

Background: The mechanisms underlying lysosome rupture-mediated inflammasome activation are not understood.
Results: Inhibition of CaMKII, TAK1, or JNK specifically suppressed lysosome rupture-induced NLRP3 inflammasome activation.
Conclusion: Activation of the Ca2+-CaMKII-TAK1-JNK pathway in lysosome rupture is necessary for complete activation of the NLRP3 inflammasome.
Significance: Our results suggest novel roles for the Ca2+-CaMKII-TAK1-JNK pathway in the regulation of the inflammasome and propose potential therapeutic targets for inflammatory diseases.
Keywords: c-Jun N-terminal Kinase (JNK), Ca2+/Calmodulin-dependent Protein Kinase II (CaMKII), Inflammasome, Innate Immunity, Mitogen-activated Protein Kinase (MAPK), Signal Transduction, NLRP3, TAK1, Lysosome Rupture, siRNA Screen
Abstract
Lysosome rupture triggers NLRP3 inflammasome activation in macrophages. However, the underlying mechanism is not fully understood. Here we showed that the TAK1-JNK pathway, a MAPK signaling pathway, is activated through lysosome rupture and that this activation is necessary for the complete activation of the NLRP3 inflammasome through the oligomerization of an adapter protein, apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC). We also revealed that the activation of the TAK1-JNK pathway is sustained through Ca2+ ions and that calcium/calmodulin-dependent protein kinase type II functions upstream of the TAK1-JNK pathway and specifically regulates lysosome rupture-induced NLRP3 inflammasome activation. These data suggest a novel role for the TAK1-JNK pathway as a critical regulator of NLRP3 inflammasome activation.
Introduction
The lysosome is an organelle responsible for the digestion and degradation of endocytosed or autophagocytosed macromolecular particles in cells. Particularly, in macrophages, the lysosome is developed and matured for immunity against infection. However, excessive phagocytosis induces lysosome destabilization, eventually leading to lysosome rupture. For example, pathogenic bacteria such as Listeria monocytogenes evade phagocytosis in macrophages through the production of toxins that destabilize the lysosome membrane. The needle-like shape of crystal structures such as silica, asbestos, and monosodium urate (MSU)2 crystals allows these molecules to physically penetrate lysosome membranes. Lysosome rupture triggers various cellular responses, such as cell death, NLRP3 inflammasome activation, and autophagy.
The NLRP3 inflammasome is a multiple-protein complex comprising NLRP3, apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), and caspase 1, and the activation of this complex, in turn, activates caspase 1, which cleaves pro-IL-1β or pro-IL-18, generating the mature forms of these inflammatory cytokines, IL-1β or IL-18 (1). The NLRP3 inflammasome regulates multiple aspects of inflammation, and the dysregulation of this complex leads to undesirable inflammatory states. Limited by lysosome rupture, NLRP3 inflammasome activation has been associated with various human inflammatory diseases such as infection, pneumonia, gout, and atherosclerosis. Although lysosome rupture-induced NLRP3 inflammasome activation is considered the primary cause of inflammation, the underlying mechanism is not fully understood.
Recent studies have demonstrated that some kinases contribute to inflammasome activation. For example, the double-stranded RNA-dependent protein kinase (PKR) is activated through inflammasome-activating stimuli and kinase activity-dependent interactions with NLRP3, NLRP1, AIM2, and NLRC4, leading to the complete activation of the inflammasome (2). In response to Salmonella typhimurium infection, PKCδ phosphorylates the Ser-533 residue of NLRC4 to activate this inflammasome (3). In addition, it has been shown recently that Syk and JNK are required for the activation of the inflammasomes NLRP3 and AIM2 through the regulation of ASC phosphorylation and oligomerization (4). There are abundant kinase inhibitor compounds available, and some kinase-targeted drugs have been used as clinical cues. Therefore, elucidating the regulatory mechanism of inflammasome activation through kinases might lead to new therapeutic developments.
The stress-responsive MAPK pathway is activated through various stresses, such as oxidative stress and infection (5, 6). Here we confirmed that JNK, a stress-responsive MAPK, is activated after lysosome rupture and that JNK inhibition suppresses NLRP3 inflammasome activation. Although the involvement of JNK in NLRP3 inflammasome activation has been verified, the mechanism underlying how lysosome rupture induces JNK activation remains poorly understood. In this study, we identified the lysosome rupture-induced Ca2+-CaMKII-TAK1-JNK pathway, which regulates NLRP3 inflammasome activation, using an siRNA screen for mitogen-activated protein kinase kinase kinases (MAP3Ks) and a screen for inhibitors. The results suggest that these inhibitors and kinases might be potential drug candidates and targets for regulating NLRP3 inflammasome activation.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
Oxozeaenol, SB202190, SP600125, Bay11-7082, KN-93 water-soluble, KN-92 (Merck Millipore, Billerica, MA), LPS (O55:B5), CA-074ME, E-64d, bafilomycin A1, ATP, poly(dA:dT), disuccinimidyl suberate, dantrolene (Sigma-Aldrich, St. Louis, MO), l-leucyl-l-leucine methyl ester (LLME) (Chem-Impex International, Wood Dale, IL), calyculin A (LC Laboratories, Boston, MA), bis(2-aminophenyl)ethyleneglycol-tetraacetic acid, tetraacetoxymethyl ester (BAPTA-AM), Hoechst 33342 (Dojindo, Kumamoto, Japan), phorbol 12-myristate 13-acetate (PMA), xestospongin C (Wako Pure Chemical Industries, Osaka, Japan), and Texas Red-Dextran (Invitrogen) were purchased.
Antibodies for p-TAK1 (Thr-184/187) (Cell Signaling Technology, catalog no. 4508), p-JNK (Thr-183/Tyr-185) (Cell Signaling Technology, catalog no. 9251), p-p38 (Thr-180/Tyr-182) (Cell Signaling Technology, catalog no. 9211), cleaved IL-1β (Cell Signaling Technology, catalog no. 2021), p38α (L53F8, Cell Signaling Technology, catalog no. 9228), cleaved caspase 1 (Asp-297, D57A2, (Cell Signaling Technology, catalog no. 4199), TAK1 (M-579, Santa Cruz Biotechnology), caspase 1 p10 (C-20, Santa Cruz Biotechnology), caspase 1 caspase recruitment domain (A-19, Santa Cruz Biotechnology), JNK (FL, Santa Cruz Biotechnology), p38α (C-20-G, Santa Cruz Biotechnology), IκBα (C-21, Santa Cruz Biotechnology), ASC ((N-15)-R, Santa Cruz Biotechnology), ASC (TMS-1, Medical and Biological Laboratories, Nagoya, Aichi, Japan), p62 CT (Progen Biotechnik GmbH, Heidelberg, Germany), LC3 (Cosmo Bio, Tokyo, Japan), FLAG (1E6, Wako), FLAG (M2, Sigma), Actin (AC-40, Sigma), CD16/32 (mouse BD Fc block, BD Pharmingen), and PE Ly-6G (1A8, BioLegend, San Diego, CA) were purchased.
