

Background: Structural information for substrate binding to human steroidogenic cytochrome P450 17A1 (CYP17A1) is unavailable.
Results: Within a common overall orientation, different steroids adopt subtly different positions.
Conclusion: Steric and hydrogen-bonding modulation of lateral/vertical orientation controls CYP17A1-mediated steroid oxidation.
Significance: Understanding the CYP17A1 mechanism provides opportunities for better targeted drug design.
Keywords: Cytochrome, Cytochrome P450, Prostate Cancer, Protein Structure, Steroid, Steroidogenesis, X-ray Crystallography
Abstract
The human cytochrome P450 17A1 (CYP17A1) enzyme operates at a key juncture of human steroidogenesis, controlling the levels of mineralocorticoids influencing blood pressure, glucocorticoids involved in immune and stress responses, and androgens and estrogens involved in development and homeostasis of reproductive tissues. Understanding CYP17A1 multifunctional biochemistry is thus integral to treating prostate and breast cancer, subfertility, blood pressure, and other diseases. CYP17A1 structures with all four physiologically relevant steroid substrates suggest answers to four fundamental aspects of CYP17A1 function. First, all substrates bind in a similar overall orientation, rising ∼60° with respect to the heme. Second, both hydroxylase substrates pregnenolone and progesterone hydrogen bond to Asn202 in orientations consistent with production of 17α-hydroxy major metabolites, but functional and structural evidence for an A105L mutation suggests that a minor conformation may yield the minor 16α-hydroxyprogesterone metabolite. Third, substrate specificity of the subsequent 17,20-lyase reaction may be explained by variation in substrate height above the heme. Although 17α-hydroxyprogesterone is only observed farther from the catalytic iron, 17α-hydroxypregnenolone is also observed closer to the heme. In conjunction with spectroscopic evidence, this suggests that only 17α-hydroxypregnenolone approaches and interacts with the proximal oxygen of the catalytic iron-peroxy intermediate, yielding efficient production of dehydroepiandrosterone as the key intermediate in human testosterone and estrogen synthesis. Fourth, differential positioning of 17α-hydroxypregnenolone offers a mechanism whereby allosteric binding of cytochrome b5 might selectively enhance the lyase reaction. In aggregate, these structures provide a structural basis for understanding multiple key reactions at the heart of human steroidogenesis.
Introduction
The cytochrome P450 superfamily of heme monooxygenases performs diverse physiological functions, ranging from drug and xenobiotic metabolism to hormone and vitamin biosynthesis. The human cytochrome P450 (P450)3 enzyme 17A1 (CYP17A1) functions specifically at a critical juncture in human steroidogenesis (1). Its initial substrates are also substrates for mineralocorticoid biosynthesis by other enzymes. CYP17A1 catalysis leads to either steroid precursors of glucocorticoids like cortisol that regulate immune response or androgens like testosterone that drive the development and maintenance of male characteristics or are converted to estrogens in females (2). In later life, however, androgens drive the development of prostate cancer, the cancer of highest incidence and the second leading cause of cancer deaths in American men, whereas estrogens are a long recognized driver of hormone-responsive breast cancer (3). Thus this enzyme has garnered substantial interest as a relatively new drug target, validated by successful use of the CYP17A1 inhibitor abiraterone in men with castration-resistant prostate cancer (4–6) and its current evaluation in breast cancer patients.
Abiraterone acetate, the Food and Drug Administration-approved prodrug form of this CYP17A1 inhibitor, improves overall survival in men with metastatic castration-resistant prostate cancer, including patients for whom the disease has progressed following chemotherapy, with compounds such as docetaxel and the androgen receptor blocker enzalutamide (5, 7). Abiraterone binds with high affinity to the CYP17A1 active site heme iron (8) that is essential for catalysis, which effectively and systemically prevents androgen production. However, by doing so, this inhibitor also increases the pool of precursors for mineralocorticoid production and halts CYP17A1-mediated production of glucocorticoids, which occurs in the same active site and also requires the heme iron. The resulting steroid imbalances in patients treated with abiraterone can frequently lead to hypertension, hypokalemia, and adrenocortical insufficiency, which must then be monitored and treated with additional drugs (9). Furthermore, there is some evidence that the increase in mineralocorticoids associated with complete inhibition of CYP17A1 may facilitate the flow of androgen precursors through a “backdoor” androgen biosynthesis pathway (10), proposed to provide a possible escape route that could permit cancer progression. Selective inhibition of the CYP17A1-mediated androgen biosynthesis proven to increase overall survival, whereas sparing CYP17A1-mediated glucocorticoid biosynthesis to prevent corticosteroid imbalances would ameliorate both of these clinically relevant issues for prostate cancer patients. CYP17A1 impairment has also been associated with Cushing's syndrome (11), some forms of congenital adrenal hyperplasia (12), and polycystic ovary syndrome (13–15). Substantial potential for improving prostate cancer treatment and therapies for these other diseases thus lies in an improved understanding of the mechanisms whereby CYP17A1 performs catalysis.
In general, CYP17A1 hydroxylates the mineralocorticoid precursor steroids pregnenolone and progesterone to yield 17α-hydroxypregnenolone and 17α-hydroxyprogesterone, respectively (see Fig. 1). These resulting C17-hydroxylated steroids can serve as substrates for glucocorticoid biosynthesis or for the subsequent CYP17A1-mediated 17,20-lyase reaction to yield the androgens dehydroepiandrosterone or androstenedione (see Fig. 1). Functional variations on this general pathway for different substrates provide clues to key protein/small molecule interactions that direct catalysis.
FIGURE 1.

Summary of human cytochrome P450 17A1 reactions. Progesterone is hydroxylated at either C16 (minor, red) or C17 (major, blue) via Groves hydrogen abstraction and rebound mediated by the typical P450 iron (IV)-oxo intermediate (21). The resulting 17α-hydroxyprogesterone metabolite undergoes the 17,20-lyase reaction to androstenedione (pink) with very low efficiency. Conversely, pregnenolone is hydroxylated only at C17 (blue). The resulting 17α-hydroxypregnenolone metabolite efficiently undergoes the 17,20-lyase reaction to form the 19-carbon androgen dehydroepiandrosterone (pink), proposed to occur via an iron-peroxy intermediate (22). The latter reaction is facilitated as much as 10-fold by the presence of cytochrome b5 (25). CYP17A1 hydroxylase substrates progesterone and pregnenolone are also converted to mineralocorticoids and 17α-hydroxy products to glucocorticoids, largely by other cytochrome P450 enzymes.
First, although human CYP17A1 hydroxylates pregnenolone (a Δ5,3-ol steroid) only at carbon 17, progesterone (the corresponding Δ4,3-keto steroid) is hydroxylated at C17 as the major product, but also at C16 to generate an additional minor product (see Fig. 1) (14, 16). It is known that in other species CYP17A1 with Leu at position 105 generates much less 16α-hydroxyprogesterone (14) and that an A105L mutation in the human CYP17A1 enzyme decreases progesterone 16α-hydroxylation and increases its 17α-hydroxylation (17). Suggested to somehow alter substrate position in the CYP17A1 active site or alter protein flexibility, the structural basis for the effects of the A105L mutation on substrate and regioselectivity of hydroxylation have not been elucidated experimentally.
Second, CYP17A1 also performs a carbon-carbon bond cleavage, which is unusual for cytochrome P450 enzymes (18). For human CYP17A1, this 17,20-lyase reaction proceeds far less efficiently for the Δ4,3-keto 17α-hydroxyprogesterone substrate than for its counterpart, the Δ5,3-ol 17α-hydroxypregnenolone (see Fig. 1). As a result, the Δ5,3-ol 17α-hydroxypregnenolone 17,20-lyase product dehydroepiandrosterone (see Fig. 1) is the physiologically relevant intermediate in the formation of all human androgens and estrogens (19). Although hydroxylation is well established to occur by the Groves hydrogen abstraction/oxygen rebound mechanism mediated by an Fe(IV)-oxo catalytic intermediate called Compound I (20, 21) (see Fig. 1), a mechanism for cleavage of the bond between carbons 17 and 20 has been an ongoing debate in the literature. Recent data, however, favors a ferric peroxy anion intermediate (see Fig. 1) (22, 23), and spectroscopic evidence has suggested that the two 17α-hydroxy steroids might interact differentially with this peroxy intermediate (24).
Third, although the presence of cytochrome b5 has relatively little effect on the hydroxylation reactions, the presence of this small heme protein (25–28) substantially and selectively facilitates the 17,20-lyase reaction. Compartmentalization of b5 and developmental changes in b5 levels control the tissue specificity and timing of androgen production in humans. Individuals with nonfunctional b5 are unable to perform the lyase reaction and produce sex steroids, although the hydroxylase reaction required for glucocorticoid synthesis is operational (29, 30). Facilitation of the lyase reaction by b5 occurs without electron delivery (25). Thus it has been suggested that b5 might selectively stabilize the intermediate in the lyase reaction or cause substrates to assume orientations in the CYP17A1 active site more favorable for the lyase chemistry, but the mechanism remains unresolved (31).
The structural basis for each of these effects—hydroxylase substrate regioselectivity, lyase reaction selectivity for the Δ4,3-keto versus Δ5,3-ol 17-hydroxylated substrate, and cytochrome b5 facilitation of the lyase versus hydroxylase reaction—is unknown. Homology models and docking studies have suggested that substrates were likely to orient essentially parallel to the plane of the heme (32) and proposed a “bi-lobed” active site to carry out the separate hydroxylase and lyase reactions (33–36). The only known structures of CYP17A1, in the presence of the steroidal inhibitors abiraterone or TOK-001, were published recently (8). These steroidal inhibitors both orient more nearly perpendicular to the heme, evoking the prediction of a similar binding mode for substrates (8), but the actual binding of CYP17A1 substrates is unknown.
