

Background: Loss of Krev-interaction trapped 1 (KRIT1) drives the activation of endothelial cells.
Results: Blocking vascular endothelial growth factor (VEGF) signaling reverses the endothelial cell changes after KRIT1 depletion and permeability in KRIT1-deficient mice.
Conclusion: Increased VEGF signaling downstream of KRIT1 loss alters endothelial cell behavior and is responsible for permeability in vivo.
Significance: These studies describe a novel regulation of endothelial barrier function by KRIT1.
Keywords: Adherens Junction, β-Catenin (B-Catenin), Permeability, Phosphotyrosine Signaling, Vascular Endothelial Growth Factor (VEGF), CCM, Krit1
Abstract
Disruption of endothelial cell-cell contact is a key event in many cardiovascular diseases and a characteristic of pathologically activated vascular endothelium. The CCM (cerebral cavernous malformation) family of proteins (KRIT1 (Krev-interaction trapped 1), PDCD10, and CCM2) are critical regulators of endothelial cell-cell contact and vascular homeostasis. Here we show novel regulation of vascular endothelial growth factor (VEGF) signaling in KRIT1-depleted endothelial cells. Loss of KRIT1 and PDCD10, but not CCM2, increases nuclear β-catenin signaling and up-regulates VEGF-A protein expression. In KRIT1-depleted cells, increased VEGF-A levels led to increased VEGF receptor 2 (VEGFR2) activation and subsequent alteration of cytoskeletal organization, migration, and barrier function and to in vivo endothelial permeability in KRIT1-deficient animals. VEGFR2 activation also increases β-catenin phosphorylation but is only partially responsible for KRIT1 depletion-dependent disruption of cell-cell contacts. Thus, VEGF signaling contributes to modifying endothelial function in KRIT1-deficient cells and microvessel permeability in Krit1+/− mice; however, VEGF signaling is likely not the only contributor to disrupted endothelial cell-cell contacts in the absence of KRIT1.
Introduction
Endothelial cell-cell contact is a tightly regulated feature of endothelial monolayers and is in itself a key regulator of endothelial behavior and vascular homeostasis (1). The importance of maintaining endothelial cell-cell contact within a finite yet plastic range is reflected in the ubiquitous nature of permeability defects in vascular diseases, including atherosclerosis, chronic inflammatory disease, and pulmonary edema (2–5). In addition, increased vascular permeability correlates with neo-angiogenesis, particularly in injured tissues (6) and in hereditary/developmental vascular malformations such as hereditary hemorrhagic telangiectasia and hereditary small vessel disease (7, 8). Despite the clear prevalence of endothelial cell-cell contact defects in vascular disease, it is still unclear how these defects are translated into disease pathogenesis.
Cerebral cavernous malformation (CCM)2 is a relatively common neurovascular disease marked by focal areas of dilated, leaky microvessels, abnormal basement membrane, and a lack of smooth muscle cells (9). This disease is commonly caused by loss of one of three genes, Krev-interaction trapped 1 (Krit1) (10), cerebral cavernous malformations 2 (Ccm2) (11), and programmed cell death 10 (Pdcd10) (12). Of these we have shown that KRIT1 is an important regulator of endothelial cell-cell junctions (13). Heterozygous loss of KRIT1 drives the normally stable, quiescent endothelium toward an active, disrupted state marked by lost barrier function, increased stress fiber formation, and loss of adherens junction integrity both in vitro (13, 14) and in vivo (15, 16). We previously showed that KRIT1 is a component of the adherens junction complex, where it functions downstream of Rap1 GTPase to stabilize the interaction of VE-cadherin and β-catenin (13). Loss of KRIT1 triggers a loss of β-catenin from sites of cell-cell contact, leading to both loss of VE-cadherin adhesion and increased nuclear β-catenin (14). Although the destabilization of cell-cell contacts can be extrapolated to explain the observed changes in endothelial behavior after loss of KRIT1, the exact mechanisms that underlie these changes remain unclear.
Previously we demonstrated that knockdown of KRIT1 in endothelial cells increases nuclear β-catenin localization and activation of β-catenin-dependent transcription (14). β-Catenin is a key regulator of vascular endothelial growth factor (VEGF) expression (17), a major regulator of vascular homeostasis. Correspondingly, KRIT1 depletion increased Vegfa mRNA and also increased serum levels of VEGF-A in Krit1+/− mice (14, 15). Predicting the contribution of VEGF overexpression to CCM pathogenesis is difficult as VEGF overexpression in transgenic mice exhibits tissue- or cell type-dependent phenotypic variability. However, increased vessel density and endothelial barrier disruption are common outcomes (18–21). Some VEGF overexpression models also demonstrate altered basement membrane composition and vessel lumen enlargement, which have also been reported in CCM (19, 21). Nevertheless, mice overexpressing VEGF have not been reported to develop CCM-like vascular lesions. These data suggest that increased VEGF expression is unlikely to be the sole cause of CCM formation, yet the correlation between VEGF overexpression and the corresponding endothelial and vascular changes after KRIT1 reduction in vitro and in vivo strongly support examination of the signaling interplay between KRIT1 and VEGF. Thus, the goal of the present study was to investigate whether VEGF signaling contributes to KRIT1 depletion-dependent phenotypes and, if so, to examine the underlying mechanism. This information would improve our understanding of the role of KRIT1 in the endothelium and provide new insights into vascular homeostasis and CCM development.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfection
HPAEC (Invitrogen) were cultured in 1:1 Dulbecco's modified Eagle's medium (DMEM):F-12 supplemented with 5% fetal bovine serum (FBS), 1% endothelial cell growth supplement (ECGS, ScienCell, Carlsbad, CA), 1% antimycotic/antibiotic solution (Invitrogen), and 50 μm heparin (Calbiochem) at 37 °C with 5% CO2. HPAEC were grown on 2 μg/cm2 gelatin-coated tissue culture plates and only passage 3 to passage 6 were used in experiments. Bovine aortic endothelial cells (BAEC, a gift from A. Smrcka, University of Rochester) and wild type (WT), Krit1 knock-out (−/−), and reconstituted (9/6) mouse embryonic fibroblasts (MEFs; a gift from F. Retta, University of Sienna) were cultured in DMEM high glucose with 10% FBS, 1% penicillin/streptomycin/l-glutamine, and 1% nonessential amino acids (Invitrogen) at 37 °C with 5% CO2. BAEC and HPAEC were transfected with 30 ng of siRNA using the HiPerfect transfection reagent (Qiagen, Valencia, CA) as reported previously (13). Alternatively, HPAEC were transfected with siRNA using siPort Amine (Ambion/Invitrogen) according to the manufacturer's instructions. Transfection efficiencies ranged from 80 to 95% based on transfection of fluorescently labeled siRNAs (data not shown). Activity of this anti-KRIT1 siRNA against bovine and human protein has been reported previously (13). Co-transfection of siRNA and cDNA was performed using Amaxa nucleofection (Lonza, Basel, Switzerland) as previously described (14). KRIT1 siRNA-conditioned media were harvested from KRIT1 siRNA-transfected HPAE 24h after transfection and spun briefly. Clarified conditioned media was added directly to cultures of negative control (NC) siRNA-transfected cells and incubated at 37 °C for the indicated times. Recombinant human VEGF and VEGFR2/Fc were obtained from R&D Biosystems (Dallas, TX). SU6656 was obtained from (Tocris/R&D Biosystems).
siRNA and Plasmids
Non-targeting negative control siRNA #1 and anti-KRIT1 siRNA (AM16708, Ambion/Invitrogen) were used as reported previously (13, 14). For TOPFlash reporter assays and VEGF enzyme-linked immunoassay (ELISA), Krit1, Ccm2, and Pdcd10 expression was knocked down using Dharmacon ON-TARGETplus siRNAs (J-003825-06, HALJF-000001, and J-004436-05, respectively) and compared with Dharmacon ON-TARGETplus non-targeting control #1 (D-001810-01, Fisher). pCDNA3.1 (−) was obtained from Invitrogen and FLAG-tagged WT β-catenin and dominant-negative TCF (dn-TCF) constructs were a gift from Dr. Eric Fearon (University of Michigan).
TOPFlash Reporter Assay
Reporter assays were performed using the Dual-Glo luciferase assay system (Promega, Madison, WI) as previously described (14). Super 8x TOPFlash and FOPFlash reporter constructs were obtained from the Randall Moon lab via Addgene (Cambridge, MA). Data were analyzed using one-way ANOVA with Dunn's multiple comparisons post-hoc testing.
VEGF ELISA
ELISA high binding plates (Fisher/Thermo Scientific) were coated with 1 μg/ml of mouse anti-VEGF (R&D Biosystems) in PBS overnight at room temperature. Plates were then blocked with 300 μl of blocking buffer (1% BSA in PBS) for 1 h at room temperature. After washing with wash buffer (PBS with 0.2% Tween 20), 100 μl samples of conditioned medium from siRNA-transfected 60-mm plates were added to appropriate wells and incubated at room temperature for 2 h. A standard curve of recombinant VEGF from 0 to 2000 pg/ml was run alongside experimental samples. Plates were washed again and incubated with 1.2 μg/ml goat anti-VEGF (R&D Biosystems) for 2 h at room temperature. After washing, streptavidin-HRP (Thermo Fisher Scientific) was added for 1 h followed by detection with 3,3′,5,5′-tetramethylbenzidine (TMB; eBioscience, San Diego, CA). Data were analyzed using one-way ANOVA with Tukey's post-hoc testing.
