

Background: LRRK2 kinase activity is linked to neurodegeneration.
Results: Novel small molecule inhibitors provide insight into the structure and function of the LRRK2 kinase domain.
Conclusion: A unique ATP-binding pocket structure in LRRK2 allows for potent and specific activity-selective and mutant-selective small molecules.
Significance: Novel structure-activity relationships can be exploited for the development of new classes of kinase inhibitors.
Keywords: High-throughput Screening (HTS), Leucine-rich Repeat Kinase 2 (LRRK2), Neurodegeneration, Protein Kinase, Recombinant Protein Expression, Alphascreen, Park8, Kinase Inhibitor
Abstract
Pathogenic mutations in the LRRK2 gene can cause late-onset Parkinson disease. The most common mutation, G2019S, resides in the kinase domain and enhances activity. LRRK2 possesses the unique property of cis-autophosphorylation of its own GTPase domain. Because high-resolution structures of the human LRRK2 kinase domain are not available, we used novel high-throughput assays that measured both cis-autophosphorylation and trans-peptide phosphorylation to probe the ATP-binding pocket. We disclose hundreds of commercially available activity-selective LRRK2 kinase inhibitors. Some compounds inhibit cis-autophosphorylation more strongly than trans-peptide phosphorylation, and other compounds inhibit G2019S-LRRK2 more strongly than WT-LRRK2. Through exploitation of structure-activity relationships revealed through high-throughput analyses, we identified a useful probe inhibitor, SRI-29132 (11). SRI-29132 is exquisitely selective for LRRK2 kinase activity and is effective in attenuating proinflammatory responses in macrophages and rescuing neurite retraction phenotypes in neurons. Furthermore, the compound demonstrates excellent potency, is highly blood-brain barrier-permeant, but suffers from rapid first-pass metabolism. Despite the observed selectivity of SRI-29132, docking models highlighted critical interactions with residues conserved in many protein kinases, implying a unique structural configuration for the LRRK2 ATP-binding pocket. Although the human LRRK2 kinase domain is unstable and insoluble, we demonstrate that the LRRK2 homolog from ameba can be mutated to approximate some aspects of the human LRRK2 ATP-binding pocket. Our results provide a rich resource for LRRK2 small molecule inhibitor development. More broadly, our results provide a precedent for the functional interrogation of ATP-binding pockets when traditional approaches to ascertain structure prove difficult.
Introduction
Mutations in the Leucine-rich repeat kinase 2 (LRRK2) gene were identified in familial Parkinson disease in late 2004 (1, 2), and in the next year, the most common LRRK2 mutation G2019S was found to increase LRRK2 kinase activity in autophosphorylation assays (3). Kinetic kinase assays that measure phosphorylation of generic peptide substrates also show enhanced kinase activities resulting from the G2019S mutation (4).
In addition to the protein kinase domain, LRRK2 is comprised of a unique arrangement of conserved protein domain motifs that also harbor pathogenic mutations (5). Although no protein substrate of LRRK2 has been demonstrated convincingly to be dependent on LRRK2 kinase activity in vivo, measurements of LRRK2 autophosphorylation in vitro, or directly from cell lysates, using autophosphorylation-specific antibodies show that pathological mutations, whether in the GTPase domain (e.g. R1441C) or kinase domain (e.g. G2019S or I2020T), activate, or otherwise enhance, the proportion of autophosphorylated LRRK2 (6–8). However, apart from the G2019S mutation that seems to enhance all aspects of kinase activity, other pathogenic mutations have variable effects on kinase activity, for example the R1441C and I2020T mutations that enhance the proportion of enzyme in an active state without affecting other kinetic parameters (9, 10).
Overexpression of mutant (e.g. G2019S) LRRK2 protein in mature neurons in culture results in toxicity that can be blocked with mutations in conserved residues in the kinase domain that abolish kinase activity (8, 11, 12). Complete ablation of LRRK2 expression in rats and mice is overall well tolerated, although some pathologies have been noted (13–15). Nevertheless, inhibition of LRRK2 kinase activity for prolonged periods of time may be tolerated poorly so that small molecule inhibitors that selectively target mutant LRRK2 (e.g. G2019S) may be desired as potential therapeutics for Parkinson disease and related disorders.
The first wave of LRRK2 kinase inhibitors discovered with in vitro assays includes several non-selective kinase inhibitors that all showed excellent potency. They include staurosporine (16, 17) sunitinib (18), CZC-25146 (19), and TAE684 (20) Lower-potency and non-selective LRRK2 kinase inhibitors have also been used to block toxicities caused by LRRK2 overexpression in model systems (21). More selective inhibitors have been discovered on several distinct small molecule scaffolds that include LRRK2 inhibitor 1(LRRK2-IN-1) (22), GSK2578215A (23), and HG-10-102-01 (24). However, none of these molecules have desirable pharmacokinetics for in vivo use.
A major limitation in the field is the lack of structural information on the LRRK2 kinase domain that would otherwise guide the refinement of these molecules into more useful compounds for preclinical discovery. We and others have devoted significant resources to purify and crystallize the human LRRK2 kinase domain, so far without success. A high-resolution structure of the ameba LRRK2 kinase domain homolog (ROCO4) has been described and proposed as a platform for understanding the human LRRK2 kinase domain (25). However, selective LRRK2 inhibitors have not been demonstrated to interact with ROCO4, thereby limiting the utility of the structure for LRRK2 small molecule inhibitor campaigns.
Besides the lack of high-resolution structures of the LRRK2 protein kinase, another major limitation in small molecule design is deciding which LRRK2-associated kinase activity should be utilized to test efficacy. LRRK2 possesses significant intrinsic cis-autophosphorylation (and not trans-autophosphorylation) (26) activities and only weakly phosphorylates other substrate proteins in vitro, such as generic substrates and interacting proteins (3, 27). In designing LRRK2 small molecule kinase inhibitors, it may be desirable to preferentially target trans over cis LRRK2 kinase activities for therapeutic effects.
There are no studies that systematically compare the efficacies of different LRRK2 kinase activities with respect to small molecule inhibitors. The kinase domain may be fluidic in conformation, adopting different structural configurations depending on the substrate. Small molecule inhibitors would interact differently depending on the conformation of the ATP pocket and could, theoretically, be used as probes to understand the conformations critical for catalysis.
Here we use novel high-throughput screening assays to systematically compare small molecules that bind to the LRRK2 ATP pocket. On the basis of our results, it is possible to identify activity-selective and mutant-selective small molecule inhibitors. Although we find that the ameba LRRK2 homolog is a poor model to understand the human LRRK2 ATP pocket, we show the feasibility of a mutagenesis approach to correct aspects of the structure to converge toward the human LRRK2 ATP pocket.
Finally, we disclose the identity of hundreds of structurally diverse molecules that likely bind to the LRRK2 ATP pocket, along with the discovery of a novel and efficacious brain-permeable LRRK2 inhibitor, SRI-29132 (11). These findings should provide a resource for structural interrogation of the LRRK2 kinase domain as well as potential leads for small molecule inhibitor programs.
EXPERIMENTAL PROCEDURES
Recombinant Proteins and Peptide Substrate, Protein Expression, and Purifications
All proteins used for experiments were of >95% purity, as assessed by Coomassie stain and/or mass spectrometry. Human recombinant WT and G2019S-LRRK2 protein purified from SF9 insect cells was purchased from Invitrogen (Δ970, N-terminal GST tag). The LRRK2 peptide substrate H-RLGAWRFTTLRRARQGNTKQR-OH was purchased from GenScript. Proteins were assessed to at least 95% purity by Coomassie stain and/or mass spectrometry.
For bacterial expression vectors, GST tags were cloned from pGEX (GE Healthcare Life Science) into pET21a(+) plasmids (Novagen). Human, ameba, zebrafish, frog, and “mosaic” LRRK2 kinase domains were synthesized by GenScript with codon optimization and cloned into the pet21a(+)-GST vector. The entropic bristle tags N250 and C120 were subcloned from plasmids made available by Molecular Kinetics Inc. Plasmids were expressed in BL21 codon plus Escherichia coli (Agilent Technologies) by induction with 0.2 mm isopropyl β-d-1-thiogalactopyranoside for 16 h (h) at 16 °C. Cells were harvested by centrifugation and lysed by sonication or French press in buffer containing 50 mm Tris (pH7.5), 150 mm NaCl, 10 mm MgCl2 and 10% Glycerol. Insoluble fractions were derived from supernatants isolated after ultracentrifugation (100 thousand relative centrifugal force, kRCF), and pellets were collected into 2× Laemmli buffer and sonicated.
Supernatants were loaded onto GST resins (Genscript), washed with lysis buffer (described above), and eluted with lysis buffer containing 10 mm reduced glutathione (Sigma). The eluted protein was loaded onto a Superdex 200 size exclusion column (GE Healthcare) in lysis buffer. The purified protein was then concentrated with modified Amicon filtration units with 3 kDa molecular weight cut-off. The human LRRK2 ROC (GTPase) domain was purified as described previously (28).
Kinase Assays
For high-throughput trans-peptide kinase phosphorylation assays, the ADP-Hunter assay kit (DiscoverRx) was used in 2.5-μl assay format (1536 well plates) or 10-μl assay format (384 well plates) in kinase assay buffer (50 mm Tris (pH 7.5), 10 mm MgCl2, 1 mm EGTA, 0.01% Tween 20, and 2% DMSO2). Reactions were initiated at room temperature by adding LRRK2 protein (30 nm) together with 100 μm ATP. LRRK2 peptide substrate was included, as indicated, at 5 μm. Background wells included all reagents except enzyme. Reactions were carried out for 4 h and stopped by addition of ADP-Hunter Plus reagents A and B according to the instructions of the manufacturer. Fluorescence was determined with an EnVision multilabel reader (PerkinElmer Life Sciences) with excitation set at 530 nm and emission at 590 nm. The compound concentration used in the initial high-throughput screen (25,856 compounds) was 10 μg/ml.
