

Background: CGI-58 plays an essential role in lipid homeostasis with poorly defined mechanisms.
Results: CGI-58 promotes autophagy as a novel lysophosphatidylglcyerol acyltransferase.
Conclusion: CGI-58 regulates lipid homeostasis in part by promoting autophagy.
Significance: Our findings provide potential molecular mechanisms by which CGI-58 mutations cause dyslipidemia.
Keywords: Autophagy, Lipid Droplet, Lipid Metabolism, Mammalian Target of Rapamycin (mTOR), Mitochondria
Abstract
CGI-58 is a lipid droplet-associated protein that, when mutated, causes Chanarin-Dorfman syndrome in humans, which is characterized by excessive storage of triglyceride in various tissues. However, the molecular mechanisms underlying the defect remain elusive. CGI-58 was previously reported to catalyze the resynthesis of phosphatidic acid as a lysophosphatidic acid acyltransferase. In addition to triglyceride, phosphatidic acid is also used a substrate for the synthesis of various mitochondrial phospholipids. In this report, we investigated the propensity of CGI-58 in the remodeling of various phospholipids. We found that the recombinant CGI-58 overexpressed in mammalian cells or purified from Sf9 insect cells catalyzed efficiently the reacylation of lysophosphatidylglycerol to phosphatidylglycerol (PG), which requires acyl-CoA as the acyl donor. In contrast, the recombinant CGI-58 was devoid of acyltransferase activity toward other lysophospholipids. Accordingly, overexpression and knockdown of CGI-58 adversely affected the endogenous PG level in C2C12 cells. PG is a substrate for the synthesis of cardiolipin, which is required for mitochondrial oxidative phosphorylation and mitophagy. Consequently, overexpression and knockdown of CGI-58 adversely affected autophagy and mitophagy in C2C12 cells. In support for a key role of CGI-58 in mitophagy, overexpression of CGI-58 significantly stimulated mitochondrial fission and translocation of PINK1 to mitochondria, key steps involved in mitophagy. Furthermore, overexpression of CGI-58 promoted mitophagic initiation through activation of 5′-AMP-activated protein kinase and inhibition of mTORC1 mammalian target of rapamycin complex 1 signaling, the positive and negative regulators of autophagy, respectively. Together, these findings identified novel molecular mechanisms by which CGI-58 regulates lipid homeostasis, because defective autophagy is implicated in dyslipidemia and fatty liver diseases.
Introduction
In most cells of vertebrates, energy is stored as TAG3 in the lipid droplets. The hydrolysis of these lipids provides a source of substrates for the synthesis of phospholipids, which are used for membrane structure, mitochondrial respiration, and ATP production (1). The complete hydrolysis of TAG is catalyzed by several lipases, including adipose TAG lipase (ATGL), hormone-sensitive lipase, and monoacylglycerol lipase (2). The initial step in TAG hydrolysis in various tissues is mediated by ATGL. However, the maximal activation of adipocyte lipolysis requires the CGI-58 protein, also known as α/β hydrolase domain 5. CGI-58 was shown to interact with perilipin and ATGL on the surface of lipid droplets (3, 4). In the absence of PKA activation, CGI-58 binds to perilipin and is sequestered at the surface of the lipid droplets. Activation of PKA promotes the release of CGI-58 from perilipin, allowing it to bind to ATGL, which stimulates the activity of ATGL to hydrolyze TAG (4, 5).
CGI-58 is a member of the lipase subfamily of α/β-hydrolase fold enzymes (6) and plays an important role in the maintenance of TAG homeostasis. Mutations in the CGI-58 gene cause Chanarin-Dorfman syndrome, a neutral lipid storage disorder in humans. Clinical manifestations include ichthyosis, hepatic steatosis, cardiomyopathy, ataxia, and mental retardation (7). CGI-58 mutant proteins are unable to induce ATGL activity, which results in cellular lipid accumulation in the form of TAG (4). The mutations that affect this association hinder both TAG hydrolysis (4) and recycling of neutral lipids to phospholipids (8).
In addition to supporting ATGL activation, CGI-58 has been reported to catalyze the resynthesis of phosphatidic acid as an acyl-CoA-dependent lysophosphatidic acid acyltransferase (LPAAT) that prefers arachidonoyl-CoA and oleoyl-CoA as acyl donors (1, 9). This function is consistent with the presence of a consensus sequence of HXXXXD at the C terminus of the CGI-58 protein, which is conserved in homologs of CGI-58 from all vertebrate and some invertebrate species (1). However, CGI-58 mutations that cause Chanarin-Dorfman syndrome were shown to have normal LPAAT activity (9). Thus the biochemical role of CGI-58 on phospholipids biosynthesis remains unclear.
Although both ATGL and CGI-58 mutations in humans cause a neutral lipid storage disease (10, 11), phenotypic differences exist between CGI-58 and ATGL mutations in humans and rodents. For example, ATGL mutations in humans cause no ichthyosis, whereas CGI-58 mutations in humans always cause ichthyosis (6, 11). Additionally, whole body ATGL knock-out mice are viable, whereas global deletion of CGI-58 leads to neonatal lethality caused by a defect in skin barrier function (12). These observations suggest that CGI-58 may possess additional functions beyond ATGL activation. In this study, we identified CGI-58 as a novel lysophosphatidylglycerol acyltransferase (LPGAT) that catalyzes the resynthesis of PG using acyl-CoA as acyl donor. Moreover, CGI-58 plays an important role in regulating mitochondrial dynamics and autophagy, linking lipid droplets to the mitochondrial quality control process.
MATERIALS AND METHODS
Reagents
Monolysocardiolipin (MLCL), lysophosphatidic acid, lysophosphatidylcholine (LPC), lysophosphatidylethanolamine (LPE), lysophosphatidylserine (LPS), lysophosphatidylinositol (LPI), and lysophosphatidylglycerol (LPG) were purchased from Avanti Polar Lipids (Alabaster, AL). [14C]Oleoyl-CoA (50 mCi/mmol) and [14C]palmitoyl-CoA were purchased from American Radio Labeled Chemicals (St. Louis, MO). Iodine, PEG 1500, rapamycin, bafilomycin A1, and Mdivi-1 were purchased from Sigma. Monoclonal antibodies to MFN2 and OPA1 were purchased from Abcam (Cambridge, MA). Anti-LC3 and anti-CGI-58 antibodies were purchased from Novus Biologicals (Littleton, CO). Anti-p62 antibody was from American Research Products Inc. (Belmont, MA). Monoclonal anti-FLAG antibody (M2) and anti-FLAG agarose resin were from Sigma. Anti-PINK1 polyclonal antibody (A01) was purchased from Abnova (Walnut, CA). Anti-NRF2 and anti-GAPDH antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Donkey anti-rabbit and donkey anti-mouse IgG horseradish peroxidase-conjugated antibodies were purchased from GE Healthcare.
Expression of CGI-58 in Mammalian Cells
The FLAG epitope (DYKDDDDK) was attached to the N terminus of CGI-58 by PCR amplification using a forward primer that carried the FLAG sequence fused in-frame with the start codon of the CGI-58 coding region. A FLAG-tagged CGI-58 expression vector was generated by subcloning the cDNA into the BamHI and NotI sites of pcDNA3.1(+)/Hygro mammalian expression vector (Invitrogen). 293T cells were transiently transfected with pcDNA3.1-FLAG-CGI-58 for 48 h. Then the cells were harvested, and the cell lysates were aliquoted and stored at −80 °C. The expression level of FLAG-CGI-58 was measured by Western blot using anti-FLAG antibody. The cell lysates were subsequently used in in vitro acyltransferase assay.
Expression of CGI-58 in Insect Cells
Expression of CGI-58 in insect cells was performed by using a Bac-to-Bac baculovirus expression system (Invitrogen) according to the manufacturer's instructions. The FLAG-tagged CGI-58 full-length coding sequence was subcloned from pcDNA3.1-FLAG-CGI-58 into BamHI and NotI sites of pFastbac1 vector, which was subsequently transformed into DH10BacTM Escherichia coli cells to generate a recombinant bacmid that carries the insertion of the CGI-58 cDNA. High titer recombinant baculovirus was generated by transfecting the bacmid DNA into Sf9 insect cells, followed by several rounds of amplification to increase viral titer. Sf9 cells were typically infected with virus for 3 days and then harvested in ice-cold PBS. The FLAG-tagged CGI-58 protein was purified using anti-FLAG-agarose resin (Sigma). Purified CGI-58 protein was confirmed by Coomassie Blue staining and subsequently used in in vitro acyltransferase assay.