Cell Culture
THP-1, HEK293A, and HEK293FT cells were obtained from RIKEN, the ATCC, and Invitrogen, respectively. THP-1 cells were maintained in RPMI 1640 medium supplemented with 10% FBS. The HEK293A and HEK293FT cells were maintained in DMEM (4500 mg/liter glucose) supplemented with 10% FBS. THP-1 cells were infected with lentivirus carrying ASC-FLAG and selected in the presence of 0.5 μg/ml puromycin for at least 2 weeks to generate ASC-FLAG-stable THP-1 cells. THP-1 macrophages were differentiated using 10 ng/ml PMA for 3 days, and the cells were changed to fresh medium containing 10 ng/ml PMA 1 day after treatment.
Plasmid Construction
Expression plasmids containing the NLRP3 inflammasome components were generated through PCR amplification using cDNA from THP-1 cells and ligated into the pcDNA3/GW vector. The FLAG-MKK7-FLAG-JNK1 plasmid was a gift from Dr. Yukiko Gotoh (University of Tokyo, Tokyo, Japan) (7). The lentivirus vector for ASC-FLAG expression was generated through recombination from ASC-FLAG pcDNA3/GW into the pRRL with a puromycin resistance gene through an LR reaction via the pENTR vector for lentivirus packaging.
miRNA Construction of JNK and Lentivirus Transduction
The constructs were prepared using the BLOCK-iT Pol II miR RNAi expression vector kit with EmGFP (Invitrogen). Pre-miRNA and the reverse complement (Hmi413688_top_MAPK8 and Hmi413688_bot_MAPK8, referred to as #1; Hmi413689_top_MAPK8 and Hmi413689_bot_MAPK8, referred to as #2; Hmi413696_top_MAPK9 and Hmi413689_bot_MAPK9, referred to as #1; and Hmi413697_top_MAPK9 and Hmi413697_bot_MAPK9, referred to as #2; Invitrogen) were annealed and ligated into the pcDNA6.2-GW/EmGFP-miR vector. The lentivirus vectors (pLenti6.4/Em3-miRNA) were generated through recombination from the control plasmid (pcDNA6.2-GW/EmGFPmiR-neg) or each pcDNA6.2-GW/EmGFP-miR for JNK1 and JNK2 into pLenti6.4/R4R2/V5-DEST with pENTR-pCMV through a BP reaction with pDONR221, followed by an LR reaction. The plasmids pLenti6.4/EmGFP-miRNA, pLP1, pLP2, and pLP/VSVG were transfected into HEK293FT cells using polyethylenimine (PEI)-MAX, followed by a medium change to remove the transfection reagents. The virus-containing medium was harvested and concentrated using PEG 6000. The virus was subsequently injected into THP-1 cells cultured in medium containing 10 ng/ml PMA and 5 μg/ml Polybrene for 5–6 days. Fresh medium containing 10 ng/ml PMA was added daily 2 days after transduction.
Establishment of JNK1 and JNK2 Knockout THP-1 Cells
THP-1 cells were transduced with fusiogenic envelope G glycoprotein of the vesicular stomatitis virus (VSV-G) pseudotyped retrovirus containing Cas9-P2A-puro and selected by 0.5 μg/ml puromycin for 2 weeks. THP-1 cells stably expressing Cas9-P2A-puro were transduced with a lentivirus containing U6-gRNA for MAPK8 or MAPK9-EFS-DsRed. DsRed(+) cells were sorted and cultured further. Cas9-P2A-puro (Addgene, catalog no. 49535) was recombined into modified pMXs-GW vectors. MAPK8-gRNA or MAPK9-gRNA was generated by annealing of the oligos CACCGAATTTTTATAGTGTAGAGAT and AAACATCTCTACACTATAAAAATTC or CACCGTCAGTTTTATAGTGTGCAAG and AAACCTTGCACACTATAAAACTGAC, respectively, and then ligated into BsmBI-digested guide RNA expression vectors.
siRNA Transfection
For the MAP3K knockdown experiments, THP-1 macrophages were transfected with 20 nm siRNA oligonucleotides (Qiagen, Venle, Netherlands) or Stealth RNAi siRNA negative control med GC duplex #2 and #3 (Invitrogen) using Lipofectamine RNAiMAX (Invitrogen), followed by treatment with 10 ng/ml PMA. The cells were changed into fresh medium containing 10 ng/ml PMA 1 day after seeding.
Immunoblot Analysis
The cells were lysed using immunoprecipitation lysis buffer (50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 10 mm EDTA (pH 8.0), 1% sodium deoxycholate, 1% Triton X-100, 5 μg/ml leupeptin, and 1 mm PMSF) supplemented with PhosSTOP (Roche Applied Science) or PPase inhibitor mixture (8 mm NaF, 12 mm β-glycerophosphate, 1 mm Na3VO4, 1.2 mm Na2MoO4, 5 μm cantharidin, and 2 mm imidazole). The cell lysates were centrifuged, and the supernatants were mixed with the same amount of SDS sample buffer (80 mm Tris-HCl (pH 8.8), 80 μg/ml bromphenol blue, 28.8% glycerol, 4% SDS, and 20 mm dithiothreitol). The culture supernatants were collected and centrifuged. The supernatants were mixed with 4 volumes of acetone, followed by incubation at −20 °C and subsequent sedimentation through centrifugation. The samples were dissolved in SDS sample buffer. After boiling, the samples were separated through electrophoresis and transferred to PVDF membranes. The membranes were probed with the appropriate antibodies and detected using an ECL system (GE Healthcare).
Immunofluorescence Microscopy Analysis of ASC Speckle
Thioglycollate-elicited peritoneal macrophages were harvested from mice by peritoneal lavage 3 days after intraperitoneal injection of 4% thioglycollate medium and seeded on cover glass. The culture medium was replaced to remove non-adherent cells within 2 h. The next day, adherent cells were primed with 100 ng/ml LPS for 3 h, followed by stimulation with 400 μg/ml MSU crystals and inhibitors for 3 h. After stimulation, cells were fixed, permeabilized, and then stained with anti-ASC ((N-15)-R) antibody, followed by Alexa Fluor 488-conjugated anti-rabbit IgG and Hoechst 33342. Microscopy analysis was performed using BIOZERO (Keyence). Eight optical fields per sample were acquired, and ASC speckle-containing cells were counted manually.