RNA Isolation and Semi-quantitative RT-PCR
RNA was isolated using an RNeasy Purification kit (Qiagen) or Trizol (Invitrogen) extraction according to the manufacturer's instructions. Complementary DNA was obtained with Transcriptor reverse transcriptase (Roche Applied Science) using Oligo(dT) primers (Invitrogen). Amplifications were run in a 7000 real-time PCR system (Applied Biosystems/Invitrogen) using previously published bovine, mouse, and human primer sets (14, 15) or Taqman gene expression assays (Krit1, Hs00184988; Ccm2, Hs00230191; Pdcd10, Hs00200578; Gapdh, Hs99999905, Invitrogen). Each value was calculated using the comparative Ct method (22) and normalized to Gapdh or Actb internal controls. All samples were run in at least triplicate. Data were analyzed using one-way ANOVA with Tukey's post-hoc testing.
Immunoprecipitation and Western Blotting
Lysates were prepared as reported previously (13). As indicated, cells were treated with either VEGFR2/Fc or SU6656 for 8 h. Unless otherwise indicated, cells were treated with recombinant human VEGF (rhVEGF) for 15 min immediately before lysis. Monoclonal anti-phosphotyrosine (PTyr100), monoclonal rabbit anti-human VE-cadherin, and monoclonal rabbit anti-VEGF receptor 2 (VEGFR2) were obtained from Cell Signaling (Danvers, MA) and used at a dilution of 1:100 for immunoprecipitation and 1:1000 for Western blotting. Control lysates were immunoprecipitated with either mouse or rabbit non-immune IgG (Santa Cruz, Dallas, TX). VE-cadherin immunoprecipitated lysates were additionally probed with polyclonal rabbit anti-β-catenin or mouse anti-chicken β-catenin (Sigma). Phosphotyrosine precipitates were also probed with antibodies against phospho-VE cadherin Tyr-731 and Tyr-658 and phospho-β-catenin Tyr-654 and Tyr-142 (Abcam, Cambridge, MA). The secondary antibodies used were donkey anti-mouse Alexa-680 (Invitrogen) and goat anti-rabbit Dylight-800 (Fisher). An Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE) was used to image membranes and for densitometry. Data were analyzed using one-way ANOVA with Tukey's post-hoc testing.
Endothelial Monolayer Leak Assay
BAECs were plated onto 3-μm pore polyester transwell filters (Corning Life Sciences, Corning, NY) coated with 10 μg/ml human plasma fibronectin (gift from Dr. Denise Hocking, University of Rochester). Cells were transfected at the time of plating, or for co-transfections, transfection occurred before cells were re-plated on transwell inserts. Once in the transwell, the cells were allowed to grow for 48 h at 37 °C to full confluence. Cells were then incubated in DMEM with 0.1% FBS for 2 h then treated with or without 50 ng/ml VEGFR2/Fc for 8 h. Horseradish peroxidase (HRP; Sigma) was added to a concentration of 1.5 μg/ml in upper wells and incubated at 37 °C for an additional 2 h. The HRP content of the lower chamber medium was then measured using a TMB colorimetric assay. Briefly, 10-μl samples of the lower chamber medium were transferred in triplicate to a 96-well plate. 100 μl of TMB was added to each sample, and the reaction was allowed to proceed for 1 min. A standard curve of HRP from 0 to 0.5 μg/ml was run alongside experimental samples. The reaction was stopped by adding 100 μl of 2 n H2SO4 to each well. Absorbance at 450 nm was acquired, and raw absorbance values were converted into concentrations using the standard curve. After samples were removed, the transwell filters were fixed in 4% formaldehyde (Fisher), stained with 0.25% Coomassie Blue (Bio-Rad), and examined to confirm the integrity of each cell monolayer. Data were analyzed using one-way ANOVA with Tukey's post-hoc testing.
Intravital Microscopy
KRIT1 heterozygous (Krit1+/−) mice were obtained from Dr. Dean Li (University of Utah). These mice are on a clean C57BL/6 background after having been backcrossed 10 generations to C57BL/6NCrl. Mice were bred and maintained under standard conditions in the University of Rochester animal facility, which is accredited by the American Association for Accreditation of Laboratory Animal Care. All protocols were approved by the institutional review board. Mice were used between 8 and 12 weeks of age; littermate controls were used in all experiments.
Male wild type (WT, Krit1+/+) or KRIT1 heterozygous (Krit1+/−) mice were anesthetized with sodium pentobarbital (65 mg/kg intraperitoneal) and maintained on supplemental anesthetic via a jugular catheter. Preparation and visualization of the cremaster muscle as well as measurement of permeability to Alexa-488 BSA (Invitrogen) was performed as previously described (15, 23). The vascular wall Ps (cm/s) is calculated from the measured flux of fluorescent label across the vessel wall over time normalized to microvessel surface area at a known BSA concentration gradient (24). Corrections are made for the source volume and surface area due to the confocal slice (23). Tail vein injection of 3 mg/kg SU5416 (TOCRIS/R&D Biosystems; diluted in 4% DMSO/96% corn oil; Sigma) was performed 1 h before surgery. Images were acquired from at least 17 vessel sites from 3 or more animals. Vessel sites are the appropriate determinant for n in these experiments and are treated as statistically independent. Data are given as combined microvessel permeability (arteriole and venule). Individual arteriole and venule means ± S.E. are given in Table 1. Data were analyzed using Kruskal-Wallis testing (non-parametric ANOVA) with Dunn's post-hoc testing.
TABLE 1.
Mean arteriole and venule permeability in the presence or absence of SU5416
Permeability (Ps) was calculated from the increase in fluorescent intensity immediately outside of the microvessel over time adjusted for microvessel surface area, initial Alexa-488-BSA concentration gradient, and source volume.
| Genotype/treatment | Arteriole Ps ± S.E. × 10−6 cm/s | Venule Ps ± S.E. × 10−6 cm/s | Number of vessel sites | Number of mice |
|---|---|---|---|---|
| Krit1+/+ | 0.53 ± 0.13 | 0.66 ± 0.16 | 20 | 5 |
| Krit1+/+ +SU5416 | 0.8563 ± 0.1198 | 0.78 ± 0.09 | 17 | 3 |
| Krit1+/− | 1.65 ± 0.25 | 1.38 ± 0.24 | 27 | 5 |
| Krit1+/− +SU5416 | 0.68 ± 0.08 | 0.55 ± 0.06 | 24 | 4 |
| Krit1+/− +vehicle | 1.24 ± 0.34 | 1.16 ± 0.14 | 20 | 3 |
Immunofluorescence and F-actin Quantitation
Cells were transfected at 80% confluence on 10 μg/ml fibronectin-coated glass coverslips and allowed to grow to confluence. Cells were then fixed with 4% formaldehyde for 20 min followed by permeabilization for 5 min with 0.2% Triton X-100 and blocking with 10% normal goat serum for 1 h. Cells were then incubated with rhodamine-phalloidin (Invitrogen, 14 μm) for 30 min on ice and counterstained with 1 μg/ml Hoechst (Invitrogen). Images were captured at room temperature using Metamorph software (Molecular Devices, Sunnyvale, CA) with an UPLSAPO 20x (n.a. 0.75) objective on an Olympus IX70 microscope and an ORCA-ER digital camera (Hamamatsu, Japan). Images were imported into ImageJ for further analysis. F-actin fluorescence intensity was calculated along a line scan drawn perpendicular to the main cell axis. Cortical actin along the cell edge was excluded from the analysis. Intensity values were obtained by integrating the area under the curve of the intensity histogram generated by the line scan. The area calculated was then divided by length of the line scan, yielding an average intensity value. Data were analyzed using one-way ANOVA with Tukey's post-hoc testing.
Migration Assay
An in vitro wound healing migration assay was performed as previously described (25). Briefly, BAEC were plated on tissue culture plastic and transfected with anti-KRIT1 or negative control siRNA. A second transfection was performed 48 h later. 24 h after the second transfection an area was denuded by a rubber policeman at the center of the plate, and cells were treated with 25 ng/ml VEGFR2/Fc for 24 h. Images were taken at the time of wounding (0 h) and after 24 h, and the relative distance traveled by the cells at the acellular front was determined using ImageJ. Mitomycin C (0.5 μg/ml, Sigma) was added to control wells to account for interference from the mitogenic response to wounding, which was negligible (data not shown). Data were analyzed using one-way ANOVA with Tukey's post-hoc testing.
Subcellular Fractionation
Subcellular fractionation was performed as previously described (14). Briefly, BAEC transfected with negative control or anti-KRIT1 siRNA were suspended in ice-cold hypotonic lysis buffer (20 mm HEPES-KOH, pH 7.5, 1.5 mm MgCl2, 5 mm KCl) for 10 min followed by needle homogenization. A portion of the resulting total cell lysate was reserved, and the remaining lysates were centrifuged at 750 × g for 10 min to pellet the nuclei. The supernatant was then removed (cytosolic). The pellet was laid over a 1 m sucrose gradient, spun at 500 × g, then lysed in buffer containing 1% Nonidet P-40. The total, cytosolic, and nuclear fractions were analyzed for β-catenin via Western blot. Nuclear and cytosolic markers (lamin A/C and RhoGDI) were analyzed to ensure fractions were not contaminated (data not shown).