To probe the binding orientations of the physiologically relevant substrates and the structural basis of CYP17A1 function, a series of experimental x-ray structures were generated for CYP17A1 in complexes with both hydroxylase and both lyase substrates with the mutation A105L. Comparisons among these structures identify steric and hydrogen bonding interactions between CYP17A1 and distal portions of the substrate that play key roles in modulating spatial relationships between sites of metabolism on the opposite end of substrates and the catalytic heme iron. A steric rationale is provided for hydroxylase regioselectivity, whereas differences observed for hydrogen bonding and substrate positioning may form the basis for substrate selectivity of the lyase reaction. This new information informs a working hypothesis for how cytochrome b5 might selectively facilitate the lyase reaction.
EXPERIMENTAL PROCEDURES
Mutagenesis
The CYP17A1 gene in the pCWori+ vector has an N-terminal truncation of residues 1–19, a slight modification of the new N terminus, and a C-terminal 4× histidine tag, as described (8). The A105L mutation was incorporated using the QuikChange Lightning site-directed mutagenesis kit (Agilent).
Expression
The resulting pCW17A1Δ19H plasmid containing either the wild type or A105L sequence was transformed into Escherichia coli JM109 cells by heat shock at 42 °C, spread on lysogeny broth agarose plates containing 50 μg/ml ampicillin and incubated overnight at 37 °C. To select for the pCWori+ vector, all expression media were supplemented with 50 μg/ml ampicillin. Lysogeny broth (5 ml) was inoculated with a single colony from the aforementioned plate and incubated at 37 °C with shaking at 250 rpm for 6 h. Overnight cultures (200 ml of lysogeny broth) were inoculated with 50 μl of the initial culture and incubated at 37 °C for 18 h with 250 rpm shaking. Terrific Broth (1 liter/2.8-liter Fernbach flask) was inoculated with 10 ml of the overnight culture. This culture was grown at 37 °C with shaking at 250 rpm until reaching an optical density of 0.5 at 600 nm. Overexpression was then induced by the addition of isopropyl β-d-1-thiogalactopyranoside (0.5 mm). The heme precursor δ-aminolevulinic acid was added to 0.61 mm. Cultures were grown at 28 °C with shaking at 140 rpm for an additional 72 h. The cells were then collected by centrifugation at 6300 × g for 10 min. Cells were resuspended in 50 mm Tris-HCl, pH 7.4, 20% (v/v) glycerol, and 300 mm NaCl and stored at −80 °C until purification.
Purification
Cells were thawed and lysed by sonication (six times for 30 s at 1-min intervals on ice). The resulting lysate was centrifuged at 9900 × g for 15 min. Membrane proteins were extracted by stirring this lysate in the presence 2% (v/v) Emulgen 913 (Desert Biologicals) for 90 min, followed by ultracentrifugation (100,000 × g for 1 h). The resulting supernatant was loaded on nickel-nitrilotriacetic acid-agarose resin (Qiagen) pre-equilibrated with Ni buffer (50 mm Tris-HCl, pH 7.4, 20% (v/v) glycerol, 300 mm NaCl, and 0.2% (v/v) Emulgen 913). The resin was subsequently washed with 2 column volumes of Ni buffer, 6 column volumes of Ni buffer supplemented with 100 mm glycine, and eluted using 4 column volumes of Ni buffer supplemented with 100 mm glycine and 80 mm histidine. Elution fractions were pooled based on absorbance of the heme Soret peak, diluted 4.2-fold in CM buffer (50 mm Tris-HCl, pH 7.4, 20% (v/v) glycerol, and 100 mm glycine), supplemented with 0.2% (v/v) Emulgen 913, and loaded onto a 5-ml carboxymethyl-Sepharose fast-flow column (GE Healthcare) previously equilibrated with CM buffer. The column was washed with 10 column volumes of CM buffer and eluted with CM buffer supplemented with 500 mm NaCl. Fractions were pooled based on the heme Soret absorbance and concentrated to ∼1 ml. Concentrated protein was injected onto a Superdex 200 gel filtration column (GE Healthcare) pre-equilibrated with buffer containing 50 mm Tris-HCl, pH 7.4, 20% (v/v) glycerol, 100 mm glycine, and 500 mm NaCl. The major peak with absorbance for the heme was collected and concentrated. All purification was performed at 4 °C. CYP17A1 A105L protein generated for crystallization studies was purified with 50 μm of one of the ligands present in all purification buffers.
Protein Crystallization, Data Collection, and Structure Determination
CYP17A1 A105L was crystallized by hanging drop vapor diffusion. Purified protein (∼29 mg/ml) containing either 50 μm of one of the substrates or 10 μm abiraterone and 0.5% (v/v) Emulgen 913 was mixed 1:1 to form 2-μl drops that were then equilibrated against precipitant solution at 20 °C. The precipitant solution used to crystallize CYP17A1 A105L with progesterone, pregnenolone, 17α-hydroxypregnenolone, and abiraterone consisted of 100 mm Tris-HCl, pH 8.5, 25% (v/v) PEG 4000 (Hampton Research), 150 mm magnesium chloride hexahydrate, and 4–6% (v/v) glycerol. The precipitant solution used to crystallize CYP17A1 A105L with 17α-hydroxyprogesterone contained 175 mm Tris-HCl, pH 8.5, 30% (v/v) PEG 3350 (Hampton Research), 250 mm lithium sulfate, and 3% (v/v) glycerol. Crystals were cryoprotected in a 7:3 mixture of mother liquor and 80% (v/v) glycerol and flash cooled in liquid nitrogen. Diffraction data were collected on Beamlines 14-1 and 12-2 of the Stanford Synchrotron Radiation Lightsource and processed using XDS (37). Data collection and refinement statistics and deposition codes in the Protein Data Bank are given in Table 1. Structures were solved by molecular replacement using Phaser (38) and the structure of wild type CYP17A1 (Protein Data Bank codes 3RUK or 3SWZ (8)) as a search model using data to the respective resolution cutoff for the different structures (2.5–3.0 Å), yielding log likelihoods of 9,094–13,090. Model building and refinement were performed iteratively with Coot (39) and PHENIX (40), respectively, using data to a cutoff of <I/σ(I)> of 1.5 or higher in the outer shell. Hydrogens were modeled in the riding positions for protein and substrates. Reference model restraints were incorporated only for the 3.0 Å 17α-hydroxypregnenolone data set, to avoid overfitting. After the protein structures were essentially completed, ligands were added. The ligand omit 2Fo − Fc maps shown were calculated in PHENIX. Ligand models were obtained from the Hetero-compound Information Center at Uppsala (HIC-Up) or constructed using the PRODRG2 server. Superpositions between CYP17A1 molecules were generated using the secondary structure matching algorithm in COOT (39). Superposition between P450cam and CYP17A1 was generated by the same process, then optimized using least squares fit for the hemes, also in COOT (39). Probe-occupied active site volumes were generated using VOIDOO (41) (probe radius = 1.4 Å; grid spacing = 0.33). The figures were prepared in PyMOL (42).
TABLE 1.
Statistics for x-ray data collection and refinement
| CYP17A1 A105L abiraterone | CYP17A1 A105L pregnenolone | CYP17A1 A105L progesterone | CYP17A1 A105L 17α-hydroxy-progesterone | CYP17A1 A105L 17α-hydroxy-pregnenolone | |
|---|---|---|---|---|---|
| Protein Data Bank code | 4NKV | 4NKW | 4NKX | 4NKY | 4NKZ |
| Data collection | |||||
| Beamline | SSRL 12-2 | SSRL 14-1 | SSRL 12-2 | SSRL 12-2 | SSRL 12-2 |
| Space group | P212121 | P212121 | P212121 | P212121 | P212121 |
| Cell dimensions: a, b, c, (Å) | 90.7, 153.3, 167.7 | 86.1, 152.6, 173.8 | 85.9, 153.0, 173.2 | 91.3, 151.8, 168.0 | 85.8, 151.1, 170.3 |
| Molecules/a.u. | 4 | 4 | 4 | 4 | 4 |
| Resolution (Å)a | 39.31-2.65 (2.79-2.65) | 39.11-2.50 (2.63-2.50) | 39.13-2.79 (2.94-2.79) | 39.15-2.55 (2.69-2.55) | 38.70-3.00 (3.17-3.00) |
| Total reflectionsa | 450,626 (62,230) | 557,941 (64,186) | 380,303 (52,751) | 511,008 (71,923) | 287,854 (36,646) |
| Unique reflectionsa | 68,380 (9,523) | 79,365 (11,000) | 56,987 (7,891) | 76,008 (10,667) | 44,496 (6,106) |
| Redundancya | 6.6 (6.5) | 7.0 (5.8) | 6.7 (6.7) | 6.7 (6.7) | 6.5 (6.0) |
| Rpima | 0.044 (0.556) | 0.060 (0.717) | 0.074 (0.579) | 0.060 (0.666) | 0.080 (0.650) |
| <I/σ(I)>a | 6.8 (1.5) | 11.0 (2.1) | 9.7 (1.5) | 12.3 (1.5) | 8.9 (1.5) |
| Completeness (%)a | 99.3 (96.1) | 99.3 (95.6) | 99.2 (95.5) | 99.1 (96.3) | 98.9 (94.3) |
| Refinement | |||||
| Resolution (Å) | 38.59-2.65 | 39.11-2.50 | 39.13-2.80 | 39.15-2.55 | 38.70-3.00 |
| No. reflections | 68,166 | 79,236 | 56,820 | 75,825 | 43,862 |
| R/Rfree (%) | 19.1/24.3 | 18.9/24.5 | 18.4/25.7 | 17.8/24.4 | 19.1/26.1 |
| Ramachandran (%) | |||||
| Favored | 96.44 | 95.50 | 95.63 | 96.46 | 96.78 |
| Allowed | 3.45 | 4.39 | 4.26 | 3.54 | 3.06 |
| Outliers | 0.11 | 0.11 | 0.11 | 0 | 0.16 |
| Wilson B factor | 61.4 | 52.8 | 56.9 | 51.4 | 68.6 |
| No. atoms/B factors | |||||
| Protein | 14913/71.6 | 14852/71.8 | 14925/59.3 | 14832/56.8 | 14806/80.1 |
| Ligand | 104/36.7 | 92/61.1 | 92/46.3 | 96/52.3 | 96/65.7 |
| Heme | 172/50.5 | 172/50.4 | 172/41.9 | 172/44.1 | 172/63.8 |
| Water | 139/50.4 | 87/52.6 | 46/37.4 | 161/44.5 | 8/65.9 |
| RMSD bond (Å) | 0.011 | 0.008 | 0.008 | 0.007 | 0.010 |
| RMSD angle (°) | 1.2 | 1.2 | 1.1 | 1.2 | 1.3 |
| Coordinate error (maximum-likelihood based) | 0.41 | 0.34 | 0.41 | 0.33 | 0.39 |
| Key bond distances and anglesb | |||||
| N/ON202-O3 (Å) | 2.7 ± 0.1 | 2.8 ± 0.1 | 2.7 ± 0.3 | 2.9 ± 0.05 | 2.8, 2.5, 3.8, 3.0 |
| ∠C3-O3-N/ON202 (°) | 102 ± 3 | 121 ± 5 | 121 ± 7 | 137 ± 4 | 144, 132, 135, 136 |
| C16-iron (Å) | 5.9 ± 0.2 | 4.9 ± 0.2 | 4.8 ± 0.2 | 4.7 ± 0.1 | 5.1, 5.1, 4.6, 4.7 |
| C17-iron (Å) | 5.7 ± 0.1 | 4.8 ± 0.1 | 4.8 ± 0.1 | 5.0 ± 0.1 | 4.8, 4.8, 4.4, 4.6 |
| C20-iron (Å) | 4.6 ± 0.2 | 4.6 ± 0.1 | 4.6 ± 0.1 | 4.6 ± 0.2 | 4.6, 4.6, 4.4, 4.4 |
a Statistics corresponding to the highest resolution shell are in parentheses.
