

Abstract
Natural proteins can be versatile building blocks for multimeric, self-assembling structures. Yet, creating protein-based assemblies with specific geometries and chemical properties remains challenging. Highly porous materials represent particularly interesting targets for designed assembly. Here we utilize a strategy of fusing two natural protein oligomers using a continuous alpha-helical linker to design a novel protein that self assembles into a 750 kDa, 225 Å diameter, cube-shaped cage with large openings into a 130 Å diameter inner cavity. A crystal structure of the cage showed atomic level agreement with the designed model, while electron microscopy, native mass spectrometry, and small angle x-ray scattering revealed alternate assembly forms in solution. These studies show that accurate design of large porous assemblies with specific shapes is feasible, while further specificity improvements will likely require limiting flexibility to select against alternative forms. These results provide a foundation for the design of advanced materials with applications in bionanotechnology, nanomedicine and material sciences.
Keywords: Porous protein nanoparticles, protein design, self-assembly, symmetry, synthetic biology and bioengineering
Large and diverse supramolecular assemblies can arise from repeated interactions between multiple copies of a single molecular building block1. Many natural proteins have acquired self-assembling properties during evolution, thereby endowing their hosts with novel functionalities and increased fitness2. Such naturally occurring protein assemblies have not only been extensively studied for their native functions, but have also been widely exploited for synthetic functionalities3, 4, 5. Various natural protein assemblies, such as ferritins, bacterial S-layers and virus capsids, have for some time been points of focus for developments in nanotechnology6. However, engineering de novo protein assemblies with similar forms and pre-determined capabilities has been difficult until very recently7, 8, 9, 10, 11, 12.
One method to engineer large self-assembling protein structures, articulated several years ago13, is by creating a fusion of two different oligomeric proteins arranged in a particular orientation. In the simplest scenario, fusing a dimeric domain to a trimeric domain brings together two symmetry elements (e.g. a 2-fold and a 3-fold axis of symmetry) whose repeated application leads to large, highly symmetric assemblies. Diverse symmetries and architectures are possible, depending on the specific geometric arrangement between the two symmetry axes. A solution to the problem of predictably orienting the separate oligomeric domains is to use a short alpha-helical linker to join the two protein components, which are themselves required to have alpha helical termini so that an unbroken helix spans the two parts. Variations on that linking strategy have also been described14. So far, there are two literature reports demonstrating the production of fusion-based materials that are sufficiently well-ordered to establish design success in atomic or near-atomic detail. These include a 12-subunit tetrahedral cage15, 16 and extended arrays of molecules14. A distinct approach for designing self-assembling protein cages or shells has been developed recently by King et al.17, 18, wherein computational amino acid sequence design is used to create novel interfaces between naturally oligomeric proteins. In contrast to that method, the oligomer fusion approach relies entirely on natural protein interfaces, thereby avoiding the challenging problem of designing new protein-protein interfaces. Here we sought to extend the oligomer fusion method to the construction of larger protein assemblies than have been achieved to date. Specifically, we undertook work to engineer a highly porous, 24-subunit protein cage in the shape of a cube, with perfect octahedral symmetry.
Porosity is an attractive feature of self-assembled supramolecules19. The design and engineering of molecular materials with high porosity has been actively pursued by organic and inorganic chemists. Porous materials, such as those exemplified by metal-organic frameworks (MOFs) have been widely studied20; they have been explored for applications in gas storage, molecular separations, and drug delivery21. The largest diameter openings reported so far in MOF's are on the order of 100 Å20. Biological macromolecules provide alternate strategies. A DNA-based cage in the shape of a cube, with a diameter somewhat smaller than 100 Å, was reported more than two decades ago22 and became an iconic figure for the structural DNA nanotechnology field23. An octahedrally shaped DNA cage with an internal cavity of 140 Å was reported nearly ten years ago24. Very large DNA cubes and polyhedra, with opening in the range of several hundred nanometers, have been realized recently25, after some thirty years of development in the field. Small RNA cubes were designed very recently and shown to form particles of the intended size26. Open cages based on polypeptides or proteins have recently been demonstrated by the oligomer-fusion method15, 16, by the design of novel subunit interfaces17, by the design of two-component mixtures18, and using coiled-coil peptide sequences10 or peptide-inspired foldamers27. In the case of molecular cages designed from protein subunits, the largest internal openings reported so far are around 45 Å15, 17, which is comparable in size to a single protein molecule of intermediate size. Protein assemblies with larger openings could be useful as frameworks for assisted protein crystallization, as reaction vessels28, or as vehicles for delivering large cargos29, as well as agents for nanomedicine30.