ASC Pyroptosome Assay
An ASC pyroptosome assay was performed as described previously, with slight modifications (8). Briefly, the cells were washed with PBS and harvested in buffer A (20 mm HEPES-KOH (pH 7.5), 10 mm KCl, 1.5 mm MgCl2, 1 mm EDTA, 1 mm EGTA, 320 mm sucrose, 5 μg/ml leupeptin, and 1 mm PMSF) supplemented with PPase inhibitor mixture. The cells were lysed by shearing 20 times through a 27-gauge needle, and, subsequently, the cell lysates were centrifuged at 600 × g to remove the bulk nuclei and unbroken cells. The resulting supernatants were centrifuged at 17,700 × g to pellet the ASC pyroptosomes. The pellets were resuspended in CHAPS buffer (20 mm HEPES-KOH (pH 7.5), 5 mm MgCl2, 0.5 mm EGTA, 0.1% CHAPS, 5 μg/ml leupeptin, and 1 mm PMSF) and reacted with 2 mm disuccinimidyl suberate for 30 min, followed by quenching in SDS sample buffer.
Lactate Dehydrogenase (LDH) Assay
Lactate dehydrogenase release was measured using the lactate dehydrogenase cytotoxic test (Wako). Lactate dehydrogenase release was defined as [culture supernatant/(culture supernatant + 0.1% Triton-X100 lysed cells)].
MSU-induced Peritonitis Mouse Model
Eight-week-old male mice were given an intraperitoneal injection of 5 mg/kg 5-Z-Oxozeaenol or 20 mg/kg SP600125 for 1 h prior to challenge with 3 mg MSU crystals through intraperitoneal injection. Oxozeanol was dissolved in dimethyl sulfoxide and diluted in PBS. SP600125 was dissolved in dimethyl sulfoxide and diluted in 10% PPCES buffer (30% PEG-400, 20% polypropylene glycol, 15% Cremophor EL, 5% ethanol, and 30% saline). The mice were sacrificed 6 h after injection, and the peritoneal cavities were lavaged using 5 ml of PBS supplemented with 5 mm EDTA. The peritoneal cells were blocked with CD16/CD32 antibodies and subsequently stained with PE Ly-6G antibody. The number of neutrophils (Ly-6G+) in the lavage was assessed using the flow cytometry FL2 channel. MSU crystals were prepared as described previously with slight modifications (9). Briefly, 1.0 g of uric acid was added to 200 ml of sterile water containing 0.24 g of NaOH and subsequently adjusted to pH 7.2 using HCl. After dilution through heating, the solution was cooled to room temperature, and the recrystallized MSU crystals were harvested and autoclaved. The MSU crystals were diluted in PBS and sonicated prior to use in subsequent experiments.
Statistical Analysis
All values are presented as the mean ± S.D., except the values obtained from the mouse model, which are presented as the mean ± S.E. Prism software (GraphPad) was used for statistical analysis. Analysis of variance followed by Dunnett's test or unpaired Student's t test was used. p < 0.05 was considered significant.
RESULTS
Lysosome Rupture Activates JNK, Leading to NLRP3 Inflammasome Activation
First, we investigated whether lysosome rupture activates stress-responsive MAPKs. LLME, a lysosomotropic agent, is typically used to induce pinocytosis-dependent lysosome rupture. Di-peptidases in the lysosome cleave LLME, and the cleaved form of this molecule destabilizes the lysosome membrane, resulting in lysosome rupture (10). In THP-1 macrophages treated with LLME, we detected the activation of stress-responsive MAPKs such as p38 MAPK and JNK. Inhibitors of cathepsin B or cysteine proteases attenuate the activation of MAPKs, although we observed the same degree of LC3-II conversion (an indicator of lysosome rupture) between control and inhibitor-treated cells (Fig. 1A). Moreover, we also observed a similar attenuation in cells pretreated with bafilomycin A1 (an inhibitor of lysosome maturation) cells (Fig. 1B). These results suggest that lysosome rupture activates stress-responsive MAPKs and that cathepsin B activity is necessary for the activation of these enzymes. Next we examined the physiological relevance of stress-responsive MAPKs using various inhibitors. Both lysosome rupture-induced cell death and lysosome rupture-induced NLRP3 inflammasome activation require cathepsin activity (11–15). Therefore, we investigated the effects of MAPK inhibitors on cell death and NLRP3 inflammasome activation. On the basis of an assay monitoring lactate dehydrogenase release, no clear effects of MAPK inhibitors on cell death were observed (data not shown). However, we revealed that the JNK inhibitor suppressed the release of cleaved-IL-1β or the caspase 1 p10 subunit (the active form of caspase 1) (Fig. 1C), suggesting NLRP3 inflammasome activation. Furthermore, this attenuation was also observed in JNK1 or JNK2 knockdown THP-1 macrophages (Fig. 1D). Moreover, we established the JNK1 and JNK2 knockout THP-1 cell lines by using the clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 system and confirmed that NLRP3 inflammasome activation, indicating the amount of the active form of caspase 1 (caspase 1 p20) in the culture supernatant, was reduced in JNK1 and JNK2 knockout THP-1 cell lines (Fig. 1E). Furthermore, recent studies suggest that Syk and JNK regulate NLRP3 and AIM2 inflammasomes through ASC phosphorylation (4). These results suggest that lysosome rupture activates JNK, which promotes NLRP3 inflammasome activation.
FIGURE 1.

Lysosome rupture-activated JNK is required for the complete activation of the NLRP3 inflammasome. A and B, lysates of THP-1 macrophages pretreated with CA074-ME (CA) or E-64d (A) and bafilomycin A1 (B), followed by stimulation with LLME, were immunoblotted with antibodies against p-MAPKs and total MAPKs. Dimethyl sulfoxide (DMSO, D) and ethanol (EtOH) were used as solvents. C, THP-1 macrophages were unprimed or primed with LPS for 2 h, followed by LLME stimulation for 3 h with the indicated inhibitors. The culture supernatants were collected and immunoblotted with antibodies against caspase 1 p10 and cleaved IL-1β. D, THP-1 macrophages infected with a lentivirus expressing negative control or JNK1 or JNK2 shRNA-miR were primed with LPS for 2 h, followed by stimulation with LLME for 2 h. Culture supernatants and lysates were collected and immunoblotted with antibodies against caspase 1 p10 and cleaved IL-1β or procaspase 1 and JNK1. N.C., negative control siRNA; L, 1 mm LLME stimulation for 3 h. E, human genome sequences of MAPK8 exon2 and MAPK9 exon2 containing the transcription start site, first Met, are indicated. The underline indicates the target sequence of CRISPR gRNA (top panel). Non-transduced and MAPK8- or MAPK9-gRNA-transduced THP-1 cells were differentiated by treatment with 30 ng/ml PMA. THP-1 macrophages were primed with LPS for 2 h, followed by LLME stimulation for 3 h. The culture supernatants were collected and immunoblotted with an antibody against cleaved caspase 1 (caspase 1 p20) (bottom panel). The results are representative of at least three (A, C, and D) and two (B and E) independent experiments.
