

Background: Altered sarco-endoplasmic reticulum Ca2+ ATPase 2b (SERCA2b) expression and activity contributes to β cell dysfunction in diabetes.
Results: SERCA2b deficiency occurs secondary to loss of pancreatic and duodenal homeobox 1 (Pdx-1)-mediated transcriptional regulation.
Conclusion: Pdx-1 maintains SERCA2b expression and endoplasmic reticulum (ER) calcium levels in the β cell.
Significance: These findings elucidate a novel pathway that contributes to β cell ER stress.
Keywords: β Cell, Calcium ATPase, Diabetes, Endoplasmic Reticulum Stress (ER Stress), Pancreatic Islet, Pancreatic And Duodenal Homeobox 1, Sarco-endoplasmic Reticulum Calcium ATPase
Abstract
Although the pancreatic duodenal homeobox 1 (Pdx-1) transcription factor is known to play an indispensable role in β cell development and secretory function, recent data also implicate Pdx-1 in the maintenance of endoplasmic reticulum (ER) health. The sarco-endoplasmic reticulum Ca2+ ATPase 2b (SERCA2b) pump maintains a steep Ca2+ gradient between the cytosol and ER lumen. In models of diabetes, our data demonstrated loss of β cell Pdx-1 that occurs in parallel with altered SERCA2b expression, whereas in silico analysis of the SERCA2b promoter revealed multiple putative Pdx-1 binding sites. We hypothesized that Pdx-1 loss under inflammatory and diabetic conditions leads to decreased SERCA2b levels and activity with concomitant alterations in ER health. To test this, siRNA-mediated knockdown of Pdx-1 was performed in INS-1 cells. The results revealed reduced SERCA2b expression and decreased ER Ca2+, which was measured using fluorescence lifetime imaging microscopy. Cotransfection of human Pdx-1 with a reporter fused to the human SERCA2 promoter increased luciferase activity 3- to 4-fold relative to an empty vector control, and direct binding of Pdx-1 to the proximal SERCA2 promoter was confirmed by chromatin immunoprecipitation. To determine whether restoration of SERCA2b could rescue ER stress induced by Pdx-1 loss, Pdx1+/− mice were fed a high-fat diet. Isolated islets demonstrated an increased spliced-to-total Xbp1 ratio, whereas SERCA2b overexpression reduced the Xbp1 ratio to that of wild-type controls. Together, these results identify SERCA2b as a novel transcriptional target of Pdx-1 and define a role for altered ER Ca2+ regulation in Pdx-1-deficient states.
Introduction
Diabetes mellitus is a worldwide epidemic affecting an estimated 285 million people, and the prevalence of this disorder is expected to rise to 439 million by the year 2030 (1). Altered pancreatic β cell function and survival are central components of the pathophysiology of type 1 (T1D)4 and type 2 diabetes (T2D). As secretory cells, β cells are dependent upon a highly developed endoplasmic reticulum (ER) to ensure that proteins are produced robustly and folded efficiently. Under conditions of metabolic stress, proteins may fail to fold correctly within the β cell ER lumen, initiating an unfolded protein response (UPR). The UPR limits the delivery of new proteins to the ER and, ultimately, increases protein folding capacity through increased transcription and activity of protein chaperones and foldases. However, if the inciting stress is unresolved, continual stimulation of the UPR leads to the activation of apoptotic pathways and β cell death, a transition referred to as ER stress (2–4). Data from rodent and human studies illustrate a pivotal role for β cell ER stress during the progression of both major forms of diabetes mellitus (5–9).
β Cell ER stress is induced in response to a number of insults, including hyperglycemia and downstream oxidative stress, increased saturated free fatty acids, and proinflammatory cytokines. One unifying consequence of these insults and accumulation of unfolded proteins is thought to be altered ER calcium (Ca2+) regulation (10). Under normal conditions, ER Ca2+ is estimated to be at least three orders of magnitude higher than cytosolic Ca2+, and Ca2+ within the ER lumen serves as a required cofactor for a number of steps involved in protein processing and maturation (11–14). The integrity of this gradient is actively maintained by the sarco-endoplasmic reticulum calcium ATPase (SERCA) pump, which transports two Ca2+ ions into the ER at the expense of one ATP molecule (15). At least 14 different SERCA isoforms have been identified to date (16, 17), and we have previously shown SERCA2b to be the most prevalent isoform expressed in the mouse pancreatic islet. We and others have also shown that β cell SERCA2b levels are diminished in human and rodent models of T1D and T2D (18–21), resulting in Ca2+ dysregulation, impaired insulin secretion, and ER stress (19). However, the specific transcriptional pathways that regulate SERCA2b expression in the β cell remain largely uncharacterized.
The pancreatic duodenal homeobox 1 (Pdx-1) transcription factor is known to play an indispensable role in β cell development and secretory function, and recent data also implicate Pdx-1 in the maintenance of ER health (22–27). Islets isolated from Pdx-1 haploinsufficient mice demonstrate ER stress when animals are exposed to a high-fat diet, and Pdx-1 has been shown to enhance the transcription of a number of proteins involved in UPR signaling and ER function, including activating transcription factor 4 (Atf4) and Wolframin (Wfs1) (9, 28). Furthermore, reductions in total Pdx-1 levels as well as impaired nuclear localization of Pdx-1 have been described under hyperglycemic and diabetic conditions (29–31). Interestingly, our in silico analysis of the human SERCA2b promoter revealed multiple putative binding sites for Pdx-1. Given an emerging role for Pdx-1 in the maintenance of an ER-specific subgenome, we hypothesized that Pdx-1 may serve as a transcriptional regulator of the SERCA2 gene and that loss of Pdx-1 may underlie decreased SERCA2b expression, altered Ca2+ regulation, and activation of β cell ER stress signaling pathways in diabetes.
EXPERIMENTAL PROCEDURES
Animals and Islet Preparations
Animals were maintained under protocols approved by the Indiana University Institutional Animal Care and Use Committee, the United States Department of Agriculture Animal Welfare Act (9 CFR, Parts 1, 2, and 3), and the Guide for the Care and Use of Laboratory Animals (32). C57BLKs/J-db/db mice and heterozygote littermate controls were obtained from The Jackson Laboratory (Bar Harbor, ME) at 12 weeks of age. Pdx-1 haploinsufficient mice on a mixed background were a gift from Chris Wright (Vanderbilt University School of Medicine, Nashville, TN). Male Pdx-1 haploinsufficient and wild-type littermates were weaned at 4 weeks of age and the following week, begun on either a normal chow diet (17% kcal from fat) or a high-fat diet (45% kcal from fat) for 8 weeks. Mouse cages were kept in a standard light-dark cycle with ad libitum access to food and water. Intraperitoneal glucose tolerance tests were performed at 11 weeks of age using a protocol described previously (33, 34). Blood glucose was measured using an AlphaTRAK glucometer from Abbott Laboratories (Abbott Park, IL). After 8 weeks of diet treatment, mouse pancreatic islets were isolated by collagenase digestion as described previously (35). Human islets isolated from cadaveric non-diabetic and diabetic donors were obtained from the National Disease Research Interchange or the Integrated Islet Distribution Program. On arrival, islets were immediately placed in DMEM containing 5.5 mm glucose, 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen) and incubated overnight at 37 °C with 5% CO2. Our analysis included islets from five non-diabetic donors and three donors with an established diagnosis of T2D. The average age of non-diabetic donors was 45.2 ± 6.1 years (S.E.), and the average body mass index was 27.6 ± 2.8 kg/m2. The average age of the donors with T2D was 53.0 ± 4.4 years, and the average body mass index was 22.7 ± 2.7 kg/m2.
Cell Culture and in Vitro Islet Treatment
INS-1 832/13 rat insulinoma cells were cultured as described previously (36, 37). NIH-3T3 immortalized mouse fibroblast cells were cultured at 37 °C and 5% CO2 in DMEM supplemented with 10% fetal bovine serum and 100 units/ml penicillin (38). For in vitro viral overexpression and knockdown experiments, viruses were replicated and purified using HEK-293T cells. For adenoviral transduction in cell lines, culture medium was replaced with fresh medium containing adenovirus-expressing mouse Pdx-1 (39), mouse SERCA2b (40), Pdx-1 siRNA (41), random siRNA (42), or a LacZ control virus, followed by overnight incubation. The anti-Pdx-1 siRNA sequence used was 5′-GAAAGAGGAAGATAAGAAA-3′, and the random siRNA sequence was 5′-GAGACCCTATCCGTGATTA-3′. After 24–48 h, RNA or protein was isolated for the indicated analyses, or cells were used for Ca2+ imaging. For viral transduction of isolated mouse islets, groups of 50–100 islets were hand-picked within 1 h of isolation, incubated with adenovirus for 24 h, and washed with PBS followed by Ca2+ imaging or RNA or protein isolation. To simulate hyperglycemic and proinflammatory conditions in vitro, INS-1 cells were treated with 25 mm glucose and 5 ng/ml IL-1β for 16–24 h using a protocol established previously (19).