For cis-autophosphorylation high-throughput assays, AlphaScreen components were combined into kinase reaction buffer (50 mm HEPES (pH 7.5), 10 mm MgCl2, 2 mm DTT, 1 mm EGTA, 0.01% Tween 20, and 2% DMSO). LRRK2 enzyme was used at a final concentration of 10 nm together with 10 μm ATP (∼Km ATP) for a total volume of 2.5 μl in 1536-well plates. Enzymatic reactions were carried out for 30 min at room temperature and then stopped by adding AlphaScreen protein A acceptor beads (20 μg/ml) together with the rabbit anti-p1503 LRRK2 antibody, generated in-house as described and used at a final concentration of 0.625 nm. Following a 30-min incubation at room temperature, GSH donor beads were added to 20 μg/ml. Background readings were generated from reactions including all reagents except for ATP. The fluorescence signal was determined on an EnVision multilabel reader (PerkinElmer Life Sciences) using the factory-preset AlphaScreen settings.
For recombinant kinase proteins isolated from E. coli, reactions were mixed with 1 μm purified kinase in buffer containing 50 mm Tris (pH 7.5), 150 mm NaCl, 10 mm MgCl2 with 100 μm ATP, and 3 μm purified human LRRK2 ROC (GTPase) domain. Kinase reactions were collected and run on an SDS-page gel, followed by Western blot analysis using phosphorylation-specific antibodies.
Commercially Available LRRK2 Inhibitors
The Kinase Inhibitor Library (25,856 compounds) designed around known ATP competitive and allosteric kinase inhibitors was purchased from Life Chemicals. Because these chemicals likely assume different modes of inhibition (e.g. allosteric, ATP-competitive, mixed competitive) and we utilized functional (substrate-based) kinase assays, half-maximal inhibitory concentrations were plotted and are presented in this manuscript versus calculations of Ki that might incorrectly assume a binding mode. Stocks (10 mg/ml) of chemicals were maintained in DMSO at −80 °C. Known LRRK2 inhibitors were purchased from Xcessbio, Sigma, or Calbiochem.
In Silico Analysis of ATP-binding Pockets
Three-dimensional models were generated by CHEMSKETCH. Three-dimensional model human LRRK2 ATP-binding pockets were generated with the SWISS-MODEL homology modeling server on the basis of the ameba LRRK2 crystal structure (PDB code 4F0F). Surface electrostatic composition was modeled using PyMOL. Compound 11 (SRI-29132) docking models were generated by accurate docking functions in iGEMDOCK and interrogated further with PyMOL. Two-dimensional ligand interaction diagrams were generated with LigPlus on the basis of the iGEMDOCK docking model. Sequence alignments were made using SPEM (Sequence and Secondary Structure Profile Enhanced Multiple Alignment, Laboratory of Biophysics and Bioinformatics, University of Buffalo).
Macrophage Culture
Thioglycollate-elicited macrophages, rich in endogenous LRRK2, were harvested from 8- to 12-week-old bacterial artificial chromosome transgenic mice overexpressing G2019S LRRK2 (Jackson strain B6.Cg-Tg(Lrrk2*G2019S)2Yue/J), their non-transgenic litter-mates, or LRRK2 knockout animals (animals obtained courtesy of Matthew Farrer and Heather Melrose, Mayo Jacksonville). 1.5 ml of 4% brewer's yeast thioglycollate medium (Fluka) was injected into the peritoneal cavity.
72 h later, the peritoneal cavity was gavaged by injecting 10 ml of ice-cold PBS followed by withdrawing the PBS containing the cell suspension. Macrophages (5- to 10-μm diameter) were counted with a Z2 particle counter (Beckman Coulter). 80,000 cells/well were plated into 24-well tissue culture-treated dishes (Nunclon) in DMEM supplemented with 10% FBS, 100 μg/ml streptomycin, and 5 mm Glutamax (Invitrogen).
Cells were allowed to rest overnight before treatment with the indicated kinase inhibitor or DMSO control. Cells were maintained in the presence of the indicated concentration of inhibitor for 3 days. Cells were then treated with 100 ng/ml of ultrapure LPS (Invivogen) for 6 h in the presence of the inhibitor. Medium was removed and snap-frozen until analysis for TNF through ELISA.
Cells were lysed from the bottom of the well with 2× Laemmli buffer and sonicated. TNF was assayed through Ready-Set-Go anti mouse TNF ELISA (eBioscience) according to the instructions of the manufacturer. Dilutions were determined empirically. For Western blot analysis of cell lysates, 10 μg of protein/lane was loaded onto 10 cm of 7.5% Tris-Glycine Extended Gels (Bio-Rad) and electrophoresed at 120 V for 65 min.
Primary Neuron Culture and Assay
Dissociated hippocampal neurons were plated onto poly-d-lysine-coated coverslips in a 24-well plate at 5 × 105 cells/dish, as described previously (29). Neurons were cultured in neuronal media (Neurobasal, B27, and glutamax, Invitrogen). Neurons were transfected with LRRK2 and eGFP plasmids, as described previously (30), using Lipofectamine 2000 (Invitrogen) according to the instructions of the manufacturer. Neurons were transfected 5 days after plating cells. Four hours after adding the Lipofectamine 2000/DNA mixture, the entire medium was exchanged for fresh neuronal medium.
Seventy-two hours post-transfection, images were recorded on a Carl Zeiss Cell Observer microscope with a ×10 objective in a PeCon incubation chamber with a cool LED light source (Colibri, Carl Zeiss). Approximately 100 neurons/condition were imaged across three coverslips, and neurite length was quantified in an unbiased semiautomated manner using the HCA-vision Neurite analysis module as described previously (31).
Western Blots
Protein was transferred to PVDF overnight. Membranes were blocked with LiCor blocking buffer for 1 h at room temperature and probed with N241/A (anti-LRRK2, Neuromab), 1:2000 UDD2 (anti-p935 LRRK2, Epitomics), or anti β-actin (Santa Cruz Biotechnology) overnight. LiCor secondary antibodies included IRDye 800CW donkey anti-mouse and anti-rabbit and IRDye 680RD donkey anti-mouse and donkey anti-rabbit. Membranes were scanned on a LiCor Odyssey system according to the manufacturer's specifications.
Medicinal Chemistry
Chemicals
All chemicals used, unless noted otherwise, were purchased from Sigma-Aldrich.
Statistics
The high-throughput data were imported into ActivityBase (IDBS) for analysis and reporting. Results of the single-concentration screen were expressed as percent inhibition, which was calculated as 100 × (1 − (test well − median background control) / (median full reaction control − median background control). Any compound with an inhibition ≥50.75% was designated as active and pursued for concentration-response analysis. The concentration-response data were analyzed using a four-parameter logistic fit (Excel Fit equation 205) with the maximum and minimum locked at 100 and 0, respectively. Experiments other than high-throughput were analyzed with non-linear regression analyses and comparisons of means with one-way analysis of variance tests with Tukey's post hoc test using GraphPad software.
RESULTS
Identification of G2019S-selective LRRK2 Inhibitors
The G2019S-LRRK2 mutation enhances LRRK2 kinase activity and is the most common mutation in LRRK2 that leads to Parkinson disease. Several high-throughput assays have been developed to identify small molecule LRRK2 inhibitors that generally revolve around tracking phosphate incorporation into generic peptide substrates.
We developed a novel high-throughput screen insensitive to the nature of the substrate by tracking ADP formation (directly proportional to kinase activity) in LRRK2 kinase reactions in 1536-well plates using ADP-Hunter technology. This assay showed a Z′ score of 0.75 ± 0.07 with typical signal-to-background ratios of 6.5. WT-LRRK2 demonstrated a Km for ATP of 12.7 μm, with the pathogenic mutant G2019S-LRRK2 showing a similar but slightly increased affinity for ATP (Km = 10.4 μm) (Fig. 1, A and B). Evaluation of a known ATP-competitive multikinase inhibitor, sunitinib, showed a 10-fold greater potency for G2019S-LRRK2 compared with WT-LRRK2 (Fig. 1, C and D). Sunitinib, therefore, shows a higher affinity for G2019S-LRRK2 because the affinity of ATP for WT-LRRK2 and G2019S-LRRK2 is similar. This result is consistent with past studies using different peptide-based LRRK2 kinase assays (18).
FIGURE 1.

Identification of G2019S-selective inhibitors. Measurements of LRRK2 kinase activity through quantification of ADP formation. 5 μm of LRRK2 peptide substrate was included in all conditions. A and B, determination of ATP affinity for WT (A) and G2019S-LRRK2 protein (B) with respect to LRRK2-mediated trans-peptide phosphorylation. C and D, potency of the non-selective kinase inhibitor sunitinib for WT (C) and G2019S-LRRK2 kinase (D). E, the IC50 was calculated from compounds that demonstrated inhibition of either WT or G2019S-LRRK2 kinase activity. A list of these compounds is provided in supplemental Table 1. F, examples of commercially available triazolopyridazine compounds that demonstrate selectivity for G2019S-LRRK2 kinase compared with WT-LRRK2 kinase activities. The indicated values were derived from at least three independent assays, and data are mean ± S.D.
The strong preference of sunitinib for G2019S-LRRK2 inhibition shows that selectivity in more potent and specific compounds might be possible. The ADP-Hunter assay we developed was applied to a large commercial library of small molecules designed primarily around known efficacious ATP-competitive kinase inhibitors in addition to allosteric inhibitors. In the first-pass screen of more than 25,000 compounds, 620 demonstrated significant inhibition of G2019S-LRRK2 activity (supplemental Table 1). These were followed into a 10-point concentration-response assay ranging from 0.1 nm to 30 μm in both WT-LRRK2 and G2019S-LRRK2 experiments.
IC50 values plotted for each active compound showed a slight correlation for inhibition between WT and G2019S-LRRK2 (R2 = 0.33), but, overall, there was a strong tendency for more potent inhibition of G2019S-LRRK2 compared with WT-LRRK2 (Fig. 1E, p < 0.0001, slope = 0.32 ± 0.02).