Densitometry and Statistics
Densitometry was performed using the Odyssey Infrared Imaging System using within-lane background subtraction. For quantitation, blots from at least 3 individual experiments (from separate transfections) were analyzed. Each lane was then normalized to negative control siRNA values. As appropriate, phospho-VEGFR2, phospho-β-catenin, or co-immunoprecipitated β-catenin values were further standardized to total immunoprecipitated VEGFR2, β-catenin, or VE-cadherin, respectively. Error was calculated for the ratio of experimental to negative control and subsequently for the ratio of experimental to total protein using multivariate Taylor expansion analysis.
Statistical analysis (i.e. one way ANOVA with appropriate post-hoc testing) was performed using PRISM software (Version 4.0, GraphPad Software Inc., La Jolla, CA). Significance was set at α = 0.05.
RESULTS
Loss of KRIT1 and PDCD10, but Not CCM2, Increases Nuclear β-Catenin Signaling
Ablation of KRIT1 expression in endothelial cells causes a shift of β-catenin localization to the nucleus and the activation of β-catenin-dependent transcription (14). To examine whether these events occurred universally downstream of loss of CCM family proteins, we transfected HPAEC with siRNAs targeting Krit1, Ccm2, and Pdcd10 (Fig. 1A). siRNA-mediated depletion of KRIT1 and PDCD10, but not CCM2, increased activity of the β-catenin reporter TOPFlash (26) but not the activity of the inactive FOPFlash reporter, indicating an increase in β-catenin-dependent transcription (Fig. 1B). The increase in nuclear β-catenin signaling corresponded with significant increases in Vegfa mRNA in both KRIT1 and PDCD10-depleted HPAEC (Fig. 1C) but not CCM2 siRNA-transfected cells. To determine whether increased Vegfa mRNA corresponded with an increase in secreted VEGF-A protein, we performed an ELISA on the conditioned medium of negative control and KRIT1 siRNA-transfected endothelial cells. Compared with negative control, medium from KRIT1-depleted cells contained significantly more VEGF-A (Fig. 1D). Co-transfection with dn-TCF, a suppressor of β-catenin-dependent gene transcription (26), was able to reduce the concentration of VEGF-A protein (Fig. 1D) and Vegfa mRNA (Fig. 1E) in KRIT1-depleted cells down to control levels, indicating that the increase in VEGF expression was due to activation of nuclear β-catenin signaling.
FIGURE 1.

Loss of KRIT1 and PDCD10, but not CCM2, stimulates nuclear β-catenin-dependent expression of Vegfa. A, knockdown efficiency of anti-KRIT1, CCM2, and PDCD10 siRNA on Krit1, CCM2, PDCD10, and Ctnnb1 (β-catenin) mRNA expression. B, β-catenin-dependent reporter activity in HPAEC transfected with NC, anti-KRIT1, -CCM2, and -PDCD10 siRNA. TOPFlash activity is shown relative to NC siRNA-transfected cells (±S.E.) and compared with cells overexpressing β-catenin [βcat]. n = 4, p = 0.0011 by ANOVA; *, p < 0.05 by post-hoc test versus NC-transfected cells. C, expression of Vegfa mRNA in siRNA-transfected HPAEC. Data shown are the average expression from three experiments, ± S.E., p = 0.0002 by ANOVA; *, p < 0.001 by post-hoc test versus NC-transfected cells. D, concentration of VEGF-A secreted from NC and KRIT1 siRNA-transfected cells. n = 9, p = 0.003 by ANOVA; *, p < 0.05 by post-hoc test versus NC-transfected cells; **, p < 0.01 versus KRIT1 siRNA-transfected cells. E, expression of Vegfa mRNA in siRNA-transfected cells co-expressing dn-TCF ±S.E; n = 3, p = 0.003 by ANOVA; *, p < 0.05 by post-hoc test versus NC-transfected cells; **, p < 0.01 versus KRIT1 siRNA-transfected cells.
VEGF Secreted from KRIT1-depleted Endothelial Cells Leads to VEGFR2 Activation
Secreted VEGF-A acts as an autocrine and paracrine ligand for VEGFR2 (also called KDR), a key mediator of angiogenesis and endothelial cell behavior (27–29). As KRIT1-depleted endothelial cells chronically secrete VEGF-A, we looked to see whether KRIT1-depleted endothelial cells also exhibited increased VEGFR2 activation. VEGFR2 tyrosine phosphorylation was increased ∼2-fold in KRIT1-depleted HPAEC versus negative control-transfected cells, an increase half that seen in control cells treated with 25 ng/ml VEGF (Fig. 2, A–C). This increase could be inhibited in a dose-dependent manner by pretreatment with a recombinant fusion of the VEGFR2 extracellular domain and the Fc portion of human IgG (VEGFR2/Fc). Treatment with 25 ng/ml VEGFR2/Fc returned VEGFR2 phosphorylation (normalized to total VEGFR2) to near background levels, whereas enhanced VEGFR2 phosphorylation was completely blocked at 50 ng/ml (Fig. 2B). The recombinant soluble VEGFR2/Fc chimera binds VEGF with high affinity and is a potent VEGF antagonist (30) but was unable to reverse VEGFR2 phosphorylation below background levels (NC), suggesting that the low level of VEGFR2 phosphorylation seen in control cells is not mediated by paracrine signaling. Interestingly, 25 ng/ml VEGFR2/Fc appeared to reduce both KRIT1 depletion-dependent and VEGF-dependent VEGFR2 phosphorylation to similar levels, although the initial activation was much higher in VEGF-treated cells.
FIGURE 2.

Expression of VEGF by KRIT1-depleted cells activates VEGFR2. A, tyrosine phosphorylation of VEGFR2 in NC- and KRIT1-siRNA-transfected cells treated with 5, 25, or 50 ng/ml VEGFR2/Fc ± rhVEGF (25 ng/ml). IB, immunoblot. Blots are representative, n = 4. B, densitometric quantification of VEGFR2 phosphorylation from four independent experiments treated as in A. Tyr(P) (P-Tyr) signal was normalized to VEGFR2 immunoprecipitation, shown relative to untreated NC-transfected cells, and plotted versus VEGFR2/Fc ± S.E. n = 4 independent experiments. C, densitometric quantification of VEGFR2 tyrosine phosphorylation after VEGF treatment from four independent experiments treated as in A. Tyr(P) signal was normalized to VEGFR2 immunoprecipitation and shown relative to untreated NC-transfected cells ± S.E. p < 0.0001 by ANOVA; *, p < 0.05 by post-hoc testing versus untreated NC-transfected cells. D, expression of Vegfa mRNA in WT, Krit1 null (−/−), and Krit1 reconstituted (9/6) MEF; ±S.E; n = 3; p = 0.008 by ANOVA. *, p < 0.05 by post-hoc test versus WT. E, tyrosine 1175 phosphorylation (pY1175) in WT, −/−, and 9/6 MEF. WB, Western blot. n = 7. F, densitometric quantification of VEGFR2 Tyr(P)-1175 for all experiments treated as in E. Data shown are Tyr(P)-1175 normalized to total VEGFR2, ± S.E; n = 7; p = 0.02 by ANOVA; *, p < 0.05 by post-hoc testing versus WT. G, tyrosine phosphorylation of VEGFR2 in NC siRNA-transfected cells treated with conditioned media from KRIT1-siRNA-transfected cells. CM, conditioned media. Blots are representative, n = 3. IP, immunoprecipitation. H, densitometric quantification of VEGFR2 tyrosine phosphorylation from 3 independent experiments treated as in G. Tyr(P) signal was normalized to VEGFR2 immunoprecipitation and plotted versus time, ±S.E; p = 0.0023 by ANOVA; *, p < 0.01 by post-hoc testing versus untreated.
These data suggest that increased VEGF expression following depletion of KRIT1 is sufficient to activate VEGFR2. However, as these experiments were performed using transient transfection of KRIT1 siRNA, we wondered whether long term KRIT1 deficiency would cause the same effect given that receptor-tyrosine kinases such as VEGFR2 are known to employ feedback mechanisms that down-regulate long term signaling (31). We obtained WT, Krit1 knock-out (−/−), and reconstituted (9/6) MEF cell lines previously characterized by Retta and co-workers (32). These undifferentiated cells expressed modest levels of VEGF-A and VEGFR2 (not shown and Fig. 2D). Krit1−/− MEFs expressed 1.5-fold higher levels of Vegfa mRNA than WT or 9/6 MEF lines (Fig. 2D), which was similar to the increase we observed in KRIT1-depleted endothelial cells (Fig. 1C). Importantly, Krit1−/− MEFs also demonstrated higher VEGFR2 activation, as measured using anti-VEGFR2 phosphotyrosine 1175 antibody (Fig. 2, E and F). We then asked whether secretion of VEGF by KRIT1-depleted cells was sufficient to initiate sustained activation of VEGFR2. Cell culture media on negative control siRNA-transfected cells was replaced with conditioned media from KRIT1 siRNA-transfected cells. VEGFR2 phosphorylation was measured over a time course from 15 min to 4 h after media replacement. Conditioned media from KRIT1-deficient cells stimulated a robust and acute activation of VEGFR2 as shown by tyrosine phosphorylation (Fig. 2G). This activation decreased over time, but notably even 4 h post-exchange, VEGFR2 phosphorylation was still elevated over untreated control (1.57 ± 0.159; Fig. 2H). Thus, these data confirm that chronic VEGF-A overexpression can stimulate sustained VEGFR2 activation in KRIT1-deficient cells.