A MAP3K siRNA Screen Identified TAK1 as an Upstream Kinase of JNK
Although JNK regulates NLRP3 inflammasome activation, how lysosome rupture induces the activation of JNK remains unknown. To identify a lysosome rupture-activated kinase upstream of JNK, we performed an siRNA screen focusing on MAP3Ks by monitoring JNK activation. The results showed that JNK activation is attenuated in MAP3K7 (TAK1) siRNA-transfected THP-1 macrophages (Fig. 2, A and B). TAK1, a member of the MAP3K family, is activated downstream of the TGFβ receptor (16), the IL-1 receptor (17), Toll-like receptors (TLRs) (18), and the TNF-α receptor (19). TAK1 activation induces the activation of MKK3/MKK6, MKK4/MKK7, or IκB kinase (IKK), leading to the activation of p38, JNK, or the NF-κB pathway, respectively (16, 19).
FIGURE 2.

MAP3K siRNA screening identified TAK1 as an upstream kinase of LLME-activated JNK. A, lysates from MAP3K siRNA- or negative control (NC) siRNA-transfected THP-1 macrophages, followed by stimulation with LLME, were immunoblotted with antibodies against p-JNK and total JNK. As a positive control, the lysates were pretreated with an inhibitor (In, E-64d). B, the band intensity of p-JNK (54 kDa) relative to total JNK (54 kDa) was measured and plotted using ImageJ software. The values represent the mean ± S.D. (n = 3). *, p < 0.05. The results are representative of at least three independent experiments. AU, arbitrary units.
TAK1 Is Necessary for the Activation of JNK and the NLRP3 Inflammasome
Indeed, we observed that TAK1 is activated in response to LLME stimuli (Fig. 3A), and the strong activation of TAK1 was also detected in cells treated with calyculin A (a phosphatase inhibitor) (Fig. 3B). Because PP2A or PP6 rapidly dephosphorylates TAK1, calyculin A suppressed this dephosphorylation (20, 21). Furthermore, 5-Z-Oxozeaenol, a TAK1 inhibitor (22), strongly suppressed the LLME-induced activation of JNK and p38 and partially suppressed the activation of NF-κB (Fig. 3C). It has been reported that TAK1 is involved in NLRP3 inflammasome activation in an NF-κB activity-dependent manner to facilitate classical NLRP3 agonist stimulation (23) or hypotonic stress-induced cell volume change (24). However, although Bay11-7082 is an inhibitor of the NF-κB pathway, this compound has recently been proposed as a direct NLRP3 inhibitor independent of the NF-κB pathway (25). Therefore, how TAK1 is involved in NLRP3 inflammasome activation remains unclear. Indeed, we confirmed that the inhibition of TAK1, NF-κB, or JNK suppresses NLRP3 inflammasome activation in response to LLME stimuli (Fig. 3D). Furthermore, the same results were obtained with MSU crystal (another lysosome rupture inducer)-induced NLRP3 inflammasome activation (Fig. 3E). In addition, in an MSU crystal-induced peritonitis mouse model, preadministration of the TAK1 inhibitor suppressed neutrophil migration into the peritoneal cavity (Fig. 3F). It has been reported that the same result was observed in JNK1- or JNK2-deficient mice (4). These data suggest that the TAK1-JNK pathway might be a potent therapeutic target for immune diseases associated with NLRP3 inflammasome activation, such as gout and pneumonia.
FIGURE 3.

The TAK1-JNK pathway regulates NLRP3 inflammasome activation. A and B, lysates from THP-1 macrophages stimulated with LLME for the indicated times in the absence (A) or presence of calyculin A (B) were immunoblotted with antibodies against p-TAK1 and TAK1. C, lysates from THP-1 macrophages stimulated with LLME for the indicated times in the absence or presence of Oxozeaenol were immunoblotted with antibodies against p-MAPKs, MAPKs, and IκBα. DMSO, dimethyl sulfoxide. D and E, LPS-primed THP-1 macrophages were subjected to LLME stimulation for 3 h with the indicated inhibitors, such as Oxozeaenol (0.5 μm, Oxo), SP600125 (10 μm, SP), SB202190 (10 μm, SB), and Bay11-7082 (10 μm, Bay). The culture supernatants were collected and immunoblotted with antibodies against caspase 1 p10 and cleaved IL-1β. F, mice were challenged intraperitoneally with 3 mg of MSU crystals at 1 h after TAK1 inhibitor administration. The number of neutrophils in the peritoneal cavities at 6 h after injection of the MSU crystals was plotted. *, p < 0.05. The results are representative of at least three (A, C, D, and F) and two (B and E) independent experiments.
JNK Promotes the Activation of NLRP3 and AIM2 Inflammasomes through the Regulation of ASC Oligomerization
Previous studies have shown that JNK induces the phosphorylation of ASC and that this phosphorylation is required for the oligomerization of ASC (4). To confirm the mechanism by which the TAK1-JNK pathway regulates the NLRP3 inflammasome, we compared the effects of TAK1 and JNK inhibitors on ATP (another classical NLRP3 agonist)-triggered NLRP3 inflammasome activation and poly(dA:dT) (an AIM2 agonist)-triggered AIM2 inflammasome activation. Interestingly, the JNK inhibitor suppressed caspase 1 cleavage mediated through both NLRP3 and AIM2 agonists, but the TAK1 inhibitor only suppressed caspase 1 cleavage through the NLRP3 agonist (Fig. 4, A and B). These results suggest that the components of the inflammasome, such as ASC or caspase 1, are targets of the JNK pathway. Stimulation through poly(dA:dT) transfection revealed that other MAP3Ks most likely function upstream of JNK. Therefore, we examined the granulation of the adaptor protein ASC. Endogenous ASC forms speckles (granulation) in lysosome-ruptured primary peritoneal macrophages treated with MSU crystals. This MSU crystal-induced formation of ASC speckles was reduced significantly in TAK1 or JNK inhibitor-treated cells (Fig. 4C). Moreover, LLME stimuli induced ASC oligomerization in cross-linked pellets, containing heavy membranes, such as mitochondria, whereas the TAK1 or JNK inhibitor suppressed the dimerization and oligomerization of ASC (Fig. 4D). Although the data could not reach statistical significance, the reproducibility of this experiment was sufficient. Considered together with the data in Fig. 4C, TAK1 and JNK are probably involved in ASC oligomerization. Accordingly, the constitutively active form of JNK accelerates IL-1β cleavage in HEK293A cells, reconstituting the NLRP3 inflammasome. However, accelerated IL-1β cleavage does not occur without the coexpression of ASC in the NLRP3 inflammasome (Fig. 4E). Taken together, these results suggest that the JNK pathway regulates ASC oligomerization.