For LRRK2 trans-peptide phosphorylation, many compounds failed to inhibit WT-LRRK2 but showed strong inhibition of G2019S-LRRK2. One series of structurally related commercially available compounds on the basis of a triazolopyridazine motif demonstrated a strong preference for inhibition of G2019S-LRRK2. Four examples are shown in Fig. 1F.
LRRK2 Autophosphorylation-selective Inhibitors
Pathogenic mutations in LRRK2, such as R1441C and G2019S, enhance the proportion of autophosphorylated LRRK2 to total LRRK2 in kinase assays using recombinant protein derived from cell lines. This has been demonstrated at two autophosphorylation sites in particular, Thr(P)-1503 and Ser(P)-1292 (6, 7). In the ADP-Hunter assay, we noticed that significant ADP formation occurred over time in the absence of any peptide substrate (Fig. 2A). This can be explained by autophosphorylation activity because no ADP formation could be observed in this assay with kinase-dead LRRK2 (D1994A) recombinant protein.
FIGURE 2.

Identification of activity-selective LRRK2 inhibitors. A, measurement of ADP formation with the addition of 5 μm of LRRK2 peptide substrate or without (Autophos). B and C, determination of ATP affinity for WT (B) and G2019S-LRRK2 protein (C) with respect to LRRK2 cis-autophosphorylation. D, IC50 values were calculated from compounds that demonstrated measurable inhibition of either WT or G2019S-LRRK2 autophosphorylation activity. E, cis-autophosphorylation inhibition plotted against trans-peptide inhibition for the same set of compounds. A list of compound identities is given in supplemental Table 1. F and G, examples of commercially available compounds selective for inhibition of cis-autophosphorylation (F) or trans-peptide phosphorylation (G). The indicated values were derived from at least three independent assays, and data are mean ± S.D.
The autophosphorylation signal represented only ∼10% of total ADP formed at the 30-min time point (Fig. 2A). Because of the reduced amount of ADP generated, the overall signal-to-background ratio was insufficient to generate favorable reliability in a high-throughput assay.
We previously described the development and characterization of a highly specific LRRK2 autophosphorylation antibody directed to the Thr(P)-1503 residue. The Thr(P)-1503 residue is localized to an α helix peripheral to the guanosine-binding pocket of the ROC (GTPase) domain. We used this antibody to design a novel high-throughput assay to track autophosphorylation using AlphaScreen technology (Fig. 2). In this assay, the affinity for ATP (with respect to autophosphorylation) was slightly higher than that for trans-peptide phosphorylation but overall comparable at 9.5 μm ATP for WT-LRRK2 and 7.4 μm ATP for G2019S-LRRK2 (Fig. 2, B and C).
In 1536-well plates, the Z′ score of the AlphaScreen autophosphorylation assay was 0.77 ± 0.07, with typical signal-to-background ratios of 6.0. Each of the 620 LRRK2 trans-peptide inhibitors (Fig. 1E and supplemental Table 1) was evaluated in a 10-point concentration response curve for both WT and G2019S-LRRK2 cis-autophosphorylation inhibition.
As opposed to trans-peptide phosphorylation inhibition, these LRRK2 inhibitor compounds demonstrated markedly uniform inhibition potencies for both WT and G2019S-LRRK2 kinase activity (Fig. 2D, R2 = 0.91, slope = 0.96 ± 0.01. Therefore, many inhibitors that show G2019S-LRRK2 preference in trans-peptide phosphorylation had no such selectivity in LRRK2 cis-autophosphorylation activity. The LRRK2 inhibitor sunitinib, which demonstrated a 10-fold preference for G2019S-LRRK2 inhibition in trans-peptide phosphorylation assays (Fig. 1), demonstrated IC50 values of 266 and 231 nm for WT and G2019S-LRRK2 inhibition of cis-autophosphorylation activity, respectively.
These results suggest that the ATP pocket where the inhibitor compounds interact may be structurally unique, depending on trans or cis phosphosubstrates, modified further by the presence of the pathogenic G2019S mutation localized to the activation loop. To test this hypothesis, inhibition potencies for all active kinase inhibitors (Fig. 1) were compared for LRRK2 trans-peptide or cis-autophosphorylation (Fig. 2E).
A poor correlation could be observed for cis versus trans inhibition (R2 = 0.13), demonstrating that potency of inhibition of trans-peptide phosphorylation did not predict well potency of cis-autophosphorylation inhibition (Fig. 2E). Fig. 2F shows examples of three commercially available compounds that show a strong preference for inhibition of cis-autophosphorylation activity, whereas Fig. 2G shows examples of compounds highly selective for trans-peptide inhibition.
Optimization of the Triazolopyridazine (TPZ) LRRK2 Inhibitor Series
Most compounds showed reduced potency as well as a loss of preference for G2019S-LRRK2 inhibition in cis-autophosphorylation assays. However, the TPZ series showed good potency in blocking both trans-peptide and cis-autophosphorylation assays and retained the preference for G2019S-LRRK2 inhibition. The major potency determinant of the series, on the basis of evaluation of commercially available compounds, revolves around modifications in the 6-thioether side chain and the pendant 3-heteroaryl or phenyl ring linked to the [1,2,4]triazolo[4,3-b]pyridazine scaffold. During the course of our experiments, another group working independently reported the discovery of a similar chemical series (32).
Fig. 3 illustrates 20 synthesized compounds with a range of activities for LRRK2 inhibition that exemplify structure-activity relationships. All synthesized compounds are presented in supplemental Table 2. Systematic modification of the alkyl substituent of the thioether side chain altered the inhibition potency for both trans-phosphorylation and cis-autophosphorylation and modified the selectivity toward G2019S-LRRK2 (Fig. 3A).
FIGURE 3.

Optimization of potency for G2019S-selective TPZ-LRRK2 inhibitors. G2019S-LRRK2 kinase inhibition (IC50) for trans-peptide phosphorylation (peptide phos) or cis-autophosphorylation (Autophos) activities. Ratios for G2019S-LRRK2 to WT-LRRK2 autophosphorylation inhibition are given. A and B, side chain (A) and pendant (B) heterocycle structure-activity-relationships with respect to potency and G2019S-LRRK2 selectivity. The indicated values were derived from at least three independent assays. Chemical synthesis schemes are given in the supplemental information and Figs. 1 and 2.
Amide analogs 5-12 give 9, 10 (SRI-29451) and 11 (SRI-29132) as the most effective compounds in this series. Amides 8-12 retain some of the selectivity seen in original TPZ compounds (Fig. 1F). In general, a 2-fold preference for G2019S-LRRK2 over WT-LRRK2 is retained as potency is enhanced. Efforts to replace the 3-thiophene with substituted thiophenes (13), another heterocycle (14-16), or a substituted phenyl (17-20) had no significant effect on altering the selectivity toward G2019S-LRRK2 but decreased overall potency (Fig. 3B).
Modeling TPZ Compounds in the LRRK2 ATP Pocket
Recently, a high-resolution structure was identified on the basis of the LRRK2 kinase homolog in ameba (25). The structure has been proposed as a model for small molecule design and the human LRRK2 ATP pocket. Superimposing the human LRRK2 kinase sequence into this structure revealed a docking model for 11 where the polarizable sulfur atom of the thioether side chain may interact with the positively charged γ-amine of the catalytic lysine residue at 1906 in LRRK2 (Fig. 4).
FIGURE 4.
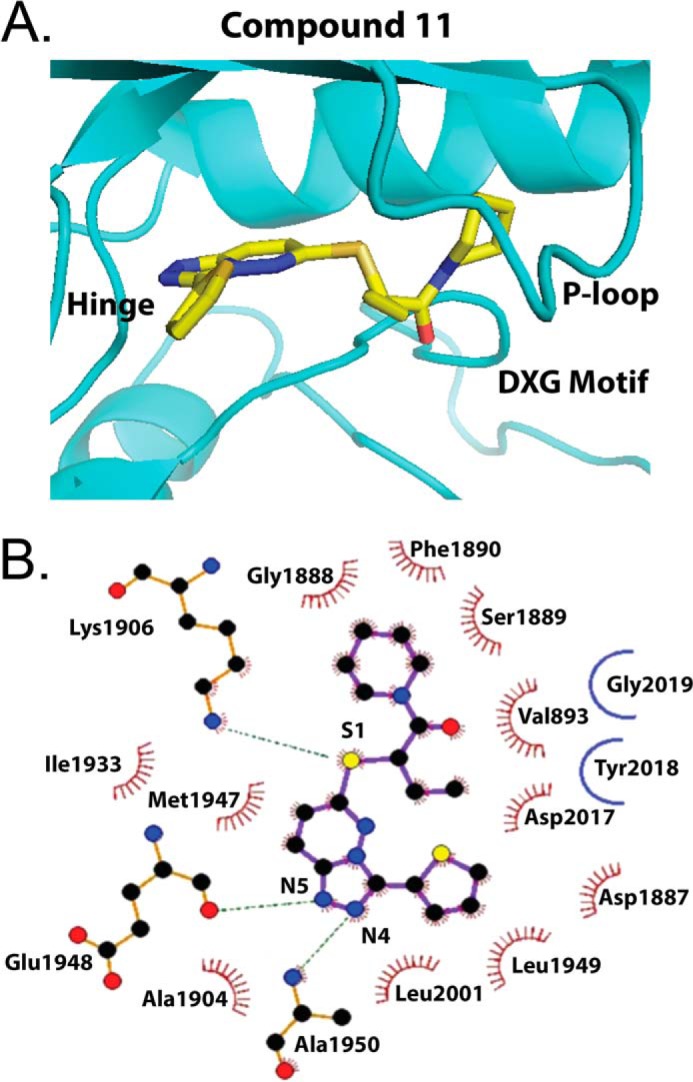
Homology docking model of TPZ compound 11 in the LRRK2 ATP pocket on the basis of the ameba LRRK2 crystal structure (PDB code 4F0F). Shown are a three-dimensional model (A) and ligand interaction diagram (B) highlighting compound 11 interaction between the piperidine ring and P loop, the thioether interaction with the conserved ATP pocket Lys-1906, and interaction of the [1,2,4]triazolo ring with the hinge alanine and glutamate residues. The atoms are color coded (gray, carbon; red, oxygen; blue, nitrogen; yellow, sulfur). Residues in hydrophobic contact with 11 are represented by red semicircles with radiating spokes. Hydrogen-bonding is represented by green dashed lines. Gly-2019 and Tyr-2018 do not interact directly with 11 in this model, but the pathogenic Gly-to-Ser-2019 mutation may stabilize a DXG in conformation to promote 11 docking (25).