VEGF Signaling Contributes to KRIT1 Depletion-dependent Changes in Barrier Function, Cytoskeletal Organization, and Migration
KRIT1-deficient endothelial cells exhibit loss of barrier function (13, 16, 33), which may reflect the pro-angiogenic potential of KRIT1 deficient cells in vivo. VEGF is known to increase the permeability of endothelial cells in vitro (Refs. 34–36 and Fig. 3A); however, only under pathological conditions does VEGF/VEGFR2-driven angiogenesis occur with signs of edema or hemorrhaging (1, 37). If KRIT1 depletion-mediated loss of barrier function were dependent on the activation of VEGFR2, we would predict that we could restore barrier function by blocking activation of VEGFR2 at the receptor level. KRIT1 siRNA-transfected endothelial monolayers were treated with 50 ng/ml VEGFR2/Fc for 8 h before performing a transwell endothelial leak assay. Treatment with VEGFR2/Fc blocked the ability of HRP (44 kDa) to cross the endothelial barrier compared with vehicle alone (Fig. 3A). In addition, we could reverse KRIT1-siRNA-mediated permeability by co-transfecting cells with dn-TCF (Fig. 3B), suggesting that β-catenin-dependent VEGF-A expression is required for the observed loss of barrier function.
FIGURE 3.

Loss of KRIT1 leads to VEGF-dependent barrier disruption and migration. A, HRP leak through BAEC monolayers transfected with NC- or KRIT1-siRNA ± 25 ng/ml VEGFR2/Fc or recombinant human VEGF (50 ng/ml). Data shown are the mean HRP concentration, ± S.E. n = 3, p = 0.0005 by ANOVA; *, p < 0.05 by post-hoc testing versus NC-transfected cells; **, p < 0.01 versus NC-transfected cells; ***, p < 0.01 versus KRIT1. B, HRP leak through BAEC monolayers transfected with NC- or KRIT1-siRNA, ± co-expression of dn-TCF. Data shown are the mean HRP concentration ± S.E; n = 3; p = 0.001 by ANOVA; *, p < 0.01 by post-hoc test versus NC; **, p < 0.01 versus KRIT1 siRNA-transfected cells. C, cremaster microvessel permeability in WT or Krit1+/− (+/-) mice treated ± 3 mg/kg SU5416 or with vehicle alone. Data shown are the mean Ps ± S.E. n ≥ 17 vessel sites; p < 0.0001 by non-parametric ANOVA; *, p < 0.001 by post-hoc testing versus WT; **, p < 0.001 versus +/−. D, epifluorescence images of rhodamine-phalloidin-stained NC and KRIT1-siRNA-transfected HPAE-treated ± 25 ng/ml VEGFR2/Fc or 50 ng/ml rhVEGF. Images are representative of three separate experiments; scale bar = 50 μm. E, fluorescent intensity quantification of D. n ≥ 40 cells from 10 fields of view ±S.E. p < 0.0001 by ANOVA; *, p < 0.001 by post-hoc test versus NC-transfected cells; **, p < 0.001 versus KRIT1 siRNA-transfected cells. F, percent wound closure of NC- and KRIT1-siRNA-transfected cells treated ± 25 ng/ml VEGFR2/Fc or 25 ng/ml rhVEGF. Data shown are the mean ± S.E. n = 8; p < 0.0001 by ANOVA; *, p < 0.001 by post-hoc test versus NC-transfected cells; **, p < 0.001 versus KRIT1-siRNA-transfected cells.
As increased VEGF-A expression and VEGFR2 activation was required for increased endothelial monolayer leak after KRIT1 depletion in vitro, we asked whether blocking VEGF signal transduction would also reverse the increase in microvascular permeability observed in Krit1+/− mice in vivo. Because VEGFR2/Fc contains human IgG sequences and would elicit an immune response in our mice, we substituted it with the VEGFR2 small molecule inhibitor SU5416 (38) for our in vivo studies. Using intravital microscopy, we examined microvessel permeability in wild type and Krit1+/− male mice treated with 3 mg/kg SU5416 or vehicle control. SU5416 had no statistical effect on wild type microvessel permeability, although it did trend slightly upward in the presence of the inhibitor (0.6 ± 0.1 versus 0.82 ± 0.07; Fig. 3C). In contrast, SU5416 completely reversed the increase in permeability in Krit1+/− mice (1.54 ± 0.17 versus 0.65 ± 0.07; Fig. 3C). Although vehicle alone exhibited some ability to decrease permeability, the mean was not significantly different from that in untreated heterozygous animals and came nowhere near the amount of inhibition caused by the presence of SU5416 (Fig. 3C). Thus, blocking VEGF signaling is sufficient to prevent the disruption of the endothelial barrier in vitro and in vivo after loss of KRIT1.
Next we examined the number and intensity of stress fibers formed in KRIT1-depleted cells using rhodamine-phalloidin staining of F-actin, which has been linked to the loss of barrier function in KRIT1-depleted cells (13, 16). As previously reported, KRIT1-siRNA transfection significantly increased the proportion of F-actin fluorescence as quantified using Image J. VEGF is also known to elicit cytoskeletal arrangement into stress fibers through Rho/Rho kinase signaling (39). Treatment of control cells with 25 ng/ml VEGF for 10 min recapitulated the actin stress fiber phenotype seen in KRIT1-depleted cells (Fig. 3D). This prompted us to test whether the increased stress fiber formation after loss of KRIT1 is dependent on VEGF. VEGFR2/Fc treatment blocked the increase in stress fiber formation in KRIT1-depleted endothelial cells (Fig. 3, D and E) but had no effect in control cells.
VEGF/VEGFR2 activation is a potent stimulator of endothelial cell migration (Refs. 39–41 and Fig. 3F); thus the activation of this signaling pathway in KRIT1-deficient endothelial cells could promote migration and contribute to the proto-angiogenic phenotype of these cells. However, the effect of KRIT1 knock-out on cell migration remains controversial, as both increased and decreased β1 integrin activation, a major modulator of endothelial migration, have been described in KRIT1-deficient cells (42, 43). We utilized the well characterized in vitro wound-healing assay to examine whether loss of KRIT1 promoted the migration of endothelial cells. Cells were transfected with anti-KRIT1 siRNA 24 and 48 h before wounding, and wound images were taken at time of wounding (0 h) and after 24 h. Control siRNA-transfected endothelial cells migrated to cover ∼40% of the original wound space over the 24-h span (Fig. 3F). However, KRIT1-depleted cells migrated to cover >50% of the wound space, a 25% increase in migration similar to that stimulated by the addition of VEGF (Fig. 3F). VEGFR2/Fc added at the time of wounding reduced migration in KRIT1-siRNA-transfected cells to control levels (Fig. 3F), suggesting that VEGF signaling contributes to the increase in cell migration under these conditions.
VEGFR2 Activation Downstream of KRIT1 Depletion Leads to Increased β-Catenin Phosphorylation
Having established that VEGF signaling contributes to changes in endothelial behavior downstream of loss of KRIT1, we then wanted to explore the underlying molecular mechanism. VEGFR2 activation can disrupt the adherens junction complex by mediating the phosphorylation of adherens junction proteins (36, 44). For example, phosphorylation of β-catenin at Tyr-654 disrupts the interaction of the β-catenin armadillo motifs with the VE-cadherin cytoplasmic domain (45, 46), whereas phosphorylation at Tyr-142 disrupts the β-catenin/α-catenin interaction (47). Similarly, phosphorylation of the VE-cadherin cytoplasmic domain at Tyr-731 or Tyr-658 prevents binding of p120 catenin and β-catenin (48). Thus we hypothesized that VEGFR2 activation could increase phosphorylation of some or all of these residues in KRIT1-depleted cells. KRIT1 siRNA transfection modestly increased the tyrosine phosphorylation of β-catenin on residues Tyr-654 and Tyr-142 to levels comparable to VEGF stimulation (Fig. 4) while eliciting no change in the tyrosine phosphorylation of Tyr-731 and Tyr-658 of VE-cadherin (data not shown). Subsequently, Wessel et al. (49) published that VEGF is unable to increase Tyr(P)-731 in human umbilical vein endothelial cells, suggesting that this residue is not significantly affected by VEGFR2 signaling. Surprisingly, pretreatment with 25 ng/ml VEGFR2/Fc reversed tyrosine phosphorylation of Tyr-654 and Tyr-142 on β-catenin (Fig. 4). As Tyr-654 is phosphorylated by Src downstream of VEGFR2 activation (36, 46), we also treated KRIT1-depleted cells with the Src family kinase inhibitor SU6656 (1 μm). SU6656 was also able to reverse Tyr-654 phosphorylation (Fig. 4, A and B), suggesting that phosphorylation of this residue in KRIT1-depleted cells occurs via an Src-mediated mechanism.