FIGURE 4.

The TAK1-JNK pathway is required for ASC oligomerization, and JNK activity is sufficient to reconstitute NLRP3 inflammasome activation. A, LPS-primed THP-1 macrophages were pretreated with the indicated inhibitors for 10 min prior to stimulation with 5 mm ATP for 1 h. The culture supernatants were collected and immunoblotted with antibodies against caspase 1 p10 and cleaved IL-1β. Oxo, Oxozeaenol; SP, SP600125; SB, SB202190; Bay, Bay11-7082. MilliQ (MQ) and DMSO (D) were used as solvents. B, THP-1 macrophages were stimulated through poly(dA:dT) transfection and treated with the indicated inhibitors for 6 h. The culture supernatants were collected and immunoblotted with antibodies against caspase 1 p10 and cleaved IL-1β. C, thioglycollate-elicited peritoneal macrophages were primed with LPS and stimulated by MSU crystals with the indicated inhibitors. The macrophages were stained for anti-ASC antibody (green, top row) and Hoechst 33342 (blue, center row). Scale bars = 20 μm. The white arrows indicate ASC speckle-containing cells. The percentage of cells with ASC speckles was measured and plotted. DMSO, dimethyl sulfoxide. D, immunoblot analysis of ASC pyroptosomes in THP-1 macrophages subjected to LLME stimulation with the indicated inhibitors for 90 min. The band intensity of the ASC dimer was measured and plotted using ImageJ software. AU, arbitrary unit. E, HEK293A cells were cotransfected with NLRP3 inflammasome components and a constitutive active form of JNK in the presence or absence of an ASC plasmid. Medium without serum was replaced at 1 day after transfection, and the culture supernatants were collected 6 h later and immunoblotted with antibodies for cleaved IL-1β. The values represent the mean ± S.D. (n = 3). *, p < 0.05. The results are representative of at least two (A and B) and three (C, D, and E) independent experiments.
Ca2+-CaMKII Functions Upstream of the TAK1-JNK Pathway and Regulates NLRP3 Inflammasome Activation
Recent studies have demonstrated that the Ca2+ ion is a potent regulator of NLRP3 inflammasome activation (26). The results of a recent study indicate that the intracellular Ca2+ ion chelator BAPTA-AM suppresses the activation of TAK1 and the NLRP3 inflammasome, particularly in response to hypotonic stimulation (24). Consistently, the reduction of TAK1-JNK pathway activation was observed in BAPTA-AM-treated cells (Fig. 5A). We also explored sources of Ca2+ ions using an extracellular Ca2+ chelator, an inositol trisphosphate 3 channel blocker, and a ryanodine receptor blocker. However, these inhibitors did not suppress JNK activation (Fig. 5, B–D), suggesting that Ca2+ ions directly from the lysosome are sufficient to evoke the signaling pathway, leading to lysosome rupture. CaMKII is indicated as an upstream kinase of TAK1 in the Wnt or Toll-like receptor signaling pathway (27, 28). Hence, we also assessed the involvement of CaMKII in lysosome rupture-induced signal transduction. A CaMKII inhibitor suppressed the LLME-induced activation of the TAK1-JNK pathway and the NLRP3 inflammasome (Fig. 5, E and F). However, this attenuation was not observed when using ATP as a stimulus (a different stimulus from lysosome rupture) to induce NLRP3 inflammasome activation (Fig. 5G), indicating that CaMKII specifically responds to lysosome rupture. These data suggest that lysosome rupture induces Ca2+ influx from the lysosome to the cytosol and activates the CaMKII-TAK1-JNK pathway to promote NLRP3 inflammasome activation and that this signaling pathway is specifically activated in response to lysosome rupture (Fig. 6).
FIGURE 5.

Ca2+-CaMKII regulates the TAK1-JNK pathway and the NLRP3 inflammasome in lysosome rupture. A, lysates from THP-1 macrophages pretreated with BAPTA-AM, followed by stimulation with LLME, were immunoblotted with antibodies against p-JNK and total JNK or p-TAK1 and total TAK1. DMSO, dimethyl sulfoxide. B, C, and D, lysates from THP-1 macrophages stimulated using LLME with EDTA or EGTA (B) and lysates from THP-1 macrophages pretreated with xestospongin C (C) or dantrolene (D), prior to stimulation with LLME, were immunoblotted with antibodies against p-JNK and total JNK. MilliQ (MQ) was used as a solvent. E, lysates from THP-1 macrophages pretreated with KN-93 or KN-92, followed by stimulation with LLME, were immunoblotted with antibodies against p-JNK and total JNK or p-TAK1 and total TAK1. F, LPS-primed THP-1 macrophages were stimulated with LLME and treated with the indicated inhibitors for 3 h. The culture supernatants were collected and immunoblotted with antibodies against caspase 1 p10 and cleaved IL-1β. Oxo, Oxozeaenol; SP, SP600125. G, LPS-primed THP-1 macrophages were pretreated with the indicated inhibitors for 10 min prior to stimulation with 5 mm ATP for 1 h. The culture supernatants were collected and immunoblotted with antibodies against caspase 1 p10 and cleaved IL-1β. SB, SB202190. The results are representative of at least three (A, B, D, F, and G) and two (C and E) independent experiments.
FIGURE 6.