The triazolopyridazine ring forms a hydrogen-bonding network through H-bond acceptor interactions by N4 and N5 with the backbone amide NH of the Glu-1948 and Ala-1950 residues in the hinge region, whereas the cyclic amide terminus of the 6-thioether side chain packs squarely against Phe-1890 in a hydrophobic interaction with the highly mobile P loop. Compound 12, which has an oxygen atom replaced for the carbon atom on the piperidine ring, interrupts this hydrophobic interaction.
Many protein kinases have a strongly evolutionarily conserved DFG motif critical for “in” or “out” catalytic conformations important for kinase activation (33). LRRK2 has a unique DYG motif that is further modified to DYS by the pathogenic G2019S mutation (3). G2019S-LRRK2 has been hypothesized to have a hydrogen-bond-stabilized magnesium binding motif in an out conformation (9, 25, 34).
The piperidine ring of 10 and 11 may be better stabilized in the hydrophobic pocket introduced by the more rigid LRRK2-specific DYS motif (Fig. 4). WT-LRRK2, on the other hand, has a more flexible magnesium binding motif (DYG) that might interfere with piperidine ring stabilization.
Evaluation of TPZ Compounds as Candidate LRRK2 Inhibitors
Two of the more potent TPZ compounds, 10 (SRI-29451) and 11 (SRI-29132), were selected for further evaluation. Both compounds demonstrated exceptional solubility in water and have good predicted gut and blood-brain barrier permeability in a parallel artificial membrane permeation assay (Fig. 5A), although compound 11 has substantially higher permeability values than 10. Both compounds are poor P-glycoprotein substrates in MDCK-MDR-1 assays.
FIGURE 5.

Drug properties, selectivity, and potency of TPZ compounds. A, absorption, distribution, metabolism, and excretion (ADME) characteristics of two selected TPZ compounds (Comp) that demonstrate superior solubility (Sol) but poor rat and human liver microsome stability, with percent unmodified compound given after 1 h in the presence of NADPH+. In NADPH− reactions, 100% of compound remained after 1 h in all experiments. Corresponding mass spectrometry analyses of NADPH+ metabolites are given in supplemental Fig. 3. Apparent membrane permeability constants (Papp) are given from MDR1-MDCK and Caco-2 permeability assays (gut) and blood-brain barrier (BBB) parallel artificial membrane permeation assays (PAMPA). B, compound 11 was evaluated at 1 μm (shown) and 10 μm in commercial kinase assays involving the entire constituencies of the kinase collections available through EMD Millipore and Invitrogen. All assays were performed at Km(ATP) of the respective kinase. C, three kinases other than WT or G2019S-LRRK2 (glycogen synthase kinase 3, aurora kinase A, and phosphatidylinositol 3 kinase γ) were inhibited more than 50% at 10 μm as indicated. D–G, relative potencies of compounds 10 and 11 in LRRK2 cis-autophosphorylation kinase assays. The indicated values were derived from at least three independent assays.
Although both 10 and 11 were completely stable in microsomes to 1-h incubation time points, in the presence of the NADPH cofactor with the microsomes, both compounds were rapidly oxidized and inactivated. Mass spectroscopy analysis of time courses of resultant metabolites from these assays show oxidation of the sulfur atom of the critical 6-thioether side chain, potentially creating diastereomers of the sulfoxide (supplemental Fig. 3).
To determine kinome selectivity, compound 11 was submitted to kinase activity assays in commercial panels of protein and lipid kinases that represent most of the known kinome. Despite docking models suggesting the critical interaction of 11 with residues conserved in nearly every protein kinase (e.g. the conserved catalytic lysine), the selectivity profile at 1 μm concentration of 11 was exquisite because only WT and G2019S-LRRK2 kinases were fully inhibited in a screen that involved over 600 kinases (Fig. 5B). At the 10 μm dose, three other kinases were significantly (but not completely) inhibited: glycogen synthase kinase 3, aurora kinase A, and phosphatidylinositol 3 kinase γ.
Compounds 10 and 11 demonstrate an approximate 2-fold selectivity for G2019S-LRRK2 over WT-LRRK2, with 10 being twice as potent as 11 in vitro. Both compounds have a striking preference (∼30-fold) for the inhibition of autophosphorylation activity (Fig. 5, E and G) compared with trans-peptide phosphorylation activity (Fig. 5, D and F). Because the Km for ATP is comparable in both the trans and cis phosphorylation assays, these compounds highlight conformational differences in the ATP pocket with respect to cis-autophosphorylation and trans-peptide kinase activity.
Previously, we and others found that primary macrophages express high levels of endogenous LRRK2 protein (35–37). Macrophages are usually challenging cells because they express high levels of P-glycoprotein (38), but MDCK-MDR-1 assay results suggest that 10 and 11 are poor P-glycoprotein substrates (Fig. 5A). To determine TPZ compound efficacy in vitro in targeting macrophage-derived endogenous LRRK2, several concentrations of 10 and 11 were incubated with cultured cells derived from mBAC-transgenic (WT or G2019S-LRRK2, Fig. 6B) or knockout-LRRK2 mice (Fig. 6C) for 3 days, and then these cells were stimulated with LPS (100 ng/ml) for 6 h to drive a proinflammatory response.
FIGURE 6.
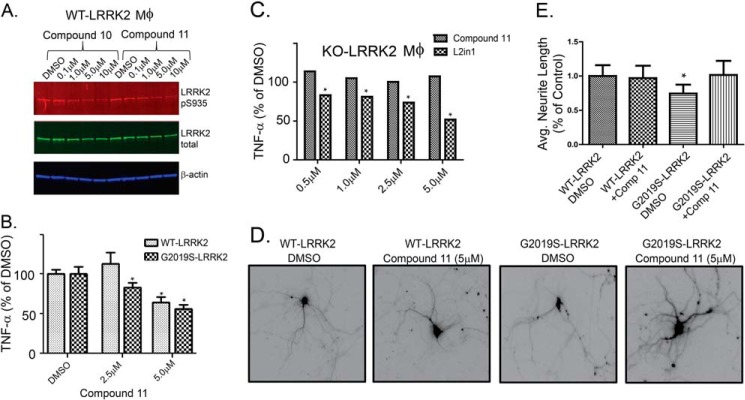
Efficacy of TPZ compounds 10 and 11 in vitro. A, the indicated concentration of compound was applied to primary macrophages for 72 h and stimulated with LPS (100 ng/ml) for 6 h, and then lysates were evaluated by Western blot analysis for Ser(P)-935-LRRK2 and total LRRK2 levels. B, supernatants from LPS-exposed macrophages also treated with the indicated dose of compound 11 were evaluated for TNF release. Macrophages were derived from WT mice and G2019S-BAC mice (B) or knockout LRRK2 mice (C). Compound 11 demonstrated efficacy (*, p < 0.05) in reducing TNF release at a 2.5 μm dose in G2019S-LRRK2 macrophages but not in knockout LRRK2 macrophages. D, representative images of epifluorescence emitted by transfected primary hippocampal neurons at 10 days in vitro. Compound 11 was applied to the cultures, as indicated, for a 72-h period of time prior to image acquisition. E, neurite lengths were calculated from >100 neurons/condition and expressed as a percentage of the control (DMSO) average. *, p < 0.05, given by one-way analysis of variance with Tukey's post hoc corrections, and error bars represent mean ± S.E.
First, we determined whether compounds 10 and 11 diminished LRRK2 phosphorylation at Ser-935. The link between LRRK2 autophosphorylation activity and Ser(P)-935 levels is not clear, but this site has been demonstrated to measure the potency of known selective LRRK2 inhibitors (22). Two main mechanisms that explain Ser(P)-935 as a marker for LRRK2 inhibition include the hypothesis that LRRK2 inhibitors block LRRK2 activation of an unidentified kinase that phosphorylates Ser-935 or that LRRK2 inhibitors change the solubility profiles of LRRK2 so that kinases that phosphorylate at Ser-935 can no longer access this site. Both compounds 10 and 11 are effective in reducing pSer935 levels for endogenous LRRK2 in low micromolar but not nanomolar concentrations (Fig. 6A). The IC50 values for inhibition of endogenous Ser(P)935 formation by 10 and 11 are ∼2 μm.
We have demonstrated in the past that blocking LRRK2 in brain-derived macrophages inhibits proinflammatory responses to the Toll-like receptor 4 agonist LPS (35). To test whether the TPZ inhibitor series is likewise effective in both WT and G2019S-LRRK2 primary macrophage cultures, we applied several concentrations of 11 to cultured cells for 3 days, followed by LPS stimulation for 6 h, and TNF release was measured by ELISA. Compound 11 was effective in reducing TNF release at a 5 μm dose (a concentration of ∼IC90 with respect to Ser(P)-935 reduction) in both WT and G2019S-LRRK2 macrophage cultures (Fig. 6B).
Compound 11 has a 2-fold preference for inhibition of G2019S-LRRK2, and, consistent with this observation, TNF release was inhibited at a 2.5 μm dose in G2019S-LRRK2 but not WT-LRRK2 (Fig. 6B). At 5 μm concentrations of 11, few protein kinases were affected in selectivity screens, but partial inhibition of multiple kinases could have the net effect of reducing TNF concentrations during LPS stimulation.