FIGURE 4.

KRIT1 depletion leads to VEGF-dependent tyrosine phosphorylation of β-catenin. A, left panel, tyrosine 654 phosphorylation (pY654) of β-catenin in NC- and KRIT1-siRNA-transfected BAEC-treated ± 25 ng/ml VEGFR2/Fc or ± 1 μm SU6656. IgG, mouse IgG; IP, immunoprecipitation; IB, immunoblot. Right panel, comparison of KRIT1 siRNA-induced and VEGF-induced (50 ng/ml) Tyr-654 phosphorylation. Blots are representative, n = 4. B, densitometric quantification of β-catenin Tyr-654 phosphorylation from four independent experiments treated as in A. Tyr(P)-654 signal was normalized to β-catenin immunoprecipitation and displayed relative to NC-transfected cells, ±S.E. p = 0.002 by ANOVA; *, p < 0.01 by post-hoc testing versus NC-transfected cells; **, p < 0.05 versus KRIT1 siRNA-transfected cells. C, tyrosine 142 phosphorylation (pY142) of β-catenin in NC- and KRIT1-siRNA-transfected BAEC-treated ± 25 ng/ml VEGFR2/Fc or 50 ng/ml rhVEGF. The line indicates removal of intervening lanes. Blots are representative, n = 3. D, densitometric quantification of β-catenin Tyr-142 phosphorylation from three independent experiments treated as in C. The Tyr(P)-142 signal was normalized to β-catenin immunoprecipitation and displayed relative to NC-transfected cells ±S.E. p = 0.0003 by ANOVA; *, p < 0.05 by post-hoc testing versus NC-transfected cells; **, < 0.01 versus NC-transfected cells; ***, p < 0.05 versus KRIT1 siRNA-transfected cells.
VEGFR2 Activation Is Not Sufficient for β-Catenin Dissociation from VE-cadherin
Phosphorylation of β-catenin at Tyr-654 and/or Tyr-142 is commonly thought to disrupt cadherin adhesion complexes (36), although tyrosine phosphorylation of β-catenin can also be observed in intact cell-cell contacts (50). In addition, VEGFR2-mediated RhoA activation is considered to be a key mediator of VEGF-induced junction disassembly (4). Previously, we had shown that loss of KRIT1 destabilized the interaction between β-catenin and VE-cadherin (14); thus we clearly wanted to test the dependence of this effect on VEGF signaling. We examined the association of VE-cadherin and β-catenin in NC and KRIT1 siRNA-transfected cells treated with and without VEGFR2/Fc. As previously shown, KRIT1 depletion reduced the association of β-catenin with VE-cadherin by ∼50%, similar to the decrease observed in VEGF-treated cells (Fig. 5, A and B). This was accompanied by a corresponding increase in nuclear-localized β-catenin (Fig. 5C). VEGFR2/Fc did not significantly rescue the decreased co-immunoprecipitation of β-catenin and VE-cadherin in KRIT1-depleted cells, although the percent of dissociation was slightly lower (Fig. 5, A and B). β-Catenin translocation to the nucleus was also only partially rescued by VEGFR2/Fc treatment (Fig. 5C). Interestingly, inhibition of Rho kinase (ROCK) by H-1152 was also unable to reverse the loss of β-catenin/VE-cadherin association in KRIT1-depleted cells (Fig. 5D) despite the well established requirement for ROCK signaling in KRIT1 depletion-dependent endothelial leak (16). These data suggest that VEGFR2 activation is only partially responsible for the loss of β-catenin from KRIT1-depleted cell-cell contacts and imply that stabilization of β-catenin/VE-cadherin binding may be the key function of KRIT1 at adherens junctions.
FIGURE 5.
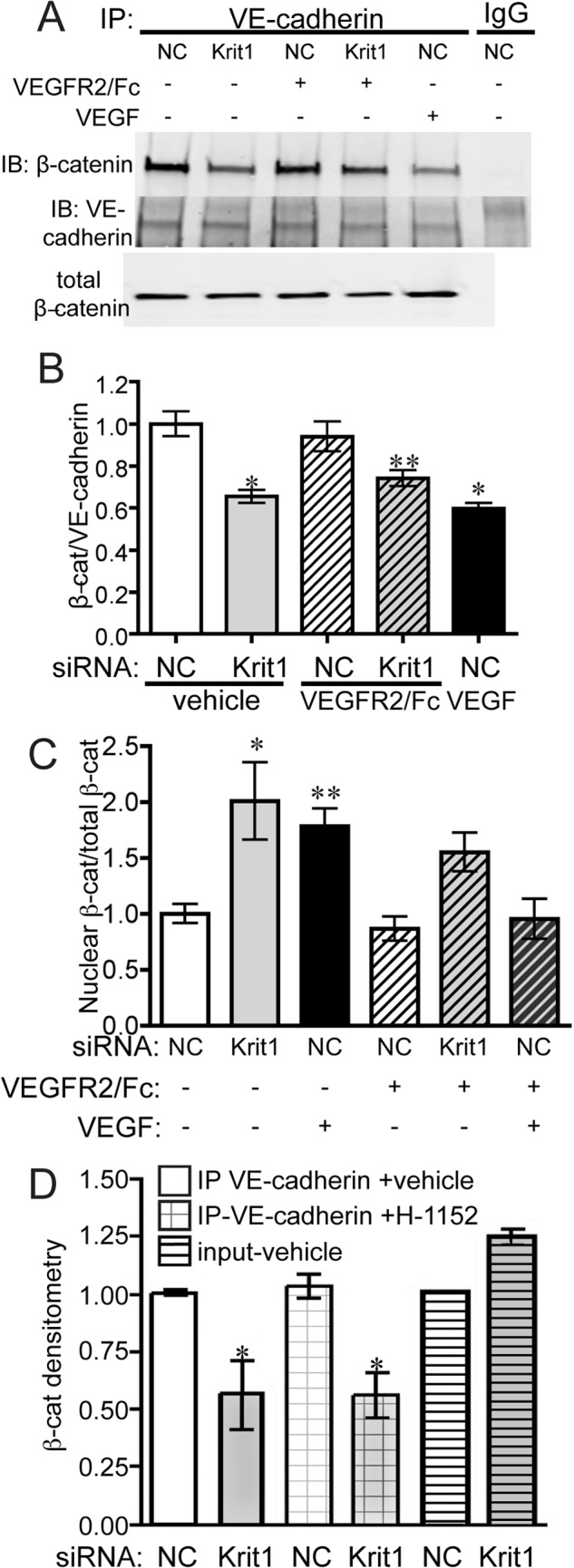
KRIT1 depletion-dependent dissociation of β-catenin and VE-cadherin is not reversed by inhibition of VEGFR2. A, co-immunoprecipitation of VE-cadherin and β-catenin in NC- and KRIT1-siRNA-transfected HPAEC-treated ± 25 ng/ml VEGFR2/Fc or rhVEGF (50 ng/ml). IgG, rabbit IgG; IP, immunoprecipitation; IB, immunoblot. Blots are representative, n = 5. B, densitometric quantification of β-catenin co-immunoprecipitation from five independent experiments treated as in A. Data shown are β-catenin-normalized to VE-cadherin immunoprecipitation ±S.E. p = 0.0004 by ANOVA; *, p < 0.001; **, p < 0.05 by post-hoc testing versus untreated NC-transfected cells. C, densitometric quantification of nuclear β-catenin in NC- and KRIT1-siRNA-transfected HPAEC-treated ± 50 ng/ml VEGFR2/Fc or rhVEGF (50 ng/ml). Nuclear fractions were isolated as described under “Experimental Procedures.” Data shown are nuclear β-catenin-normalized to total β-catenin for each condition from 5 independent experiments. p = 0.0013 by ANOVA; *, p < 0.01; **, p < 0.05 by Dunnett's multiple comparison test versus untreated NC-transfected cells. D, densitometric quantification of β-catenin co-immunoprecipitation with VE-cadherin in NC and KRIT1 siRNA-transfected HPAE treated with 100 nm H-1152. Data are combined from five independent experiments. Data shown are β-catenin-normalized to VE-cadherin immunoprecipitation ±S.E. p = 0.0082 by ANOVA; *, p < 0.05 by post-hoc testing versus vehicle-treated NC-transfected cells.