In Vitro Acyltransferase Assay
The lysophospholipids acyltransferase activities of recombinant CGI-58 were determined by measuring the incorporation of radiolabeled acyl moieties of acyl-CoAs (acyl donors) into phospholipids in the presence of relevant lysophospholipids (acyl acceptors) as described previously (13). MLCL, lysophosphatidic acid, LPE, LPC, LPI, LPS, and LPG were used in the assay. The reaction mixture contained 80 mm Tris/HCl, pH 7.4, 200 μm lysophospholipids, 20 μm [14C]oleoyl-CoA (50 mCi/mmol) or [14C]palmitoyl-CoA (50 mCi/mmol) and enzyme preparations (50 μg of total protein from 293T cell lysates or 500 ng of purified protein from Sf9 cells) in a total volume of 200 μl. For CGI-58 LPGAT kinetics analysis, the indicated concentration of LPG was used as substrate. The reactions were incubated at room temperature for 30 min. The lipids were extracted using a method as described previously (13). The extracted lipids were dried and separated by TLC with chloroform:methanol:water (65:25:4, v/v/v) or chloroform:ethanol:water:triethylamine (30:35:7:35, v/v/v/v). After separation, the TLC plates were exposed to a PhosphorImager screen to visualize the radiolabeled products with a Molecular Dynamics STORM860 Scanner (Sunnyvale, CA).
Lipid Extraction
Lipids in C2C12 cells were extracted using chloroform/methanol (2:1, v/v). In brief, cells pellets were resuspended in chloroform/methanol (2:1, v/v) and incubated at room temperature for 1 h. After addition of 0.9% KCl, phase separation was carried out by centrifugation. The organic phase was dried, and the lipids were finally diluted in chloroform/methanol (2:1, v/v), separated on TLC, resolved by chloroform/methanol/water (65:25:4, v/v), and visualized by iodine vapor. The product from an in vitro LPGAT assay was used as PG marker. The PG content in cells was quantified using ImageJ.
Subcellular Fractionation Analysis
Subcellular fractionation analysis was carried out to localize CGI-58 stably expressed in C2C12 cells as described previously (14, 15). Briefly, C2C12-CGI-58 overexpression cells were homogenized with a Dounce homogenizer in 10 volumes (w/v) of solution consisting of 225 mm mannitol, 75 mm sucrose, 0.1 mm EGTA, and 30 mm Tris/HCl, pH 7.4. The homogenate was first centrifuged at 600 × g for 10 min to remove cell debris and nuclear fractions. The crude mitochondrial fraction was obtained by centrifuging the supernatant at 8,000 × g for 10 min. For isolation of the pure mitochondrial fraction and mitochondria-associated membranes, the crude mitochondrial pellet was resuspended in mitochondrial suspension buffer consisting of 250 mm mannitol, 5 mm HEPES, pH 7.4, and 0.5 mm EGTA and then fractionated by Percoll gradient ultracentrifuge at 95,000 × g for 30 min. Microsomal fraction was prepared from the postmitochondrial supernatant by sedimentation at 100,000 × g for 1 h. The mitochondrial, mitochondria-associated membrane, and microsomal fractions were resuspended in PBS buffer and analyzed by Western blot analysis using anti-FLAG antibody. Tom20 and calnexin were used as mitochondria and endoplasmic reticulum biomarkers, respectively.
Immunofluorescence
COS-7 cells growing on a coverslip were transiently co-transfected with mitochondria-targeted GFP (mito-EGFP) together with pcDNA3.1-FLAG-CGI-58 or empty pcDNA3.1 vector for 48 h. Cells were then fixed in 4% (v/v) paraformaldehyde for 10 min, washed twice with PBS, and then permeabilized with 0.1% Triton X-100 in PBS for 10 min. Fixed cells were preincubated for 30 min in PBS containing 1% BSA at room temperature to block nonspecific binding. Cells were incubated with anti-FLAG monoclonal antibody (1:200 dilution in 1% BSA/PBS) for 3 h at room temperature. After three brief washes with PBS, the cells were incubated for 1 h at room temperature with Cy3-conjugated donkey anti-mouse IgG (Jackson ImmunoResearch Laboratories Inc., West Grove, PA). The cells were also counterstained with DAPI (Molecular Probes, Eugene, OR) to visualize nucleus. Cells were then washed four times with PBS and analyzed with a confocal fluorescence microscope (Leica TCS SP8 AOBS).
Generation of C2C12-CGI-58 Overexpression and shRNA Knockdown Cell Lines
C2C12 cells were transfected with pcDNA3.1-FLAG-CGI-58 or empty vector as a control. The stable transfectants were screened for hygromycin resistance by culturing in DMEM (Invitrogen) supplemented with hygromycin B (400 μg/ml), 10% FBS, 1% penicillin, and streptomycin and maintained in 95% air plus 5% CO2 at 37 °C. For generation of C2C12-CGI-58 knockdown cell line, a CGI-58 specific shRNA was engineered into pLKO.1 vector, which was used to generate recombinant lentiviruses in 293T cells. The C2C12 cells were infected with blank virus or virus containing CGI-58 specific shRNA. The stable infectants were screened for puromycin resistance by culturing DMEM supplemented with puromycin (4 μg/ml), 10% FBS, 1% penicillin, and streptomycin and maintained in 95% air plus 5% CO2 at 37 °C.
Mitochondrial Staining
For mitochondrial morphology assay, C2C12 cells stably expressing CGI-58 or vector control were stained with MitoTracker Red (Invitrogen) according to the manufacturer's instructions. In brief, the cells were stained with 25 nm MitoTracker Red for 15 min at 37 °C, followed by three washes with PBS. For some experiments, C2C12-CGI-58 cells were pretreated with 25 μm Mdivi-1 for 4 h and then stained with MitoTracker Red. The stained cells were immediately analyzed with a confocal fluorescence microscope.
Mitochondrial Fusion Assay
Mitochondrial fusion assay was described previously (16). Briefly, C2C12 cells stably expressing CGI-58 or vector control were seeded with 5 × 105 cells in 6-cm dishes and transfected with mito-EGFP or with mitochondria-targeted DsRed (mito-DsRed), respectively. After 30 h, individual pools of cells expressing mito-EGFP and mito-DsRed were mixed and co-plated at a 1:1 ratio onto 13-mm round coverslips. Fusion was then induced after 6 h by a 60-s treatment with a 50% (w/v) solution of PEG 1500 in PBS, followed by extensive washes in DMEM supplemented with 10% (v/v) FCS. To inhibit protein synthesis, cycloheximide (20 μg/ml) was added 30 min before fusion and kept in all solutions, and cell culture medium was used subsequently until cells were fixed for 10 min with ice-cold 4% (v/v) paraformaldehyde in PBS. After three washes with PBS, coverslips were mounted on slides and kept in a dark box at 4 °C overnight.
Statistical Analysis
Statistical comparisons were done using two-tailed nonpaired Student t tests to evaluate the difference between the two groups. For comparisons among more groups, one-way analysis of variance was used, and values were considered statistically significant at p value of 0.05. The data were expressed as means plus standard errors of the means.
RESULTS
CGI-58 Is a Novel LPGAT
Previous studies indicated that CGI-58 catalyzes the synthesis of phosphatidic acid as a LPAAT enzyme. However, neither study investigated its role in the remodeling of PG and CL, although phosphatidic acid is a substrate for the synthesis of both phospholipids. Using 293T cells transiently transfected with expression vector for human CGI-58 or vector control, we analyzed the acyltransferase activity of the recombinant CGI-58 toward various lysophospholipids, including MLCL, LPG, LPC, LPE, LPI, and LPS. The result showed that overexpression of CGI-58 significantly increased LPGAT activity when compared with vector control. In contrast, the recombinant CGI-58 failed to recognize other lysophospholipids as substrates (Fig. 1A). In accordance with the previous studies, the recombinant CGI-58 also exhibited the LPAAT activity (Fig. 1B). To confirm the LPGAT activity was not caused by a compensatory response of the endogenous LPGAT enzymes, we next analyzed LPGAT activity of the purified recombinant CGI-58, which was overexpressed in Sf9 insect cells and purified using anti-FLAG antibody agarose resin. The results show that the purified CGI-58 protein exhibited strong LPGAT activity when compared with vehicle (Fig. 1C). The quality of the purified protein was verified by Coomassie Blue staining using BSA as the mass marker (Fig. 1D).
FIGURE 1.