To determine whether the effects of 11 are specific to LRRK2, we applied LPS to macrophage cultures from KO-LRRK2 animals and treated these cells with 11. No significant reduction of TNF release was observed in cells lacking LRRK2 (Fig. 6C). In contrast, LRRK2-IN-1, a selective LRRK2 inhibitor used widely in the literature, was effective in reducing TNF release in the KO-LRRK2 cells at nanomolar concentrations, suggesting there is off-target inhibition activity for LRRK2-IN-1 that evokes similar phenotypes as on-target effects.
Transient overexpression of G2019S-LRRK2 in primary neurons results in the shortening of neurite outgrowth, and this phenotype is dependent on G2019S-LRRK2 kinase activity (8, 11, 12, 39). Consistent with these results, the most reduction in neurite length could be observed with G2019S-LRRK2 overexpression (Fig. 6, D and E). Exposing these neurons to compound 11 at the time of transfection resulted in the normalization of neurite length to control (WT) levels (Fig. 6E).
Because of the exceptional solubility and permeability of compound 11 in vitro (Fig. 5A), we administered this compound via tail vein injection to mice and obtained the intravenous clearance profile (Fig. 7A). Consistent with the poor oxidative stability of this compound in microsome assays, active 11 is removed rapidly from the circulation. However, the brain and plasma levels of drug were comparable at all time points measured, indicating exceptional permeability across the blood-brain barrier (Fig. 7B). Inhibition of endogenous brain LRRK2 (reduced Ser(P)-935 levels) could be detected only at the 5-min time point post-injection (Fig. 7C), potentially consistent with the rapid oxidation and clearance of 11 from the brain and periphery.
FIGURE 7.
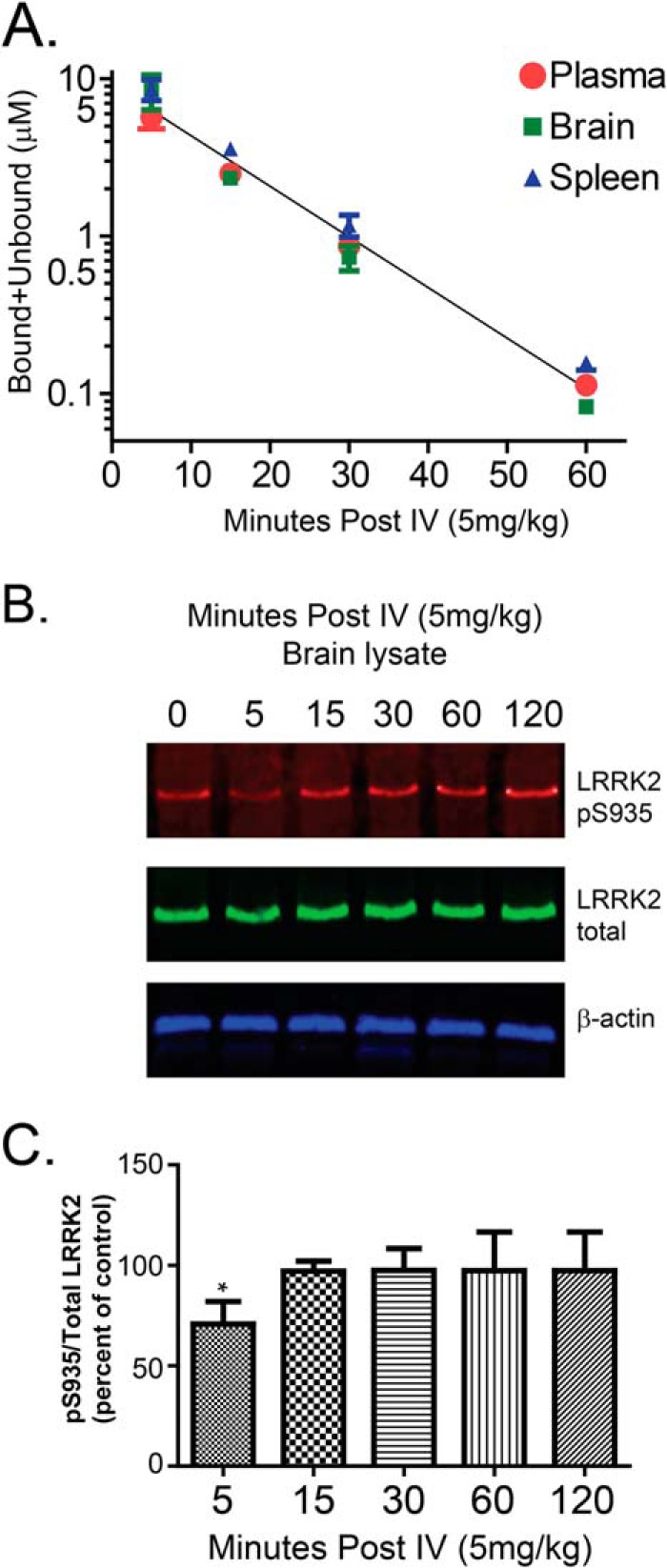
Efficacy of TPZ compound 11 in vivo. A, bound and unbound drug concentrations were measured by mass spectrometry in plasma, brain, and spleen at the indicated time point post-intravenous (tail vein) injection. B, measurement of whole-brain p935-LRRK2 levels from mice compared with total LRRK2. C, a significant reduction in Ser(P)-935 levels can be seen at the 5-min post-injection (*, p < 0.05) time point but not at subsequent time points. Immunoblots and calculated average values are representative of at least three mice per group. One-way analysis of variance with Tukey's post hoc corrections was used to calculate significance, and error bars represent mean ± S.E.
Structural Analysis of the LRRK2 ATP-binding Pocket
Compound 11 is predicted to interact with residues highly conserved across most protein kinases (e.g. the Lys-1906 that forms a salt bridge with the αC helix) and yet displays an exquisite selectivity for LRRK2. Thus, LRRK2 arranges canonical structural motifs in the ATP pocket in a different orientation than other kinases. Unfortunately, the 6-thioether moiety in compound 11 is critical for both exceptional potency and oxidative liability, precluding this compound from further development.
A limiting factor in the effort to find a successful substitute for the sulfur atom of the 6-thioether side chain is the imperfect structural knowledge of the human LRRK2 ATP-binding pocket. The ameba homolog of the LRRK2 protein kinase is the closest known high-resolution structure to the human kinase domain, with ∼31% overall sequence homology (Fig. 8A).
FIGURE 8.

Poor conservation and insolubility of the mammalian LRRK2 ATP-binding pocket. A, kinase domain similarity between the human and frog (88% ATP pocket identity), zebrafish (72% kinase domain similarity), and ameba (31% ATP pocket identity) LRRK2 kinase domains. B, hydrophobicity space fill of the ameba LRRK2 kinase domain crystal structure compared with the imposed LRRK2 human kinase domain sequence on this structure. Arrows indicate the ATP-binding pocket. C and D, representative comparison of solubility profiles of the isolated kinase domains expressed by E. coli. for ameba and human (C) and for the zebrafish and frog LRRK2 kinase domains (D). E and F, recovery of the human LRRK2 kinase domain solubility with the inclusion of either an N-terminal (Asn-144) or C-terminal (Cys-250) entropic bristle (EB) tag (E) but failure of this protein or higher-order LRRK2 kinase domain proteins (frog and zebrafish) (F) to demonstrate any kinase activity.
We were able to purify and crystallize ameba LRRK2 according to published protocols (25), but compound 11 and all other LRRK2-selective compounds we tested did not bind or inhibit ameba LRRK2 at high micromolar concentrations (Fig. 9B), showing that any cocrystal structures with this enzyme will not be useful for further refinement of our inhibitor series by structure-based design. Overall, LRRK2 has a similar primary amino acid sequence from ameba to human, and similar hydrophobicity profiles, which is important for successful purification (Fig. 8B). However, the isolated human LRRK2 kinase domain is completely insoluble in E. coli or insect cell expression systems, in contrast to the ameba LRRK2 homolog (Fig. 8C).
FIGURE 9.

Creation of a humanized LRRK2 ATP-binding pocket from the ameba LRRK2 kinase domain. A, sequence alignment of the human and ameba LRRK2 kinase domains together with the selected artificial mosaic sequence that substitutes human LRRK2 ATP pocket residues into the ameba scaffold. B, kinase assays measuring autophosphorylation as detected by immunoblot analysis with a Thr(P) antibody. Autophosphorylation of the ameba LRRK2 kinase domain is not impaired by 10 μm treatment of the indicated kinase inhibitor. The mosaic protein is also kinase-active and becomes sensitive to LRRK2 kinase inhibitors, with staurosporine inhibiting the largest amount of activity. C, stereotypical binding of a promiscuous ATP-competitive small molecule, staurosporine (green), to the ZAP-70 protein kinase domain (purple). Residues involved in hydrogen bonding are colored by elements (red, oxygen; blue, nitrogen). Hydrogen bonds are shown as dashed lines. D, best fit model of staurosporine interaction in the ameba LRRK2 homolog. Docking to the ATP pocket demonstrates a critical steric clash of the Phe equivalent to human LRRK2 Leu-2001. The conserved Asp-2017 and Lys-1906 show a steric clash with staurosporine in ameba LRRK2 and, to a lesser extent, mosaic LRRK2. E, inhibition profiles of staurosporine in blocking autophosphorylation activity of human LRRK2, mosaic LRRK2, and ameba LRRK2, with IC50 values given.
To test the hypothesis that aggregation and insolubility may be blocking the expression of active human LRRK2 kinase enzyme, we employed entropic bristles at either the N- or C-terminal domains (Fig. 8D). Entropic bristle domains have been used to rescue the solubility of kinase proteins (40).
The addition of an N- or C-terminal entropic bristle to the human LRRK2 kinase domain confers exceptional solubility profiles, similar to the ameba homolog (Fig. 8D). However, the soluble protein showed no kinase activity (Fig. 8F), and cleavage of the bristle tags resulted in rapid instability and aggregation of the protein. Furthermore, frog and zebrafish homologs of LRRK2, which have much more similarity to active ameba LRRK2, also show insoluble profiles (Fig. 8E) and are inactive (Fig. 8F).