Immunoblot Analysis
Isolated islets or cultured cells were washed with PBS and lysed with buffer containing 50 mm Tris (pH 8.0), 150 mm NaCl, 0.05% desoxycholate, 0.1% IGEPAL CA-630 (Sigma-Aldrich, St. Louis, MO), 0.1% SDS, 0.2% sarcosyl, 10% glycerol, 1 mm DTT, 1 mm EDTA, 10 mm NaF, EDTA-free protease inhibitors (Roche Applied Science), phosphatase inhibitors (Roche Applied Science), 2 mm MgCl2, and 0.05% v/v benzonase nuclease (Sigma-Aldrich). Lysate from isolated islets was further disrupted by mechanical shearing using a 20-gauge needle and syringe. Protein concentration was measured using the Bio-Rad DC protein assay (Bio-Rad) and a SpectraMax M5 multiwell plate reader (Molecular Devices, Sunnyvale, CA). Equal concentrations of proteins were suspended in 10% SDS solution prior to electrophoresis using a 4–20% Mini-Protean TGX gel in a Mini-Protean Tetra apparatus (Bio-Rad). Proteins were transferred to a PVDF membrane and blocked with Odyssey blocking buffer (LI-COR, Lincoln, NE) prior to incubation with primary antibodies. The primary antibodies used included the following: Pdx-1 (catalog no. 07-696 Millipore, Billerica, MA), SERCA2 (catalog no. sc-8095, Santa Cruz Biotechnology, Dallas, TX), Actin (catalog no. 691002, MP Biomed, Santa Ana, CA), and GAPDH (catalog no. ab9484, Abcam, Cambridge, UK). Of note is that the SERCA2 antibody does not distinguish between the SERCA2a and SERCA2b isoforms. However, we have shown previously that SERCA2a is expressed at very low levels in mouse islets (19). Immunoblots were scanned using a LI-COR Odyssey 1828 scanner and analyzed with LI-COR Image Studio software, with the densitometry of the scanned images calculated by ImageJ software (National Institutes of Health, Bethesda, MD).
Real-time quantitative RT-PCR
Cultured cells or isolated islets were processed for total RNA using RNeasy Mini plus or Micro plus kits (Qiagen, Valencia, CA), according to the instructions of the manufacturer and as described previously (43, 44). Total RNA was reverse-transcribed at 37 °C for 1 h using random hexamers, 0.5 mm dNTPs, 5× first-strand buffer, 0.01 mm DTT, and Moloney murine leukemia virus reverse transcriptase (all from Invitrogen). Quantitative RT-PCR was performed using either TaqMan proprietary primers (Invitrogen) or SYBR Green I dye and primer sequences and methods published previously(37)
Fura-2/AM Cytoplasmic Calcium Imaging
Intracellular cytosolic Ca2+ was measured using the ratiometric calcium indicator Fura-2/AM) from Invitrogen and a modification of methods published previously (37). INS-1 832/13 cells were seeded in a glass bottom 50-mm plate. After 24 h, cells were transduced with siPdx-1 or random siRNA adenovirus, and isolated mouse islets were transduced with SERCA2 or LacZ adenovirus as described. Prior to imaging, INS-1 cells and islets were incubated at 37 °C and 5% CO2 in 4 μm Fura-2/AM and 0.02% pluronic F127 (Invitrogen) for 1 h and then washed and incubated with Hanks' balanced salt solution (Invitrogen) supplemented with 0.1% BSA.
Islets were next transferred to a small volume chamber (Warner Instruments, Hamden, CT) mounted on the microscope stage. INS-1 cells and islets were perifused with a gradient pump (Bio-Rad) and maintained at 37 °C. For INS-1 cells, 10 mm caffeine or 1 μm thapsigargin were used to activate ryanodine receptors or inhibit SERCA activity, respectively. Fura-2/AM fluorescence was measured with excitation at 340 and 380 nm and emission at 510 nm. Images were captured using a Zeiss Z1 microscope with a ×10 or ×20 objective, and results were analyzed with Zen Blue software (Zeiss, Oberkochen, Germany).
Fluorescence Lifetime Imaging Microscopy (FLIM)
A D4ER adenovirus described previously was used for direct analysis of ER Ca2+ levels (45, 46). The probe was created by replacing the Ca2+ binding domain of the D1ER construct with D4 to provide lower Ca2+ affinity, with the new construct placed downstream of the rat insulin promoter. Briefly, INS-1 cells were cultured for 18 h with the D4ER adenovirus and siPdx-1 adenovirus or random siRNA adenovirus. Cells were then allowed to recover for 6 h. FLIM was carried out in accordance with a protocol published previously (47). In brief, the Alba FastFLIM system (ISS Inc., Champaign, IL) was coupled to an Olympus IX71 microscope using a ×60 water-immersion lens (Olympus, Tokyo, Japan). Confocal scanning was controlled by Build 143 VistaVision software (ISS Inc.) at 530/43 nm acceptor and 480/40 nm receptor wavelengths. Regions of interest were selected with >75 count averages. The lifetime determination was obtained by analyzing the first 11–12 modulation frequencies (10–120 MHz). Efficiency of FRET was estimated next using the following equation: EFRET = 1 − (τDA / τD), where τD and τDA are the donor fluorescence lifetime obtained in the absence and presence of the acceptor, respectively.
Luciferase Reporter Assays
Our previous publication utilized luciferase constructs incorporating different lengths of the human SERCA2 promoter (19), and the same constructs were used for this series of experiments. Approximately 2.0 × 104 NIH-3T3 mouse fibroblast cells were seeded in 12-well plates 24 h before transfection. Next, 100 μg of plasmid was transfected into cells using Metafectene Pro transfection reagent (Biotex, Munich, Germany) according to the instructions of the manufacturer. Total luminescence was measured using a luciferase assay system kit (Promega, Madison, WI) using a SpectraMax M5 plate reader (Molecular Devices). Luminosity results were normalized to total protein content as measured with the same BCA assay technique described for immunoblotting.
A luciferase construct with a deleted proximal Pdx-1 binding region was created by mutagenesis using the Stratagene QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA) according to the instructions of the manufacturer, and the site-specific deletion was confirmed by automated DNA sequencing (Genewiz, South Plainfield, NJ).
ChIP
Approximately 3.25 × 106 INS-1 cells were fixed in 1% formaldehyde for 10 min, sonicated to shear DNA fragments into the size range of 800–2000 bp and then subjected to ChIP as detailed previously (19, 48). Cross-linked protein and promoters were incubated for ∼35 h under nutation at 4 °C using anti-Pdx-1 antibody (Santa Cruz Biotechnology) with normal rabbit immunoglobulin G (Santa Cruz Biotechnology) as a control. Samples were quantitated in triplicate by SYBR Green I-based quantitative real-time PCR as described previously (44) using forward and reverse primer sequences for the rat SERCA2 promoter with the sequences 5′-CGCTTTTGGCTGTGTGGGAAG-3′ (forward) and 5′-TGGTGTCCTTGGCTTGCCTC-3′ (reverse) and the rat insulin 1 (INS1) promoter with the sequences 5′-TCAGCCAAAGATGAAGAAGGTCTC-3′ (forward) and 5′-GCATTTTCCACATCATTCCCC-3′ (reverse).
Statistical Analysis
Differences between groups were analyzed for significance using unpaired Student's t test, one-way analysis of variance with multiple comparisons and Tukey-Kramer post test, or multiple t tests with Sidak-Bonferroni correction (49), as calculated by GraphPad Prism 6.01 statistics software. p < 0.05 was considered to indicate a significant difference between groups.