CGI-58 catalyzes the synthesis of PG as a LPG acyltransferase. A, FLAG-tagged CGI-58 or vector control was transiently expressed in 293T cells, and we analyzed acyltransferase activities toward different lysophospholipids, including MLCL, LPG, LPC, LPE, LPI, and LPS. The acyltransferase assays were conducted by incubating 20 μm [14C]oleoyl-CoA with 200 μm each of the lysophospholipids in the presence of 50 μg of cell homogenates, followed by lipids extraction and TLC analysis. The position of PG is highlighted by an arrow. B, analysis of lysophosphatidic acid acyltransferase activity of recombinant FLAG-CGI-58 using the same assay condition as in A. C, analysis of LPGAT activity of recombinant FLAG-CGI-58 protein overexpressed in 293T cells and purified from Sf9 cells using the same assay condition as in A. D, SDS-PAGE analysis recombinant FLAG-CGI-58 overexpression and purified from Sf9 insect cells by Coomassie Blue staining using BSA as a protein mass marker.
We next analyzed acyl selectivity of CGI-58 toward different acyl-CoAs using palmitoyl-CoA and oleoyl-CoA as the acyl donors and various concentrations of LPG as the substrate. As shown in Fig. 2A, the recombinant FLAG-tagged CGI-58 protein transiently overexpressed in 293T cells exhibited a clear preference for oleoyl-CoA as the acyl donor, which is supported by significantly higher Km and Vmax values (99.05 ± 10.57 μm and 7.78 ± 0.61 nmol/min/mg) when compared with palmitoyl-CoA (63.33 ± 8.37 μm and 5.133 ± 0.38 nmol/min/mg) (quantified in Fig. 2B).
FIGURE 2.
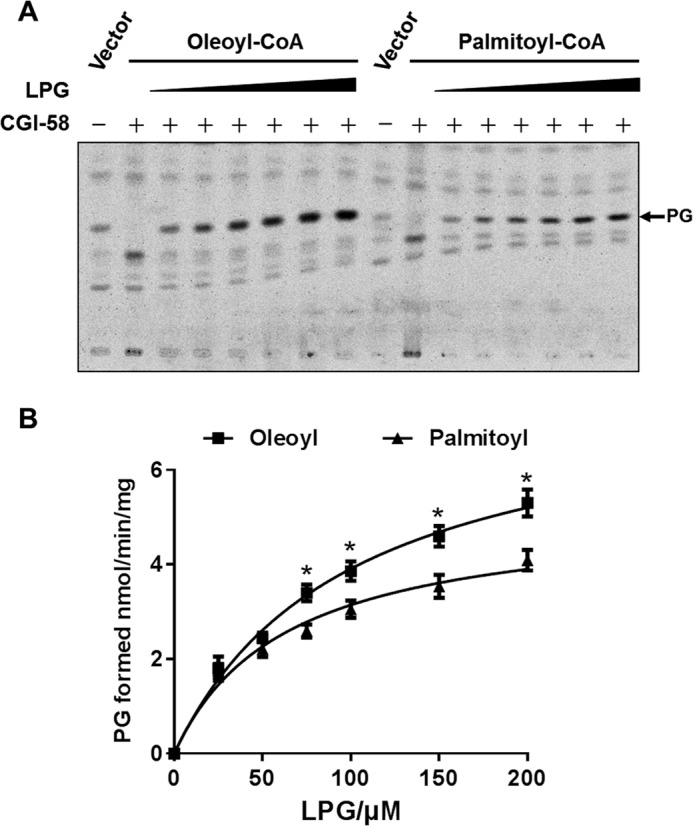
Analysis of enzymatic kinetics and substrate specificity of CGI-58 toward different acyl-CoAs. A, recombinant FLAG-CGI-58 transiently expressed in the 293T cells were analyzed for LPGAT activity using [14C]oleoyl-CoA and [14C]palmitoyl-CoA as the acyl donors and increasing doses of LPG (0, 25, 50, 75, 150, and 200 μm) as the substrate. The position of PG was highlighted by an arrow. B, analysis of enzymatic kinetics of CGI-58 toward [14C]oleoyl-CoA and [14C]palmitoyl-CoA. The Km and Vmax were 99.05 ± 10.57 μm and 7.78 ± 0.61 nmol/min/mg for oleoyl-CoA and 63.33 ± 8.37 μm and 5.133 ± 0.38 nmol/min/mg for palmitoyl-CoA, respectively.
The strong LPGAT activity of purified CGI-58 promoted us to investigate whether cellular PG content was regulated by CGI-58 overexpression or depletion in C2C12 cells. C2C12 is a metabolically active mouse skeletal muscle cell line that exhibits a very poor transfection rate for plasmid expression vector. To carry out the studies, we generated C2C12 cells stably transfected with FLAG-tagged CGI-58 or shRNA targeted to CGI-58, which resulted in 3-fold CGI-58 overexpression and 70% depletion of the endogenous CGI-58 protein, respectively, as evidenced by results from Western blot analyses using anti-CGI-58 antibody (Fig. 3, A and B). In further support for the role of CGI-58 in the resynthesis of PG as a novel LPGAT enzyme, CGI-58 overexpression and deficiency adversely changed the endogenous PG levels relative to the vector control (Fig. 3C; quantified in Fig. 3D).
FIGURE 3.
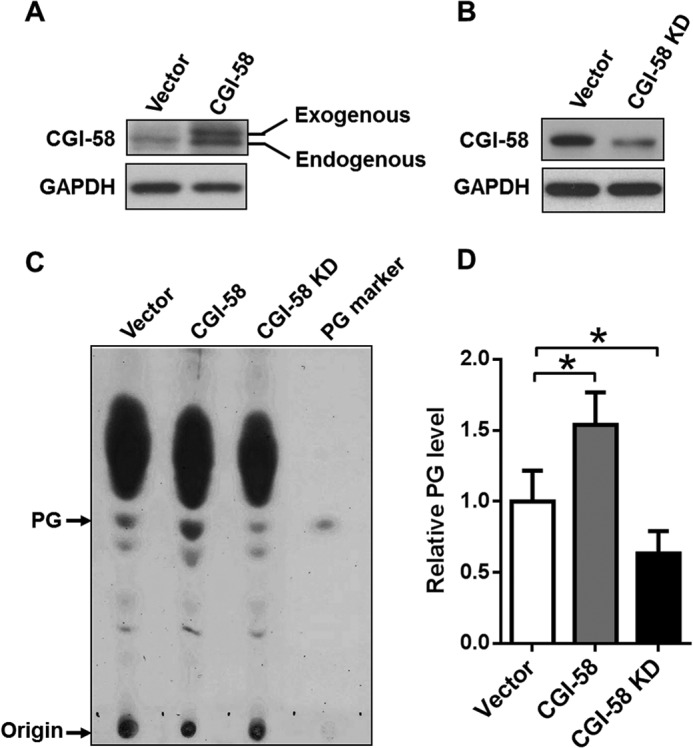
Overexpression and depletion of CGI-58 significantly changed the endogenous PG content in C2C12 cells. A and B, Western blot analysis of CGI-58 level in C2C12 cells stably overexpress CGI-58 (A) or shRNA targeted to the endogenous CGI-58 gene (B). C, C2C12 cells stably overexpressing CGI-58 or shRNA targeted to the endogenous CGI-58 were analyzed for endogenous PG content by TLC analysis. The total lipids were extracted from the cells, separated on TLC, and visualized by iodine vapor. Exogenous PG was used as the molecular marker. D, quantitative analysis of relative PG content in C2C12 cells from three independent experiments by ImageJ analysis. *, p < 0.05.
CGI-58 Is Localized in Mitochondria and Microsomes
CGI-58 was previously reported to localize at the surface of lipid droplets. The identification of LPGAT activity from CGI-58 prompted us to examine its subcellular localization by subcellular fractionation analysis. Total cell lysate from C2C12 cells stably expressing CGI-58 was fractionated into cytosol, microsomal fraction, and crude mitochondrial fraction, which was subsequently fractionated to pure mitochondria and mitochondria-associated membranes, followed by Western blot analysis using Tom20 and calnexin as mitochondrial and endoplasmic reticulum markers, respectively. Consistent with the newly identified role in PG synthesis, a significant portion of CGI-58 is localized in mitochondria in addition to previously reported localization in microsomes (Fig. 4A).
FIGURE 4.