We hypothesized that, although the overall composition of the ATP-binding pocket is similar between human and ameba, the collective differences in structure may be due to unique amino acids comprising the ATP pocket (Fig. 9A, blue segments). We created a mosaic LRRK2 construct that substitutes the human amino acid ATP pocket sequence into the ameba LRRK2 kinase domain, with the resultant protein dubbed mosaic LRRK2. Mosaic LRRK2 is both soluble and active and became sensitive to small molecules known to bind and inhibit human LRRK2 (Fig. 9B).
The binding of staurosporine to the ATP-binding pockets of protein kinases is one of the most well described interactions between a protein kinase domain and a small molecule inhibitor. A docking example with ZAP-1 protein kinase is shown in Fig. 9C, highlighting the critical interactions. Superposition of the canonical staurosporine docking model to the known ameba LRRK2 structure confirms the prediction that staurosporine would not bind to ameba LRRK2. The Leu-2001 of human LRRK2 is a phenylalanine in the ameba homolog that would be predicted to sterically inhibit staurosporine (Fig. 9D, Phe shown in yellow).
Further, the conserved and critical Asp-2017 and Lys-1906 are oriented toward the ATP-binding pocket in ameba but not in human LRRK2 predictions, which would cause an additional steric clash with staurosporine for ameba LRRK2. However, models of mosaic LRRK2 still show that the pocket forms differently than human LRRK2 because of the structure peripheral to the ATP pocket-forming residues. Steric hindrance is likely to happen in mosaic LRRK2 at residues Asp-2017 and Lys-1906, although the critical Leu-2001 residue would no longer be blocking staurosporine.
The prediction that mosaic LRRK2 has a higher sensitivity to staurosporine than ameba LRRK2 in kinase assays, but less sensitivity compared with human LRRK2, was confirmed experimentally in kinase assays (Fig. 9E). However, for unknown reasons, the mosaic LRRK2 was not potently inhibited by compound 11, suggesting that further iterative refinement using site-directed mutagenesis is warranted.
The major difference between the human and ameba LRRK2 kinase domains driving flexible ATP pocket conformations could lie with the hydrophobic spine of the kinase domain (34, 41). The kinase domain spine assembles two non-consecutive hydrophobic structures important for shaping parts of the ATP pocket. Human LRRK2 kinase harbors a unique protein sequence in an otherwise extremely conserved spine motif, whereas ameba LRRK2 is much more similar to typical protein kinases. For example, the tyrosines on the magnesium binding motif (DXG motif) and catalytic motif (XRD motif) are unique to LRRK2 and evolutionarily conserved, with the exception of the ameba homolog (Fig. 10). The human LRRK2 spine has a predicted unusual curvature that may introduce flexibility into the kinase domain, for example to accept a variety of protein substrates.
FIGURE 10.
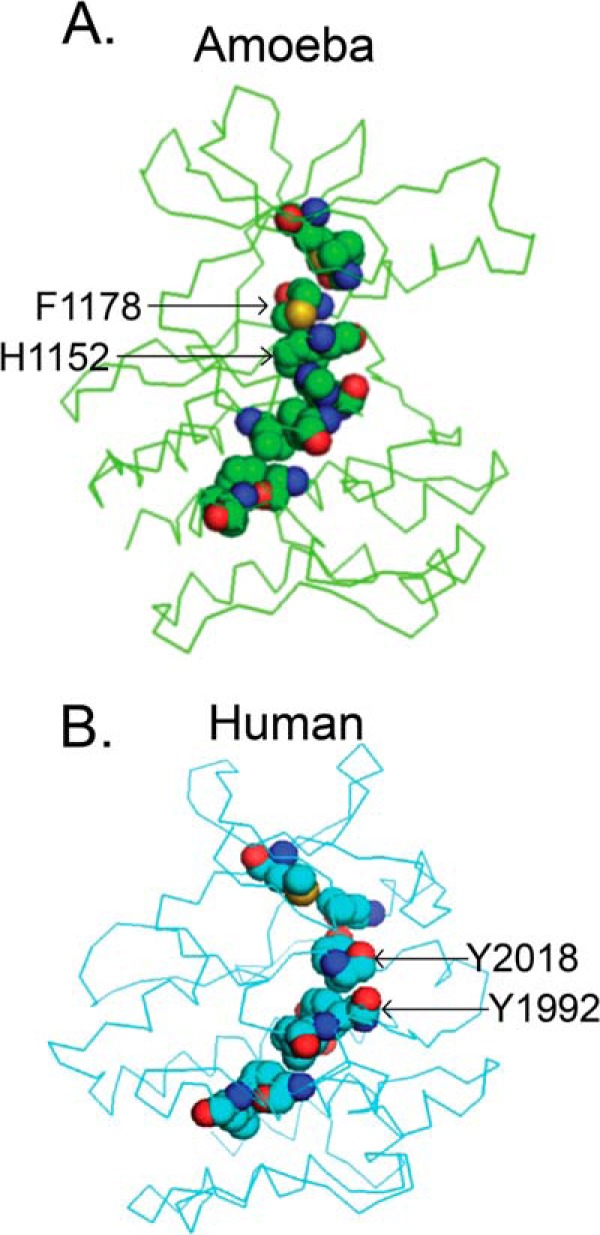
Lack of conservation of the hydrophobic spine structure. Shown are the hydrophobic spines of ameba (A, green) and human LRRK2 kinase domain models (B, cyan). Residues involved in the hydrophobic spine are represented by spheres. Key residues on the conserved DXG and XRD motif are labeled by arrows. Elements are color-coded (light blue, carbon; red, oxygen; dark blue, nitrogen; yellow, sulfur).
We modified the mosaic LRRK2 hydrophobic spine by changing the magnesium binding loop phenylalanine and catalytic loop histidine to tyrosines, as found with human LRRK2. These alterations to the mosaic protein along the hydrophobic spine resulted in a completely unstable and insoluble protein. Tertiary interactions with other LRRK2 domains might be required to stabilize the human LRRK2 ATP-binding pocket and hydrophobic spine. Overall, these results suggest that the substrate phosphorylation and inhibitor binding specificity of human LRRK2 is conducted by both the unique ATP-binding pocket and hydrophobic spine.
DISCUSSION
The specific biochemical process that links LRRK2 to neurodegeneration is not known. However, kinase activity is hypothesized to underlie neurotoxicity in model systems. Efforts to identify efficacious small molecule inhibitors have yielded several impressively potent and selective compounds. These advancements have been made despite the imperfect knowledge of LRRK2 structure. However, it is not clear which kinase substrates need to be blocked for therapeutic benefit.
To our knowledge, this is the first systematic study of how small molecules differentially target trans- and cis-phosphorylation activities. The multidimensional aspect of LRRK2 kinase activity may strongly influence the success or failure of a particular compound in preclinical models. For example, it may be desirable to inhibit only LRRK2 protein that harbors the pathogenic mutation G2019S or to inhibit one aspect of activity, such as trans-phosphorylation of a particular substrate, but leave LRRK2 cis-autophosphorylation activity untouched. Previous studies generally used a single assay to measure LRRK2 kinase activity. This has the potential to exclude rich amounts of data for dissecting the precise mechanism of LRRK2 enzyme function.
Here we developed two novel high-throughput assays and provide the structures of commercially available LRRK2 kinase inhibitors (supplemental Table 1). These compounds were also evaluated for cellular toxicities in cells that express high levels of endogenous LRRK2 (THP-1 cells (35)) in addition to Hep2G cells. Nearly all of these compounds were well tolerated (supplemental Table 1). In trans-peptide assays, most of these inhibitors have more potency toward G2019S-LRRK2 compared with WT-LRRK2. This can be explained by in silico predictions and homology modeling suggesting that G2019S induces a more stable magnesium binding loop and activation loop that may accommodate a wider variety of substrates and inhibitors (42).
Unexpectedly, these compounds did not show a preferential trend for enhanced potency of G2019S-LRRK2 cis-autophosphorylation relative to trans-peptide phosphorylation. Given this result, it is not surprising that there is a poor correlation of potency between cis-autophosphorylation and trans-peptide phosphorylation. The catalytic pocket may adopt different conformations, depending on the nature of the substrate, and the small molecule inhibitors may show different binding affinities for these different conformations (or not bind at all).
Functional IC50 values for inhibitor compounds also depend on the Km for the ATP cofactor, but in all assays we used, the values for Km ATP are similar for trans-phosphorylation and cis-autophosphorylation and for G2019S-LRRK2 and WT-LRRK2. Despite ATP binding in a similar manner without respect for the target of phosphorylation, adjacent areas of the catalytic pocket show enough structural heterogeneity to suggest that small molecules can be developed to target a selected type of kinase activity.
Of the 620 molecules we disclose that inhibit LRRK2 (supplemental Table 1), we pursued the TPZ series because of a high representation in the initial screening set, reasonable potency, and good predicted drug-like properties. Many other series may be pursued as hit-to-lead scaffolds. Structure-activity relationships, such as those defined by the TPZ series, become all the more important when a high-resolution structure is unavailable and homology models with low overall sequence conservation to the human LRRK2 kinase domain have questionable utility in fine-tuning pharmacophores. Others have recently reported the discovery of similar chemical structures during the course of our study (32).
TPZ compound 11 (SRI-29132), scrutinized here, shows interactions with conserved residues found in most ATP-binding pockets, usually invariant in orientation with respect to bound ATP. Surprisingly, 11 has remarkable specificity in inhibiting only LRRK2 from nearly all kinases in the human kinome at 1 μm concentration. Strikingly, this is not the first disclosed compound with this profile. Compound GSK 2578215A has similar specificity (23).