Here we present the design, engineering and structural characterization of a protein cage, in the shape of a cube, with openings of about 100 Å to a central cavity of 130 Å in diameter. Its outside diameter (225 Å) makes it the largest protein assembly to date that has been designed and validated in atomic detail.
Results and Discussion
Design and construction of protein cage ATC-HL3
To design a porous cube-shaped protein assembly, we employed the helix-based oligomer fusion strategy13 to computationally model a series of possible protein cages, each intended to assemble from 24 identical subunits arranged in cubic (octahedral) symmetry, and having central cavities of various diameters. These computational constructions were all fusions between various known dimeric and trimeric protein domains, selected according to the criterion that, when joined end-wise by a continuous alpha helical linker, their symmetry axes would obey a particular geometric requirement; in order to form a 24-subunit cube, their 2-fold and 3-fold symmetry axes were required to intersect at an angle nearly equal to 35.3° (the angle between a face diagonal and a body diagonal of a cube). One of these computationally constructed models was taken forward for experimental testing and is described here. The designed protein molecule is a genetic fusion of the trimeric E. coli 2-keto-3-deoxy-6-phosphogalactonate (KDPGal) aldolase31 (PDB ID: 2V82) and the dimeric N-terminal domain of E. coli FkpA protein32 (PDB ID: 1Q6U). The KDPGal aldolase constitutes the N-terminal domain of the designed fusion protein, while the N-terminal domain of the native FkpA protein constitutes the C-terminal domain of the designed fusion protein (Fig. 1). To achieve the geometry required for a cubic cage with octahedral symmetry (i.e. 2-fold and 3-fold symmetry axes intersecting at an angle of 35.3°), the design protocol determined that the requirement would be closely matched by linking these two domains with a four-residue alpha-helical linker. In the computed model, the angle between the trimeric and dimeric axes (of the N-terminal and the C-terminal domains of the fusion protein, respectively) was 36.5° (within about 1° of the ideal angle), and the axes were within 0.3 Å of intersecting. For the sake of simplicity, this fusion system will be referred to as the ATC cage (Aldolase-Three-helix Cubic protein cage).
Figure 1. Models of the engineered fusion protein and its assembled cage structure.
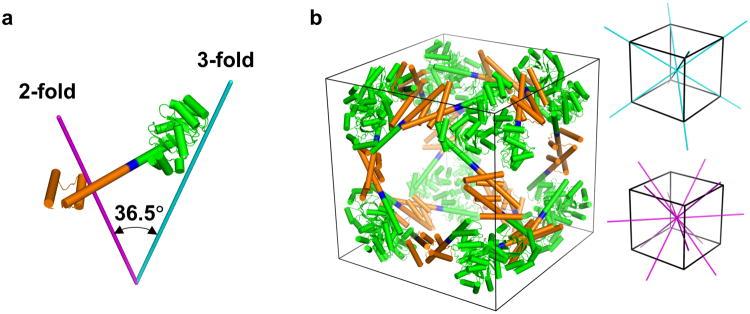
a, The designed fusion protein, with trimeric KDPGal aldolase (green), the 4-residue helical linker (blue), and the dimeric domain of FkpA protein (orange) shown with lines for the 3-fold (cyan) and 2-fold (magenta) symmetry axes. b, A model of the intended 24 subunit cage with octahedral symmetry in a bounding box (left). The three-fold symmetry axes (cyan) and two-fold symmetry axes (magenta) of a cube are shown on the right.