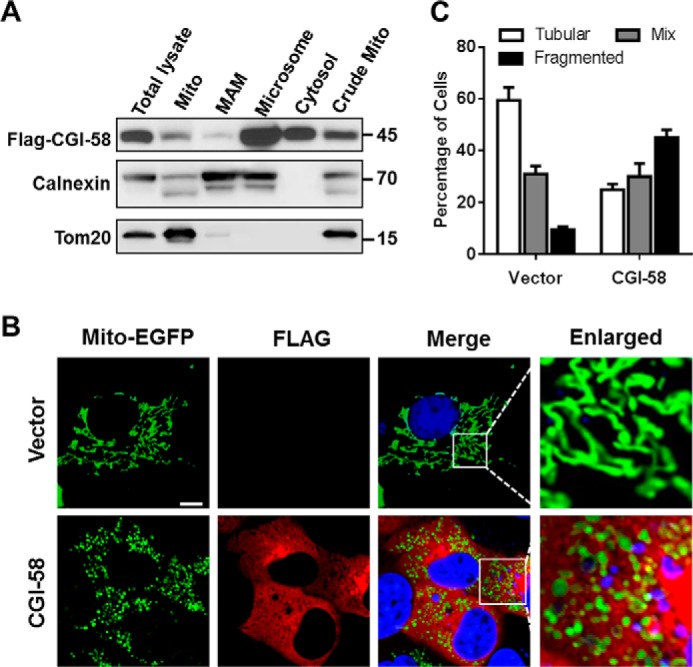
CGI-58 regulates mitochondrial dynamics as a mitochondrial protein. A, subcellular fractionation analysis of CGI-58 stably expressed in C2C12 cell. Total cell lysate from C2C12 cells were subjected to differential centrifugation to separate into the crude mitochondrial fraction (crude mito), mitochondria-associated membranes (MAM), pure mitochondria (Mito), microsomal fraction, and cytosol, followed by Western blot analysis using antibodies against FLAG, calnexin, and Tom20, which were used as markers for CGI-58, endoplasmic reticulum, and mitochondria, respectively. B, confocal image analysis of mitochondrial dynamics. COS-7 cells were transiently co-transfected with expression vectors for mito-EGFP and CGI-58 or empty vector. After 48 h of transfection, the cells were analyzed for mitochondrial dynamics and the expression pattern of CGI-58 by confocal image analysis using anti-FLAG antibody and Cy3-conjugated donkey-anti-mouse IgG. Scale bar, 10 μm. C, quantitative analysis of mitochondrial morphology in COS-7 cells overexpressing CGI-58 or vector control in three categories: tubular, mix, and fragmented from three independent experiments (n = 150 for each independent experiment).
PG is predominantly synthesized and localized in the mitochondria where it supports mitochondrial membrane structure and function as a substrate for CL synthesis (17). To determine a role for CGI-58 in mitochondrial function, we next analyzed the effect of CGI-58 overexpression in COS-7 cells on mitochondrial dynamics. COS-7 cells were co-transfected with FLAG-CGI-58 expression vector and mito-EGFP, which was used as a mitochondrial marker, followed by confocal imaging analysis of mitochondrial network. Strikingly, transient expression of CGI-58 in COS-7 cells led to mitochondrial fragmentation (Fig. 4B), which was further supported by the results from quantitative analysis of mitochondrial dynamics (Fig. 4C). Similar results were also obtained in C2C12 cells stably expressing CGI-58 (data not shown).
CGI-58 Promotes Mitochondrial Fission through Up-regulation of DRP1 Expression
To identify molecular mechanisms by which CGI-58 causes mitochondrial fragmentation, we next determined the effect of stable expression of CGI-58 in C2C12 cells on key regulators of mitochondrial fusion and fission, including MFN2 (mitofusion 2), OPA1 (optic atrophy 1), and DRP1 (dynamin-related protein 1). MFN2 and OPA1 are required for the fusion of outer and inner mitochondrial membranes, respectively, whereas DRP1 uses GTP hydrolysis to power the constriction and division of mitochondria. Consistent with increased mitochondrial fission, CGI-58 significantly increased the expression of DRP1 (Fig. 5A). In contrast, neither MFN2 nor OPA1 expression was affected by CGI-58 overexpression in C2C12 cells (Fig. 5A), suggesting that the mitochondrial fragmentation is primarily caused by increased fission. To confirm this concept, we examined whether inhibition of mitochondrial fission would restore mitochondrial dynamics in C2C12 cells stably expressing CGI-58. As shown in Fig. 5B, treatment with Mdivi-1, a mitochondrial fission inhibitor, led to restoration of tubule structures of mitochondria in C2C12 cells stably expressing CGI-58. To provide further evidence that CGI-58 did not cause a fusion defect that could also lead to mitochondrial fragmentation, we next carried out mitochondrial fusion analysis. As shown in Fig. 5C, there were no significant differences between C2C12 cells stably expressing CGI-58 and vector control in mitochondrial fusion (Fig. 5C, boxed in third panels and enlarged in fourth panels), suggesting that the mitochondrial fragmentation in the CGI-58 stable cell line is primarily caused by increased mitochondrial fission.
FIGURE 5.
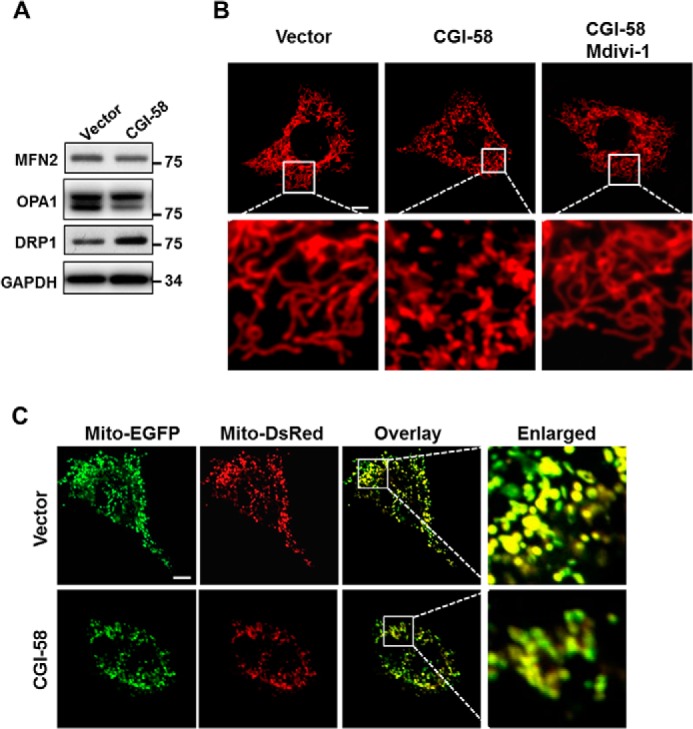
Overexpression of CGI-58 promotes mitochondrial fission by up-regulating the expression of DRP-1. A, Western blot analysis of MFN2, OPA1, and DRP1 protein levels in C2C12 cells stably expressing CGI-58 or vector control. GAPDH was used as an internal control. B, confocal microscopic analysis of mitochondrial network in C2C12 cells stably expressing CGI-58 or vector control. To identify a role of CGI-58 in mitochondrial fission, C2C12 cells stably expressing CGI-58 were also treated for 4 h with 25 μm Mdivi-1, a mitochondrial division inhibitor, and analyzed for mitochondrial dynamics after staining with MitoTracker Red, a red fluorescent dye that stains mitochondria in live cells. Scale bar, 10 μm. C, analysis of the role for CGI-58 in mitochondrial fusion in C2C12 cells. C2C12 cells stably expressing CGI-58 or vector control were transiently transfected with expression plasmids for mito-EGFP or mito-DsRed, co-plated, and fused with PEG 1500, followed by confocal microscopic imaging analysis. Scale bar, 10 μm.
CGI-58 Overexpression Enhances Autophagy in C2C12 Cells
CL was recently reported to regulate autophagosome biogenesis by directly binding to LC3 (18). Using recombinant adenoviruses overexpressing LC3-GFP fusion protein, we examined the effect of CGI-58 excess and deficiency on autophagosome biogenesis in C2C12 cells. As shown in Fig. 6A, C2C12 cells stably overexpressing CGI-58 exhibited significantly higher basal level of autophagy relative to vector controls, as evidenced by increased number of autophagosomes that exhibited as puncta. The difference was diminished in response to starvation, which also stimulated autophagy in vector control cells. Consistent with the findings, CGI-58 depletion mediated by shRNA knockdown of the endogenous CGI-58 mRNA (CGI-58 KD) significantly inhibited autophagosome biogenesis in C2C12 cells both under basal conditions and in response to stimulation with starvation (Fig. 6A). Consistent with the findings, CGI-58 overexpression also significantly depleted the expression of p62, NRF2, and increased the LC3II/LC3-I ratio (Fig. 6B). p62 and NRF2 are consumed during autophagic process. Upon initiation of autophagy, the C-terminal glycine of LC3-I is modified by addition of a PE to form LC3-II, which translocates rapidly to nascent autophagosomes in a punctate distribution. Therefore, the increased ratio of LC3-II to LC3-I indicates enhanced autophagy. Furthermore, overexpression of CGI-58 significantly increased the expression of PINK1 (Fig. 6B), a mitochondrial kinase required for the initiation of mitophagy. The translocation of PINK1, but not PARKIN, to mitochondria was stimulated by CGI-58 overexpression in C2C12 stable cell line cells (Fig. 6C).