b Atomic elements refer to steroid or heme (iron) atoms unless an amino acid designation is provided as a subscript.
Progesterone Hydroxylation Assays
Hydroxylation of progesterone (0–70 or 200 μm) by CYP17A1 was determined after HPLC separation by UV absorbance detection as described (8). A modification to this assay was the use of human NADPH-cytochrome P450 reductase bearing an N-terminal truncation (43), in addition to a K59Q mutation that prevents proteolysis. Metabolites 17α-hydroxyprogesterone, 21-hydroxyprogesterone, and 16α-hydroxyprogesterone were detected at 248 nm with retention times of ∼5.5, ∼4.1, and ∼2.2 min, respectively.
Pregnenolone Hydroxylation and 17,20-Lyase Assays
CYP17A1 (50 pmol) was incubated at 4 °C with the human P450 oxidoreductase described above and rat cytochrome b5 in either a 1:4:4 ratio (for 17,20-lyase reactions) or 1:4:0 ratio (for pregnenolone hydroxylation reactions) for 20 min. This reconstituted system was added to buffer (50 mm Tris-HCl, pH 7.4, and 5 mm magnesium chloride) containing substrate (0–7 μm pregnenolone for hydroxylation or 0–20 μm 17α-hydroxypregnenolone for 17,20-lyase) to a total volume of 480 μl. The reactions were carried out at 37 °C and were initiated by the addition of NADPH (20 μl of 25 mm). After 10 min, reactions were quenched by the addition of 20% (w/v) trichloroacetic acid (300 μl) containing 8.66 μg/ml estriol as an internal standard. Both samples and standards (650 μl) were loaded onto SPE columns (HLB cartridge, 1 ml, 30 mg of packing) pre-equilibrated with 2× 1 ml of methanol, followed by 2× 1 ml of H2O. Following sample loading, the columns were washed with 1 ml of H2O and steroids eluted using 4× 1 ml of 1:2 dichloromethane:methanol. Eluent was dried and resuspended in 100 μl of derivitizing reagent prepared as a 500:4:2 (v/w/w) mixture of N-methyl-N-(trimethylsilyl)-trifluoroacetamide/ammonium iodide/dithiothreitol and heated to 60 °C for 40 min. Derivatized samples were injected onto GC/MS (3-μl injection for lyase samples and 5-μl injections for hydroxylase samples) and analyzed in selected ion monitoring mode using instrument parameters as described (44) except that the column was Agilent DB-5MS, 15 × 0.25-mm inner diameter with 0.25-micron film. Silylated derivatives of products 17α-hydroxypregnenolone (retention time ∼16.5 min; m/z = 548) and dehydroepiandrosterone (retention time, ∼7.9 min; m/z = 432) were quantified based on their molecular ion with respect to that of silylated estriol internal standard (retention time ∼15.7 min; m/z = 504).
Ligand Binding Assays
Ligands were titrated into CYP17A1 protein and binding monitored by changes in UV-visible absorbance as described previously (45) except that 100 nm protein was in 50 mm Tris-HCl, pH 7.4, 20% (v/v) glycerol, and 100 mm glycine, and the cuvettes had path lengths of 5 cm. Changes in absorbance were fit to the tight binding equation using Prism (GraphPad Software) to determine the dissociation constants.
RESULTS
Characterization of CYP17A1 Ligand Binding and Substrate Metabolism
To compare the interactions of CYP17A1 with its various substrates and products, upon A105L mutation, and with structural findings, both dissociation constants and steady-state kinetic parameters were measured (Table 2). CYP17A1 affinities for steroid substrates and products were evaluated by monitoring the spectral shift associated with displacement of an active site water from the heme iron that occurs as ligands bind adjacent to the heme. Steady-state kinetic parameters were determined for all three substrates with appreciable product formation (progesterone and pregnenolone 17α-hydroxylation reactions and the 17α-hydroxypregnenolone 17,20-lyase reaction).
TABLE 2.
Substrate binding and steady-state kinetic parameters for the metabolism of physiologically relevant steroid substrates by CYP17A1 wild type and the A105L mutant
| Wild type | A105L | |
|---|---|---|
| Steroid substrate and product Kd values (nm) | ||
| Pregnenolone | <100 (8) | <100 |
| Progesterone | 230 ± 14 (8) | <100 |
| 17α-Hydroxypregnenolone | 210 ± 7 | <100 |
| 17α-Hydroxyprogesterone | 330 ± 22 | 390 ± 90 |
| 16α-Hydroxyprogesterone | 8700 ± 600 | 8900 ± 600 |
| Progesterone 17α-hydroxylation kinetics | ||
| kcat (min−1) | 1.01 ± 0.05 | 1.16 ± 0.01 |
| Km (μm) | 10.5 ± 1.7 | 1.96 ± 0.097 |
| Pregnenolone 17α-hydroxylation kinetics | ||
| kcat (min−1) | 0.39 ± 0.03 | 0.39 ± 0.03 |
| Km (μm) | 0.93 ± 0.24 | 0.38 ± 0.09 |
| 17α-Hydroxypregnenolone 17,20-lyase kinetics | ||
| kcat (min−1) | 0.24 ± 0.02 | 1.2 ± 0.08 |
| Km (μm) | 1.2 ± 0.3 | 1.2 ± 0.3 |
Progesterone binds more tightly to wild type CYP17A1 than either its major (17α-) or minor (16α-) hydroxylation products, with the 16α-hydroxyl conferring a notable >80-fold decrease in affinity (Table 2). Similarly, pregnenolone binds wild type CYP17A1 more tightly than its 17α-hydroxylated product. Higher affinities are observed for all pregnenolone-based (Δ5,3-ol) ligands than for the corresponding progesterone (Δ4,3-keto) compounds. The effect of the A105L mutation is to increase progesterone affinity at least 2-fold but has little effect on the already lower affinity for either progesterone hydroxylation product. Pregenenolone binds with such high affinity to both wild type and A105L that any changes in affinity caused by mutation are not apparent, but the decrease in affinity that the 17α-hydroxy substituent confers on pregnenolone is offset by the A105L mutation.
Significant catalytic differences are also observed for different substrates and upon mutation (Table 2). The wild type CYP17A1 17-hydroxylates pregnenolone with a Km that is 10-fold lower than for progesterone 17-hydroxylation, consistent with the rank order of the dissociation constants. Although the kcat is ∼3-fold lower for pregnenolone 17-hydroxylation than for progesterone 17-hydroxylation, the overall catalytic efficiency (kcat/Km) is still 4.4-fold higher for pregnenolone 17-hydroxylation. The A105L mutation does not alter kcat for the 17-hydroxylation of either substrate but does substantially decrease the Km for both reactions, consistent with general increases observed in the dissociation constants reported above.
Although pregnenolone is only hydroxylated at C17, human CYP17A1 is reported to also convert progesterone to the minor 16α-hydroxylated metabolite (Fig. 1), which can compose 10–30% of total hydroxylated metabolites depending on the system under study (17, 46). For wild type CYP17A1 in the current purified enzyme system at saturating progesterone concentrations, 16α-hydroxyprogesterone comprised 12% of the total progesterone hydroxylase activity, with 17α-hydroxyprogesterone accounting for the remainder. The A105L mutation was reported to increase 17-hydroxylation and decrease 16-hydroxylation in a cell-based system (17). In agreement, under the same conditions used to evaluate the wild type enzyme herein, CYP17A1 A105L yielded only ∼5% of the total hydroxy metabolites as the 16α-hydroxyprogesterone minor product, with 95% being the 17α-hydroxyprogesterone metabolite. Thus these functional results agree that the A105L mutation increases the affinity of CYP17A1 for progesterone and that it does so by preferentially orienting progesterone for hydrogen abstraction at C17 over C16.