RESULTS
Pdx-1 and SERCA2b Levels Are Decreased in Parallel in the β Cell under Diabetic Stress Conditions
Our previous work has shown significantly decreased expression of SERCA2b mRNA and protein in islets isolated from C57BLKs/J-db/db (db/db) mice that worsened with advancing age and increasing hyperglycemia (19, 37). To define the relationship between both Pdx-1 and SERCA2b expression in this model, islets were isolated from 12-week-old db/db mice and heterozygote littermate controls, and Pdx-1 and SERCA2b protein and mRNA were found to be decreased in parallel (Fig. 1, A and B). Previous work has also demonstrated altered SERCA2b expression in INS-1 832/13 rat insulinoma cells treated with 25 mm glucose (high glucose, or HG) combined with 5 ng/ml of IL-1β, a model that is intended to mimic the proinflammatory milieu of diabetes (19).
FIGURE 1.

Pdx-1 and SERCA2b levels are decreased in parallel in the β cell under diabetic stress conditions. A and B, protein and RNA were isolated from 12-week-old C57BLKs/J-db/db (db/db) and littermate heterozygous control (db+) mouse islets. Immunoblot analysis was performed using antibodies against SERCA2, Pdx-1, and actin, and reverse-transcribed RNA was subjected to real-time quantitative RT-PCR to measure SERCA2b and Pdx-1 transcript levels. C–E, INS-1 832/13 rat insulinoma cells were treated with 25 mm glucose and 5 ng/ml IL-1β (HG + IL-1β) for 16 and 24 h. C, immunoblot analysis was performed using antibodies against SERCA2, Pdx-1, and actin. D, quantitative protein levels are shown graphically. E, reverse-transcribed RNA was subjected to real-time PCR for quantification of SERCA2b, Pdx-1, preinsulin, and GAPDH transcript levels. F, Pdx-1 and SERCA2b mRNA levels in cadaveric human islets were graphed using a best fit line. The indicated comparisons are significantly different. *, p < 0.05; **, p < 0.01 (n = at least 6, except in A, where n = 2 samples of 100 islets from three biological replicates). Results are displayed as mean ± S.E.
Using this model, time course experiments were performed. At both 16 and 24 h post-HG + IL-1β treatment, Pdx-1 and SERCA2b protein levels were decreased coordinately (Fig. 1, C and D), whereas reductions in Pdx-1 mRNA levels preceded the loss of SERCA2 mRNA (Fig. 1E). Because the insulin gene is a known transcriptional target of Pdx-1 (50), measurement of preinsulin mRNA was measured as a positive control to confirm the transcriptional effects of Pdx-1 loss. As anticipated, decreased preinsulin mRNA expression was observed with HG + IL-1β treatment. In contrast, GAPDH has not been described to be a Pdx-1 target (51). Therefore, GAPDH mRNA was measured as a negative control to rule out nonspecific changes in gene expression under inflammatory conditions. No change in GAPDH expression was observed with HG + IL-1β treatment (Fig. 1E).
Levels of SERCA2b and Pdx-1 mRNA were next quantitated in cadaveric human islets from a cohort that included donors with no previous diagnosis of T2D (n = 5) and three donors with established T2D. Levels of SERCA2b and Pdx-1 mRNA were graphed as a line, with the x axis corresponding to SERCA2b and the y axis corresponding to Pdx-1 levels, respectively. A significant linear relationship (p = 0.038) was observed with a slope of 2.760 ± 1.044 and a coefficient of determination (R2) value of 0.5380, suggesting a positive correlation between Pdx-1 and SERCA2b levels of expression in cadaveric human islets from diabetic and non-diabetic donors (Fig. 1F).
SERCA2b Is Decreased in Islets Isolated from Pdx-1 Haploinsufficient Mice
Homozygous deletion of Pdx-1 leads to pancreatic agenesis and perinatal death (25). As such, Pdx-1 haploinsufficient mice were used to study the in vivo relationship between Pdx-1 and SERCA2b. Pdx-1 haploinsufficient mice and wild-type littermates were fed a normal chow diet containing 17% of calories from fat. At 11 weeks of age, intraperitoneal glucose tolerance tests were performed. As observed previously by other groups, Pdx-1 haploinsufficient mice were glucose-intolerant compared with wild-type littermates (Fig. 2, A and B) (28, 52–54). Similar to results obtained in db/db islets and HG + IL-1β-treated INS-1 cells, SERCA2b protein and mRNA were decreased significantly in islets isolated from Pdx-1 haploinsufficient mice compared with controls (Fig. 2, C–E).
FIGURE 2.

SERCA2b is decreased in islets isolated from Pdx-1 haploinsufficient mice. Pdx-1 haploinsufficient mice (Pdx-1+/−) and wild-type littermates (WT) were fed a normal chow diet containing 17% of kilocalories from fat. A and B, intraperitoneal glucose tolerance tests (IPGTTs) were performed at 11 weeks of age in Pdx-1+/− and WT mice, and the area under the curve analysis is shown graphically. C–E, protein and RNA were isolated from 13-week-old Pdx-1 haploinsufficient and WT littermate control mouse islets. C, immunoblot analysis was performed using antibodies against SERCA2, Pdx-1, and GAPDH. D, quantitative protein levels are shown graphically. E, reverse-transcribed islet RNA was subjected to real-time quantitative RT-PCR for quantification of SERCA2b and Pdx-1. The indicated comparisons are significantly different. *, p < 0.05; **, p < 0.01 (n = 3–10 for the intraperitoneal glucose tolerance test and n = at least 4 for immunoblot and quantitative RT-PCR). Results are displayed as mean ± S.E.
Pdx-1 Overexpression Increases SERCA2 Expression, whereas Pdx-1 Knockdown Decreases SERCA2b Levels in Vitro
NIH-3T3 mouse fibroblast cells express SERCA2b but do not natively express Pdx-1. To test the relationship between Pdx-1 and SERCA2b expression directly, NIH-3T3 fibroblasts were transduced with Pdx-1 adenovirus, and the change in SERCA2 expression was assessed. Pdx-1 overexpression in NIH-3T3 cells resulted in a nearly 2-fold increase in SERCA2 protein levels (Fig. 3, A and B). Next, adenoviral siRNA-mediated knockdown of Pdx-1 was performed in INS-1 832/13 cells that express Pdx-1 at high levels under normal conditions (55, 56). In cells treated with siRNA, Pdx-1 levels were reduced by ∼90%, resulting in a 50% decrease in SERCA2b protein and mRNA expression (Fig. 3, C–E). Taken together, these results demonstrate that islet Pdx-1 and SERCA2b levels are altered in parallel in rodent models of diabetes, whereas direct modulation of Pdx-1 levels using overexpression and knockdown strategies results in reciprocal changes in SERCA2b expression.
FIGURE 3.

Pdx-1 overexpression increased SERCA2 expression, whereas Pdx-1 knockdown decreased SERCA2b levels in vitro. A and B, Pdx-1 was overexpressed via adenoviral transduction in NIH-3T3 mouse fibroblast cells that lack native expression of Pdx-1, and immunoblot analysis was performed using antibodies against SERCA2, Pdx-1, and actin. C–E, INS-1 cells were transduced with an adenovirus that expressed siRNA against Pdx-1. C, immunoblot analysis was performed using antibodies against SERCA2, Pdx-1, and actin. D, quantitative protein levels are shown graphically. E, RNA was subjected to real-time quantitative RT-PCR for quantification of SERCA2b and Pdx-1 transcript levels. The indicated comparisons are significantly different. *, p < 0.05; **, p < 0.01 (n = at least 4). Results are displayed as mean ± S.E.
Pdx-1 Knockdown Alters β Cell Calcium Homeostasis
The anticipated effect of diminished SERCA2b expression or activity is a reduction in ER Ca2+. To directly assess the impact of Pdx-1 loss on ER Ca2+ levels, INS-1 cells were transduced with an ER-targeted D4ER adenovirus in combination with an siPdx-1 or random siRNA adenovirus. FLIM experiments were then performed as outlined under “Experimental Procedures.” Using this strategy, an increase in the lifetime of the enhanced cyan fluorescent protein (ECFP) donor indicates less FRET efficiency and, therefore, lower ER Ca2+.
Representative micrographs of each treatment group are shown in Fig. 4A. ECFP donor lifetime increased significantly from 1.72 ± 0.02 ns under control conditions to 1.83 ± 0.02 ns with Pdx-1 knockdown, consistent with reduced ER Ca2+ in the setting of Pdx-1 deficiency (Fig. 4, A and B). In separate experiments, random siRNA-treated INS-1 cells were treated with the SERCA inhibitor thapsigargin. As expected, within 2 min, thapsigargin treatment resulted in a significantly increased donor life time, and changes were comparable to those observed with Pdx-1 knockdown (Fig. 4C).
FIGURE 4.