FIGURE 6.
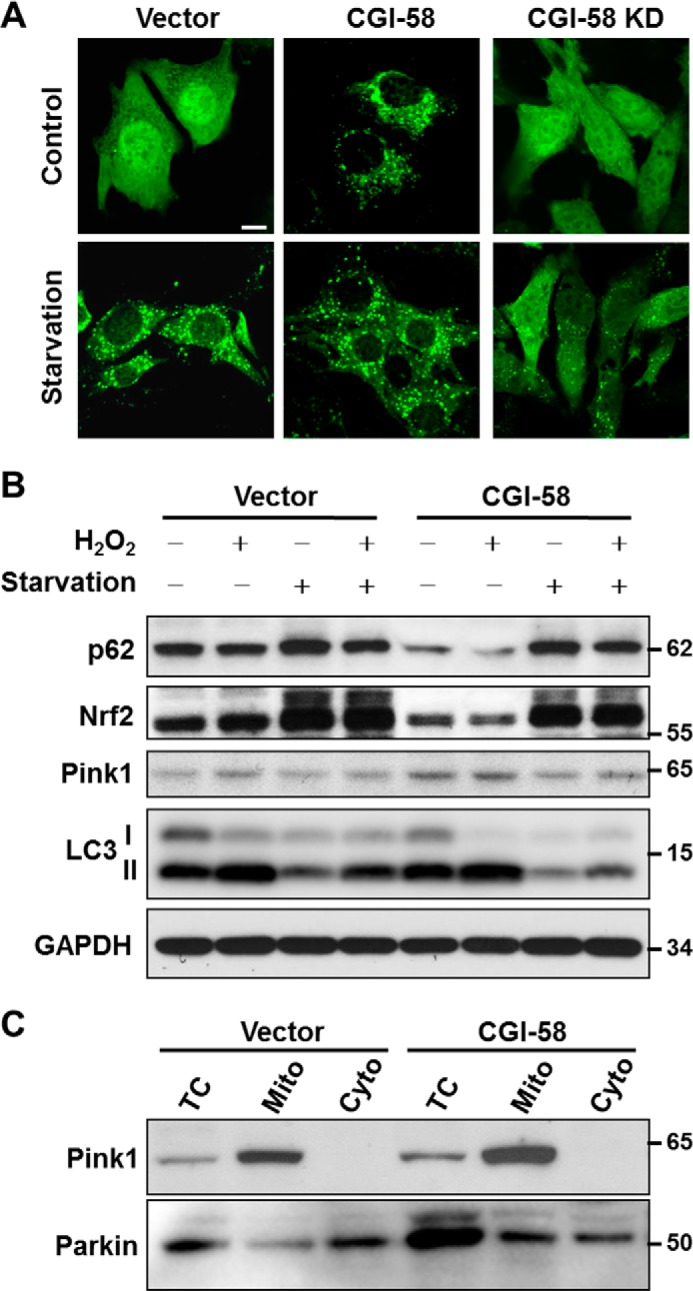
CGI-58 overexpression in C2C12 cells enhances autophagy. A, confocal microscopic analysis of autophagosomes in C2C12 cells with stable overexpression or knockdown (KD) of CGI-58 and vector control with or without starvation. C2C12 cells were infected with LC3-GFP adenovirus for 36 h, starved in EBSS buffer for another 4 h, and then fixed with 4% paraformaldehyde, followed by confocal microscopic imaging analysis. Scale bar, 10 μm. B, Western blot analysis of autophagic biomarkers, including p62, NRF2, PINK1, and LC3 in C2C12 cells stably overexpressing CGI-58 or vector control using GAPDH as internal control. C, subcellular fractionation analysis of PINK1 and PARKIN in C2C12 cells stably overexpressing CGI-58 or vector control. TC, total cell lysate; Mito, mitochondria; Cyto, cytosol.
The increased number of autophagosomes by CGI-58 could either be caused by an increased autophagy or by a reduced removal of autophagosomes as a consequence of reduced autophagic flux. To address this issue, we next examined the effects of CGI-58 overexpression on autophagic biomarkers in response to treatment with rapamycin and bafilomycin A1. Rapamycin induces autophagy by inhibiting mTORC1 signaling, whereas bafilomycin A1 inhibits the fusion of autophagosome with lysosome. The results show that the LC3-II/I ratio was significantly increased by CGI-58 overexpression under basal condition and in response to stimulation with rapamycin (Fig. 7A), suggesting an increased basal autophagy. Additionally, CGI-58 overexpression significantly decreased p-S6K1 and p-4E-BP1 levels under both basal condition and in response to the treatments, suggesting that CGI-58 promotes autophagy in part by inhibiting mTORC1 signaling. In contrast, bafilomycin A1 completely reversed the changes in LC3II/I ratio caused by CGI-58 overexpression, implicating that the increased number of autophagosomes by CGI-58 was primarily caused by an enhanced level of autophagic initiation rather than inhibition of autophagic consumption.
FIGURE 7.
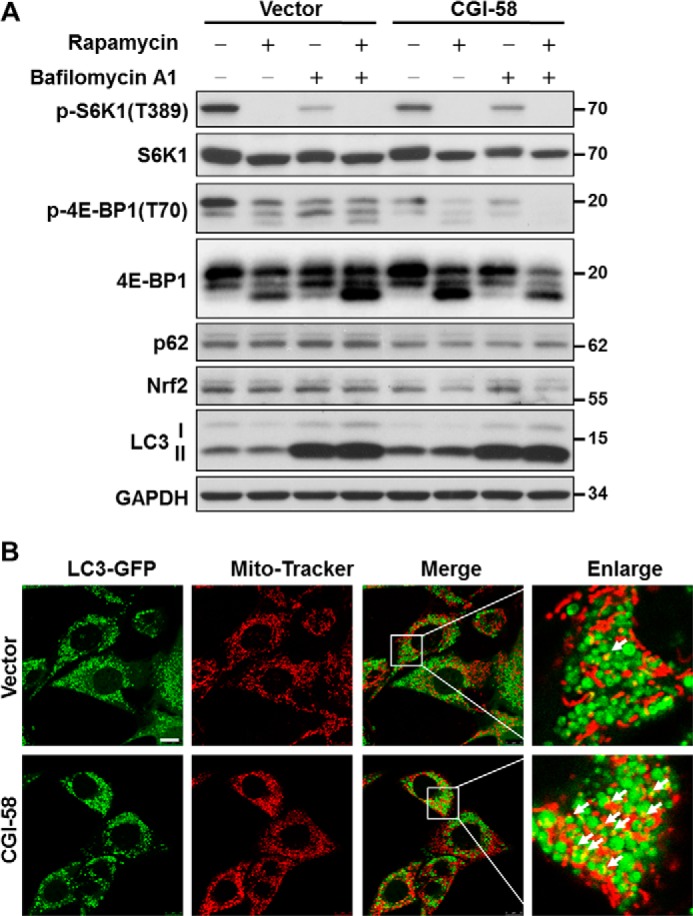
CGI-58 overexpression in C2C12 cells enhances mitophagy. A, analysis of the effect of CGI-58 overexpression in C2C12 stable cell lines on autophagic biomarkers and mTORC1 signaling in response to treatment with rapamycin, bafilomycin A1, or both by Western blot analysis. B, confocal microscopic analysis of mitophagy in C2C12 cells stably expression CGI-58 and vector control cells. The C2C12 cells stably overexpressing CGI-58 or vector cells were infected with LC3-GFP adenovirus, treated with bafilomycin A1, stained with MitoTracker Red, and then followed by confocal microscopic imaging analysis. Scale bar, 10 μm. The mitophagy puncta were highlighted by white arrows.