Human CYP17A1 has also been noted to produce very small amounts of 21-hydroxyprogesterone, also called 11-deoxycorticosterone, which is increased with the A105L mutation (46). Although the current purified system yielded no detectable 21-hydroxyprogesterone at the highest substrate concentrations used for determining steady-state kinetic parameters (70 μm), at much higher progesterone concentrations (200 μm), trace amounts of 21-hydroxyprogesterone were inconsistently detected for the A105L mutant but never for the wild type enzyme. Thus the A105L mutation appears to slightly increase the production of 21-hydroxyprogesterone.
In contrast to hydroxylation reactions, where the A105L mutation altered only the Km, in the 17,20-lyase reaction the mutation had no effect on the Km. Instead the mutation resulted in a 5-fold increase in kcat, suggesting that the A105L mutation contributes to interactions that more readily permit the lyase reaction while having little affect on affinity.
Global Overview of CYP17A1 A105L Structures
Introduction of the A105L mutation increased general CYP17A1 stability during purification (and likely during expression as well), as evaluated by overall protein yield. This consistently higher yield, perhaps in combination with an increased affinity for at least two of the steroid substrates, facilitated determination of a series of CYP17A1 A105L liganded x-ray structures for side by side comparison. All crystallized in the same space group with the same packing. Comparison of the four molecules composing the asymmetric unit for one substrate complex with any of the other substrate complexes or for a complex with the inhibitor abiraterone reveals they are all very similar to each other (average root mean square deviation (RMSD) over all Cα of 0.42 ± 0.10) and to previous structures of wild type CYP17A1 bound to abiraterone and another steroidal inhibitor (average RMSD over all Cα 0.41 ± 0.10) (8). However, for all CYP17A1 structures to date, two of the four molecules in the asymmetric unit (molecules A and B) differ slightly from the other two (molecules C and D), resulting in a higher Cα RMSD of 1.29 ± 0.06. Structural differences between these two conformations are observed at the backbone level for the N terminus, a loop between β1 strands, and the F-G region (Fig. 2). These regions all lie on one face of the molecule that was proposed to be involved in membrane binding. As is true for almost all membrane P450 structures, this surface also constitutes a packing interaction with neighboring molecules in the crystalline lattice and is known to be relatively flexible. Therefore it is difficult to determine whether these differences between molecules are functionally significant. In all structures, a short loop region between helices H and I was not modeled (Fig. 2) because of poor density, suggesting that this region is disordered. In two molecules of the structure with 17α-hydroxyprogesterone and three molecules of the structure with 17α-hydroxypregnenolone, several residues between the C and D helices were also not modeled because of ambiguous electron density.
FIGURE 2.
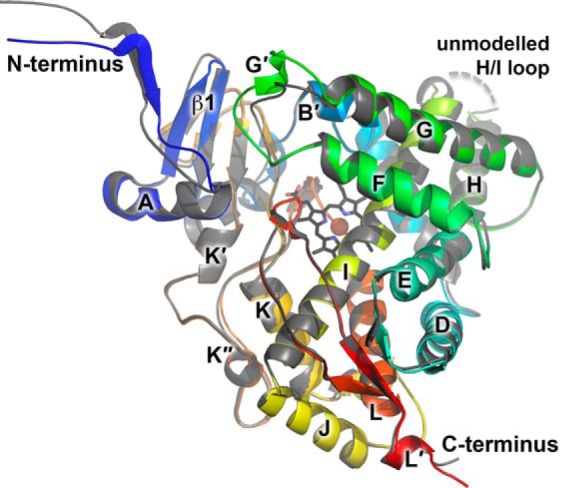
The overall structure of CYP17A1 A105L molecules A/B (shown in rainbow colors) and C/D (gray) have small variations in the backbone structure on one face of the enzyme, including the N terminus, the residues between the F and G helices, and a small portion of the adjacent β1 sheet. Dashed line, HI turn residues not modeled because of weak density.
Structure of CYP17A1 A105L with Abiraterone
To determine the effects of the A105L mutation alone on enzyme structure, the CYP17A1 A105L structure was determined in complex with the steroidal inhibitor abiraterone and compared with that of the wild type enzyme containing this same ligand (Protein Data Bank code 3RUK (8)). The 2.65 Å structure of the mutant enzyme (Table 1) is essentially identical to the wild type enzyme at the backbone level and in all other respects, save in the local region of the single site mutation (Fig. 3). The major difference between wild type CYP17A1 and the A105L mutant abiraterone structures is that the additional bulk caused by substitution of leucine for alanine projects directly into what is part of the active site for the wild type enzyme. Reduction in overall active site volume from 677 Å3 for the wild type enzyme to 642 Å3 for the A105L mutant is due almost entirely to this substitution. This reduction in active site volume does not alter the orientation of abiraterone or any of its interactions with the CYP17A1 active site. The steroid still lies with the unsubstituted α face against the I helix with the steroid plane forming a 60° angle from the plane of the heme. The only intermolecular hydrogen bond, between the 3β-hydroxyl group of abiraterone and Asn202, is present in both structures. Thus in this structure the impact of the A105L mutation appears to be only steric in nature.
FIGURE 3.
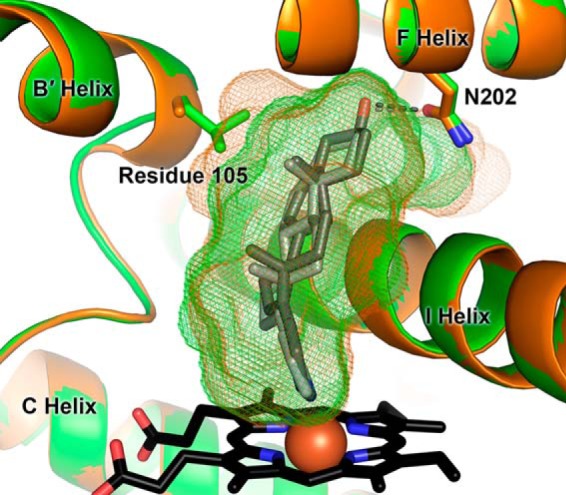
Active site of wild type CYP17A1 (orange) bound to abiraterone (Protein Data Bank code 3RUK) (8) overlaid with the active site of CYP17A1 A105L (green) bound to abiraterone. Mutation of residue 105 does not affect the orientation of the abiraterone inhibitor (shown in gray sticks) but reduces the active site volume specifically in the local region of the substituted side chain (wild type and A105L active site voids in orange and green mesh, respectively).
Structure of CYP17A1 A105L with Hydroxylase Substrates
Structures of the CYP17A1 A105L mutant with the 17α-hydroxylase substrates progesterone and pregnenolone were determined at resolutions of 2.80 and 2.50 Å, respectively (Table 1). In all four molecules of the asymmetric unit of both hydroxylase substrate complexes, the ligand steroids assume the same orientation (Fig. 4, A and B). This general orientation is also almost identical to that previously observed for abiraterone (Fig. 4C), with the α-face of the steroid packed against the I helix and the cyclopentanophenanthrene steroid nucleus forming a ∼60° angle from the heme plane. Abiraterone forms a hydrogen bond to the side chain of Asn202 in the F helix in both the wild type structure (8) and in the A105L mutant (Fig. 4C). Abiraterone and pregnenolone are both Δ5,3-ol steroids. Thus it is not surprising that the C3-OH of pregnenolone is also oriented to form a 2.8 ± 0.1 Å (average; Table 1) hydrogen bond to the Asn202 side chain (Fig. 4A). The corresponding C3-keto substituent of progesterone is located to form a hydrogen bond of similar length (2.7 ± 0.3 Å average) to Asn202 (Fig. 4B). Although progesterone must be the hydrogen bond acceptor and the amide nitrogen of Asn202 must be the hydrogen bond donor, the donor/acceptor relationship is less clear for pregnenolone. The pregnenolone C3 hydroxyl could be donor or acceptor, and the side chain nitrogen/oxygen could be donor/acceptor respectively depending on the side chain torsion. As a result of their similar hydrogen bonding interactions with Asn202 (distances and angles; Table 1) and similar packing against the I helix, the distances from C17 that is hydroxylated to the iron that would compose the catalytic iron (IV) oxo intermediate are essentially identical (4.8 ± 0.1 Å; Table 1) between the two hydroxylase substrates. Similarly, C16 (hydroxylated when progesterone is the substrate, but not when pregnenolone is the substrate) is also essentially same distance from the heme iron for both substrates (Table 2) but is located at a greater angle from the iron than C17 (Fig. 4, A and B).
FIGURE 4.

X-ray structures of CYP17A1 A105L with all four physiological substrates demonstrate overall similar orientations, with the steroid nucleus at an ∼60° angle with respect to the heme plane and the steroid α-face flat against a peptide bond in the I helix. Hydroxylase substrates pregnenolone (A, green) and progesterone (B, orange) form hydrogen bonds between their respective C3 alcohol and keto substituents and the side chain amide of Asn202, an orientation termed position 1 and similar to that observed for the steroidal inhibitor abiraterone (C, cyan). The poor 17,20-lyase substrate 17α-hydroxyprogesterone (D, magenta) is oriented similar to the hydroxylase substrates in position 1 with its C3 keto group also hydrogen bonding to Asn202. However, in different molecules of the structure with the efficient lyase substrate 17α-hydroxypregnenolone, this substrate is found either in position 1 with its C3 hydroxyl hydrogen bonding to Asn202 (E, purple) or in a position closer to the heme which is too distant from Asn202 to allow hydrogen bonding (F, yellow, position 2). For clarity, in A–F, the brackets only indicate the distances between C17 and iron. Other distances and angles are provided in Table 1. Electron density is shown as a 2Fo − Fc ligand omit map contoured at 1.0 σ. In G, overlays of hydroxylase substrates pregnenolone (green) and progesterone (orange) demonstrate the similarity of their binding modes, whereas an overlay of 17α-hydroxypregnenolone in its two observed positions illustrates important differences in hydrogen bonding (H).