Pdx-1 knockdown reduced β cell ER calcium levels. INS-1 cells were transduced with a D4ER Ca2+ reporter adenovirus in combination with adenovirus-expressing siPdx-1 or random siRNA. FLIM was used to measure endoplasmic reticulum Ca2+. A, representative micrograph of the average donor lifetime in random siRNA and Pdx-1 siRNA-treated cells. B, amplitude-weighted donor fluorescence lifetime in random siRNA and siPdx-1-treated INS-1 cells was quantified as described under “Experimental Procedures.” C, amplitude-weighted donor fluorescence lifetime in random siRNA-treated INS-1 cells was quantitated at baseline and after 2 min of treatment with thapsigargin (n = at least 26 regions of interest quantitated per treatment in three independent experiments for B and C). Results are displayed as mean ± S.E. The indicated comparisons are significantly different. **, p < 0.01).
Next, INS-1 cells that had been transduced with siPdx-1 adenovirus or control siRNA were incubated with Fura-2/AM Ca2+ dye to measure cytosolic Ca2+ levels. Fura-2/AM imaging revealed that Pdx-1 knockdown significantly increased basal Ca2+ levels within the cytosolic compartment (Fig. 5A). To assess changes in Ca2+ transit following ER Ca2+ depletion, cells were next treated with either caffeine or thapsigargin. Changes in cytosolic Ca2+ were analyzed by calculating the change in the Fura-2/AM ratio between the peak and basal measurements (ΔF) and dividing by the average of the basal ratio measured over 30 s (F0) according to the formula ΔF/F0, as indicated in Fig. 5B. Indeed, rapid Ca2+ release following caffeine and thapsigargin treatment was detected (Fig. 5, C and E). Notably, the ΔF/F0 was significantly lower in Pdx-1 siRNA-transduced cells (Fig. 5, D and F). Together, FLIM and Fura-2/AM imaging experiments suggest that loss of Pdx-1 alters β cell Ca2+ compartmentalization, leading to decreased ER Ca2+, increased basal cytosolic Ca2+ levels, and decreased Ca2+ transit following ER store depletion.
FIGURE 5.

Pdx-1 knockdown altered β cell calcium homeostasis. A, to assess basal cytosolic Ca2+ levels, the Fura-2/AM fluorescence ratio was measured as described under “Experimental Procedures” in untreated INS-1 cells and INS-1 cells transduced with adenovirus-expressing siPdx-1 or random siRNA. B–F, siPdx-1 or random siRNA-transduced INS-1 cells were treated with 10 mm caffeine or 1 μm thapsigargin to determine the effect of Pdx-1 knockdown on ER Ca2+ storage and cytosolic Ca2+ transit following ER Ca2+ depletion. B, data are described as a normalized ratio according to the formula ΔF/F0. C and E, representative Fura-2/AM Ca2+ recording with caffeine (C) and thapsigargin (E) treatment in siPdx-1- or random siRNA-transduced INS-1 cells. The calculated ΔF/F0 for each group is shown graphically in D and F. The Fura-2/AM ratio was collected from at least 120 regions of interest over three independent experiments. Results are displayed as mean ± S.E., and the indicated comparisons are significantly different. **, p < 0.01.
Pdx-1 Enhances SERCA2 Promoter Activity
Pdx-1 is known to bind to TA-rich sequences, including TAAT, ATTA, and TAAAT, in the promoters of target genes (41, 57). In silico analysis demonstrated five putative Pdx-1 binding regions in the SERCA2 promoter (Fig. 6A). To determine whether Pdx-1 is a transcriptional regulator of the SERCA2 gene, reporter assays were undertaken using different lengths of the human promoter fused to the luciferase coding region. NIH-3T3 cells were cotransfected with SERCA2 promoter constructs and a human Pdx-1 plasmid. Luciferase activity was measured 24 h after transfection and normalized to total protein content. Cotransfection of Pdx-1 increased luciferase expression 3- to 4-fold over the empty vector control in all constructs tested (Fig. 6B), suggesting that the binding region closest to the transcriptional start site might serve as a key regulatory region for Pdx-1-mediated transcriptional regulation of the SERCA2 gene. This region of the SERCA2 promoter maintains close homology between several species of mammals, including human, mouse, and rat (Fig. 6C). To confirm these findings, luciferase assays were performed following site-directed mutagenesis of this most proximal binding region. Eight base pairs were deleted, as depicted in the schematic in Fig. 6C. No increase in luciferase activity was observed using this mutant construct, suggesting that deletion of this element in the proximal promoter was sufficient to block Pdx-1 mediated transcriptional activation of the SERCA2 promoter (Fig. 6D).
FIGURE 6.

Pdx-1 enhances SERCA2 promoter activity. A, five putative Pdx-1 binding regions were identified in the human SERCA2 promoter. B, NIH-3T3 mouse fibroblast cells were cotransfected with deletion constructs of the human SERCA2 promoter placed upstream of a luciferase (Luc) coding region and also transfected with human Pdx-1 or empty vector control plasmids. Reporter assays were performed as outlined under “Experimental Procedures.” C, depiction of the mutation strategy undertaken to delete the most proximal Pdx-1 binding element. Homology between human, rat, and mouse promoters is indicated. D, NIH-3T3 cells were cotransfected with wild-type or mutated constructs combined with the Pdx-1 plasmid or the empty vector control, and reporter assays were performed (n = at least 5 independent experiments with samples analyzed in triplicate). Results are displayed as mean ± S.E. The indicated comparisons are significantly different. **, p < 0.01; n.s., not significant.
Pdx-1 Directly Binds the Proximal SERCA2 Promoter
For in vivo confirmation, ChIP experiments were performed using whole cell extract isolated from INS-1 cells. Results showed a 2-fold increase in recovery of the proximal SERCA2 promoter following immunoprecipitation with anti-Pdx-1 antibody compared with immunoprecipitation with rabbit IgG (Fig. 7A). Because the INS1 gene is known to be a transcriptional target of Pdx-1 (50), recovery of the INS1 promoter was quantitated to confirm successful pulldown, and a 3-fold increase in INS1 promoter recovery was observed. In aggregate, results from luciferase and ChIP experiments identify SERCA2 as a novel transcriptional target of Pdx-1 in the β cell.
FIGURE 7.
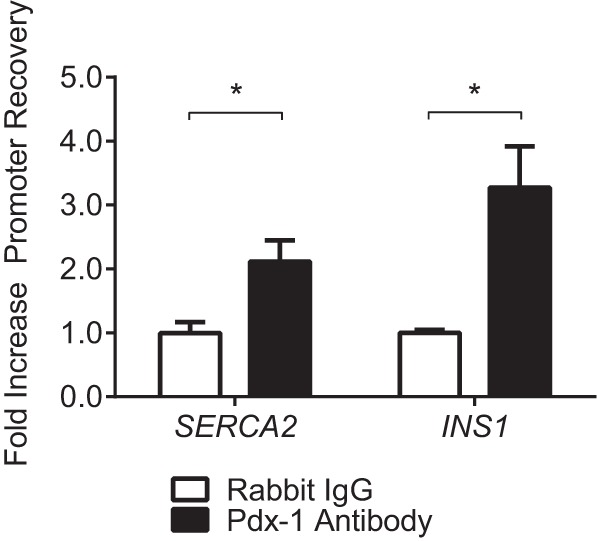
Pdx-1 directly binds the proximal SERCA2 promoter. INS-1 cells were harvested and subjected to ChIP analysis. Quantitative PCR was used to measure recovery of the SERCA2 or INS1 promoters following immunoprecipitation with anti-Pdx-1 antibody or normal rabbit IgG. Results are expressed as fold increase in percent recovery of the target gene compared with rabbit IgG and represent the mean ± S.E. for three independent experiments. The indicated comparisons are significantly different. *, p < 0.05.
Reconstitution of SERCA2 Expression Ameliorates ER Stress in Islets Isolated from Pdx-1 Haploinsufficient Mice Fed a High-fat Diet
Results from Fig. 2 demonstrated decreased SERCA2b levels in islets isolated from Pdx-1+/− mice fed a normal diet (Fig. 2, B–D). A previous publication by Sachdeva et al. (28) revealed the presence of ER stress in islets isolated from Pdx-1 haploinsufficient mice challenged with a high-fat diet.