CGI-58 Overexpression Promotes Initiation of Mitophagy in C2C12 Cells
CL is required for the initiation of mitophagy (18). To determine a role for CGI-58 in regulating mitochondrial autophagy, we next analyzed the effect of CGI-58 overexpression on mitophagic initiation by confocal imaging analysis. The C2C12 cells were infected with adenoviruses overexpressing LC3-GFP fusion protein, treated with bafilomycin A1, and stained with MitoTracker Red. The results show that CGI-58 overexpression significantly increased the number of mitophagosomes in response to treatment with bafilomycin A1 (Fig. 7B, arrows). The results support a role for CGI-58 initiation of mitophagy, which is further supported by increased PINK1 expression and translocation to mitochondria in CGI-58 overexpression stable cell line cells, as shown by Fig. 6 (B and C).
CGI-58 Controls Autophagy through Inhibition of mTORC1 Signaling
We next investigated the cellular mechanisms by which CGI-58 stimulates autophagy by analyzing the effect of CGI-58 on the signaling of AMPK and mTORC1, the activator and inhibitor of autophagy, in response to insulin stimulation (19). Insulin stimulates protein synthesis and inhibits autophagy through activation of mTORC1 signaling. In further support for a role of CGI-58 in autophagy, overexpression of CGI-58 greatly stimulated AMPK activation both under basal condition and in response to insulin stimulation, as evidenced by increased AMPK phosphorylation at Thr-172, a major activation site of the kinase (Fig. 8A). Likewise, CGI-58 overexpression also significantly inhibited mTORC1 signaling, which is supported by decreased phosphorylation of S6K1 at Thr-389 and 4E-BP1 at Thr-70, the downstream effectors of mTORC1 signaling (Fig. 8A). In contrast, CGI-58 did not significantly affect insulin-stimulated Akt phosphorylation (Fig. 8A), suggesting that CGI-58 does not directly regulate insulin sensitivity. Conversely, CGI-58 knockdown in C2C12 cells significantly inhibited AMPK signaling and stimulated mTORC1 signaling both under basal condition and in response to insulin stimulation (Fig. 8B). Taken together, these data indicated that CGI-58 controls autophagy through activation of AMPK signaling and inhibition of mTORC1 signaling.
FIGURE 8.
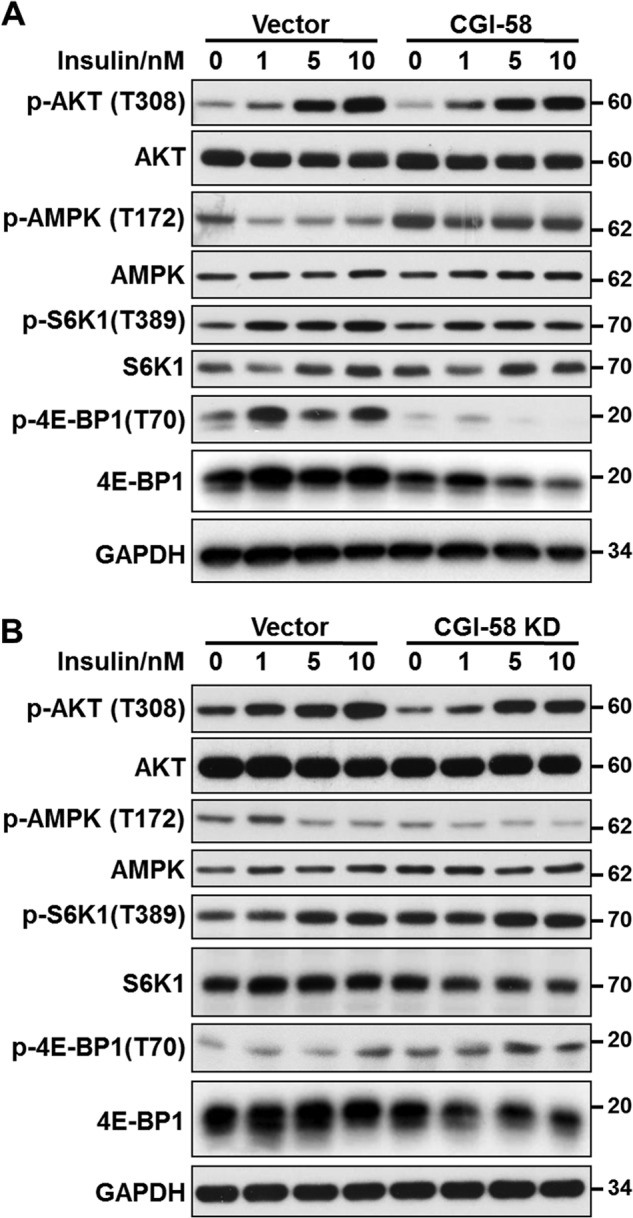
CGI-58 overexpression and depletion conversely regulated AMPK and mTORC1 signaling in C2C12 cells. A, C2C12 cells stably expressing CGI-58 or vector control were stimulated with indicated doses of insulin for 15 min, followed by analysis of phosphorylation of Akt, AMPK, S6K1, and 4E-BP1 by Western blotting using antibodies for indicated proteins and their phosphorylated forms. GAPDH was used as an internal control. B, C2C12 cells stably expressing CGI-58 shRNA or vector control were stimulated with indicated doses of insulin for 15 min, followed by a similar analysis as indicated in A.
DISCUSSION
Although CGI-58 promotes cellular TAG hydrolysis through activation of ATGL (4, 12), mice with liver-specific inactivation of ATGL exhibited a much less severe phenotype than that of liver-specific CGI-58 knock-out mice (20, 21). For example, the hepatic TAG content elevated only ∼3-fold (20), whereas mice deficient in CGI-58 expression in liver exhibited 8-fold. These differences suggest that CGI-58 must have additional functions beyond activating ATGL. Indeed, Yang et al. (22) reported a new alternative splicing isoform of the murine CGI-58 gene that was incapable of activating adipose TAG lipase. Overexpression of this isoform did not promote lipid droplet turnover or loss of intracellular TAG but retained the capacity to acylate lysophosphatidic acid. These results suggest that this splicing event may be involved in the regulation of lipid homeostasis through other unidentified mechanisms.
In this study, we investigated a role for CGI-58 in regulating the synthesis of phospholipids as a novel LPGAT enzyme. Using recombinant CGI-58 transiently expressed in mammalian cells, we demonstrated that CGI-58 possesses strong acyltransferase activity toward LPG, but not other lysophospholipids, including LPI, LPS, LPE, LPC, and MLCL. The results are further confirmed by using partially purified CGI-58 overexpressed in Sf9 insect cells. Consistent with the findings, C2C12 cells stably overexpressing CGI-58 exhibited significantly higher levels of endogenous PG relative to the vector control, whereas CGI-58 knockdown reduced the endogenous PG level. Our findings are consistent with previous reports that CGI-58 is a acyl-CoA-dependent LPAAT but exhibits no acyltransferase activity toward LPC, LPE, LPS, and LPI (1, 9), although neither report investigated CGI-58 for its acyltransferase activity toward LPG or MLCL.
PG is an important precursor for the synthesis of CL (14, 23, 24). Consequently, disruption of the PGS1 gene in yeast causes PG and CL deficiency and inhibition of growth on nonfermentable carbon sources (25). PG deficiency in mammalian cells also leads to CL deficiency, mitochondrial dysfunction, and reduced ATP production (26). Like CL, PG is subjected to remodeling subsequent to its de novo biosynthesis in mitochondria to incorporate appropriate acyl content for its biological functions and to prevent the harmful effect of LPG accumulation. Consequently, defective PG remodeling contributes to the onset of Barth syndrome, an X-linked recessive disease caused by mutations of the tafazzin gene encoding a transacylase involved in CL remodeling (27). In cultured skin fibroblasts from patients with Barth syndrome, both PG and CL remodeling is defective, as evidenced by decreased linoleic acid content both in PG and CL (28). In support of CGI-58 in PG remodeling, we demonstrate in this study that CGI-58 protein is also localized in mitochondria in addition to its previously identified role as a lipid droplet protein.
Mitochondria go through frequent cycles of fusion and fission, a process required for mitochondrial quality control by eliminating damaged mitochondria through mitophagy (29). Mitochondrial fission in mammals is mediated by DRP1, a mitochondrial GTPase that uses GTP hydrolysis to power the constriction and division of mitochondria. DRP1 is a predominantly cytosolic protein that is recruited to mitochondria during fission. Fission may help to isolate damaged segments of mitochondria and thus promote their autophagy (29). Hence, DRP1-mediated mitochondrial fission correlated with increased autophagy, and inhibition of DRP1 reduced autophagy (30). In support for a role of CGI-58 in PG biosynthesis, we identified a novel function of CGI-58 in regulating mitochondrial dynamics. We showed that overexpression of CGI-58 dramatically stimulated mitochondrial fragmentation. The fragmentation is primarily caused by increased fission caused by increased expression of DRP1, rather than fusion defect, as evidenced by the results form mitochondrial fusion analysis. Furthermore, treatment of C2C12 cells stably expressing CGI-58 with Mdivi-1 completely restores mitochondrial dynamics.