Structure of CYP17A1 A105L with 17,20-Lyase Substrates
Structures of CYP17A1 A105L co-crystallized with the efficient lyase substrate 17α-hydroxypregnenolone and the poor lyase substrate 17α-hydroxyprogesterone were determined to resolutions of 3.0 Å and 2.55 Å, respectively (Table 1). In the 17α-hydroxyprogesterone complex, the ligand binds very similarly in all four molecules of the crystal and very similar to the orientation observed for the hydroxylase substrates (Fig. 4D). The hydrogen bond between the C3-keto and side chain amide of Asn202 is maintained with similar distances and angles (Table 1). The substrate position with respect to the I helix and heme iron (e.g. C17-iron, 5.0 ± 0.1 Å; Table 1) is also consistent with the hydroxylase substrates, a state that will be referred to as position 1. In the 17α-hydroxypregnenolone structure, the general substrate position relative to the I helix is maintained, but the substrate is observed to occupy distinct positions that vary by their relative position between Asn202 and the heme. For two of the molecules of this structure, 17α-hydroxypregnenolone is in position 1 as observed for 17α-hydroxyprogesterone (Fig. 4E), with its C3 alcohol forming a hydrogen bond to Asn202 (Table 1). However, in the other two molecules of this structure, 17α-hydroxypregnenolone is found farther from Asn202 and closer to the heme iron. In its closest approach to the iron, a state referred to as position 2 (Fig. 4F), the substrate C3 alcohol is much too far removed from Asn202 to participate in even a weak hydrogen bond (3.8 Å), and the C17-iron distance is reduced to 4.4 Å. The fourth molecule shows 17α-hydroxypregnenolone in an intermediate position. The differences in position are perhaps better illustrated by comparing the electron density associated with ligand and heme for these various complexes. For 17α-hydroxypregnenolone in position 1 and all other CYP17A1 substrates, the substrate electron density is distinct from that of the heme (Fig. 4, A, B, D, and E), whereas for 17α-hydroxypregnenolone in position 2, closer to the heme, the substrate electron density is continuous with the heme (Fig. 4F), more similar to that of abiraterone (Fig. 4C). Thus overlays of the progesterone and pregnenolone complexes illustrate the high degree of similarity in their binding (Fig. 4G) in a position and orientation also shared with 17α-hydroxyprogesterone (not shown). However, an overlay of the two binding positions observed for 17α-hydroxypregnenolone illustrates differences in the hydrogen binding to Asn202 and a shift toward the heme.
Further Variation in Active Site Activity Topography
The amino acid side chains surrounding the active site, the observable waters, and the hydrogen bonding of the I helix that the steroids pack against are not significantly different between complexes (even when 17α-hydroxypregnenolone is positioned nearer to the heme) or between molecules in each structure. Very minor differences reflect small changes in torsion angles or slightly favored rotamers to best fit the density in each structure. However, in two regions these small differences result in substantial variation in active site size and topography (Fig. 5). Rather than being correlated with a particular substrate, this appears to result primarily from the relatively small differences in the backbone for molecules A and B versus molecules C and D. Like the wild type structure with abiraterone, the active site cavity extends beyond the C3 substituent, and this varies somewhat in size, even between molecules of a particular substrate complex, because of slight differences in side chain torsions. Additionally, several molecules have extensions from the main active site extending from the region of the ligand C19 substituent, which can consist of substantial extra volume as a result of the slight differences in the F-G′ loop backbone and very slight side chain torsions. In a very few molecules, one or both of these active site extensions reach the protein surface, at the intersection of the F/G′ loop, β4–1, and the first turn of the A helix or between the bases of the F and G helices, respectively. If expanded by breathing motions or conformational changes in the protein, these channels could play a role in ligand entry/exit. The cavity over the I helix exiting at the base of the F and G helices would be exposed to solvent, whereas the cavity extension exiting in the region of the F/G′ loop would be expected to be buried in the membrane.
FIGURE 5.
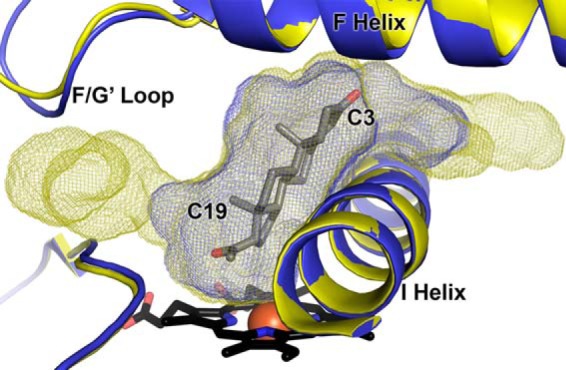
Probe-occupied cavities for the active sites of CYP17A1 A105L bound to progesterone (molecule B and D, yellow and blue, respectively) exemplify the range of volumes among the present structures. Although abiraterone always exhibits a minimal active site volume similar to that shown in blue mesh, for substrates slight modifications of backbone and side chain torsions in the F-G′ region and/or at the base of the F and G helices can result in a significant extension near C19 and/or enlargement of the extension off C3. In a few cases, these cavities extend to the protein surface.
DISCUSSION
The reactions catalyzed by CYP17A1 have long been of substantial interest because they occur at a key juncture in human steroidogenesis, controlling the biosynthesis of mineralocorticoids, glucocorticoids, and sex steroid androgens and estrogens. Originally thought to be performed by two different enzymes, a steroid 17α-hydroxylase and a 17,20-lyase or -desmolase, both catalytic functions were later shown to be accomplished by CYP17A1 (47). In part to explain how both reactions might occur in the single CYP17A1 active site, the concept of a “bi-lobed” active site was advanced (36), in which the different lobes would carry out the separate hydroxylase and lyase reactions. Homology models and docking studies suggested that substrates were likely to orient essentially parallel to the plane of the heme (32–34). In contrast, the first available structures of CYP17A1, which were complexes of steroidal inhibitor analogs of pregnenolone, did not support a bi-lobed cavity and demonstrated ligand orientation more nearly perpendicular to the heme than parallel (8). However, both of these inhibitors contained nitrogen heterocycles as substituents to the steroid scaffold at C17 and coordinated to the heme iron. In the absence of further data, subsequent work has been interpreted with differing models of steroid substrate orientation either parallel (46) or more perpendicular to the heme (8, 24) within the CYP17A1 active site. Herein experimental structures of CYP17A1 with each of the four substrates clearly demonstrate that steroid substrates adopt a binding orientation generally consistent with that of the steroidal inhibitors in the original x-ray structures (Fig. 4).
One possible caveat might be that all of the current structures were obtained in the presence of the A105L mutation. However, the only structural difference between the wild type and A105L structures with the common ligand abiraterone was a steric difference in active site volume as the larger leucine filled active site space unoccupied by abiraterone (Fig. 3). The mutation did not alter the orientation of the steroidal inhibitor in the CYP17A1 active site. The reduction in the excess width of the active site engendered by the alanine to leucine substitution is consistent with decreases in Kd for most substrates and decreases in hydroxylation Km values (Table 2). This evidence and concordance with functional evidence discussed below suggests that the A105L mutation does not significantly alter the primary orientation of substrate steroids important for C17 hydroxylation.
Thus the structural results herein are first integrated with the functional hydroxylase data to propose an internally consistent hypothesis for CYP17A1 hydroxylase selectivities. The orientations observed for CYP17A1 with progesterone or pregnenolone in the current structures are consistent with production of the major 17α-hydroxy metabolites observed. In both cases, C17 is oriented toward the heme iron. The C17α hydrogens of both hydroxylase substrates are located such that they would be clearly most susceptible to abstraction by the iron (IV) oxo intermediate in the hydroxylase reaction (Fig. 4, A and B). The C17 hydrogen is not located directly over the heme iron, but a few degrees off center, consistent with the moderate reported kinetic isotope effect for progesterone 17α-hydroxylation, interpreted as a bent transition state (46). Wild type and the A105L mutant have essentially the same kinetic isotope effect for 17α-hydroxylation, 4.1 and 3.8, respectively (46), further supporting the idea that ligand orientation for 17α-hydroxylation is unperturbed by the mutation.
Combination of the current structural and functional data suggests a mechanism underlying the generation of minor progesterone metabolites. Depending on the system under study, wild type CYP17A1 also hydroxylates progesterone to yield 10–30% (17, 46) of the total product as 16α-hydroxyprogesterone and has also been reported to produce trace amounts of 21-hydroxyprogesterone (46). The C16 and C21 hydrogens flank the C17 hydrogen, and the current structure positions them such that they would be the next most susceptible for hydrogen abstraction by the iron oxo intermediate (Fig. 6A, experimental ligand position in gray sticks). In the purified system, wild type CYP17A1 produced 88% of the total progesterone metabolites with hydroxylation at C17, whereas the remaining 12% was the minor C16-hydroxylated product, and 21-hydroxyprogesterone was not detected. Consistent with previous reports (17, 46), the A105L mutation decreased the percentage of 16-hydroxylated product, increased 17-hydroxylated product, and could produce trace amounts of the 21-hydroxylated metabolite 11-deoxycorticosterone. Thus the A105L mutation, whose nearest atom is some 8–11 Å away from the site of metabolism, substantially reduces 16-hydroxylation on one side of C17 and slightly increases C21 hydroxylation on the opposite side of C17, suggesting a rotation of the substrate in the active site (Fig. 6A, proposed minor orientation in pink sticks).