SERCA2b and Pdx-1 mRNA levels were measured in islets isolated from wild-type mice fed a normal chow or diet containing 45% of calories from fat (HFD) for 8 weeks. No significant difference in SERCA2b or Pdx-1 gene expression was observed between wild-type mice fed either diet (Fig. 8A). Next, Pdx-1 haploinsufficient mice and wild-type mice were fed an HFD for 8 weeks. SERCA2b levels were again found to be significantly lower in Pdx-1+/− mice (Fig. 8, B–D). Interestingly, compared with Pdx-1+/− mice fed normal chow (Fig. 2, C and D), a further reduction in SERCA2b and Pdx-1 mRNA levels was observed (Figs. 8, B—D, and 2, C and D). Consistent with the activation of ER stress signaling, the spliced-to-total X-box binding protein 1 (Xbp1) ratio was increased in islets from Pdx-1+/− mice fed an HFD compared with WT littermate controls (Fig. 8E).
FIGURE 8.

Reconstitution of SERCA2 expression ameliorates ER stress in islets isolated from Pdx-1 haploinsufficient mice fed a high-fat diet. Pdx-1 haploinsufficient (Pdx-1+/−) mice or wild-type littermates were fed normal chow (NC) or an HFD containing 45% kilocalories from fat for 8 weeks prior to islet isolation. A, real-time RT-PCR was used to quantitate Pdx-1 and SERCA2b transcript levels in islets isolated from WT littermate control mice fed NC or an HFD. B–E, protein and RNA were isolated from 13-week-old Pdx-1+/− and WT littermate control islets. B, immunoblot analysis was performed using antibodies against SERCA2, Pdx-1, and GAPDH. C, quantitative protein levels are shown graphically. D–E, reverse-transcribed RNA was subjected to real-time PCR for quantification of Pdx-1, SERCA2b, and total and spliced Xbp1 transcript levels. The ratio of spliced-to-total Xbp1 is indicated in D. E, immunoblot analysis demonstrating successful adenoviral overexpression of SERCA2b in islets isolated from Pdx-1+/− mice and WT littermate controls fed an HFD. G, quantification of the ratio of spliced to unspliced Xbp1 mRNA in isolated islets from Pdx-1+/− and WT mice fed an HFD and treated ex vivo with no virus or transduced with SERCA2b adenovirus or LacZ-expressing control virus (n = 12–18). The results are displayed as mean ± S.E. The indicated comparisons are significantly different. *, p < 0.05; **, p < 0.01; n.s., not significant.
To determine whether reconstitution of islet SERCA2b in this model was sufficient to rescue components of ER stress or Ca2+ signaling, isolated islets from both HFD-fed Pdx-1+/− mice and HFD-fed WT controls were transduced with SERCA2b adenovirus or a LacZ control adenovirus. Successful transduction was verified by immunoblot analysis (Fig. 8F). Notably, SERCA2b reconstitution in HFD-Pdx-1+/− islets significantly decreased the spliced Xbp1 ratio (Fig. 8G).
Finally, to assess overall glucose-stimulated Ca2+ responses, Fura-2/AM imaging was performed in HFD-Pdx-1+/− islets that had been transduced with either SERCA2b or a LacZ control adenovirus. Interestingly, in response to 20 mm glucose, the ΔF/F0 ratio was increased significantly in HFD-Pdx-1+/− islets transduced with SERCA2b, compared with LacZ control HFD-Pdx-1+/− islets (Fig. 9, A and B).
FIGURE 9.
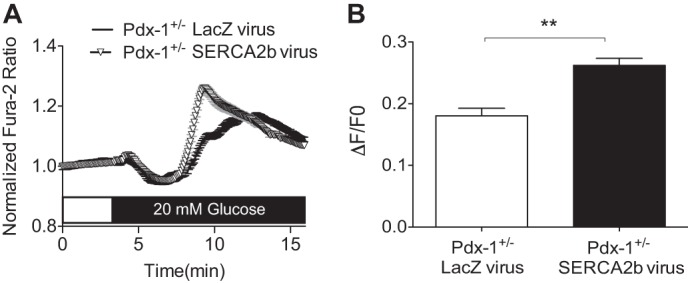
SERCA2b overexpression leads to an improved glucose-stimulated Ca2+ response in islets from Pdx-1 haploinsufficient mice fed a high-fat diet. Islets isolated from Pdx-1+/− mice fed a high fat diet for 8 weeks were transduced ex vivo with either SERCA2b or LacZ-expressing adenovirus. A, representative Fura-2/AM Ca2+ imaging recording performed at 2.5 mm glucose and following stimulation with 20 mm glucose. B, data were analyzed according to the formula ΔF/F0, and results are shown graphically. The Fura-2/AM ratio was measured from at least two regions of interest per islet, and islets were isolated from at least three biological replicates. The results are displayed as the means ± S.E. The indicated comparisons are significantly different. **, p < 0.01.
DISCUSSION
A single β cell synthesizes approximately 1 million insulin molecules/min, an arduous task that requires an extremely well developed and highly functional ER (58). ER stress in the pancreatic β cell is well appreciated in the context of obesity and type 2 diabetes (59, 60), whereas more recent and emerging data suggest an expanding role for ER stress in the development and progression of T1D (5, 8, 9). A key determinant of ER homeostasis is the maintenance of a robust intraluminal Ca2+ pool (13, 14), and this pool serves as an important store for Ca2+ release, leading to activation of a variety of signaling pathways, including incretin-induced insulin secretion (61, 62). Moreover, ER Ca2+ plays a central role in protein processing and maturation through the regulation of protein chaperone activity and the formation of chaperone complexes (13), whereas depletion of ER Ca2+ inhibits protein synthesis and facilitates protein degradation (63, 64). Studies also illustrate a strict requirement for ER-derived Ca2+ in proinsulin processing in secretory granules, where Ca2+ is needed for activity of the endopeptidases prohormone convertases 1 and 2 (65, 66).
The SERCA family of ion pumps serves as a primary gatekeeper of this ER calcium gradient. At least three SERCA isoforms are known to be expressed in the pancreatic β cell: SERCA2a, SERCA2b, and SERCA3, and expression of SERCA2 and SERCA3 isoforms has been shown to be regulated independently in the β cell (67). Although we previously observed decreased expression of all three isoforms in islets isolated from diabetic rodents (37), this series of experiments was focused on SERCA2b because it is the most highly expressed isoform in mouse islets (19). Furthermore, SERCA2b is structurally unique because it possesses an extra transmembrane domain providing this isoform with the highest relative calcium affinity (68). Our previous work has shown that altered SERCA2b expression leads to altered insulin secretion, activation of ER stress signaling pathways, and decreased β cell survival (19). The goal of this work was to identify additional transcriptional pathways that underlie dysregulated SERCA2b expression under inflammatory and diabetic conditions.
The homeobox protein Pdx-1 plays an essential role in pancreatic and β cell development and the maintenance of postnatal β cell function. Importantly, Pdx-1 is a transcriptional regulator of the insulin gene as well as other key genes involved in stimulus-secretion coupling (25, 54, 69). Pdx-1 is also recognized to play a role in β cell adaptation to metabolic stress, and Pdx-1 haploinsufficiency superimposed on a background of severe insulin resistance leads to impaired compensatory β cell mass expansion, diabetes, and premature mortality (54). Sachdeva et al. (28) recently examined the effects of diet-induced obesity in Pdx-1+/− mice and similarly found that Pdx-1 deficiency significantly limits β cell mass expansion under HFD conditions. Interestingly, these effects were not secondary to decreased proliferation but, rather, due to impaired β cell survival. Similarly, increased β cell apoptosis in Pdx-1+/− islets has also been observed by Johnson et al. (53). Interestingly, islets from HFD-fed Pdx-1+/− mice demonstrated activation of specific components of ER stress signaling, whereas similar results were seen in Pdx-1-deficient MIN6 cells. Microarray studies performed in MIN6 cells transduced with an shRNA against Pdx-1 further revealed alterations in a large subset of genes with well defined roles in the maintenance of ER function and UPR signaling, including Atf4, Wfs1, ER oxidoreductin 1b, heat shock 70-kDa protein 5/Bip/GRP78, and neuronatin. Pdx-1 was confirmed to directly bind the promoters of Atf4 and Wfs1, further confirming its role in maintenance of a specific β cell ER subgenome (28).