We examined 11 in vitro and in vivo because this series represents an unusual kinase inhibitor scaffold dissimilar from any described clinical compounds or kinase inhibitors in wide use. Many LRRK2 inhibitor programs rely on cell lines that express high levels of exogenous proteins to define cellular potencies in cells that do not normally express LRRK2. Our recent observations of high levels of endogenous LRRK2 in macrophages make it unnecessary to use engineered cell lines that are potentially fraught with artifacts.
Macrophages are one of only a few cell types that express high levels of the P-glycoprotein 1 transporter, responsible for clearing foreign substances from cells (38, 43). At low micromolar concentrations, compound 11 is effective in reducing Ser(P)-935 LRRK2 levels. Although this activity is indicative of LRRK2 inhibition (22), whether it represents inhibition of LRRK2 cis-autophosphorylation or trans-peptide phosphorylation of some substrate(s) is not clear.
Besides Ser(P)-935 measurements, corresponding reductions in TNF levels elicited by LPS exposure support our past reports of LRRK2 involvement in proinflammatory mechanisms (35). Given our recent data, we and others have published observations with the compound LRRK2-IN-1 that are probably unrelated to LRRK2 activity (35). Also, in contrast to our previous observations with LRRK2-IN-1, we could not detect a change in TNF levels with acute exposures to 11. Rather, a 3-day compound 11 exposure protocol attenuated TNF levels in response to LPS, potentially consistent with RNAi experiments in primary macrophages (35) and the reduced proinflammatory macrophage responses observed in vivo in the LRRK2 knockout rat (44). Furthermore, these results are consistent with a role for LRRK2 in the maturation of myeloid cells (e.g. monocytes and macrophages) as hypothesized previously by others (36).
The overexpression of G2019S-LRRK2 is not well tolerated in maturing (>7 days of culture in vitro) primary neurons, with reduced neurite outgrowth apparent in transient transfection experimental paradigms (6, 8, 11, 12, 39). In these past studies, the effects of G2019S-LRRK2 with respect to neurite shortening are dependent on LRRK2 kinase activity and can be rescued with LRRK2 inhibitors. We found that compound 11 also rescues this phenotype. Combining 11 with other highly specific LRRK2 inhibitors, such as GSK 2578215A and HG-10-102-01, novel LRRK2-kinase-dependent in vitro phenotypes can be discovered with very high confidence.
Despite promising data from cell cultures, compound 11 is a high-clearance compound in vivo because of the rapid oxidative metabolism of a thioether group. The oxidation clearly abolishes binding to the LRRK2 ATP pocket. Unfortunately, our efforts (and those of others (32)) to find a metabolically stable replacement for the thioether with preservation of potency were not successful. Part of the problem is the lack of a high-resolution structure of this domain to guide hypothesis-driven efforts.
We are not aware of another protein kinase with such a wealth of selective and potent inhibitors but a lack of structural knowledge of the ATP pocket. We, and many other groups, have attempted to generate crystals of the human LRRK2 kinase domain with the primary goal of developing a detailed model to enable a more effective structure-based design of inhibitor candidates. For example, the high-resolution structure derived from a crystal could guide the creation of appropriate modifications to compound 11 to eliminate oxidative metabolism potential but retain potency toward inhibition of LRRK2 kinase activity.
Although the solubility of the isolated human LRRK2 kinase domain can be enhanced dramatically using the recently described entropic bristle approaches to protect the protein from self-association (40), this modified protein has no kinase activity despite intensive biochemical testing. Targeting zebrafish LRRK2 expression during embryonic development results in dopaminergic neurodegeneration and is hypothesized as a disease model relevant to Parkinson disease (45). However, we find that both the zebrafish and frog LRRK2 kinase domains are soluble but without kinase activity (Fig. 7C). Overall, these results suggest intrinsic protein folding problems of the LRRK2 kinase domain and ATP pocket in higher-order species.
Here we are able to confirm independently that the ameba homolog of LRRK2 is readily soluble, is active, and forms crystals that can be analyzed. But we also show that this enzyme is not inhibited by any of the known selective LRRK2 kinase inhibitors or even non-selective inhibitors with extremely strong potency to human LRRK2, like staurosporine. Major differences in the ATP pocket residues preclude the utility of this structure as a model for human LRRK2.
Despite our efforts to correct the ATP pocket residues of the ameba LRRK2 protein to the human sequence, selective LRRK2 inhibitors still interacted with very low potency. We hypothesize that the unique hydrophobic spine of the kinase domain may be a critical factor in the ATP pocket forming the flexible structure necessary for a wide variety of functions, like cis-autophosphorylation and trans-peptide phosphorylation of a variety of protein substrates. The human LRRK2 protein kinase domain spine may form critical tertiary interactions with other parts of the human LRRK2 protein to stabilize and form the structural basis that allows protein kinase activity.
Human LRRK2 protein in cells has the propensity to oligomerize and inactivate, and some small molecules that bind to the LRRK2 kinase domain can destabilize the entire LRRK2 protein (16). Therefore, the same structures that lead to a unique conformation of the LRRK2 ATP pocket may also directly hinder efforts to resolve a high-resolution structure of the kinase domain.
Despite the aforementioned challenges, the novel high-throughput assays, libraries of small molecules that inhibit LRRK2, and the new functional assays reported here are useful tools to pursue targeted approaches to the development of clinical candidate LRRK2 inhibitors. LRRK2 represents one of the most promising targets for therapeutic intervention in neurodegenerative diseases, and our findings here may provide new perspectives to bring the best compounds to the clinic in the future.
Supplementary Material
Acknowledgments
We thank Tonia Tse and Damon Papac for technical assistance. We also thank the staff at the Southern Research High Throughput Screening Center for technical support encompassing compound and data management and assay execution.
This work was supported, in whole or in part, by NINDS, National Institutes of Health Grant R01NS064934. This work was also supported by the Alabama Drug Discovery Alliance, by the UAB Center for Clinical and Translational Science Grant UL1R00165, by John A. and Ruth R. Jurenko, by the American Parkinson Disease Association, and by the Southern Research Strategic Investment Fund.

This article contains supplemental Experimental Procedures, Figures 1–3, and Tables 1 and 2.
- DMSO
- dimethyl sulfoxide
- TPZ
- triazolopyridazine
- ROC
- ras-of-complex.
REFERENCES
- 1. Zimprich A., Biskup S., Leitner P., Lichtner P., Farrer M., Lincoln S., Kachergus J., Hulihan M., Uitti R. J., Calne D. B., Stoessl A. J., Pfeiffer R. F., Patenge N., Carbajal I. C., Vieregge P., Asmus F., Müller-Myhsok B., Dickson D. W., Meitinger T., Strom T. M., Wszolek Z. K., Gasser T. (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607 [DOI] [PubMed] [Google Scholar]
- 2. Paisán-Ruíz C., Jain S., Evans E. W., Gilks W. P., Simón J., van der Brug M., López de Munain A., Aparicio S., Gil A. M., Khan N., Johnson J., Martinez J. R., Nicholl D., Carrera I. M., Pena A. S., de Silva R., Lees A., Martí-Massó J. F., Pérez-Tur J., Wood N. W., Singleton A. B. (2004) Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron 44, 595–600 [DOI] [PubMed] [Google Scholar]
- 3. West A. B., Moore D. J., Biskup S., Bugayenko A., Smith W. W., Ross C. A., Dawson V. L., Dawson T. M. (2005) Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. U.S.A. 102, 16842–16847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jaleel M., Nichols R. J., Deak M., Campbell D. G., Gillardon F., Knebel A., Alessi D. R. (2007) LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson's disease mutants affect kinase activity. Biochem. J. 405, 307–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cookson M. R. (2010) The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson's disease. Nat. Rev. Neurosci. 11, 791–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sheng Z., Zhang S., Bustos D., Kleinheinz T., Le Pichon C. E., Dominguez S. L., Solanoy H. O., Drummond J., Zhang X., Ding X., Cai F., Song Q., Li X., Yue Z., van der Brug M. P., Burdick D. J., Gunzner-Toste J., Chen H., Liu X., Estrada A. A., Sweeney Z. K., Scearce-Levie K., Moffat J. G., Kirkpatrick D. S., Zhu H. (2012) Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Sci. Transl. Med. 4, 164ra161. [DOI] [PubMed] [Google Scholar]
- 7. Webber P. J., Smith A. D., Sen S., Renfrow M. B., Mobley J. A., West A. B. (2011) Autophosphorylation in the leucine-rich repeat kinase 2 (LRRK2) GTPase domain modifies kinase and GTP-binding activities. J. Mol. Biol. 412, 94–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. West A. B., Moore D. J., Choi C., Andrabi S. A., Li X., Dikeman D., Biskup S., Zhang Z., Lim K. L., Dawson V. L., Dawson T. M. (2007) Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum. Mol. Genet. 16, 223–232 [DOI] [PubMed] [Google Scholar]
- 9. Ray S., Bender S., Kang S., Lin R., Glicksman M. A., Liu M. (2014) The Parkinson disease-linked LRRK2 protein mutation I2020T stabilizes an active state conformation leading to increased kinase activity. J. Biol. Chem. 289, 13042–13053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liao J., Wu C. X., Burlak C., Zhang S., Sahm H., Wang M., Zhang Z. Y., Vogel K. W., Federici M., Riddle S. M., Nichols R. J., Liu D., Cookson M. R., Stone T. A., Hoang Q. Q. (2014) Parkinson disease-associated mutation R1441H in LRRK2 prolongs the “active state” of its GTPase domain. Proc. Natl. Acad. Sci. U.S.A. 111, 4055–4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Smith W. W., Pei Z., Jiang H., Dawson V. L., Dawson T. M., Ross C. A. (2006) Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat. Neurosci. 9, 1231–1233 [DOI] [PubMed] [Google Scholar]