FIGURE 6.
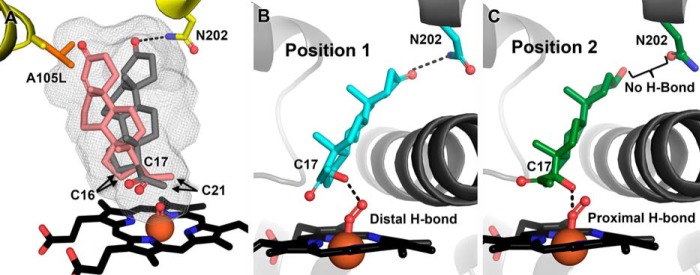
Combination of structural and functional data supports the following models for CYP17A1 catalysis. A, regioselectivity and substrate selectivity of hydroxylation. Whereas the major binding modes for progesterone and pregnenolone are oriented with C17 directed toward the iron for 17α-hydroxylation (gray sticks, orientation observed in structures), the steroid core of progesterone may also adopt an alternate plane that facilitates C16 hydroxylation (salmon sticks, proposed orientation). The A105L mutation suppresses this alternate orientation, decreasing C16 hydroxylation, increasing C17 hydroxylation, and slightly increasing hydroxylation of C21 on the opposite side of the substrate. Pregnenolone may not adopt this alternate orientation favoring C16 hydroxylation because its C3 hydroxyl is restrained by a stronger hydrogen bond with Asn202. Gray mesh is CYP17A1 wild type active site cavity. B, in the current model for substrate selectivity of the lyase reaction, 17α-hydroxyprogesterone may be a poor 17,20-lyase substrate because of its positioning in the active site. This substrate is only observed hydrogen bonding to Asn202 such that it is farther away from the iron, and its C17 hydroxyl is predicted to hydrogen bond with the distal oxygen of the peroxy intermediate, an interaction that is not conducive to the lyase reaction. C, in contrast, 17α-hydroxypregnenolone may be a more efficient substrate for the lyase reaction because it is observed to move closer to the iron, where studies suggest the C17 hydroxy may hydrogen bond with the proximal oxygen of the catalytic peroxy intermediate. This position would facilitate nucleophilic attack of the distal oxygen on C20 to perform the lyase reaction. The well established facilitation of the lyase reaction by b5 is consistent with an allosteric interaction favoring 17α-hydroxypregnenolone in position 2 versus position 1.
The present structural information demonstrates that A105L occludes part of the active site volume adjacent to the B′ helix (Fig. 3). This volume is not utilized by progesterone binding in a mode consistent with 17-hydroxylation. However, the functional data suggest that the availability of active site volume near Ala105 correlates with the production of 16α-hydroxyprogesterone. In the wild type enzyme, progesterone might take advantage of the cavity space near Ala105 to adopt a variation of the observed progesterone orientation that preferentially exposes C16 for hydroxylation (Fig. 6A, pink). Because the steroid nucleus is a relatively rigid body, reduction of volume near Ala105 would be structurally consistent with the proposition that the A105L active site topography disallows such alternative positioning of progesterone in the CYP17A1 active site, reducing production of the minor 16α-hydroxyprogesterone metabolite, increasing the catalytic efficiency of 17α-hydroxylation, and slightly increasing C21 access to the iron for hydroxylation as observed experimentally.
Thus it is proposed that in the wild type enzyme, the progesterone long axis can adopt at least two orientations in the wild type active site (Fig. 6A). The major one would pack directly against the I helix and hydrogen bond to Asn202 as observed in the crystal structure, resulting in the major 17α-hydroxy metabolite. The minor orientation would be most consistent with a shift of the long axis of the steroidal core, pivoting about C17 to move the C3 substituent into the space adjacent to Ala105 near the B′ helix, where it perhaps cannot hydrogen bond with Asn202. This proposed position for progesterone would move C16 toward the iron, increasing the likelihood of abstraction of one of the C16 hydrogens and subsequent hydroxylation to yield the minor product. This model is coincident with intramolecular kinetic isotope effect studies demonstrating that the A105L mutation affects the transition state for hydrogen abstraction at C16, with negligible effects on progesterone C17 hydroxylation (46). It is therefore likely that the proposed alternative positioning is a very modest rotation of the steroid about C17 that affects the proximity of C16 to the oxygen of compound I without substantial effects on C17. Detection of the proposed progesterone minor orientation would not be likely by x-ray crystallography, especially within the context of the A105L mutation background. In summary, the contribution of the A105L mutation to more efficient 17α-hydroxylation appears to be related to the binding of substrates and doing so in an optimal position for hydrogen abstraction at the appropriate carbon.
The current structural and functional data also suggest a potential mechanism for differential regiospecificity between hydroxylase substrates. Although the wild type active site cavity topography suggests that pregnenolone could adopt a similar minor orientation resulting in the generation of 16α-hydroxypregnenolone, no such product is observed experimentally (16). This could be due to a stronger hydrogen bonding interaction with Asn202 for pregnenolone compared with progesterone. Pregnenolone (and 17α-hydroxypregnenolone) have a C3 hydroxyl substituent with the ability to serve as either hydrogen bond donor or acceptor to the Asn202 side chain amide, which can also serve as donor or acceptor depending on the orientation. These substrates bind more tightly than either of the corresponding Δ4,3-keto progesterone-based ligands (Table 2), as reflected by at least a 2-fold decrease in the Kd and perhaps even more by the 17-hydroxylase Km, which is a full order of magnitude lower for pregnenolone than its Δ4,3-keto counterpart progesterone. This stronger interaction with Asn202 could inhibit pregnenolone from adopting the minor orientation proposed for progesterone and inhibit pregnenolone 16-hydroxylation.
A second debate in the field of steroidogenesis centers on the mechanism and substrate selectivity of the lyase reaction. Human CYP17A1 performs the 17,20-lyase reaction much more efficiently on 17α-hydroxypregnenolone to form the physiologically relevant androgen dehydroepiandrosterone, with metabolism of 17α-hydroxyprogesterone to the corresponding 17,20-lyase product androstenedione 50-fold lower (16, 25), despite similar Kd values (Table 2). However, the current structural results suggest a basis for this selectivity. Although both lyase substrates can hydrogen bond to Asn202 similar to the hydroxylase substrates, the poor lyase substrate 17α-hydroxyprogesterone was only observed in this position (called position 1). In contrast, the favored lyase substrate 17α-hydroxypregnenolone is also observed closer to the heme and far enough from Asn202 that this hydrogen bond is no longer present (termed position 2). The potential significance of this substrate repositioning was investigated by modeling the proposed intermediate for the lyase reaction into both substrate complexes. The peroxy state (22, 23) was modeled by incorporating the heme-bound oxygenated state observed in P450cam (48), wherein the proximal oxygen is bound to the iron and the distal oxygen is oriented toward a portion of the adjacent I helix with noncanonical hydrogen bonding (Fig. 6, B and C). The CYP17A1 I helix shows similar noncanonical hydrogen bonding interactions at this position, although a water molecule observed in this groove for P450cam is not apparent for CYP17A1. With the iron peroxy modeled in this manner, the distance between the distal oxygen of the peroxo thought to perform nucleophilic attack on the substrate C20 position is similar for both substrate positions (∼3.2 Å), but the distance between C17 and the proximal oxygen goes from 3.6 Å in position 1 to 3.0 Å in position 2. As a result, in position 1 (the only position observed for 17α-hydroxyprogesterone), the C17 hydroxyl is positioned to best interact with the distal oxygen of the peroxy intermediate (Fig. 6B). In contrast, position 2 (observed only for 17α-hydroxypregnenolone) brings the C17 hydroxyl within a similar distance to the proximal oxygen of the peroxy intermediate (Fig. 6C). Hydrogen bonding of the C17 hydroxyl to the distal oxygen as suggested by position 1 would tend to promote breakage of the oxygen-oxygen bond, leading to the generation of compound I and depressing the lyase reaction (24). Hydrogen bonding of the C17 hydroxyl to the proximal oxygen, as suggested by 17α-hydroxypregnenolone in position 2, would tend to preferentially stabilize the peroxy intermediate and leave the distal oxygen available to perform nucleophilic attack on C20 of the substrate, thereby facilitating the lyase reaction. Thus subtle positioning of the substrate over the heme-peroxy intermediate, as mediated by the distant interaction with Asn202, is proposed to modulate substrate selectivity for the lyase reaction. This hypothesis is consistent with several orthogonal lines of evidence. First, although the residue at Asn202 may not be the only factor, the identity of this residue appears to largely correlate with lyase specificity in various species. For CYP17A1 from human, sheep, cow, and pig, the residue at position 202 is an asparagine and these proteins preferentially perform the lyase reaction on the 17α-hydroxypregnenolone substrate to produce dehydroepiandrosterone. Conversely hamster, mouse, rat, and guinea pig have a threonine at position 202 and preferentially perform the lyase reaction on 17α-hydroxyprogesterone, yielding androstendione. Second, resonance Raman spectroscopy supports differential hydrogen bonding of the efficient and poor lyase substrates to the proximal or distal oxygens, respectively, for the ferrous dioxygen state immediately preceding the peroxy state in the P450 catalytic cycle (24). Third, resonance Raman studies with CYP17A1 bound to CO demonstrate that two distinct iron-carbon-oxygen vibrational modes exist for 17α-hydroxypregnenolone, and only a single mode exists in the presence of hydroxylase substrates and the poor lyase substrate, 17α-hydroxyprogesterone (49). Thus the recent functional and current structural evidence converges to strongly support two binding modes for 17α-hydroxypregnenolone, one of which is oriented appropriately to stabilize the peroxo intermediate and undergo the lyase reaction. Finally, repositioning of 17α-hydroxypregnenolone lower in the active site to promote the lyase reaction is consistent with the effects of the A105L mutation, where filling space in the distal part of the active site cavity results in a 5-fold increase in kcat with no change in Km.