In addition to key ER-related genes identified previously, our results suggest that altered SERCA2b expression with concomitant alterations in ER Ca2+ also contributes to ER stress observed in Pdx-1-deficient states. Here we show that Pdx-1 and SERCA2b expression are altered in parallel in db/db islets and in an in vitro model of inflammatory diabetes. Moreover, we demonstrate a significant and positive correlation between Pdx-1 and SERCA2b mRNA levels in human islets from subjects with and without T2D. To test further whether Pdx-1 directly regulates SERCA2b expression, overexpression and knockdown strategies were employed in NIH-3T3 and INS-1 cells, respectively. The results showed that Pdx-1 overexpression increases SERCA2b levels, whereas siRNA-mediated knockdown of Pdx-1 leads to a reciprocal reduction in SERCA2b expression. Luciferase assays and ChIP assays confirm that this relationship is direct and show that Pdx-1 binds a proximal region of the human SERCA2 promoter. Importantly, we recapitulated the stress paradigm employed by Sachdeva et al. (28) by treating Pdx-1+/− mice with an HFD with the goal of testing whether reconstitution of SERCA2b in islets ex vivo is sufficient to limit ER stress. Indeed, using the spliced-to-total Xbp1 ratio as a readout, we show that restoration of SERCA2b expression is capable of mitigating against ER stress in this model. Furthermore, we also show that restoration of SERCA2b improved the overall glucose-stimulated Ca2+ response in Pdx-1+/− islets from HFD-fed mice.
A potential limitation of our study is that, aside from changes in the spliced Xbp1 ratio following SERCA2b reconstitution in islets from HFD-fed Pdx-1+/− mice, we did not observe significant changes in mRNA expression of other markers of ER stress (data not shown). Although future studies will be needed to fully investigate the specific signaling arms regulated by this pathway, a novel aspect of our study is that we link modulation of Pdx-1 expression with alterations in β cell Ca2+ homeostasis. Specifically, using a genetically encoded and ER-localized cameleon reporter (45), our results show that ER Ca2+ is decreased and cytosolic Ca2+ is increased in a Pdx-1-deficient state. Although our proposed model would suggest that these changes are the result of alterations in SERCA2b expression and activity, previous literature has shown that Pdx-1 also enhances transcription of the Wfs1 gene. WFS1 is a transmembrane protein localized to the β cell ER, plasma membrane, and secretory granules (70, 71), and mutations in this gene lead to Wolfram syndrome, a disorder characterized by childhood onset of diabetes mellitus, hypoinsulinemia, diabetes insipidus, optic atrophy, and deafness in humans (72, 73). Furthermore, mouse models of diminished WFS1 also exhibit β cell apoptosis because of ER stress (74, 75). Although the precise function of WFS1 in the β cell has remained somewhat enigmatic, the function of this protein has been linked to the regulation of ER Ca2+, raising the possibility that a component of our ER Ca2+ phenotype could be secondary to WFS1 deficiency (76). In addition, we observed an increase in basal cytosolic Ca2+ levels with Pdx-1 knockdown, which could reflect a reciprocal increase in cytosolic Ca2+ resulting directly from a ER Ca2+ leak. Although not directly interrogated in our study, another possibility is that the activation of store-operated Ca2+ entry from the extracellular space also contributed to this finding (77).
Despite these caveats, our results identify SERCA2b as a new transcriptional target of Pdx-1 and identify an additional pathway through which SERCA2b expression is altered in the β cell under diabetic conditions. Moreover, we define a novel role for altered ER Ca2+ regulation in Pdx-1-deficient states.
Acknowledgments
We thank Richard N. Day for assistance with the FLIM experiments and Emily K. Sims for critical review of the manuscript. We also thank Umut Ozcan and Patrick Fueger for the SERCA2b and siRNA Pdx-1 adenoviruses, respectively, and Chris Wright for Pdx-1 haploinsufficient mice. Human islets were obtained through the NIH IIDP or from NDRI, and we thank the organ donors who participate in these programs.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 DK093954 (to C. E. M.), RO1 DK46409 (to L. S. S.), and K01 DK101683 (to M. J. M.) and by an NIDDK, National Institutes of Health research supplement to promote diversity in health-related research (to J. S. J.). This work was also supported by Veterans Affairs Merit Award I01BX001733 (to C. E. M.) and by gifts from the Sigma Beta Sorority, the Ball Brothers Foundation, and the George and Frances Ball Foundation (to C. E. M.).
- T1D
- type 1 diabetes
- T2D
- type 2 diabetes
- ER
- endoplasmic reticulum
- UPR
- unfolded protein response
- SERCA
- sarco-endoplasmic reticulum Ca2+ ATPase
- FLIM
- fluorescence lifetime imaging microscopy
- HG
- high glucose
- HFD
- high-fat diet.
REFERENCES
- 1. Shaw J. E., Sicree R. A., Zimmet P. Z. (2010) Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res. Clin. Pract. 87, 4–14 [DOI] [PubMed] [Google Scholar]
- 2. Evans-Molina C., Hatanaka M., Mirmira R. (2013) Lost in translation: endoplasmic reticulum stress and the decline of β-cell health in diabetes mellitus. Diabetes Obes. Metab. 15, 159–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cnop M., Foufelle F., Velloso L. A. (2012) Endoplasmic reticulum stress, obesity and diabetes. Trends Mol. Med. 18, 59–68 [DOI] [PubMed] [Google Scholar]
- 4. Mihailidou C., Papavassiliou A. G., Kiaris H. (2014) A crosstalk between p21 and UPR-induced transcription factor C/EBP homologous protein (CHOP) linked to type 2 diabetes. Biochimie 99, 19–27 [DOI] [PubMed] [Google Scholar]
- 5. Eizirik D. L., Cardozo A. K., Cnop M. (2008) The role for endoplasmic reticulum stress in diabetes mellitus. Endocrine Rev. 29, 42–61 [DOI] [PubMed] [Google Scholar]
- 6. Back S. H., Kaufman R. J. (2012) Endoplasmic reticulum stress and type 2 diabetes. Annu. Rev. Biochem. 81, 767–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Engin F., Nguyen T., Yermalovich A., Hotamisligil G. S. (2014) Aberrant islet unfolded protein response in type 2 diabetes. Scientific Reports 4, 4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eizirik D. L., Miani M., Cardozo A. K. (2013) Signalling danger: endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. Diabetologia 56, 234–241 [DOI] [PubMed] [Google Scholar]
- 9. Tersey S. A., Nishiki Y., Templin A. T., Cabrera S. M., Stull N. D., Colvin S. C., Evans-Molina C., Rickus J. L., Maier B., Mirmira R. G. (2012) Islet β-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes 61, 818–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bánhegyi G., Baumeister P., Benedetti A., Dong D., Fu Y., Lee A. S., Li J., Mao C., Margittai E., Ni M., Paschen W., Piccirella S., Senesi S., Sitia R., Wang M., Yang W. (2007) Endoplasmic reticulum stress. Ann. N.Y. Acad. Sci. 1113, 58–71 [DOI] [PubMed] [Google Scholar]
- 11. Meldolesi J., Pozzan T. (1998) The endoplasmic reticulum Ca2+ store: a view from the lumen. Trends Biochem. Sci. 23, 10–14 [DOI] [PubMed] [Google Scholar]
- 12. Montero M., Brini M., Marsault R., Alvarez J., Sitia R., Pozzan T., Rizzuto R. (1995) Monitoring dynamic changes in free Ca2+ concentration in the endoplasmic reticulum of intact cells. EMBO J. 14, 5467–5475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Michalak M., Robert Parker J. M., Opas M. (2002) Ca2+ signaling and calcium binding chaperones of the endoplasmic reticulum. Cell Calcium 32, 269–278 [DOI] [PubMed] [Google Scholar]
- 14. Coe H., Michalak M. (2009) Calcium binding chaperones of the endoplasmic reticulum. Gen. Physiol. Biophys. 28, F96–F103 [PubMed] [Google Scholar]