Five minor variations on the ATC cage were constructed with different helix linker sequences. The best linker sequence for the fusion protein, as judged by size exclusion chromatography and native polyacrylamide gel electrophoresis (PAGE), was used for further characterization (see Supplementary Information). The best construct, referred to here as ATC-HL3 (ATC cage with Helix-Linker version 3), was judged more promising than other variants, yet it nonetheless showed properties deemed potentially problematic for crystallization. The size-exclusion profile showed a somewhat asymmetric peak, and the main band on a native PAGE gel was diffuse (see Supplementary Fig. S3). Indeed, initial crystallization trials gave only small crystals in several conditions (see Supplementary Table S2). However, large single crystals suitable for X-ray diffraction studies were eventually obtained.
Crystal structure analysis of the ATC-HL3 cage
After a prolonged incubation time (six months to a year), we observed single crystals large enough to enable structure determination by X-ray crystallography (see Supplementary Fig. S5). Similar crystals were observed in different batches of crystallization trials, indicating that the crystallization of ATC-HL3 could be reproduced given sufficient time. The crystals yielded X-ray diffraction data only to 7 Å resolution – this was not unexpected in view of the necessarily high solvent content of the designed structure33 –yet the diffraction dataset was sufficient for an unequivocal molecular replacement solution and analysis. The limited resolution did not permit a detailed atomic refinement, but this was unnecessary in establishing the success of the design. The crystals grew in cubic space group F23, with four cube-shaped cages packed in the unit cell (Fig. 2a). Two copies of the protein subunit are contained in the asymmetric unit of the crystal, and a molecular replacement computer program automatically placed these two subunits within the unit cell in such a way that the crystal symmetry generated a complete 24-subunit cube as designed. The automatic placement of multiple subunits in a configuration that matched the design was a strong indication that the structure determination was correct, but owing to the relatively low resolution of the observed diffraction data, care was taken to validate the determined structure (see Supplementary Results and Figs. S6-S8). Calculations were performed to assess the precision with which the position of the protein molecules could be defined, and the estimated uncertainty was determined to be about 1 Å (Supplementary Fig. S7).
Figure 2. Crystal structure of the designed cubic cage ATC-HL3.
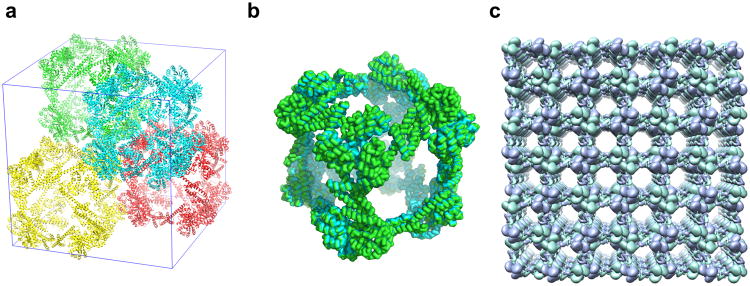
a, Four complete ATC-HL3 cages, colored differently, are packed within a unit cell (shown in blue lines). b, The cube-shaped cage observed in the crystal matches the design with high accuracy. The crystal structure is shown in green ribbon and the intended design is shown in cyan ribbon; the two are nearly perfectly overlapping. c, The packing alignment of cages in the crystal produces a highly porous protein lattice; a 3×3 block of unit cells is shown. The two independent protein chains in the asymmetric unit are colored differently (purple or green).