A third major conundrum with respect to CYP17A1 biochemistry is how the presence of cytochrome b5 promotes the 17,20-lyase reaction to increase androgen production in specific tissues and during human development. Cytochrome b5 binding is not associated with electron delivery to CYP17A1, but nonetheless increases the 17,20-lyase reaction as much as 10-fold (25). One might suggest that stabilization of the lyase iron-oxo intermediate might be accomplished if b5 binding inhibited protonation of the iron-peroxo intermediate. The peroxo intermediate operative in the lyase reaction must first be protonated on the distal oxygen, followed by a second protonation and loss of water to yield the iron-oxo compound I intermediate required for hydroxylation. However, this is incompatible with the observation that b5 does not reduce the hydroxylation reaction (25). Alternatively, b5 binding has been suggested to allosterically alter CYP17A1 conformation and/or substrate orientation to promote the lyase activity (25), but a mechanism for this has not been previously elucidated (50). The current structural results suggest that this might occur by altering the distribution of 17α-hydroxypregnenolone in position 1 (not conducive to the lyase reaction) versus position 2 (compatible with productive lyase chemistry). The anionic Glu48 and Glu49 residues of cytochrome b5 are known to interact with the cationic Arg347, Arg358, and Arg449 CYP17A1 residues based on both mutagenesis and recent solution NMR binding studies (31, 51). This means that the b5 binding site is on the opposite side of the heme and 11–21 Å away from the buried active site. Recently, however, solution NMR results established that b5 binds differentially to CYP17A1 depending on whether the hydroxylase substrate pregnenolone or the lyase substrate 17α-hydroxypregnenolone is present in the buried CYP17A1 active site (51). Cytochrome b5 binding on the proximal side of CYP17A1 has been demonstrated to change the backbone conformations for F helix residues Ile205 and Ile206 adjacent to Asn202 (52), although information on Asn202 itself is not yet available. The functional and structural data suggest that communication between the b5 binding site and the active site could allosterically favor 17α-hydroxypregnenolone binding in position 2. Optimized positioning of 17α-hydroxypregnenolone for the lyase reaction would be consistent with the observed decrease in the amount of uncoupling for the lyase reaction when b5 is present (53), increasing the lyase product generated without substantially altering CYP17A1 hydroxylase activity, which is already ∼97% coupled (53). Although we have been unable to replicate these results, phosphorylation of CYP17A1 is also reported to facilitate the lyase reaction (54, 55) and could similarly promote a conformation of CYP17A1 that increases 17α-hydroxypregnenolone in position 2 closer to the heme.
In summary, the balance of human mineralocorticoids, glucocorticoids, androgens, and estrogens depends on the interactions of CYP17A1 with its four different steroidal substrates. Although structural information has previously been lacking, the current set of x-ray structures establishes that each of these substrates binds in the CYP17A1 active site with the site(s) of metabolism oriented toward the heme, the α face packed against the I helix, and the C3 keto or alcohol substituent hydrogen bonding to Asn202 in the F helix. Complexes with the hydroxylase substrates progesterone and pregnenolone demonstrate primary binding modes consistent with production of the observed 17α-hydroxy major metabolites, whereas the structural and functional effects of the A105L mutation suggest a minor alternate orientation that is not seen in the current structure but that would be consistent with the 16α-hydroxylation observed for progesterone but not pregnenolone. Comparison of complexes between the poor lyase substrate 17α-hydroxyprogesterone and the efficient lyase substrate 17α-hydroxypregnenolone reveal that only 17α-hydroxypregnenolone is also observed positioned closer to the heme without hydrogen bonding to Asn202. This latter position, termed position 2, is consistent with spectroscopic evidence suggesting that 17α-hydroxypregnenolone hydrogen bonds differently to the proposed peroxy catalytic intermediate compared with its Δ4,3keto counterpart, resulting in C-C bond cleavage only for 17α-hydroxypregnenolone. Finally, cytochrome b5 binding to CYP17A1 is known to have an allosteric effect on the active site, which would be consistent with favoring 17α-hydroxypregnenolone localization nearer the heme in position 2 and potentially decreasing nonproductive uncoupling of the catalytic cycle. In aggregate, the set of structures herein provide a structural basis for the regioselectivity and substrate specificity of the hydroxylation reaction and the substrate specificity of the lyase reaction, as well as providing potential mechanism for the allosteric effects of cytochrome b5 on the lyase reaction. Providing such a structural basis for understanding key reactions at the crossroads of human steroidogenesis should improve our ability to modulate physiological status in diseases ranging from sexual development, fertility, and hormone-responsive breast and prostate cancer, to immune and stress responses and blood pressure.
Acknowledgments
The University of Kansas Protein Structure Laboratory was supported by National Institutes of Health Grants RR01778 and GM103420, and the Stanford Synchrotron Radiation Lightsource was supported by National Institutes of Health Grants RR00129 and GM103393.
- P450
- cytochrome P450
- b5
- cytochrome b5
- RMSD
- root mean square deviation.
REFERENCES
- 1. Miller W. L., Auchus R. J. (2011) The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 32, 81–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gilep A. A., Sushko T. A., Usanov S. A. (2011) At the crossroads of steroid hormone biosynthesis: the role, substrate specificity and evolutionary development of CYP17. Biochim. Biophys. Acta 1814, 200–209 [DOI] [PubMed] [Google Scholar]
- 3. Edwards B. K., Noone A. M., Mariotto A. B., Simard E. P., Boscoe F. P., Henley S. J., Jemal A., Cho H., Anderson R. N., Kohler B. A., Eheman C. R., Ward E. M. (2014) Annual report to the nation on the status of cancer, 1975–2010, featuring prevalence of comorbidity and impact on survival among persons with lung, colorectal, breast, or prostate cancer. Cancer 120, 1290–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ferraldeschi R., de Bono J. (2013) Agents that target androgen synthesis in castration-resistant prostate cancer. Cancer J. 19, 34–42 [DOI] [PubMed] [Google Scholar]
- 5. de Bono J. S., Logothetis C. J., Molina A., Fizazi K., North S., Chu L., Chi K. N., Jones R. J., Goodman O. B., Jr., Saad F., Staffurth J. N., Mainwaring P., Harland S., Flaig T. W., Hutson T. E., Cheng T., Patterson H., Hainsworth J. D., Ryan C. J., Sternberg C. N., Ellard S. L., Fléchon A., Saleh M., Scholz M., Efstathiou E., Zivi A., Bianchini D., Loriot Y., Chieffo N., Kheoh T., Haqq C. M., Scher H. I. (2011) Abiraterone and increased survival in metastatic prostate cancer. New Engl. J. Med. 364, 1995–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Auchus M. L., Auchus R. J. (2012) Human steroid biosynthesis for the oncologist. J. Investig. Med. 60, 495–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Loriot Y., Bianchini D., Ileana E., Sandhu S., Patrikidou A., Pezaro C., Albiges L., Attard G., Fizazi K., De Bono J. S., Massard C. (2013) Antitumour activity of abiraterone acetate against metastatic castration-resistant prostate cancer progressing after docetaxel and enzalutamide (MDV3100). Ann. Oncol. 24, 1807–1812 [DOI] [PubMed] [Google Scholar]
- 8. DeVore N. M., Scott E. E. (2012) Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001. Nature 482, 116–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pia A., Vignani F., Attard G., Tucci M., Bironzo P., Scagliotti G., Arlt W., Terzolo M., Berruti A. (2013) Strategies for managing ACTH dependent mineralocorticoid excess induced by abiraterone. Cancer Treat. Rev. 39, 966–973 [DOI] [PubMed] [Google Scholar]
- 10. Attard G., Reid A. H., Auchus R. J., Hughes B. A., Cassidy A. M., Thompson E., Oommen N. B., Folkerd E., Dowsett M., Arlt W., de Bono J. S. (2012) Clinical and biochemical consequences of CYP17A1 inhibition with abiraterone given with and without exogenous glucocorticoids in castrate men with advanced prostate cancer. J. Clin. Endocrinol. Metab. 97, 507–516 [DOI] [PubMed] [Google Scholar]
- 11. Ogo A., Haji M., Ohashi M., Nawata H. (1991) Markedly increased expression of cytochrome P-450 17α-hydroxylase (P-450c17) mRNA in adrenocortical adenomas from patients with Cushing's syndrome. Mol. Cell Endocrinol. 80, 83–89 [DOI] [PubMed] [Google Scholar]
- 12. Maitra A., Shirwalkar H. (2003) Congenital adrenal hyperplasia: biochemical and molecular perspectives. Indian J. Exp. Biol. 41, 701–709 [PubMed] [Google Scholar]
