [DOI] [PubMed] [Google Scholar]
- 33. Mitchell L., Lambert J. P., Gerdes M., Al-Madhoun A. S., Skerjanc I. S., Figeys D., Baetz K. (2008) Functional dissection of the NuA4 histone acetyltransferase reveals its role as a genetic hub and that Eaf1 is essential for complex integrity. Mol. Cell. Biol. 28, 2244–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dong J., Lai R., Nielsen K., Fekete C. A., Qiu H., Hinnebusch A. G. (2004) The essential ATP-binding cassette protein RLI1 functions in translation by promoting preinitiation complex assembly. J. Biol. Chem. 279, 42157–42168 [DOI] [PubMed] [Google Scholar]
- 35. Li S., Shogren-Knaak M. A. (2009) The Gcn5 bromodomain of the SAGA complex facilitates cooperative and cross-tail acetylation of nucleosomes. J. Biol. Chem. 284, 9411–9417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nourani A., Doyon Y., Utley R. T., Allard S., Lane W. S., Côté J. (2001) Role of an ING1 growth regulator in transcriptional activation and targeted histone acetylation by the NuA4 complex. Mol. Cell. Biol. 21, 7629–7640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Clarke A. S., Lowell J. E., Jacobson S. J., Pillus L. (1999) Esa1p is an essential histone acetyltransferase required for cell cycle progression. Mol. Cell. Biol. 19, 2515–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Grant P. A., Duggan L., Côté J., Roberts S. M., Brownell J. E., Candau R., Ohba R., Owen-Hughes T., Allis C. D., Winston F., Berger S. L., Workman J. L. (1997) Yeast Gcn5 functions in two multisubunit complexes to acetylate nucleosomal histones: characterization of an Ada complex and the SAGA (Spt/Ada) complex. Genes Dev. 11, 1640–1650 [DOI] [PubMed] [Google Scholar]
- 39. Wu P. Y., Ruhlmann C., Winston F., Schultz P. (2004) Molecular architecture of the S. cerevisiae SAGA complex. Mol. Cell 15, 199–208 [DOI] [PubMed] [Google Scholar]
- 40. Lin Y. Y., Lu J. Y., Zhang J., Walter W., Dang W., Wan J., Tao S. C., Qian J., Zhao Y., Boeke J. D., Berger S. L., Zhu H. (2009) Protein acetylation microarray reveals that NuA4 controls key metabolic target regulating gluconeogenesis. Cell 136, 1073–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Brown C. E., Howe L., Sousa K., Alley S. C., Carrozza M. J., Tan S., Workman J. L. (2001) Recruitment of HAT complexes by direct activator interactions with the AATM-related Tra1 subunit. Science 292, 2333–2337 [DOI] [PubMed] [Google Scholar]
- 42. Natarajan K., Meyer M. R., Jackson B. M., Slade D., Roberts C., Hinnebusch A. G., Marton M. J. (2001) Transcriptional profiling shows that Gcn4p is a master regulator of gene expression during amino acid starvation in yeast. Mol. Cell. Biol. 21, 4347–4368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Krogan N. J., Dover J., Wood A., Schneider J., Heidt J., Boateng M. A., Dean K., Ryan O. W., Golshani A., Johnston M., Greenblatt J. F., Shilatifard A. (2003) The Paf1 complex is required for histone H3 methylation by COMPASS and Dot1p: linking transcriptional elongation to histone methylation. Mol. Cell 11, 721–729 [DOI] [PubMed] [Google Scholar]
- 44. Hwang W. W., Venkatasubrahmanyam S., Ianculescu A. G., Tong A., Boone C., Madhani H. D. (2003) A conserved RING finger protein required for histone H2B monoubiquitination and cell size control. Mol. Cell 11, 261–266 [DOI] [PubMed] [Google Scholar]
- 45. Carrozza M. J., Li B., Florens L., Suganuma T., Swanson S. K., Lee K. K., Shia W. J., Anderson S., Yates J., Washburn M. P., Workman J. L. (2005) Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123, 581–592 [DOI] [PubMed] [Google Scholar]
- 46. Govind C. K., Qiu H., Ginsburg D. S., Ruan C., Hofmeyer K., Hu C., Swaminathan V., Workman J. L., Li B., Hinnebusch A. G. (2010) Phosphorylated Pol II CTD recruits multiple HDACs, including Rpd3C(S), for methylation-dependent deacetylation of ORF nucleosomes. Mol. Cell 39, 234–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bian C., Xu C., Ruan J., Lee K. K., Burke T. L., Tempel W., Barsyte D., Li J., Wu M., Zhou B. O., Fleharty B. E., Paulson A., Allali-Hassani A., Zhou J. Q., Mer G., Grant P. A., Workman J. L., Zang J., Min J. (2011) Sgf29 binds histone H3K4me2/3 and is required for SAGA complex recruitment and histone H3 acetylation. EMBO J. 30, 2829–2842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vermeulen M., Eberl H. C., Matarese F., Marks H., Denissov S., Butter F., Lee K. K., Olsen J. V., Hyman A. A., Stunnenberg H. G., Mann M. (2010) Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell 142, 967–980 [DOI] [PubMed] [Google Scholar]
- 49. Pokholok D. K., Harbison C. T., Levine S., Cole M., Hannett N. M., Lee T. I., Bell G. W., Walker K., Rolfe P. A., Herbolsheimer E., Zeitlinger J., Lewitter F., Gifford D. K., Young R. A. (2005) Genome-wide map of nucleosome acetylation and methylation in yeast. Cell 122, 517–527 [DOI] [PubMed] [Google Scholar]
- 50. Rao B., Shibata Y., Strahl B. D., Lieb J. D. (2005) Dimethylation of histone H3 at lysine 36 demarcates regulatory and nonregulatory chromatin genome-wide. Mol. Cell. Biol. 25, 9447–9459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Biswas D., Takahata S., Stillman D. J. (2008) Different genetic functions for the Rpd3(L) and Rpd3(S) complexes suggest competition between NuA4 and Rpd3(S). Mol. Cell. Biol. 28, 4445–4458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Smolle M., Workman J. L. (2013) Transcription-associated histone modifications and cryptic transcription. Biochim. Biophys. Acta 1829, 84–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dai J., Hyland E. M., Yuan D. S., Huang H., Bader J. S., Boeke J. D. (2008) Probing nucleosome function: a highly versatile library of synthetic histone H3 and H4 mutants. Cell 134, 1066–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Qiu H., Hu C., Zhang F., Hwang G. J., Swanson M. J., Boonchird C., Hinnebusch A. G. (2005) Interdependent recruitment of SAGA and Srb mediator by transcriptional activator Gcn4p. Mol. Cell. Biol. 25, 3461–3474 [DOI] [PMC free article] [PubMed] [Google Scholar]