- 15. Vandecaetsbeek I., Vangheluwe P., Raeymaekers L., Wuytack F., Vanoevelen J. (2011) The Ca2+ pumps of the endoplasmic reticulum and Golgi apparatus. Cold Spring Harb. Perspect. Biol. 3, a004184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Altshuler I., Vaillant J. J., Xu S., Cristescu M. E. (2012) The evolutionary history of sarco(endo)plasmic calcium ATPase (SERCA). PloS ONE 7, e52617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chemaly E. R., Bobe R., Adnot S., Hajjar R. J., Lipskaia L. (2013) Sarco (endo) plasmic reticulum calcium ATPases (SERCA) isoforms in the normal and diseased cardiac, vascular and skeletal muscle. J. Cardiovasc. Dis. Diagn. 1, 113 [Google Scholar]
- 18. Yang L., Ji W., Xue Y., Chen L. (2013) Imaging β-cell mass and function in situ and in vivo. J. Mol. Med. 91, 929–938 [DOI] [PubMed] [Google Scholar]
- 19. Kono T., Ahn G., Moss D. R., Gann L., Zarain-Herzberg A., Nishiki Y., Fueger P. T., Ogihara T., Evans-Molina C. (2012) PPAR-γ activation restores pancreatic islet SERCA2 levels and prevents β-cell dysfunction under conditions of hyperglycemic and cytokine stress. Mol. Endocrinol. 26, 257–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ramadan J. W., Steiner S. R., O'Neill C. M., Nunemaker C. S. (2011) The central role of calcium in the effects of cytokines on β-cell function: implications for type 1 and type 2 diabetes. Cell Calcium 50, 481–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cardozo A. K., Ortis F., Storling J., Feng Y.-M., Rasschaert J., Tonnesen M., Van Eylen F., Mandrup-Poulsen T., Herchuelz A., Eizirik D. L. (2005) Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic β-cells. Diabetes 54, 452–461 [DOI] [PubMed] [Google Scholar]
- 22. Jonsson J., Ahlgren U., Edlund T., Edlund H. (1995) IPF1, a homeodomain protein with a dual function in pancreas development. Int. J. Dev. Biol. 39, 789–798 [PubMed] [Google Scholar]
- 23. Kim S. K., Selleri L., Lee J. S., Zhang A. Y., Gu X., Jacobs Y., Cleary M. L. (2002) Pbx1 inactivation disrupts pancreas development and in Ipf1-deficient mice promotes diabetes mellitus. Nat. Genet. 30, 430–435 [DOI] [PubMed] [Google Scholar]
- 24. Murtaugh L. C. (2007) Pancreas and β-cell development: from the actual to the possible. Development 134, 427–438 [DOI] [PubMed] [Google Scholar]
- 25. Stoffers D. A., Zinkin N. T., Stanojevic V., Clarke W. L., Habener J. F. (1997) Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat. Genet. 15, 106–110 [DOI] [PubMed] [Google Scholar]
- 26. Hui H., Perfetti R. (2002) Pancreas duodenum homeobox-1 regulates pancreas development during embryogenesis and islet cell function in adulthood. Eur. J. Endocrinol. 146, 129–141 [DOI] [PubMed] [Google Scholar]
- 27. Al-Quobaili F., Montenarh M. (2008) Pancreatic duodenal homeobox factor-1 and diabetes mellitus type 2 (review). Int. J. Mol. Med. 21, 399–404 [PubMed] [Google Scholar]
- 28. Sachdeva M. M., Claiborn K. C., Khoo C., Yang J., Groff D. N., Mirmira R. G., Stoffers D. A. (2009) Pdx1 (MODY4) regulates pancreatic β cell susceptibility to ER stress. Proc. Natl. Acad. Sci. U.S.A. 106, 19090–19095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guo S., Dai C., Guo M., Taylor B., Harmon J. S., Sander M., Robertson R. P., Powers A. C., Stein R. (2013) Inactivation of specific β cell transcription factors in type 2 diabetes. J. Clin. Invest. 123, 3305–3316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ardestani A., Sauter N. S., Paroni F., Dharmadhikari G., Cho J.-H., Lupi R., Marchetti P., Oberholzer J., Conte J. K., Maedler K. (2011) Neutralizing interleukin-1β (IL-1β) induces β-cell survival by maintaining PDX1 protein nuclear localization. J. Biol. Chem. 286, 17144–17155 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31. Mahadevan J., Parazzoli S., Oseid E., Hertzel A. V., Bernlohr D. A., Vallerie S. N., Liu C.-Q., Lopez M., Harmon J. S., Robertson R. P. (2013) Ebselen treatment prevents islet apoptosis, maintains intranuclear Pdx-1 and MafA levels, and preserves β-cell mass and function in ZDF rats. Diabetes 62, 3582–3588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. National Research Council (2011) Guide for the Care and Use of Laboratory Animals, 8th Ed., National Academies Press, Washington, D.C. [Google Scholar]
- 33. Chaudhry Z. Z., Morris D. L., Moss D. R., Sims E. K., Chiong Y., Kono T., Evans-Molina C. (2013) Streptozotocin is equally diabetogenic whether administered to fed or fasted mice. Lab. Anim. 47, 257–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sims E. K., Hatanaka M., Morris D. L., Tersey S. A., Kono T., Chaudry Z. Z., Day K. H., Moss D. R., Stull N. D., Mirmira R. G., Evans-Molina C. (2013) Divergent compensatory responses to high-fat diet between C57BL6/J and C57BLKS/J inbred mouse strains. Am. J. Physiol. Endocrinol. Metab. 305, E1495–E1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gotoh M., Maki T., Kiyoizumi T., Satomi S., Monaco A. P. (1985) An improved method for isolation of mouse pancreatic islets. Transplantation 40, 437–438 [DOI] [PubMed] [Google Scholar]
- 36. Ogihara T., Chuang J. C., Vestermark G. L., Garmey J. C., Ketchum R. J., Huang X., Brayman K. L., Thorner M. O., Repa J. J., Mirmira R. G., Evans-Molina C. (2010) Liver X receptor agonists augment human islet function through activation of anaplerotic pathways and glycerolipid/free fatty acid cycling. J. Biol. Chem. 285, 5392–5404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Evans-Molina C., Robbins R. D., Kono T., Tersey S. A., Vestermark G. L., Nunemaker C. S., Garmey J. C., Deering T. G., Keller S. R., Maier B., Mirmira R. G. (2009) Peroxisome proliferator-activated receptor γ activation restores islet function in diabetic mice through reduction of endoplasmic reticulum stress and maintenance of euchromatin structure. Mol. Cell. Biol. 29, 2053–2067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fisher M. M., Perez Chumbiauca C. N., Mather K. J., Mirmira R. G., Tersey S. A. (2013) Detection of islet β-cell death in vivo by multiplex PCR analysis of differentially methylated DNA. Endocrinology 154, 3476–3481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Seijffers R., Ben-David O., Cohen Y., Karasik A., Berezin M., Newgard C. B., Ferber S. (1999) Increase in PDX-1 levels suppresses insulin gene expression in RIN 1046–38 cells. Endocrinology 140, 3311–3317 [DOI] [PubMed] [Google Scholar]
- 40. Park S. W., Zhou Y., Lee J., Lee J., Ozcan U. (2010) Sarco(endo)plasmic reticulum Ca2+-ATPase 2b is a major regulator of endoplasmic reticulum stress and glucose homeostasis in obesity. Proc. Natl. Acad. Sci. U.S.A. 107, 19320–19325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Iype T., Francis J., Garmey J. C., Schisler J. C., Nesher R., Weir G. C., Becker T. C., Newgard C. B., Griffen S. C., Mirmira R. G. (2005) Mechanism of insulin gene regulation by the pancreatic transcription factor Pdx-1: application of pre-mRNA analysis and chromatin immunoprecipitation to assess formation of functional transcriptional complexes. J. Biol. Chem. 280, 16798–16807 [DOI] [PubMed] [Google Scholar]
- 42. Bain J. R., Schisler J. C., Takeuchi K., Newgard C. B., Becker T. C. (2004) An adenovirus vector for efficient RNA interference-mediated suppression of target genes in insulinoma cells and pancreatic islets of Langerhans. Diabetes 53, 2190–2194 [DOI] [PubMed] [Google Scholar]