One of the major findings of the present study is a key role for CGI-58 in regulating autophagy and mitophagy. Autophagy is a lysosomal pathway by which organelles and proteins are degraded to maintain energy and cellular homeostasis when nutrients are limited (31, 32). Additionally, damaged mitochondria is usually cleared though mitophagy, a “self-eating” process required for the survival of cells. CL is a mitochondrial phospholipid required for mitochondrial bioenergetics, membrane stability, and dynamics. Recent studies suggest that CL also plays a key role in autophagy from yeast to mammals (18, 33). CL is required for autophagosome biogenesis and cargo recognition by supporting the membrane structure of autophagosomes and the activity of autophagy proteins, including LC3 and Beclin 1 (18, 34, 35). Additionally, CL mediates the cross-talk between mitochondria and lysosomes. Externalization of CL from mitochondrial inner membrane to mitochondrial surface acts as a recognition signal that directs damaged mitochondria to mitophagy (18, 36). In support for a key role of CGI-58 in mitophagy, we showed that overexpression of CGI-58 significantly enhanced autophagy, as evidenced by changes in autophagic biomarkers, including p62, NRF2, PINK1, and LC3-II. Accordingly, CGI-58 caused depletion of p62, NRF2, and LC3-I, negative indicators of autophagy. In contrast, CGI-58 overexpression significantly increased expression of PINK1 and its translocation to mitochondria. PINK1 is a key mitochondrial kinase required for the initiation of mitophagy. Consistent with the findings, C2C12 cells stably expressing CGI-58 exhibit enhanced mitophagosome biogenesis in response to treatment with bafilomycin A1.
In further support of a key role for CGI-58 in autophagy, we also identified CGI-58 as a key regulator of mTORC1 signaling. The initiation of autophagy requires the ULK complex (19). The activity of the ULK complex is negatively regulated by mTORC1 and positively regulated by AMPK (37). We showed that stable overexpression of CGI58 leads to activation of AMPK, as evidenced by increased phosphorylation of AMPK at Thr-172, a key activation phosphorylation site. Consequently, CGI-58 overexpression and deficiency adversely regulated mTORC1 signaling, which is supported by changes in phosphorylation levels of S6K1 (pS6K1 T389) and 4E-BP1 (p4E-BP1 T70) under both basal condition and in response to stimulation with insulin.
In addition to the classic pathway of lipid metabolism by cytosolic lipases, autophagic degradation of lipid droplets has recently been identified to play an essential role in hepatic lipid homeostasis by releasing free fatty acids for oxidation (31, 32). Consequently, dysregulation of autophagy is implicated by numerous studies in the pathophysiology of metabolic diseases, including obesity, dyslipidemia, and fatty liver diseases (32, 38–47). It can be envisaged from the findings of this study that CGI-58 may prevent dyslipidemia in part by promoting autophagy. Together, our current study identified novel mechanisms by which CGI-58 regulates lipid homeostasis, linking lipid droplet biology to mitochondrial quality control process through autophagy.
Acknowledgments
We thank Dr. Liqing Yu for providing us C2C12 cells stably transfected with shRNA targeted to CGI-58 and Dr. George Carman for fruitful discussion and suggestion.
This work was supported, in whole or in part, by National Institutes of Health Grant DK076685 (to Y. S.). This work was also supported by grants from NSFC of China (Grant 31271207 to Y. Z.), 973 Project (Grant 2011CB915504 to Y. Z.) and a scholarship from the Chinese National Scholarship Council (to D. X.).
- TAG
- triglyceride
- PG
- phosphatidylglycerol
- LPGAT
- lysophosphatidylglycerol acyltransferase
- MLCL
- monolysocardiolipin
- LPG
- lysophosphatidylglycerol
- LPC
- lysophosphatidylcholine
- LPE
- lysophosphatidylethanolamine
- LPI
- lysophosphatidylinositol
- LPS
- lysophosphatidylserine
- AMPK
- 5′-AMP-activated protein kinase
- ATGL
- adipose TAG lipase
- LPAAT
- lysophosphatidic acid acyltransferase
- mito-EGFP
- mitochondria-targeted GFP
- CL
- cardiolipin.
REFERENCES
- 1. Montero-Moran G., Caviglia J. M., McMahon D., Rothenberg A., Subramanian V., Xu Z., Lara-Gonzalez S., Storch J., Carman G. M., Brasaemle D. L. (2010) CGI-58/ABHD5 is a coenzyme A-dependent lysophosphatidic acid acyltransferase. J. Lipid Res. 51, 709–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Caviglia J. M., Betters J. L., Dapito D. H., Lord C. C., Sullivan S., Chua S., Yin T., Sekowski A., Mu H., Shapiro L., Brown J. M., Brasaemle D. L. (2011) Adipose-selective overexpression of ABHD5/CGI-58 does not increase lipolysis or protect against diet-induced obesity. J. Lipid Res. 52, 2032–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yamaguchi T., Omatsu N., Matsushita S., Osumi T. (2004) CGI-58 interacts with perilipin and is localized to lipid droplets. Possible involvement of CGI-58 mislocalization in Chanarin-Dorfman syndrome. J. Biol. Chem. 279, 30490–30497 [DOI] [PubMed] [Google Scholar]
- 4. Lass A., Zimmermann R., Haemmerle G., Riederer M., Schoiswohl G., Schweiger M., Kienesberger P., Strauss J. G., Gorkiewicz G., Zechner R. (2006) Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome. Cell Metab. 3, 309–319 [DOI] [PubMed] [Google Scholar]
- 5. Granneman J. G., Moore H. P., Granneman R. L., Greenberg A. S., Obin M. S., Zhu Z. (2007) Analysis of lipolytic protein trafficking and interactions in adipocytes. J. Biol. Chem. 282, 5726–5735 [DOI] [PubMed] [Google Scholar]
- 6. Lefèvre C., Jobard F., Caux F., Bouadjar B., Karaduman A., Heilig R., Lakhdar H., Wollenberg A., Verret J. L., Weissenbach J., Ozgüc M., Lathrop M., Prud'homme J. F., Fischer J. (2001) Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in Chanarin-Dorfman syndrome. Am. J. Hum. Genet. 69, 1002–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Musumeci S., D'Agata A., Romano C., Patané R., Cutrona D. (1988) Ichthyosis and neutral lipid storage disease. Am. J. Med. Genet. 29, 377–382 [DOI] [PubMed] [Google Scholar]
- 8. Igal R. A., Coleman R. A. (1998) Neutral lipid storage disease: a genetic disorder with abnormalities in the regulation of phospholipid metabolism. J. Lipid Res. 39, 31–43 [PubMed] [Google Scholar]
- 9. Ghosh A. K., Ramakrishnan G., Chandramohan C., Rajasekharan R. (2008) CGI-58, the causative gene for Chanarin-Dorfman syndrome, mediates acylation of lysophosphatidic acid. J. Biol. Chem. 283, 24525–24533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Haemmerle G., Lass A., Zimmermann R., Gorkiewicz G., Meyer C., Rozman J., Heldmaier G., Maier R., Theussl C., Eder S., Kratky D., Wagner E. F., Klingenspor M., Hoefler G., Zechner R. (2006) Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 312, 734–737 [DOI] [PubMed] [Google Scholar]
- 11. Fischer J., Lefèvre C., Morava E., Mussini J. M., Laforêt P., Negre-Salvayre A., Lathrop M., Salvayre R. (2007) The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nat. Genet. 39, 28–30 [DOI] [PubMed] [Google Scholar]
- 12. Radner F. P., Streith I. E., Schoiswohl G., Schweiger M., Kumari M., Eichmann T. O., Rechberger G., Koefeler H. C., Eder S., Schauer S., Theussl H. C., Preiss-Landl K., Lass A., Zimmermann R., Hoefler G., Zechner R., Haemmerle G. (2010) Growth retardation, impaired triacylglycerol catabolism, hepatic steatosis, and lethal skin barrier defect in mice lacking comparative gene identification-58 (CGI-58). J. Biol. Chem. 285, 7300–7311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cao J., Hawkins E., Brozinick J., Liu X., Zhang H., Burn P., Shi Y. (2004) A predominant role of acyl-CoA:monoacylglycerol acyltransferase-2 in dietary fat absorption implicated by tissue distribution, subcellular localization, and up-regulation by high fat diet. J. Biol. Chem. 279, 18878–18886 [DOI] [PubMed] [Google Scholar]