- 13. Qin K. N., Rosenfield R. L. (1998) Role of cytochrome P450c17 in polycystic ovary syndrome. Mol. Cell Endocrinol. 145, 111–121 [DOI] [PubMed] [Google Scholar]
- 14. Arlt W., Martens J. W., Song M., Wang J. T., Auchus R. J., Miller W. L. (2002) Molecular evolution of adrenarche: Structural and functional analysis of p450c17 from four primate species. Endocrinology 143, 4665–4672 [DOI] [PubMed] [Google Scholar]
- 15. Strauss J. F., 3rd. (2003) Some new thoughts on the pathophysiology and genetics of polycystic ovary syndrome. Ann. N.Y. Acad. Sci. 997, 42–48 [DOI] [PubMed] [Google Scholar]
- 16. Swart P., Swart A. C., Waterman M. R., Estabrook R. W., Mason J. I. (1993) Progesterone 16α-hydroxylase activity is catalyzed by human cytochrome P450 17α-hydroxylase. J. Clin. Endocrinol. Metab. 77, 98–102 [DOI] [PubMed] [Google Scholar]
- 17. Swart A. C., Storbeck K. H., Swart P. (2010) A single amino acid residue, Ala 105, confers 16α-hydroxylase activity to human cytochrome P450 17α-hydroxylase/17,20-lyase. J. Steroid Biochem. Mol. Biol. 119, 112–120 [DOI] [PubMed] [Google Scholar]
- 18. Sohl C. D., Guengerich F. P. (2010) Kinetic analysis of the three-step steroid aromatase reaction of human cytochrome P450 19A1. J. Biol. Chem. 285, 17734–17743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Flück C. E., Miller W. L., Auchus R. J. (2003) The 17,20-lyase activity of cytochrome p450c17 from human fetal testis favors the delta5 steroidogenic pathway. J. Clin. Endocrinol. Metab. 88, 3762–3766 [DOI] [PubMed] [Google Scholar]
- 20. Guengerich F. P. (2007) Mechanisms of cytochrome P450 substrate oxidation: MiniReview. J. Biochem. Mol. Toxicol. 21, 163–168 [DOI] [PubMed] [Google Scholar]
- 21. Groves J. T., McClusky G. A., White R. E., Coon M. J. (1978) Aliphatic hydroxylation by highly purified liver microsomal cytochrome P-450. Evidence for a carbon radical intermediate. Biochem. Biophys. Res. Commun. 81, 154–160 [DOI] [PubMed] [Google Scholar]
- 22. Akhtar M., Corina D., Miller S., Shyadehi A. Z., Wright J. N. (1994) Mechanism of the acyl-carbon cleavage and related reactions catalyzed by multifunctional P-450s: Studies on cytochrome P-450(17)α. Biochemistry 33, 4410–4418 [DOI] [PubMed] [Google Scholar]
- 23. Gregory M. C., Denisov I. G., Grinkova Y. V., Khatri Y., Sligar S. G. (2013) Kinetic solvent isotope effect in human P450 CYP17A1-mediated androgen formation: evidence for a reactive peroxoanion intermediate. J. Am. Chem. Soc. 135, 16245–16247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gregory M., Mak P. J., Sligar S. G., Kincaid J. R. (2013) Differential hydrogen bonding in human CYP17 dictates hydroxylation versus lyase chemistry. Angew. Chem. Int. Ed. Engl. 52, 5342–5345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Auchus R. J., Lee T. C., Miller W. L. (1998) Cytochrome b5 augments the 17,20-lyase activity of human P450c17 without direct electron transfer. J. Biol. Chem. 273, 3158–3165 [DOI] [PubMed] [Google Scholar]
- 26. Katagiri M., Kagawa N., Waterman M. R. (1995) The role of cytochrome b5 in the biosynthesis of androgens by human P450c17. Arch. Biochem. Biophys. 317, 343–347 [DOI] [PubMed] [Google Scholar]
- 27. Lee-Robichaud P., Wright J. N., Akhtar M. E., Akhtar M. (1995) Modulation of the activity of human 17α-hydroxylase-17,20-lyase (CYP17) by cytochrome b5: endocrinological and mechanistic implications. Biochem. J. 308, 901–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Onoda M., Hall P. F. (1982) Cytochrome b5 stimulates purified testicular microsomal cytochrome P-450 (C21 side-chain cleavage). Biochem. Biophys. Res. Commun. 108, 454–460 [DOI] [PubMed] [Google Scholar]
- 29. Idkowiak J., Randell T., Dhir V., Patel P., Shackleton C. H., Taylor N. F., Krone N., Arlt W. (2012) A missense mutation in the human cytochrome b5 gene causes 46, XY disorder of sex development due to true isolated 17,20-lyase deficiency. J. Clin. Endocrinol. Metab. 97, E465–E475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kok R. C., Timmerman M. A., Wolffenbuttel K. P., Drop S. L., de Jong F. H. (2010) Isolated 17,20-lyase deficiency due to the cytochrome b5 mutation W27X. J. Clin. Endocrinol. Metab. 95, 994–999 [DOI] [PubMed] [Google Scholar]
- 31. Naffin-Olivos J. L., Auchus R. J. (2006) Human cytochrome b5 requires residues E48 and E49 to stimulate the 17,20-lyase activity of cytochrome P450c17. Biochemistry 45, 755–762 [DOI] [PubMed] [Google Scholar]
- 32. Auchus R. J., Miller W. L. (1999) Molecular modeling of human P450c17 (17α-hydroxylase/17,20-lyase): insights into reaction mechanisms and effects of mutations. Mol. Endocrinol. 13, 1169–1182 [DOI] [PubMed] [Google Scholar]
- 33. Schappach A., Höltje H. D. (2001) Molecular modelling of 17α-hydroxylase-17,20-lyase. Pharmazie 56, 435–442 [PubMed] [Google Scholar]
- 34. Haider S. M., Patel J. S., Poojari C. S., Neidle S. (2010) Molecular modeling on inhibitor complexes and active-site dynamics of cytochrome P450 C17, a target for prostate cancer therapy. J. Mol. Biol. 400, 1078–1098 [DOI] [PubMed] [Google Scholar]
- 35. Lin D., Zhang L. H., Chiao E., Miller W. L. (1994) Modeling and mutagenesis of the active site of human P450c17. Mol. Endocrinol. 8, 392–402 [DOI] [PubMed] [Google Scholar]
- 36. Laughton C. A., Neidle S., Zvelebil M. J., Sternberg M. J. (1990) A molecular model for the enzyme cytochrome P450(17α), a major target for the chemotherapy of prostatic cancer. Biochem. Biophys. Res. Commun. 171, 1160–1167 [DOI] [PubMed] [Google Scholar]
- 37. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kleywegt G. J., Jones T. A. (1994) Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Crystallogr. D Biol. Crystallogr. 50, 178–185 [DOI] [PubMed] [Google Scholar]
- 42. DeLano W. L. (2012) The PyMOL Molecular Graphics System, version 1.5.0.1, Schroedinger, LLC, New York [Google Scholar]
- 43. Sandee D., Miller W. L. (2011) High-yield expression of a catalytically active membrane-bound protein: human P450 oxidoreductase. Endocrinology 152, 2904–2908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Moon J. Y., Kang S. M., Lee J., Cho J. Y., Moon M. H., Jang I. J., Chung B. C., Choi M. H. (2013) GC-MS-based quantitative signatures of cytochrome P450-mediated steroid oxidation induced by rifampicin. Ther. Drug Monit. 35, 473–484 [DOI] [PubMed] [Google Scholar]
- 45. DeVore N. M., Smith B. D., Wang J. L., Lushington G. H., Scott E. E. (2009) Key residues controlling binding of diverse ligands to human cytochrome P450 2A enzymes. Drug Metab. Dispos. 37, 1319–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yoshimoto F. K., Zhou Y., Peng H. M., Stidd D., Yoshimoto J. A., Sharma K. K., Matthew S., Auchus R. J. (2012) Minor activities and transition state properties of the human steroid hydroxylases cytochromes P450c17 and P450c21, from reactions observed with deuterium-labeled substrates. Biochemistry 51, 7064–7077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nakajin S., Hall P. F. (1981) Microsomal cytochrome P-450 from neonatal pig testis. Purification and properties of A C21 steroid side-chain cleavage system (17α-hydroxylase-C17,20-lyase). J. Biol. Chem. 256, 3871–3876 [PubMed] [Google Scholar]
- 48. Schlichting I., Berendzen J., Chu K., Stock A. M., Maves S. A., Benson D. E., Sweet R. M., Ringe D., Petsko G. A., Sligar S. G. (2000) The catalytic pathway of cytochrome p450cam at atomic resolution. Science 287, 1615–1622 [DOI] [PubMed] [Google Scholar]
- 49. Mak P. J., Gregory M. C., Sligar S. G., Kincaid J. R. (2014) Resonance Raman spectroscopy reveals that substrate structure selectively impacts the heme-bound diatomic ligands of CYP17. Biochemistry 53, 90–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Akhtar M. K., Kelly S. L., Kaderbhai M. A. (2005) Cytochrome b5 modulation of 17α hydroxylase and 17–20 lyase (CYP17) activities in steroidogenesis. J. Endocrinol. 187, 267–274 [DOI] [PubMed] [Google Scholar]
- 51. Estrada D. F., Laurence J. S., Scott E. E. (2013) Substrate-modulated cytochrome P450 17A1 and cytochrome b5 interactions revealed by NMR. J. Biol. Chem. 288, 17008–17018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Estrada D. F., Skinner A. L., Laurence J. S., Scott E. E. (2014) Human cytochrome P450 17A1 conformational selection: modulation by ligand and cytochrome b5. J. Biol. Chem. 289, 14310–14320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Khatri Y., Gregory M. C., Grinkova Y. V., Denisov I. G., Sligar S. G. (2014) Active site proton delivery and the lyase activity of human CYP17A1. Biochem. Biophys. Res. Commun. 443, 179–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang L. H., Rodriguez H., Ohno S., Miller W. L. (1995) Serine phosphorylation of human P450c17 increases 17,20-lyase activity: Implications for adrenarche and the polycystic ovary syndrome. Proc. Natl. Acad. Sci. U.S.A. 92, 10619–10623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tee M. K., Miller W. L. (2013) Phosphorylation of human cytochrome P450c17 by p38α selectively increases 17,20-lyase activity and androgen biosynthesis. J. Biol. Chem. 288, 23903–23913 [DOI] [PMC free article] [PubMed] [Google Scholar]