- 12. Greggio E., Jain S., Kingsbury A., Bandopadhyay R., Lewis P., Kaganovich A., van der Brug M. P., Beilina A., Blackinton J., Thomas K. J., Ahmad R., Miller D. W., Kesavapany S., Singleton A., Lees A., Harvey R. J., Harvey K., Cookson M. R. (2006) Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis. 23, 329–341 [DOI] [PubMed] [Google Scholar]
- 13. Baptista M. A., Dave K. D., Frasier M. A., Sherer T. B., Greeley M., Beck M. J., Varsho J. S., Parker G. A., Moore C., Churchill M. J., Meshul C. K., Fiske B. K. (2013) Loss of leucine-rich repeat kinase 2 (LRRK2) in rats leads to progressive abnormal phenotypes in peripheral organs. PLoS ONE 8, e80705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tong Y., Yamaguchi H., Giaime E., Boyle S., Kopan R., Kelleher R. J., 3rd, Shen J. (2010) Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of α-synuclein, and apoptotic cell death in aged mice. Proc. Natl. Acad. Sci. U.S.A. 107, 9879–9884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Andres-Mateos E., Mejias R., Sasaki M., Li X., Lin B. M., Biskup S., Zhang L., Banerjee R., Thomas B., Yang L., Liu G., Beal M. F., Huso D. L., Dawson T. M., Dawson V. L. (2009) Unexpected lack of hypersensitivity in LRRK2 knock-out mice to MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine). J. Neurosci. 29, 15846–15850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sen S., Webber P. J., West A. B. (2009) Dependence of leucine-rich repeat kinase 2 (LRRK2) kinase activity on dimerization. J. Biol. Chem. 284, 36346–36356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Covy J. P., Giasson B. I. (2009) Identification of compounds that inhibit the kinase activity of leucine-rich repeat kinase 2. Biochem. Biophys. Res. Commun. 378, 473–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nichols R. J., Dzamko N., Hutti J. E., Cantley L. C., Deak M., Moran J., Bamborough P., Reith A. D., Alessi D. R. (2009) Substrate specificity and inhibitors of LRRK2, a protein kinase mutated in Parkinson's disease. Biochem. J. 424, 47–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ramsden N., Perrin J., Ren Z., Lee B. D., Zinn N., Dawson V. L., Tam D., Bova M., Lang M., Drewes G., Bantscheff M., Bard F., Dawson T. M., Hopf C. (2011) Chemoproteomics-based design of potent LRRK2-selective lead compounds that attenuate Parkinson's disease-related toxicity in human neurons. ACS Chem. Biol. 6, 1021–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang J., Deng X., Choi H. G., Alessi D. R., Gray N. S. (2012) Characterization of TAE684 as a potent LRRK2 kinase inhibitor. Bioorg. Med. Chem. Lett. 22, 1864–1869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee B. D., Shin J. H., VanKampen J., Petrucelli L., West A. B., Ko H. S., Lee Y. I., Maguire-Zeiss K. A., Bowers W. J., Federoff H. J., Dawson V. L., Dawson T. M. (2010) Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson's disease. Nat. Med. 16, 998–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Deng X., Dzamko N., Prescott A., Davies P., Liu Q., Yang Q., Lee J. D., Patricelli M. P., Nomanbhoy T. K., Alessi D. R., Gray N. S. (2011) Characterization of a selective inhibitor of the Parkinson's disease kinase LRRK2. Nat. Chem. Biol. 7, 203–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Reith A. D., Bamborough P., Jandu K., Andreotti D., Mensah L., Dossang P., Choi H. G., Deng X., Zhang J., Alessi D. R., Gray N. S. (2012) GSK2578215A: a potent and highly selective 2-arylmethyloxy-5-substitutent-N-arylbenzamide LRRK2 kinase inhibitor. Bioorg. Med. Chem. Lett. 22, 5625–5629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Choi H. G., Zhang J., Deng X., Hatcher J. M., Patricelli M. P., Zhao Z., Alessi D. R., Gray N. S. (2012) Brain penetrant LRRK2 inhibitor. ACS Med. Chem. Lett. 3, 658–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gilsbach B. K., Ho F. Y., Vetter I. R., van Haastert P. J., Wittinghofer A., Kortholt A. (2012) Roco kinase structures give insights into the mechanism of Parkinson disease-related leucine-rich-repeat kinase 2 mutations. Proc. Natl. Acad. Sci. U.S.A. 109, 10322–10327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Greggio E., Zambrano I., Kaganovich A., Beilina A., Taymans J. M., Daniëls V., Lewis P., Jain S., Ding J., Syed A., Thomas K. J., Baekelandt V., Cookson M. R. (2008) The Parkinson disease-associated leucine-rich repeat kinase 2 (LRRK2) is a dimer that undergoes intramolecular autophosphorylation. J. Biol. Chem. 283, 16906–16914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stafa K., Tsika E., Moser R., Musso A., Glauser L., Jones A., Biskup S., Xiong Y., Bandopadhyay R., Dawson V. L., Dawson T. M., Moore D. J. (2014) Functional interaction of Parkinson's disease-associated LRRK2 with members of the dynamin GTPase superfamily. Hum. Mol. Genet. 23, 2055–2077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Deng J., Lewis P. A., Greggio E., Sluch E., Beilina A., Cookson M. R. (2008) Structure of the ROC domain from the Parkinson's disease-associated leucine-rich repeat kinase 2 reveals a dimeric GTPase. Proc. Natl. Acad. Sci. U.S.A. 105, 1499–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Volpicelli-Daley L. A., Luk K. C., Patel T. P., Tanik S. A., Riddle D. M., Stieber A., Meaney D. F., Trojanowski J. Q., Lee V. M. (2011) Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72, 57–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Smith W. W., Pei Z., Jiang H., Moore D. J., Liang Y., West A. B., Dawson V. L., Dawson T. M., Ross C. A. (2005) Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc. Natl. Acad. Sci. U.S.A. 102, 18676–18681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vallotton P., Lagerstrom R., Sun C., Buckley M., Wang D., De Silva M., Tan S. S., Gunnersen J. M. (2007) Automated analysis of neurite branching in cultured cortical neurons using HCA-Vision. Cytometry A 71, 889–895 [DOI] [PubMed] [Google Scholar]
- 32. Franzini M., Ye X. M., Adler M., Aubele D. L., Garofalo A. W., Gauby S., Goldbach E., Probst G. D., Quinn K. P., Santiago P., Sham H. L., Tam D., Truong A., Ren Z. (2013) Triazolopyridazine LRRK2 kinase inhibitors. Bioorg. Med. Chem. Lett. 23, 1967–1973 [DOI] [PubMed] [Google Scholar]
- 33. Treiber D. K., Shah N. P. (2013) Ins and outs of kinase DFG motifs. Chem. Biol. 20, 745–746 [DOI] [PubMed] [Google Scholar]
- 34. Ray S., Liu M. (2012) Current understanding of LRRK2 in Parkinson's disease: biochemical and structural features and inhibitor design. Future Med. Chem. 4, 1701–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moehle M. S., Webber P. J., Tse T., Sukar N., Standaert D. G., DeSilva T. M., Cowell R. M., West A. B. (2012) LRRK2 inhibition attenuates microglial inflammatory responses. J. Neurosci. 32, 1602–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thévenet J., Pescini Gobert R., Hooft van Huijsduijnen R., Wiessner C., Sagot Y. J. (2011) Regulation of LRRK2 expression points to a functional role in human monocyte maturation. PLoS ONE 6, e21519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hakimi M., Selvanantham T., Swinton E., Padmore R. F., Tong Y., Kabbach G., Venderova K., Girardin S. E., Bulman D. E., Scherzer C. R., LaVoie M. J., Gris D., Park D. S., Angel J. B., Shen J., Philpott D. J., Schlossmacher M. G. (2011) Parkinson's disease-linked LRRK2 is expressed in circulating and tissue immune cells and upregulated following recognition of microbial structures. J. Neural Transm. 118, 795–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Misra S., Ujházy P., Varticovski L., Arias I. M. (1999) Phosphoinositide 3-kinase lipid products regulate ATP-dependent transport by sister of P-glycoprotein and multidrug resistance associated protein 2 in bile canalicular membrane vesicles. Proc. Natl. Acad. Sci. U.S.A. 96, 5814–5819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. MacLeod D., Dowman J., Hammond R., Leete T., Inoue K., Abeliovich A. (2006) The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron 52, 587–593 [DOI] [PubMed] [Google Scholar]
- 40. Santner A. A., Croy C. H., Vasanwala F. H., Uversky V. N., Van Y. Y., Dunker A. K. (2012) Sweeping away protein aggregation with entropic bristles: intrinsically disordered protein fusions enhance soluble expression. Biochemistry 51, 7250–7262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kornev A. P., Haste N. M., Taylor S. S., Eyck L. F. (2006) Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc. Natl. Acad. Sci. U.S.A. 103, 17783–17788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu M., Kang S., Ray S., Jackson J., Zaitsev A. D., Gerber S. A., Cuny G. D., Glicksman M. A. (2011) Kinetic, mechanistic, and structural modeling studies of truncated wild-type leucine-rich repeat kinase 2 and the G2019S mutant. Biochemistry 50, 9399–9408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lemaire S., Van Bambeke F., Mingeot-Leclercq M. P., Tulkens P. M. (2007) Modulation of the cellular accumulation and intracellular activity of daptomycin towards phagocytized Staphylococcus aureus by the P-glycoprotein (MDR1) efflux transporter in human THP-1 macrophages and Madin-Darby canine kidney cells. Antimicrob. Agents Chemother. 51, 2748–2757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Daher J. P., Volpicelli-Daley L. A., Blackburn J. P., Moehle M. S., West A. B. (2014) Abrogation of α-synuclein-mediated dopaminergic neurodegeneration in LRRK2-deficient rats. Proc. Natl. Acad. Sci. U.S.A. 111, 9289–9294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sheng D., Qu D., Kwok K. H., Ng S. S., Lim A. Y., Aw S. S., Lee C. W., Sung W. K., Tan E. K., Lufkin T., Jesuthasan S., Sinnakaruppan M., Liu J. (2010) Deletion of the WD40 domain of LRRK2 in Zebrafish causes Parkinsonism-like loss of neurons and locomotive defect. PLoS Genet. 6, e1000914. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.