DISCUSSION
We previously demonstrated that loss of KRIT1 triggers dissolution of the β-catenin-VE-cadherin interaction, leading to increased nuclear β-catenin activity and increased expression of β-catenin-responsive genes, including Vegfa. We now show that the subsequent increase in VEGF signaling plays a key role in modifying endothelial function in KRIT1-deficient cells. The increase in Vegfa mRNA expression translates to an increase in secreted VEGF-A (Fig. 1) that, although modest, is sufficient to increase activation of VEGFR2 in both transiently transfected cells and stable knock-out cell lines (Fig. 2). The resulting activation of VEGFR2 contributes strongly to the loss of barrier function in both KRIT1 siRNA-transfected cells in vitro and in Krit1-deficient mice in vivo (Fig. 3). In addition, inhibition of VEGFR2 activation using VEGFR2/Fc reverses the increase in stress fiber formation commonly observed in CCM. The fact that under these conditions KRIT1 depletion-mediated endothelial functions appears to be VEGF-dependent may indicate that loss of KRIT1 tips cells past a threshold such that VEGFR2 signaling can now alter endothelial behavior. However, VEGF signaling was not sufficient for KRIT1 expression-dependent changes in β-catenin localization, as VEGFR2/Fc was unable to fully restore β-catenin-VE-cadherin association (Fig. 5). Taken together, our data define a new signaling pathway downstream of reduced KRIT1 expression and provide additional support to the idea that under quiescent conditions the main function of KRIT1 is to maintain endothelial cell-cell junctions by promoting the association of β-catenin with VE-cadherin.
Additionally, our study implies that KRIT1 normally acts as a negative regulator of VEGF signaling by limiting nuclear β-catenin. Indeed, it was recently shown that overexpression of KRIT1 limits VEGF-induced tube formation, sprout formation, and endothelial migration (51). Notably, our data indicate that loss of KRIT1 not only releases the limiting influence of KRIT1 expression but leads to VEGFR2 activation independent of exogenous stimulation. The resultant increase in VEGFR2 activation is required for KRIT1 depletion-dependent barrier disruption, migration, and β-catenin phosphorylation. This strongly suggests that activation of VEGFR2 downstream of KRIT1 depletion acts as a feed-forward mechanism to exacerbate the initial effects of loss of cell-contact integrity. Importantly, this demonstrates for the first time that activation of nuclear β-catenin signaling is directly responsible for some of the phenotypes associated with CCM.
The stimulation of VEGF signaling in KRIT1-deficient cells is perhaps not totally unexpected given the known phenotype of KRIT1 deletion. Increased VEGF in vivo leads to a leaky, tortuous vascular network with increased neovascularization, as seen in various transgenic mouse models (18, 20, 21). Similarly, in a Matrigel plug assay, transplantation of KRIT1-deficient cells led to increased vascular density in the plug (51), and leaky vessels are a hallmark of CCM lesions in both mice and humans (52, 53). On the other hand, loss of KRIT1 does not merely trigger downstream signals bearing the hallmarks of the canonical VEGF signaling pathway. Like VEGF stimulation, KRIT1 depletion promotes the activation of ERK and RhoA (16, 51, 54). However, in normal cells activation of Akt in response to VEGF is robust, yet in KRIT1-deficient cells Akt phosphorylation is actually reduced compared with control (51). This discrepancy suggests that although cross-talk with the VEGF pathway is an important consequence of loss of KRIT1, the biochemical and biological effects of loss of this key regulator likely exceed its ability to activate VEGF signaling.
Hereditary CCM has been linked to loss of one of three CCM family proteins (Krit1, CCM2, PDCD10), which vary widely in structure, binding partners, and function. Because of this diversity, it has been difficult to identify a single signaling pathway or biological function essential to CCM pathogenesis. KRIT1 and CCM2, which directly bind to each other, clearly cooperate in at least one biochemical pathway that leads to loss of cell junction integrity and activation of RhoA (16, 54). PDCD10 has been shown to bind to CCM2 in an overexpression system (55), but evidence for an interaction between the two endogenous proteins is lacking, and the activation of RhoA downstream of loss of PDCD10 remains controversial (54, 56). Animal models support an independent role for PDCD10 in the regulation of vascular morphogenesis and integrity. Krit1 and CCM2 knock-out mice are phenotypically similar (57, 58), but PDCD10-deficient mice exhibit more severe developmental vascular defects (59). In zebrafish, PDCD10 deficiency causes heart and circulation effects distinct from those seen in Krit1 and CCM2 mutants (59). Then again, loss of CCM2 or PDCD10, but not KRIT1, activate p38 and Akt (51, 60), challenging the idea that loss of PDCD10 promotes CCM formation via an independent mechanism.
Our data also show for the first time that KRIT1 and PDCD10 can activate at least one common downstream signaling pathway independent of CCM2. Furthermore, we show that loss of KRIT1 increases cell migration, which has been reported in PDCD10-, but not CCM2-, deficient cells (60). The activation of nuclear β-catenin signaling in both Krit1- and PDCD10-depleted endothelial cells led to an increase in Vegfa mRNA. However, it is unlikely that increased VEGF expression would have the same effect on PDCD10-depleted cells that we demonstrate in KRIT1-depleted cells, as it has been reported that PDCD10 is required to stabilize VEGFR2 expression (61). Thus, increased Vegfa expression in PDCD10 mutants could be limited to paracrine effects on the proliferation, migratory behavior, or barrier function of non-endothelial cell types. Nonetheless, β-catenin-dependent gene transcription is not limited to the up-regulation of Vegfa (62), leading to the possibility that KRIT1 and PDCD10 signaling can intersect in other pathways; for example, cell cycle regulation through Ccnd1 up-regulation. Clearly, there is much we still do not understand about the biochemical and cellular functions of the CCM family of proteins; thus, a unifying hypothesis regarding CCM pathogenesis remains elusive.
Finally, our study focused on the contribution of VEGF signaling to changes in endothelial behavior observed in KRIT1 depleted endothelial cells. Our work highlights the role of KRIT1 as a master regulator of endothelial cell-cell junction integrity, which includes modifying the response to growth factors such as VEGF. However, VEGF is a known regulator of pathological angiogenesis (63); thus the suggestion that VEGF/KRIT1 cross-talk could have a function in the formation of CCM is an obvious one. Intermittent reports of increased VEGF expression in lesions or serum of CCM patients (64, 65) correlate nicely with our discovery that Krit1+/− mice have higher serum levels of VEGF-A (15). However, interpretation of these clinical reports is limited by small group size and the inclusion of both sporadic and hereditary CCMs; therefore, the clinical import of VEGF signaling remains unclear.
Based on our study we envision two potential mechanisms by which enhanced VEGF signaling could contribute to CCM. First, increased VEGF/VEGFR2 activation could directly stimulate pathological angiogenesis, leading to CCM formation. This idea does not explain the apparent restriction of lesions to the central nervous system and skin (66) but correlates well with the decrease in barrier function and increase in migration seen in our study. However, Vegfa is not the only gene product affected by increasing nuclear β-catenin, and the complete consequence of nuclear β-catenin signaling on CCM formation is likely to be impacted by changes in other gene transcripts. For example, it has also been shown that loss of KRIT1 increases phosphorylation and activation of c-jun (67), a transcription factor that plays a key role in permeability in vivo (68). Nevertheless, VEGF signaling could potentiate CCM formation and/or increase symptomatology. Even slightly elevated VEGF serum levels could promote a pro-angiogenic environment, so any additional stimulus, whether normal or pathological, would produce a correspondingly amplified response. VEGF is also a potent permeability agent that could exacerbate the leakiness of blood vessels in the lesion, thus increasing risk of hemorrhage, a major cause of patient symptoms. Without additional studies, it remains unclear whether the present Food and Drug Administration-approved anti-VEGF therapies would constitute effective treatments for CCM. Future studies to determine whether inhibition of VEGFR2 signaling is sufficient to block CCM formation in vivo will be an important next step.
This work was supported, in whole or in part, by National Institutes of Health Grant HL117885-01 (to A. J. G.). This work was also supported by American Heart Association Grant 0930071N (to A. J. G.) and American Heart Association Predoctoral Fellowship 14PRE2038009 (to P. V. D.).
- CCM
- cerebral cavernous malformation
- KRIT1
- Krev interaction trapped protein 1
- PDCD10
- programmed cell death 10
- HPAEC
- human pulmonary artery endothelial cells
- BAEC
- bovine aortic endothelial cells
- MEF
- mouse embryonic fibroblast
- VEGFR2
- vascular endothelial growth factor receptor 2
- VEGFR2/Fc
- vascular VEGFR2 extracellular domain fused with Fc portion of human IgG
- dn-TCF
- dominant negative TCF
- NC
- negative control
- TMB
- 3,3′,5,5′-tetramethylbenzidine
- rhVEGF
- recombinant human VEGF
- ANOVA
- analysis of variance
- Ps
- permeability.