- 14. Yang Y., Cao J., Shi Y. (2004) Identification and characterization of a gene encoding human LPGAT1, an endoplasmic reticulum-associated lysophosphatidylglycerol acyltransferase. J. Biol. Chem. 279, 55866–55874 [DOI] [PubMed] [Google Scholar]
- 15. Chu C. T., Bayır H., Kagan V. E. (2014) LC3 binds externalized cardiolipin on injured mitochondria to signal mitophagy in neurons: implications for Parkinson disease. Autophagy 10, 376–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li J., Liu X., Wang H., Zhang W., Chan D. C., Shi Y. (2012) Lysocardiolipin acyltransferase 1 (ALCAT1) controls mitochondrial DNA fidelity and biogenesis through modulation of MFN2 expression. Proc. Natl. Acad. Sci. U.S.A. 109, 6975–6980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dowhan W. (1997) Molecular basis for membrane phospholipid diversity: why are there so many lipids? Annu. Rev. Biochem. 66, 199–232 [DOI] [PubMed] [Google Scholar]
- 18. Chu C. T., Ji J., Dagda R. K., Jiang J. F., Tyurina Y. Y., Kapralov A. A., Tyurin V. A., Yanamala N., Shrivastava I. H., Mohammadyani D., Qiang Wang K. Z., Zhu J., Klein-Seetharaman J., Balasubramanian K., Amoscato A. A., Borisenko G., Huang Z., Gusdon A. M., Cheikhi A., Steer E. K., Wang R., Baty C., Watkins S., Bahar I., Bayır H., Kagan V. E. (2013) Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 15, 1197–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lamb C. A., Yoshimori T., Tooze S. A. (2013) The autophagosome: origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 14, 759–774 [DOI] [PubMed] [Google Scholar]
- 20. Wu J. W., Wang S. P., Alvarez F., Casavant S., Gauthier N., Abed L., Soni K. G., Yang G., Mitchell G. A. (2011) Deficiency of liver adipose triglyceride lipase in mice causes progressive hepatic steatosis. Hepatology 54, 122–132 [DOI] [PubMed] [Google Scholar]
- 21. Guo F., Ma Y., Kadegowda A. K., Betters J. L., Xie P., Liu G., Liu X., Miao H., Ou J., Su X., Zheng Z., Xue B., Shi H., Yu L. (2013) Deficiency of liver comparative gene identification-58 causes steatohepatitis and fibrosis in mice. J. Lipid Res. 54, 2109–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang X., Lu X., Liu J. (2010) Identification of a novel splicing isoform of murine CGI-58. FEBS Lett. 584, 903–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen D., Zhang X.-Y., Shi Y. (2006) Identification and functional characterization of hCLS1, a human cardiolipin synthase localized in mitochondria. Biochem. J. 398, 169–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nie J., Hao X., Chen D., Han X., Chang Z., Shi Y. (2010) A novel function of the human CLS1 in phosphatidylglycerol synthesis and remodeling. Biochim. Biophys. Acta 1801, 438–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhong Q., Gohil V. M., Ma L., Greenberg M. L. (2004) Absence of cardiolipin results in temperature sensitivity, respiratory defects, and mitochondrial DNA instability independent of pet56. J. Biol. Chem. 279, 32294–32300 [DOI] [PubMed] [Google Scholar]
- 26. Kawasaki K., Kuge O., Chang S. C., Heacock P. N., Rho M., Suzuki K., Nishijima M., Dowhan W. (1999) Isolation of a chinese hamster ovary (CHO) cDNA encoding phosphatidylglycerophosphate (PGP) synthase, expression of which corrects the mitochondrial abnormalities of a PGP synthase-defective mutant of CHO-K1 cells. J. Biol. Chem. 274, 1828–1834 [DOI] [PubMed] [Google Scholar]
- 27. Claypool S. M., Koehler C. M. (2012) The complexity of cardiolipin in health and disease. Trends Biochem. Sci. 37, 32–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vreken P., Valianpour F., Nijtmans L. G., Grivell L. A., Plecko B., Wanders R. J., Barth P. G. (2000) Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem. Biophys. Res. Commun. 279, 378–382 [DOI] [PubMed] [Google Scholar]
- 29. Twig G., Elorza A., Molina A. J., Mohamed H., Wikstrom J. D., Walzer G., Stiles L., Haigh S. E., Katz S., Las G., Alroy J., Wu M., Py B. F., Yuan J., Deeney J. T., Corkey B. E., Shirihai O. S. (2008) Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee Y., Lee H. Y., Hanna R. A., Gustafsson A. B. (2011) Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 301, H1924–H1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Singh R., Cuervo A. M. (2011) Autophagy in the cellular energetic balance. Cell Metab. 13, 495–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Singh R., Kaushik S., Wang Y., Xiang Y., Novak I., Komatsu M., Tanaka K., Cuervo A. M., Czaja M. J. (2009) Autophagy regulates lipid metabolism. Nature 458, 1131–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen S., He Q., Greenberg M. L. (2008) Loss of tafazzin in yeast leads to increased oxidative stress during respiratory growth. Mol. Microbiol. 68, 1061–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hailey D. W., Rambold A. S., Satpute-Krishnan P., Mitra K., Sougrat R., Kim P. K., Lippincott-Schwartz J. (2010) Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141, 656–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huang W., Choi W., Hu W., Mi N., Guo Q., Ma M., Liu M., Tian Y., Lu P., Wang F. L., Deng H., Liu L., Gao N., Yu L., Shi Y. (2012) Crystal structure and biochemical analyses reveal Beclin 1 as a novel membrane binding protein. Cell Res. 22, 473–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen S., Tarsio M., Kane P. M., Greenberg M. L. (2008) Cardiolipin mediates cross-talk between mitochondria and the vacuole. Mol. Biol. Cell 19, 5047–5058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim J., Kim Y. C., Fang C., Russell R. C., Kim J. H., Fan W., Liu R., Zhong Q., Guan K. L. (2013) Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 152, 290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ebato C., Uchida T., Arakawa M., Komatsu M., Ueno T., Komiya K., Azuma K., Hirose T., Tanaka K., Kominami E., Kawamori R., Fujitani Y., Watada H. (2008) Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 8, 325–332 [DOI] [PubMed] [Google Scholar]
- 39. Zhao T., Huang X., Han L., Wang X., Cheng H., Zhao Y., Chen Q., Chen J., Xiao R., Zheng M. (2012) Central role of mitofusin 2 in autophagosome-lysosome fusion in cardiomyocytes. J. Biol. Chem. 287, 23615–23625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Codogno P., Meijer A. J. (2010) Autophagy: a potential link between obesity and insulin resistance. Cell Metab. 11, 449–451 [DOI] [PubMed] [Google Scholar]
- 41. Coupé B., Ishii Y., Dietrich M. O., Komatsu M., Horvath T. L., Bouret S. G. (2012) Loss of autophagy in pro-opiomelanocortin neurons perturbs axon growth and causes metabolic dysregulation. Cell Metab. 15, 247–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Goldman S., Zhang Y., Jin S. (2010) Autophagy and adipogenesis: implications in obesity and type II diabetes. Autophagy 6, 179–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yang L., Li P., Fu S., Calay E. S., Hotamisligil G. S. (2010) Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 11, 467–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhou L., Liu F. (2010) Autophagy: roles in obesity-induced ER stress and adiponectin downregulation in adipocytes. Autophagy 6, 1196–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang Y., Goldman S., Baerga R., Zhao Y., Komatsu M., Jin S. (2009) Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc. Natl. Acad. Sci. U.S.A. 106, 19860–19865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ezaki J., Matsumoto N., Takeda-Ezaki M., Komatsu M., Takahashi K., Hiraoka Y., Taka H., Fujimura T., Takehana K., Yoshida M., Iwata J., Tanida I., Furuya N., Zheng D. M., Tada N., Tanaka K., Kominami E., Ueno T. (2011) Liver autophagy contributes to the maintenance of blood glucose and amino acid levels. Autophagy 7, 727–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Komatsu M., Waguri S., Ueno T., Iwata J., Murata S., Tanida I., Ezaki J., Mizushima N., Ohsumi Y., Uchiyama Y., Kominami E., Tanaka K., Chiba T. (2005) Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 169, 425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]