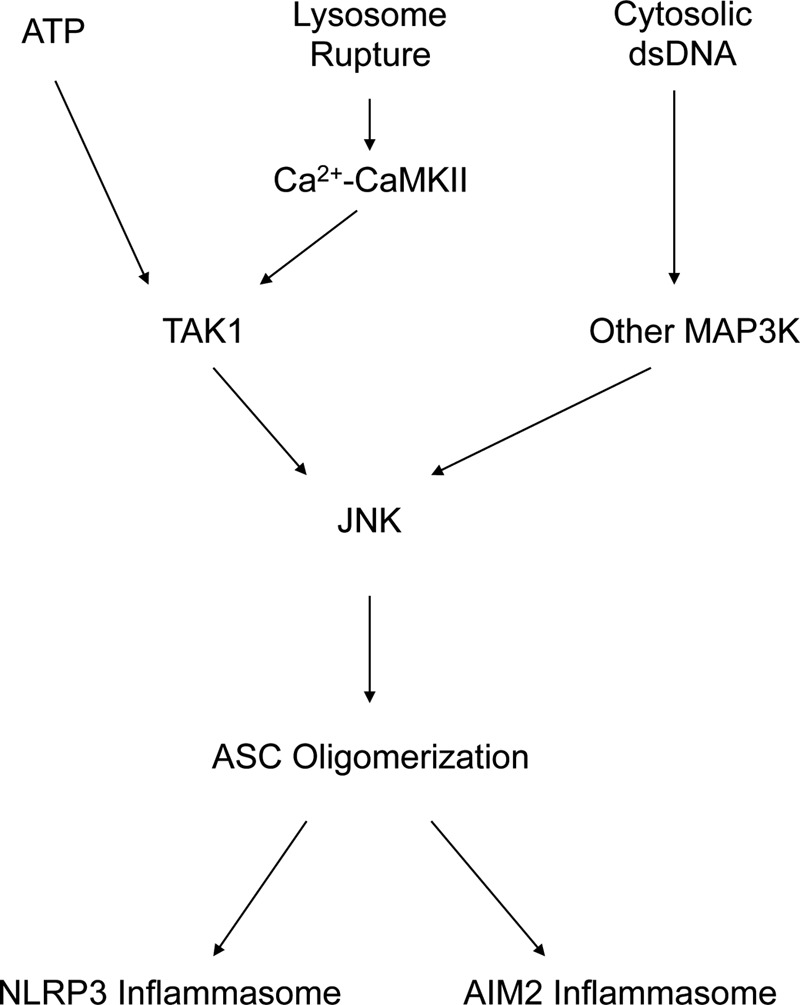
The proposed model.
DISCUSSION
The NLRP3 inflammasome is activated through various stresses, including K+ efflux, reactive oxygen species, and lysosome rupture. Additionally, various types of infectious stresses activate other inflammasomes, such as AIM2. Inflammasome activation leads to inflammatory cytokine maturation through proteolysis and tissue inflammation. However, how such diverse stresses activate inflammasomes remains unclear, and it is unknown how diverse stress stimuli integrate into a single inflammasome. The results of this study suggest that the MAP3K-JNK pathway is important for this integration. The MAPK cascade comprises the hierarchical MAP3K-MAP2K-MAPK pathway (where MAP2K mitogen-activated protein kinase kinase), with at least 18 MAP3K members. Each MAP3K responds to specific stress stimuli to some extent and eventually induces the activation of p38 MAPK and JNK. Therefore, our hypothesis is that the MAP3K-JNK pathway integrates various stress stimuli into one inflammasome. Here we showed that, in response to lysosome rupture, Ca2+-CaMKII-activated TAK1 is the responsible MAP3K for JNK activation. In the case of ATP stimulation, although Ca2+-CaMKII is not involved in this pathway, TAK1 is also responsible. However, in the case of poly(dA:dT) transfection, another MAP3K is most likely responsible for JNK activation (Fig. 6). Further studies are needed to uncover each responsive MAP3K for a specific stress that regulates NLRP3 inflammasome activation through JNK.
Certainly, JNK regulates NLRP3 and AIM2 inflammasomes through the oligomerization of ASC. However, the effect of a JNK inhibitor on NLRP3 inflammasome activation was strongly observed compared with AIM2 inflammasome activation (Figs. 3, D and E, and 4, A and B), suggesting an additional mechanism by which JNK regulates NLRP3 inflammasome activation besides oligomerization of ASC. Recently, mitochondria and mitochondrial apoptotic dysfunction were implicated in NLRP3 inflammasome activation through the production of reactive oxygen species (29), mitochondrial DNA release (30, 31), interactions with mitochondrial antiviral signaling protein (MAVS) (32, 33), and cardiolipin (mitochondrial lipid) (34) and interactions between NLRP3 and ASC on the endoplasmic reticulum and mitochondria (35). Notably, JNK promotes mitochondrial apoptosis through the phosphorylation of Bcl-2 family proteins, such as Bcl-2 (36), Bcl-xL (37), and Bax (38), and Bcl-2 regulatory proteins (7). Therefore, it is reasonable to suggest that JNK regulates NLRP3 inflammasome activation through the phosphorylation of the Bcl-2 family, which regulates the function of these proteins. Indeed, recent studies have suggested that NLRP3 inflammasome activation is attenuated in Bcl-2 (an antiapoptotic protein) transgenic macrophages (29, 31) or VDAC1 (a mitochondrial apoptotic protein) knockdown macrophages (29). Determining whether JNK-mediated mitochondrial dysfunction and the JNK-mediated apoptotic pathway are also involved in NLRP3 inflammasome activation will clarify the novel regulatory mechanism of NLRP3 inflammasome activation through JNK. Therefore, it might be important to analyze the relationship between apoptosis and inflammasome activation to elucidate the additional regulatory mechanism of inflammasome activation through JNK.
It has been suggested that lysosomal proteases released after lysosome rupture might cleave unidentified cytosol substrates, thereby triggering NLRP3 inflammasome activation. We could not identify cytosol substrates of released lysosomal proteases. However, we propose that Ca2+ is another factor for activation of the TAK1-JNK pathway through CaMKII. It has been shown previously that, even in crystal structure-induced NLRP3 inflammasome activation, Ca2+ influx is necessary for the complete activation of the inflammasome. However, the source of Ca2+ in previous studies was different from our results. It has been reported that Ca2+ is derived from the extracellular space through the TRPM2 channel (39) or the endoplasmic reticulum through the inositol trisphosphate 3 receptor (40, 41). However, we did not observed any reduction of JNK activation using an extracellular Ca2+ chelator, an inositol trisphosphate 3 receptor blocker, or a ryanodine receptor blocker. In general, the lysosome, an organelle derived from the endoplasmic reticulum, contains abundant amount of Ca2+. Therefore, we suggest that Ca2+ from the lysosome is sufficient to evoke the Ca2+-CaMKII-TAK1-JNK pathway, which activates the NLRP3 inflammasome. However, the activation of this pathway is occasional and dependent on the degree of lysosome rupture. For instance, in the early phase of lysosome rupture, the damaged lysosomes fuse with undamaged lysosomes to recover function and activate damaged lysosome-specific autophagy machinery (42, 43). However, excessive lysosome rupture induces apoptosis, cleaving the proapoptotic protein Bid through released cathepsins (11–13). Depending on the degree of lysosome rupture, the cells select an appropriate signaling pathway to induce proper cellular responses. The Ca2+-CaMKII-TAK1-JNK pathway is one of the signaling pathways activated through lysosome rupture, which induces NLRP3 inflammasome activation, leading to cytokine production and the activation of the immune system.