The cubic cage observed in the crystal structure matches the computer design with exceptionally high accuracy. The root-mean-square deviation is only 1.2 Å for all 6,480 alpha carbon atoms of the twenty-four chains in a complete assembly (Fig. 2b). The cage has nearly perfect 24-fold octahedral symmetry, and it sits on a point of tetrahedral (23) point symmetry in the crystal. In accordance with the design, the outer diameter of the cage is 225 Å and the interior cavity is 132 Å in diameter (see Supplementary Fig. S9); these parameters are comparable to the first DNA-based octahedron24. This interior diameter is considerably larger than has been achieved to date for any other designed protein cage structures (see Supplementary Fig. S10).
The structure of an individual cage is highly porous, with openings of around 100 Å as dictated by the design, and this porosity is extended further in the crystal owing to the way the cages are packed together. Adjacent cages contact each other along their edges (Fig. 2a). This gives rise to large voids in the crystal that are nearly as large as one entire cage. The orientation of the cages along principle directions of the crystal gives rise to large open windows that are aligned throughout the length of the specimen (Fig. 2c). The solvent content of this crystal form is ∼82%. Only 0.16% of all structures in the Protein Data Bank34 have such a high solvent content. Porous organic molecules have value for multiple applications21. Porous protein molecules have potential for similar yet distinct applications, such as encapsulating other protein molecules, which are too big to be encapsulated by small-molecule cages. Such highly-porous protein frameworks could also be used for assisted crystallization and X-ray analysis35. However, if this were to be done by directly attaching a guest molecule to the protein subunit, then symmetry dictates that multiple copies of the guest molecule would have to fit within an individual void space of the crystal.
Biophysical studies on the designed cage
Although the crystal structure of the ATC-HL3 cage closely matches the design, we sought to use biophysical approaches to understand the potential causes for the long time required for crystallization. First, to determine whether discrete heterogeneous assemblies were present in solution, native mass-spectrometry analysis36 was carried out. The spectra showed that, in addition to a small trimeric species, three major assembly species corresponding to 12-mer, 18-mer and 24-mer, were present in solution (Fig. 3a). It is notable that the alternate species differ from the full 24-mer cube by multiples of six subunits. Only those cases are capable of simultaneously satisfying all the natural interfaces of the dimer and trimer components. Because six is the lowest common multiple of two and three, partial structures not composed of multiples of six subunits would have to contain unsatisfied interfaces, and no such structures (larger than an individual trimer) are observed. The dominance of assembly forms that are ‘closed’, in the sense of having all their binding interfaces satisfied, signals the importance of cooperativity in the assembly studied here.
Figure 3. Native mass spectrometry of the ATC-HL3 protein cage.

a, The native mass spectrum (top) indicated three major assembly forms composed from 12, 18, and 24 subunits. The sizes of these three major assembly species were confirmed by a tandem mass spectrometry analysis (below) in which the masses were examined after stripping one subunit from each assembly form to give (n-1) subunits. A trimeric species was also evident in the native mass spectrum (top). b, Hypothetical models are shown of the three major assembly forms observed: atetrahedron (top), a triangular prism (middle), and the cube (bottom). The cube form shown is the intended computation design, while the two smaller forms were constructed by rigid body positioning of dimers and trimers allowed by minor bending at the linkers between protein domains. Trimers occupying different vertices are color-coded differently.
The 24-mer observed by native mass spectrometry must correspond to the designed cube, whose structure was validated by crystallography. We propose hypothetical models of the 18-mer and the 12-mer in which all of the dimeric and trimeric interfaces are satisfied. While the designed 24-mer is composed of 8 trimers at the vertices of a cube, our model of the 18-mer places six trimers at the vertices of a triangular prism, while the 12-mer is a tetrahedron with 4 trimers at the vertices (Fig. 3b). Under this scenario, different assemblies arise due to the intrinsic flexibility of the helix linkers between the trimeric and dimeric components. Flexibility of helix linkers, resulting in heterogeneity, has been observed before in another smaller helix-based oligomeric-fusion system15, 16. According to the mass spectrometry results here, if we account for detector efficiency and assume that the ionization probabilities of the different species are similar, the population ratios of the 12-mer, 18-mer and 24-mer (in 200 mM ammonium acetate and pH 7.3) are approximately 10%, 50% and 40% by weight.