REFERENCES
- 1. Wallez Y., Huber P. (2008) Endothelial adherens and tight junctions in vascular homeostasis, inflammation, and angiogenesis. Biochim. Biophys. Acta 1778, 794–809 [DOI] [PubMed] [Google Scholar]
- 2. Orr A. W., Stockton R., Simmers M. B., Sanders J. M., Sarembock I. J., Blackman B. R., Schwartz M. A. (2007) Matrix-specific p21-activated kinase activation regulates vascular permeability in atherogenesis. J. Cell Biol. 176, 719–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mammoto A., Mammoto T., Kanapathipillai M., Wing Yung C., Jiang E., Jiang A., Lofgren K., Gee E. P., Ingber D. E. (2013) Control of lung vascular permeability and endotoxin-induced pulmonary oedema by changes in extracellular matrix mechanics. Nat. Commun. 4, 1759. [DOI] [PubMed] [Google Scholar]
- 4. Lee Y. C. (2005) The involvement of VEGF in endothelial permeability: a target for anti-inflammatory therapy. Curr. Opin. Investig. Drugs 6, 1124–1130 [PubMed] [Google Scholar]
- 5. Middleton J., Americh L., Gayon R., Julien D., Aguilar L., Amalric F., Girard J. P. (2004) Endothelial cell phenotypes in the rheumatoid synovium: activated, angiogenic, apoptotic and leaky. Arthritis Res. Ther. 6, 60–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bryan B. A., D'Amore P. A. (2007) What tangled webs they weave: Rho-GTPase control of angiogenesis. Cell. Mol. Life Sci. 64, 2053–2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pantoni L. (2010) Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol 9, 689–701 [DOI] [PubMed] [Google Scholar]
- 8. Marchuk D. A., Srinivasan S., Squire T. L., Zawistowski J. S. (2003) Vascular morphogenesis: tales of two syndromes. Hum. Mol. Genet. 12, R97–R112 [DOI] [PubMed] [Google Scholar]
- 9. Clatterbuck R. E., Eberhart C. G., Crain B. J., Rigamonti D. (2001) Ultrastructural and immunocytochemical evidence that an incompetent blood-brain barrier is related to the pathophysiology of cavernous malformations. J. Neurol. Neurosurg. Psychiatr. 71, 188–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sahoo T., Johnson E. W., Thomas J. W., Kuehl P. M., Jones T. L., Dokken C. G., Touchman J. W., Gallione C. J., Lee-Lin S. Q., Kosofsky B., Kurth J. H., Louis D. N., Mettler G., Morrison L., Gil-Nagel A., Rich S. S., Zabramski J. M., Boguski M. S., Green E. D., Marchuk D. A. (1999) Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1). Hum. Mol. Genet. 8, 2325–2333 [DOI] [PubMed] [Google Scholar]
- 11. Liquori C. L., Berg M. J., Siegel A. M., Huang E., Zawistowski J. S., Stoffer T., Verlaan D., Balogun F., Hughes L., Leedom T. P., Plummer N. W., Cannella M., Maglione V., Squitieri F., Johnson E. W., Rouleau G. A., Ptacek L., Marchuk D. A. (2003) Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am. J. Hum. Genet. 73, 1459–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bergametti F., Denier C., Labauge P., Arnoult M., Boetto S., Clanet M., Coubes P., Echenne B., Ibrahim R., Irthum B., Jacquet G., Lonjon M., Moreau J. J., Neau J. P., Parker F., Tremoulet M., Tournier-Lasserve E. (2005) Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am. J. Hum. Genet. 76, 42–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Glading A., Han J., Stockton R. A., Ginsberg M. H. (2007) KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions. J. Cell Biol. 179, 247–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Glading A. J., Ginsberg M. H. (2010) Rap1 and its effector KRIT1/CCM1 regulate β-catenin signaling. Dis. Model Mech. 3, 73–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Corr M., Lerman I., Keubel J. M., Ronacher L., Misra R., Lund F., Sarelius I. H., Glading A. J. (2012) Decreased krev interaction-trapped 1 expression leads to increased vascular permeability and modifies inflammatory responses in vivo. Arterioscler. Thromb. Vasc. Biol. 32, 2702–2710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stockton R. A., Shenkar R., Awad I. A., Ginsberg M. H. (2010) Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J. Exp. Med. 207, 881–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Easwaran V., Lee S. H., Inge L., Guo L., Goldbeck C., Garrett E., Wiesmann M., Garcia P. D., Fuller J. H., Chan V., Randazzo F., Gundel R., Warren R. S., Escobedo J., Aukerman S. L., Taylor R. N., Fantl W. J. (2003) β-Catenin regulates vascular endothelial growth factor expression in colon cancer. Cancer Res. 63, 3145–3153 [PubMed] [Google Scholar]
- 18. Spilsbury K., Garrett K. L., Shen W.-Y., Constable I. J., Rakoczy P. E. (2000) Overexpression of vascular endothelial growth factor (VEGF) in the retinal pigment epithelium leads to the development of choroidal neovascularization. Am. J. Pathol. 157, 135–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Larcher F., Murillas R., Bolontrade M., Conti C. J., Jorcano J. L. (1998) VEGF/VPF overexpression in skin of transgenic mice induces angiogenesis, vascular hyperpermeability, and accelerated tumor development. Oncogene 17, 303–311 [DOI] [PubMed] [Google Scholar]
- 20. Wang Y., Jin K., Mao X. O., Xie L., Banwait S., Marti H. H., Greenberg D. A. (2007) VEGF-overexpressing transgenic mice show enhanced post-ischemic neurogenesis and neuromigration. J. Neurosci. Res. 85, 740–747 [DOI] [PubMed] [Google Scholar]
- 21. Veron D., Reidy K. J., Bertuccio C., Teichman J., Villegas G., Jimenez J., Shen W., Kopp J. B., Thomas D. B., Tufro A. (2010) Overexpression of VEGF-A in podocytes of adult mice causes glomerular disease. Kidney Int. 77, 989–999 [DOI] [PubMed] [Google Scholar]
- 22. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 23. Sarelius I. H., Kuebel J. M., Wang J., Huxley V. H. (2006) Macromolecule permeability of in situ and excised rodent skeletal muscle arterioles and venules. Am. J. Physiol. Heart Circ. Physiol. 290, H474–H480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huxley V. H., Curry F. E., Adamson R. H. (1987) Quantitative fluorescence microscopy on single capillaries: α-lactalbumin transport. Am. J. Physiol. 252, H188–H197 [DOI] [PubMed] [Google Scholar]
- 25. Xie H., Pallero M. A., Gupta K., Chang P., Ware M. F., Witke W., Kwiatkowski D. J., Lauffenburger D. A., Murphy-Ullrich J. E., Wells A. (1998) EGF receptor regulation of cell motility: EGF induces disassembly of focal adhesions independently of the motility-associated PLCγ signaling pathway. J. Cell Sci. 111, 615–624 [DOI] [PubMed] [Google Scholar]
- 26. Korinek V., Barker N., Morin P. J., van Wichen D., de Weger R., Kinzler K. W., Vogelstein B., Clevers H. (1997) Constitutive transcriptional activation by a β-catenin-Tcf complex in APC−/− colon carcinoma. Science 275, 1784–1787 [DOI] [PubMed] [Google Scholar]
- 27. Koch S., Tugues S., Li X., Gualandi L., Claesson-Welsh L. (2011) Signal transduction by vascular endothelial growth factor receptors. Biochem. J. 437, 169–183 [DOI] [PubMed] [Google Scholar]
- 28. Eichmann A., Simons M. (2012) VEGF signaling inside vascular endothelial cells and beyond. Curr. Opin. Cell Biol. 24, 188–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Risau W. (1997) Mechanisms of angiogenesis. Nature 386, 671–674 [DOI] [PubMed] [Google Scholar]
- 30. Kaplan J. B., Sridharan L., Zaccardi J. A., Dougher-Vermazen M., Terman B. I. (1997) Characterization of a soluble vascular endothelial growth factor receptor-immunoglobulin chimera. Growth Factors 14, 243–256 [DOI] [PubMed] [Google Scholar]
- 31. Duval M., Bédard-Goulet S., Delisle C., Gratton J.-P. (2003) Vascular endothelial growth factor-dependent down-regulation of Flk-1/KDR involves Cbl-mediated ubiquitination. Consequences on nitric oxide production from endothelial cells. J. Biol. Chem. 278, 20091–20097 [DOI] [PubMed] [Google Scholar]
- 32. Goitre L., Balzac F., Degani S., Degan P., Marchi S., Pinton P., Retta S. F. (2010) KRIT1 regulates the homeostasis of intracellular reactive oxygen species. PLoS ONE 5, e11786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu J. J., Stockton R. A., Gingras A. R., Ablooglu A. J., Han J., Bobkov A. A., Ginsberg M. H. (2011) A mechanism of Rap1-induced stabilization of endothelial cell-cell junctions. Mol. Biol. Cell 22, 2509–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lal B. K., Varma S., Pappas P. J., Hobson R. W., 2nd, Durán W. N. (2001) VEGF increases permeability of the endothelial cell monolayer by activation of PKB/akt, endothelial nitric-oxide synthase, and MAP kinase pathways. Microvasc. Res. 62, 252–262 [DOI] [PubMed] [Google Scholar]
- 35. Esser S., Lampugnani M. G., Corada M., Dejana E., Risau W. (1998) Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J. Cell Sci. 111, 1853–1865 [DOI] [PubMed] [Google Scholar]