Therefore, we propose that the Ca2+-CaMKII-TAK1-JNK pathway activated through lysosome rupture regulates NLRP3 inflammasome activation by promoting ASC oligomerization. Together with previous reports, these results suggest that JNK plays a central role in regulating inflammasome activation.
Acknowledgments
We thank the members of the Cell Signaling Laboratory for critical comments. We also thank Yukiko Gotoh (University of Tokyo, Tokyo, Japan) for plasmids.
This work was supported by KAKENHI from Japan Society for the Promotion of Science (JSPS) and Ministry of Education, Culture, Sports, Science and Technology (MEXT), by the Global Center of Education and Research for Chemical Biology of the Diseases, by the Global Center of Excellence Program (GCOE) Program, by the “Understanding of Molecular and Environmental Bases for Brain Health” study conducted under the Strategic Research Program for Brain Sciences by MEXT, by the Advanced Research for Medical Products Mining Programme of the National Institute of Biomedical Innovation, by the Funding Program for Next Generation World-leading Researchers, by the Nagase Science and Technology Foundation, by the Astellas Foundation for Research on Metabolic Disorders, and by the Takeda Science Foundation.
- MSU
- monosodium urate
- LLME
- l-leucyl-l-leucine methyl ester
- PMA
- phorbol 12-myristate 13-acetate
- CaMKII
- calcium/calmodulin-dependent protein kinase type II
- MAP3K
- mitogen-activated protein kinase kinase kinase.
REFERENCES
- 1. Latz E., Xiao T. S., Stutz A. (2013) Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 13, 397–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lu B., Nakamura T., Inouye K., Li J., Tang Y., Lundbäck P., Valdes-Ferrer S. I., Olofsson P. S., Kalb T., Roth J., Zou Y., Erlandsson-Harris H., Yang H., Ting J. P., Wang H., Andersson U., Antoine D. J., Chavan S. S., Hotamisligil G. S., Tracey K. J. (2012) Novel role of PKR in inflammasome activation and HMGB1 release. Nature 488, 670–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Qu Y., Misaghi S., Izrael-Tomasevic A., Newton K., Gilmour L. L., Lamkanfi M., Louie S., Kayagaki N., Liu J., Kömüves L., Cupp J. E., Arnott D., Monack D., Dixit V. M. (2012) Phosphorylation of NLRC4 is critical for inflammasome activation. Nature 490, 539–542 [DOI] [PubMed] [Google Scholar]
- 4. Hara H., Tsuchiya K., Kawamura I., Fang R., Hernandez-Cuellar E., Shen Y., Mizuguchi J., Schweighoffer E., Tybulewicz V., Mitsuyama M. (2013) Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat. Immunol. 14, 1247–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wagner E. F., Nebreda A. R. (2009) Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 9, 537–549 [DOI] [PubMed] [Google Scholar]
- 6. Arthur J. S., Ley S. C. (2013) Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 13, 679–692 [DOI] [PubMed] [Google Scholar]
- 7. Tsuruta F., Sunayama J., Mori Y., Hattori S., Shimizu S., Tsujimoto Y., Yoshioka K., Masuyama N., Gotoh Y. (2004) JNK promotes Bax translocation to mitochondria through phosphorylation of 14-3-3 proteins. EMBO J. 23, 1889–1899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fernandes-Alnemri T., Wu J., Yu J. W., Datta P., Miller B., Jankowski W., Rosenberg S., Zhang J., Alnemri E. S. (2007) The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 14, 1590–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Giamarellos-Bourboulis E. J., Mouktaroudi M., Bodar E., van der Ven J., Kullberg B. J., Netea M. G., van der Meer J. W. (2009) Crystals of monosodium urate monohydrate enhance lipopolysaccharide-induced release of interleukin 1 β by mononuclear cells through a caspase 1-mediated process. Ann. Rheum. Dis. 68, 273–278 [DOI] [PubMed] [Google Scholar]
- 10. Uchimoto T., Nohara H., Kamehara R., Iwamura M., Watanabe N., Kobayashi Y. (1999) Mechanism of apoptosis induced by a lysosomotropic agent, l-leucyl-l-leucine methyl ester. Apoptosis 4, 357–362 [DOI] [PubMed] [Google Scholar]
- 11. Cirman T., Oresić K., Mazovec G. D., Turk V., Reed J. C., Myers R. M., Salvesen G. S., Turk B. (2004) Selective disruption of lysosomes in HeLa cells triggers apoptosis mediated by cleavage of Bid by multiple papain-like lysosomal cathepsins. J. Biol. Chem. 279, 3578–3587 [DOI] [PubMed] [Google Scholar]
- 12. Blomgran R., Zheng L., Stendahl O. (2007) Cathepsin-cleaved Bid promotes apoptosis in human neutrophils via oxidative stress-induced lysosomal membrane permeabilization. J. Leukocyte Biol. 81, 1213–1223 [DOI] [PubMed] [Google Scholar]
- 13. Droga-Mazovec G., Bojic L., Petelin A., Ivanova S., Romih R., Repnik U., Salvesen G. S., Stoka V., Turk V., Turk B. (2008) Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J. Biol. Chem. 283, 19140–19150 [DOI] [PubMed] [Google Scholar]
- 14. Hornung V., Bauernfeind F., Halle A., Samstad E. O., Kono H., Rock K. L., Fitzgerald K. A., Latz E. (2008) Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9, 847–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Duewell P., Kono H., Rayner K. J., Sirois C. M., Vladimer G., Bauernfeind F. G., Abela G. S., Franchi L., Nuñez G., Schnurr M., Espevik T., Lien E., Fitzgerald K. A., Rock K. L., Moore K. J., Wright S. D., Hornung V., Latz E. (2010) NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yamaguchi K., Shirakabe K., Shibuya H., Irie K., Oishi I., Ueno N., Taniguchi T., Nishida E., Matsumoto K. (1995) Identification of a member of the MAPKKK family as a potential mediator of TGF-β signal transduction. Science 270, 2008–2011 [DOI] [PubMed] [Google Scholar]
- 17. Ninomiya-Tsuji J., Kishimoto K., Hiyama A., Inoue J., Cao Z., Matsumoto K. (1999) The kinase TAK1 can activate the NIK-I κB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature 398, 252–256 [DOI] [PubMed] [Google Scholar]
- 18. Irie T., Muta T., Takeshige K. (2000) TAK1 mediates an activation signal from Toll-like receptor(s) to nuclear factor-κB in lipopolysaccharide-stimulated macrophages. FEBS Lett. 467, 160–164 [DOI] [PubMed] [Google Scholar]