- 43. Sturek J. M., Castle J. D., Trace A. P., Page L. C., Castle A. M., Evans-Molina C., Parks J. S., Mirmira R. G., Hedrick C. C. (2010) An intracellular role for ABCG1-mediated cholesterol transport in the regulated secretory pathway of mouse pancreatic β cells. J. Clin. Invest. 120, 2575–2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Evans-Molina C., Garmey J. C., Ketchum R., Brayman K. L., Deng S., Mirmira R. G. (2007) Glucose regulation of insulin gene transcription and pre-mRNA processing in human islets. Diabetes 56, 827–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ravier M. A., Daro D., Roma L. P., Jonas J.-C., Cheng-Xue R., Schuit F. C., Gilon P. (2011) Mechanisms of control of the free Ca2+ concentration in the endoplasmic reticulum of mouse pancreatic β-cells: interplay with cell metabolism and [Ca2+]c and role of SERCA2b and SERCA3. Diabetes 60, 2533–2545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Palmer A. E., Jin C., Reed J. C., Tsien R. Y. (2004) Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc. Natl. Acad. Sci. U.S.A. 101, 17404–17409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Day R. N. (2014) Measuring protein interactions using Forster resonance energy transfer and fluorescence lifetime imaging microscopy. Methods 66, 200–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chakrabarti S. K., James J. C., Mirmira R. G. (2002) Quantitative assessment of gene targeting in vitro and in vivo by the pancreatic transcription factor, Pdx1: importance of chromatin structure in directing promoter binding. J. Biol. Chem. 277, 13286–13293 [DOI] [PubMed] [Google Scholar]
- 49. Abdi H. (2007) Bonferonni and Šidák corrections for multiple comparisons. in Encyclopedia of Measurement and Statistics (Salkind N., ed), pp. 103–107, Sage, Thousand Oaks, CA [Google Scholar]
- 50. Qiu Y., Guo M., Huang S., Stein R. (2002) Insulin gene transcription is mediated by interactions between the p300 coactivator and PDX-1, BETA2, and E47. Mol. Cell. Biol. 22, 412–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fernandez-Zapico M. E., van Velkinburgh J. C., Gutiérrez-Aguilar R., Neve B., Froguel P., Urrutia R., Stein R. (2009) MODY7 gene, KLF11, is a novel p300-dependent regulator of Pdx-1 (MODY4) transcription in pancreatic islet β cells. J. Biol. Chem. 284, 36482–36490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Brissova M., Shiota M., Nicholson W. E., Gannon M., Knobel S. M., Piston D. W., Wright C. V., Powers A. C. (2002) Reduction in pancreatic transcription factor PDX-1 impairs glucose-stimulated insulin secretion. J. Biol. Chem. 277, 11225–11232 [DOI] [PubMed] [Google Scholar]
- 53. Johnson J. D., Ahmed N. T., Luciani D. S., Han Z., Tran H., Fujita J., Misler S., Edlund H., Polonsky K. S. (2003) Increased islet apoptosis in Pdx1+/− mice. J. Clin. Invest. 111, 1147–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kulkarni R. N., Jhala U. S., Winnay J. N., Krajewski S., Montminy M., Kahn C. R. (2004) PDX-1 haploinsufficiency limits the compensatory islet hyperplasia that occurs in response to insulin resistance. J. Clin. Invest. 114, 828–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Asfari M., Janjic D., Meda P., Li G., Halban P. A., Wollheim C. B. (1992) Establishment of 2-mercaptoethanol-dependent differentiated insulin-secreting cell lines. Endocrinology 130, 167–178 [DOI] [PubMed] [Google Scholar]
- 56. Wang H., Iezzi M., Theander S., Antinozzi P. A., Gauthier B. R., Halban P. A., Wollheim C. B. (2005) Suppression of Pdx-1 perturbs proinsulin processing, insulin secretion and GLP-1 signalling in INS-1 cells. Diabetologia 48, 720–731 [DOI] [PubMed] [Google Scholar]
- 57. Docherty H. M., Hay C. W., Ferguson L. A., Barrow J., Durward E., Docherty K. (2005) Relative contribution of PDX-1, MafA and E47/β2 to the regulation of the human insulin promoter. Biochem. J. 389, 813–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Scheuner D., Kaufman R. J. (2008) The unfolded protein response: a pathway that links insulin demand with β-cell failure and diabetes. Endocr. Rev. 29, 317–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ozcan U., Cao Q., Yilmaz E., Lee A. H., Iwakoshi N. N., Ozdelen E., Tuncman G., Görgün C., Glimcher L. H., Hotamisligil G. S. (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461 [DOI] [PubMed] [Google Scholar]
- 60. Araki E., Oyadomari S., Mori M. (2003) Impact of endoplasmic reticulum stress pathway on pancreatic β-cells and diabetes mellitus. Exp. Biol. Med. 228, 1213–1217 [DOI] [PubMed] [Google Scholar]
- 61. Baggio L. L., Drucker D. J. (2007) Biology of incretins: GLP-1 and GIP. Gastroenterology 132, 2131–2157 [DOI] [PubMed] [Google Scholar]
- 62. Gautier J. F., Choukem S. P., Girard J. (2008) Physiology of incretins (GIP and GLP-1) and abnormalities in type 2 diabetes. Diabetes Metab. 34, S65–S72 [DOI] [PubMed] [Google Scholar]
- 63. Bedard K., Szabo E., Michalak M., Opas M. (2005) Cellular functions of endoplasmic reticulum chaperones calreticulin, calnexin, and ERp57. Int. Rev. Cytol. 245, 91–121 [DOI] [PubMed] [Google Scholar]
- 64. Görlach A., Klappa P., Kietzmann T. (2006) The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxid. Redox Signal. 8, 1391–1418 [DOI] [PubMed] [Google Scholar]
- 65. Shennan K. I., Taylor N. A., Docherty K. (1994) Calcium- and pH-dependent aggregation and membrane association of the precursor of the prohormone convertase PC2. J. Biol. Chem. 269, 18646–18650 [PubMed] [Google Scholar]
- 66. Seidah N. G. (2011) What lies ahead for the proprotein convertases? Ann. N.Y. Acad. Sci. 1220, 149–161 [DOI] [PubMed] [Google Scholar]
- 67. Váradi A., Molnár E., Ashcroft S. J. (1996) A unique combination of plasma membrane Ca2+-ATPase isoforms is expressed in islets of Langerhans and pancreatic β-cell lines. Biochem. J. 314, 663–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Clausen J. D., Vandecaetsbeek I., Wuytack F., Vangheluwe P., Andersen J. P. (2012) Distinct roles of the C-terminal 11th transmembrane helix and luminal extension in the partial reactions determining the high Ca2+ affinity of sarco(endo)plasmic reticulum Ca2+-ATPase isoform 2b (SERCA2b). J. Biol. Chem. 287, 39460–39469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. McKinnon C. M., Docherty K. (2001) Pancreatic duodenal homeobox-1, PDX-1, a major regulator of β cell identity and function. Diabetologia 44, 1203–1214 [DOI] [PubMed] [Google Scholar]
- 70. Hatanaka M., Tanabe K., Yanai A., Ohta Y., Kondo M., Akiyama M., Shinoda K., Oka Y., Tanizawa Y. (2011) Wolfram syndrome 1 gene (WFS1) product localizes to secretory granules and determines granule acidification in pancreatic β-cells. Hum. Mol. Genet. 20, 1274–1284 [DOI] [PubMed] [Google Scholar]
- 71. Fonseca S. G., Urano F., Weir G. C., Gromada J., Burcin M. (2012) Wolfram syndrome 1 and adenylyl cyclase 8 interact at the plasma membrane to regulate insulin production and secretion. Nat. Cell Biol. 14, 1105–1112 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 72. Inoue H., Tanizawa Y., Wasson J., Behn P., Kalidas K., Bernal-Mizrachi E., Mueckler M., Marshall H., Donis-Keller H., Crock P. (1998) A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome). Nat. Genet. 20, 143–148 [DOI] [PubMed] [Google Scholar]
- 73. Barrett T. G., Bundey S. E. (1997) Wolfram (DIDMOAD) syndrome. J. Med. Genet. 34, 838–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Riggs A. C., Bernal-Mizrachi E., Ohsugi M., Wasson J., Fatrai S., Welling C., Murray J., Schmidt R. E., Herrera P. L., Permutt M. A. (2005) Mice conditionally lacking the Wolfram gene in pancreatic islet β cells exhibit diabetes as a result of enhanced endoplasmic reticulum stress and apoptosis. Diabetologia 48, 2313–2321 [DOI] [PubMed] [Google Scholar]
- 75. Ishihara H., Takeda S., Tamura A., Takahashi R., Yamaguchi S., Takei D., Yamada T., Inoue H., Soga H., Katagiri H., Tanizawa Y., Oka Y. (2004) Disruption of the WFS1 gene in mice causes progressive β-cell loss and impaired stimulus-secretion coupling in insulin secretion. Hum. Mol. Genet. 13, 1159–1170 [DOI] [PubMed] [Google Scholar]
- 76. Takei D., Ishihara H., Yamaguchi S., Yamada T., Tamura A., Katagiri H., Maruyama Y., Oka Y. (2006) WFS1 protein modulates the free Ca2+ concentration in the endoplasmic reticulum. FEBS Lett. 580, 5635–5640 [DOI] [PubMed] [Google Scholar]
- 77. Smyth J. T., Hwang S. Y., Tomita T., DeHaven W. I., Mercer J. C., Putney J. W. (2010) Activation and regulation of store-operated calcium entry. J. Cell. Mol. Med. 14, 2337–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]