- 36. Chen X. L., Nam J.-O., Jean C., Lawson C., Walsh C. T., Goka E., Lim S.-T., Tomar A., Tancioni I., Uryu S., Guan J.-L., Acevedo L. M., Weis S. M., Cheresh D. A., Schlaepfer D. D. (2012) VEGF-induced vascular permeability is mediated by FAK. Dev. Cell 22, 146–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gaengel K., Genové G., Armulik A., Betsholtz C. (2009) Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler. Thromb. Vasc. Biol. 29, 630–638 [DOI] [PubMed] [Google Scholar]
- 38. Tong R. T., Boucher Y., Kozin S. V., Winkler F., Hicklin D. J., Jain R. K. (2004) Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 64, 3731–3736 [DOI] [PubMed] [Google Scholar]
- 39. van Nieuw Amerongen G. P. (2003) Involvement of RhoA/Rho kinase signaling in VEGF-induced endothelial cell migration and angiogenesis in vitro. Arterioscler. Thromb. Vasc. Biol. 23, 211–217 [DOI] [PubMed] [Google Scholar]
- 40. Bernatchez P. N., Soker S., Sirois M. G. (1999) Vascular endothelial growth factor effect on endothelial cell proliferation, migration, and platelet-activating factor synthesis is Flk-1-dependent. J. Biol. Chem. 274, 31047–31054 [DOI] [PubMed] [Google Scholar]
- 41. Zeng H., Sanyal S., Mukhopadhyay D. (2001) Tyrosine residues 951 and 1059 of vascular endothelial growth factor receptor-2 (KDR) are essential for vascular permeability factor/vascular endothelial growth factor-induced endothelium migration and proliferation, respectively. J. Biol. Chem. 276, 32714–32719 [DOI] [PubMed] [Google Scholar]
- 42. Liu W., Draheim K. M., Zhang R., Calderwood D. A., Boggon T. J. (2013) Mechanism for KRIT1 release of ICAP1-mediated suppression of integrin activation. Mol. Cell 49, 719–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Faurobert E., Rome C., Lisowska J., Manet-Dupé S., Boulday G., Malbouyres M., Balland M., Bouin A.-P., Kéramidas M., Bouvard D., Coll J.-L., Ruggiero F., Tournier-Lasserve E., Albiges-Rizo C. (2013) CCM1-ICAP-1 complex controls β1 integrin-dependent endothelial contractility and fibronectin remodeling. J. Cell Biol. 202, 545–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Monaghan-Benson E., Burridge K. (2009) The regulation of vascular endothelial growth factor-induced microvascular permeability requires Rac and reactive oxygen species. J. Biol. Chem. 284, 25602–25611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Huber A. H., Weis W. I. (2001) The structure of the β-catenin/E-cadherin complex and the molecular basis of diverse ligand recognition by β-catenin. Cell 105, 391–402 [DOI] [PubMed] [Google Scholar]
- 46. Roura S., Miravet S., Piedra J., García de Herreros A., Duñach M. (1999) Regulation of E-cadherin/catenin association by tyrosine phosphorylation. J. Biol. Chem. 274, 36734–36740 [DOI] [PubMed] [Google Scholar]
- 47. Piedra J., Miravet S., Castaño J., Pálmer H. G., Heisterkamp N., García de Herreros A., Duñach M. (2003) p120 Catenin-associated Fer and Fyn tyrosine kinases regulate β-catenin Tyr-142 phosphorylation and β-catenin-α-catenin interaction. Mol. Cell. Biol. 23, 2287–2297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Potter M. D., Barbero S., Cheresh D. A. (2005) Tyrosine phosphorylation of VE-cadherin prevents binding of p120-and β-catenin and maintains the cellular mesenchymal state. J. Biol. Chem. 280, 31906–31912 [DOI] [PubMed] [Google Scholar]
- 49. Wessel F., Winderlich M., Holm M., Frye M., Rivera-Galdos R., Vockel M., Linnepe R., Ipe U., Stadtmann A., Zarbock A., Nottebaum A. F., Vestweber D. (2014) Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-cadherin. Nat. Immunol. 15, 223–230 [DOI] [PubMed] [Google Scholar]
- 50. Lampugnani M. G., Corada M., Andriopoulou P., Esser S., Risau W., Dejana E. (1997) Cell confluence regulates tyrosine phosphorylation of adherens junction components in endothelial cells. J. Cell Sci. 110, 2065–2077 [DOI] [PubMed] [Google Scholar]
- 51. Wüstehube J., Bartol A., Liebler S. S., Brütsch R., Zhu Y., Felbor U., Sure U., Augustin H. G., Fischer A. (2010) Cerebral cavernous malformation protein CCM1 inhibits sprouting angiogenesis by activating DELTA-NOTCH signaling. Proc. Natl. Acad. Sci. U.S.A. 107, 12640–12645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Maddaluno L., Rudini N., Cuttano R., Bravi L., Giampietro C., Corada M., Ferrarini L., Orsenigo F., Papa E., Boulday G., Tournier-Lasserve E., Chapon F., Richichi C., Retta S. F., Lampugnani M. G., Dejana E. (2013) EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature 498, 492–496 [DOI] [PubMed] [Google Scholar]
- 53. Gault J., Shenkar R., Recksiek P., Awad I. A. (2005) Biallelic somatic and germ line CCM1 truncating mutations in a cerebral cavernous malformation lesion. Stroke 36, 872–874 [DOI] [PubMed] [Google Scholar]
- 54. Borikova A. L., Dibble C. F., Sciaky N., Welch C. M., Abell A. N., Bencharit S., Johnson G. L. (2010) Rho kinase inhibition rescues the endothelial cell cerebral cavernous malformation phenotype. J. Biol. Chem. 285, 11760–11764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hilder T. L., Malone M. H., Bencharit S., Colicelli J., Haystead T. A., Johnson G. L., Wu C. C. (2007) Proteomic identification of the cerebral cavernous malformation signaling complex. J. Proteome Res. 6, 4343–4355 [DOI] [PubMed] [Google Scholar]
- 56. Chan A. C., Drakos S. G., Ruiz O. E., Smith A. C., Gibson C. C., Ling J., Passi S. F., Stratman A. N., Sacharidou A., Revelo M. P., Grossmann A. H., Diakos N. A., Davis G. E., Metzstein M. M., Whitehead K. J., Li D. Y. (2011) Mutations in 2 distinct genetic pathways result in cerebral cavernous malformations in mice. J. Clin. Invest. 121, 1871–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Whitehead K. J., Plummer N. W., Adams J. A., Marchuk D. A., Li D. Y. (2004) Ccm1 is required for arterial morphogenesis: implications for the etiology of human cavernous malformations. Development 131, 1437–1448 [DOI] [PubMed] [Google Scholar]
- 58. Whitehead K. J., Chan A. C., Navankasattusas S., Koh W., London N. R., Ling J., Mayo A. H., Drakos S. G., Jones C. A., Zhu W. (2009) The cerebral cavernous malformation signaling pathway promotes vascular integrity via Rho GTPases. Nat. Med. 15, 177–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yoruk B., Gillers B. S., Chi N. C., Scott I. C. (2012) Ccm3 functions in a manner distinct from Ccm1 and Ccm2 in a zebrafish model of CCM vascular disease. Dev. Biol. 362, 121–131 [DOI] [PubMed] [Google Scholar]
- 60. Zhu Y., Wu Q., Xu J.-F., Miller D., Sandalcioglu I. E., Zhang J.-M., Sure U. (2010) Differential angiogenesis function of CCM2 and CCM3 in cerebral cavernous malformations. Neurosurg. Focus 29, E1. [DOI] [PubMed] [Google Scholar]
- 61. He Y., Zhang H., Yu L., Gunel M., Boggon T. J., Chen H., Min W. (2010) Cerebral cavernous malformation gene CCM3 is critical for vascular development by regulating VEGFR2 signaling. Sci. Signal. 3, ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Giles R. H., van Es J. H., Clevers H. (2003) Caught up in a Wnt storm: Wnt signaling in cancer. Biochim. Biophys. Acta 1653, 1–24 [DOI] [PubMed] [Google Scholar]
- 63. Takahashi H., Shibuya M. (2005) The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin. Sci. 109, 227–241 [DOI] [PubMed] [Google Scholar]
- 64. Jung K.-H., Chu K., Jeong S.-W., Park H.-K., Bae H.-J., Yoon B.-W. (2003) Cerebral cavernous malformations with dynamic and progressive course: correlation study with vascular endothelial growth factor. Arch. Neurol. 60, 1613–1618 [DOI] [PubMed] [Google Scholar]
- 65. Abe T., Morishige M., Ooba H., Kamida T., Fujiki M., Kobayashi H., Sakoda T., Kimba Y. (2009) The association between high VEGF levels and multiple probable punctuate cavernous malformations. Acta Neurochir. 151, 855–859 [DOI] [PubMed] [Google Scholar]
- 66. Fischer A., Zalvide J., Faurobert E., Albiges-Rizo C., Tournier-Lasserve E. (2013) Cerebral cavernous malformations: from CCM genes to endothelial cell homeostasis. Trends Mol. Med. 19, 302–308 [DOI] [PubMed] [Google Scholar]
- 67. Goitre L., De Luca E., Braggion S., Trapani E., Guglielmotto M., Biasi F., Forni M., Moglia A., Trabalzini L., Retta S. F. (2014) KRIT1 loss of function causes a ROS-dependent upregulation of c-Jun. Free Radic. Biol. Med. 68, 134–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Fahmy R. G., Waldman A., Zhang G., Mitchell A., Tedla N., Cai H., Geczy C. R., Chesterman C. N., Perry M., Khachigian L. M. (2006) Suppression of vascular permeability and inflammation by targeting of the transcription factor c-Jun. Nat. Biotechnol. 24, 856–863 [DOI] [PubMed] [Google Scholar]