- 19. Sakurai H., Miyoshi H., Toriumi W., Sugita T. (1999) Functional interactions of transforming growth factor β-activated kinase 1 with IκB kinases to stimulate NF-κB activation. J. Biol. Chem. 274, 10641–10648 [DOI] [PubMed] [Google Scholar]
- 20. Kajino T., Ren H., Iemura S., Natsume T., Stefansson B., Brautigan D. L., Matsumoto K., Ninomiya-Tsuji J. (2006) Protein phosphatase 6 down-regulates TAK1 kinase activation in the IL-1 signaling pathway. J. Biol. Chem. 281, 39891–39896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim S. I., Kwak J. H., Wang L., Choi M. E. (2008) Protein phosphatase 2A is a negative regulator of transforming growth factor-β1-induced TAK1 activation in mesangial cells. J. Biol. Chem. 283, 10753–10763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ninomiya-Tsuji J., Kajino T., Ono K., Ohtomo T., Matsumoto M., Shiina M., Mihara M., Tsuchiya M., Matsumoto K. (2003) A resorcylic acid lactone, 5Z-7-oxozeaenol, prevents inflammation by inhibiting the catalytic activity of TAK1 MAPK kinase kinase. J. Biol. Chem. 278, 18485–18490 [DOI] [PubMed] [Google Scholar]
- 23. Gong Y. N., Wang X., Wang J., Yang Z., Li S., Yang J., Liu L., Lei X., Shao F. (2010) Chemical probing reveals insights into the signaling mechanism of inflammasome activation. Cell Res. 20, 1289–1305 [DOI] [PubMed] [Google Scholar]
- 24. Compan V., Baroja-Mazo A., López-Castejón G., Gomez A. I., Martínez C. M., Angosto D., Montero M. T., Herranz A. S., Bazán E., Reimers D., Mulero V., Pelegrín P. (2012) Cell volume regulation modulates NLRP3 inflammasome activation. Immunity 37, 487–500 [DOI] [PubMed] [Google Scholar]
- 25. Juliana C., Fernandes-Alnemri T., Wu J., Datta P., Solorzano L., Yu J. W., Meng R., Quong A. A., Latz E., Scott C. P., Alnemri E. S. (2010) Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. J. Biol. Chem. 285, 9792–9802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee G. S., Subramanian N., Kim A. I., Aksentijevich I., Goldbach-Mansky R., Sacks D. B., Germain R. N., Kastner D. L., Chae J. J. (2012) The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 492, 123–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ishitani T., Kishida S., Hyodo-Miura J., Ueno N., Yasuda J., Waterman M., Shibuya H., Moon R. T., Ninomiya-Tsuji J., Matsumoto K. (2003) The TAK1-NLK mitogen-activated protein kinase cascade functions in the Wnt-5a/Ca2+ pathway to antagonize Wnt/β-catenin signaling. Mol. Cell. Biol. 23, 131–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu X., Yao M., Li N., Wang C., Zheng Y., Cao X. (2008) CaMKII promotes TLR-triggered proinflammatory cytokine and type I interferon production by directly binding and activating TAK1 and IRF3 in macrophages. Blood 112, 4961–4970 [DOI] [PubMed] [Google Scholar]
- 29. Zhou R., Yazdi A. S., Menu P., Tschopp J. (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 [DOI] [PubMed] [Google Scholar]
- 30. Nakahira K., Haspel J. A., Rathinam V. A., Lee S. J., Dolinay T., Lam H. C., Englert J. A., Rabinovitch M., Cernadas M., Kim H. P., Fitzgerald K. A., Ryter S. W., Choi A. M. (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shimada K., Crother T. R., Karlin J., Dagvadorj J., Chiba N., Chen S., Ramanujan V. K., Wolf A. J., Vergnes L., Ojcius D. M., Rentsendorj A., Vargas M., Guerrero C., Wang Y., Fitzgerald K. A., Underhill D. M., Town T., Arditi M. (2012) Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Subramanian N., Natarajan K., Clatworthy M. R., Wang Z., Germain R. N. (2013) The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 153, 348–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Park S., Juliana C., Hong S., Datta P., Hwang I., Fernandes-Alnemri T., Yu J. W., Alnemri E. S. (2013) The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J. Immunol. 191, 4358–4366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Iyer S. S., He Q., Janczy J. R., Elliott E. I., Zhong Z., Olivier A. K., Sadler J. J., Knepper-Adrian V., Han R., Qiao L., Eisenbarth S. C., Nauseef W. M., Cassel S. L., Sutterwala F. S. (2013) Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39, 311–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Misawa T., Takahama M., Kozaki T., Lee H., Zou J., Saitoh T., Akira S. (2013) Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 14, 454–460 [DOI] [PubMed] [Google Scholar]
- 36. Yamamoto K., Ichijo H., Korsmeyer S. J. (1999) BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G2/M. Mol. Cell. Biol. 19, 8469–8478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kharbanda S., Saxena S., Yoshida K., Pandey P., Kaneki M., Wang Q., Cheng K., Chen Y. N., Campbell A., Sudha T., Yuan Z. M., Narula J., Weichselbaum R., Nalin C., Kufe D. (2000) Translocation of SAPK/JNK to mitochondria and interaction with Bcl-x(L) in response to DNA damage. J. Biol. Chem. 275, 322–327 [DOI] [PubMed] [Google Scholar]
- 38. Kim B. J., Ryu S. W., Song B. J. (2006) JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J. Biol. Chem. 281, 21256–21265 [DOI] [PubMed] [Google Scholar]
- 39. Zhong Z., Zhai Y., Liang S., Mori Y., Han R., Sutterwala F. S., Qiao L. (2013) TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat. Commun. 4, 1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Murakami T., Ockinger J., Yu J., Byles V., McColl A., Hofer A. M., Horng T. (2012) Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. U.S.A. 109, 11282–11287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Triantafilou K., Hughes T. R., Triantafilou M., Morgan B. P. (2013) The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J. Cell Sci. 126, 2903–2913 [DOI] [PubMed] [Google Scholar]
- 42. Hung Y. H., Chen L. M., Yang J. Y., Yang W. Y. (2013) Spatiotemporally controlled induction of autophagy-mediated lysosome turnover. Nat. Commun. 4, 2111. [DOI] [PubMed] [Google Scholar]
- 43. Maejima I., Takahashi A., Omori H., Kimura T., Takabatake Y., Saitoh T., Yamamoto A., Hamasaki M., Noda T., Isaka Y., Yoshimori T. (2013) Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 32, 2336–2347 [DOI] [PMC free article] [PubMed] [Google Scholar]