To further test the assembly of the ATC-HL3 fusion protein, we carried out electron microscopy analysis. Negative-stain electron micrographs showed particle sizes in the range of 10-20 nm (Fig. 4a), consistent with the mass spectrometry results, and with an upper size limit in agreement with the designed structure. Heterogeneous forms were evident. 2D class averages resulted in both square and triangular projections consistent with the presence of a few geometrically different assemblies (Fig. 4b and Supplementary Fig. S11). The square projections are consistent with the intended cube-shaped design, or possibly with side views of the 18-mer. The triangular images likely reflect views of the 12-mer as well as top views of the 18-mer. Although definitive shape assignments were not pursued, the class averaging from negatively stained protein particles accords with the data from native mass spectrometry regarding the presence of a small number of geometrically distinct species in solution.
Figure 4. Negatively stained transmission electron microscopy of the ATC-HL3 cage.
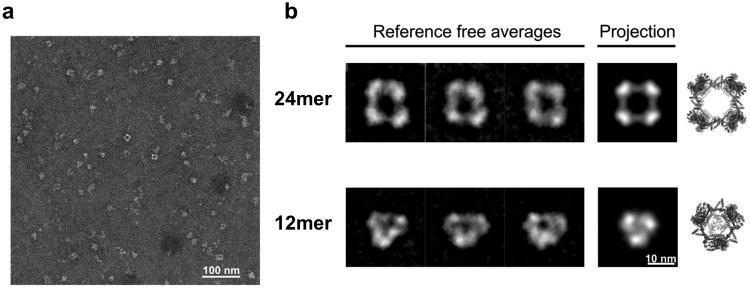
a, A wide field view showing assembled protein particles preserved in stain. b, Examples of 2D class averages (left) obtained after aligning, clustering, and averaging similar particle images. For comparison to what would be expected for EM images of the designed protein assembly, calculated projections (middle) are shown of models of the 24-subunit cube assembly (the intended design) and an alternate 12-subunit tetrahedral assembly (both calculated using a 4 nm low-pass image filter). Three dimensional atomic models are also shown (right).
Lastly, to probe the multimeric solution structure of ATC-HL3 further, we carried out small-angle X-ray scattering (SAXS)37 in various solution conditions. We focused on conditions similar to those in which crystals formed (see Methods). In conditions with well-defined Guinier regions, SAXS profiles corresponded to average radii of gyration (Rg) ranging from 70 Å to 81 Å relative to 92 Å calculated from the crystal structure of the cube (Fig. 5a and supplementary Table S4). The SAXS data showed oscillations about a q4 decay (indicative of globular particles with limited heterogeneity), but the oscillations were not nearly as dramatic as those calculated38 from the crystal structure (Supplementary Material). The SAXS profiles calculated from the idealized models of the 12-mer and 18-mer likewise showed sharp features not reflected in the observed data. Indeed it was not possible to obtain a good fit to the experimental data with those models. We determined that the sharp features in the calculated SAXS profiles were characteristic of highly symmetric assemblies. We therefore created slightly flexed (and less perfectly symmetric) versions of our assembly models using normal mode analysis of an elastic network model (Supplementary Material). By using a collection of flexed models for the 24-mer, 18-mer, and 12-mer, we were able to obtain an excellent fit to the observed scattering curve (Fig. 5 & Supplementary Fig. S14). According to the SAXS fitting, the weight percentages of the trimer, 12-mer, 18-mer, and 24-mer were about 17%, 35%, 17%, 31%, with estimated uncertainties of about 10%. Those estimates differ from those from native mass spectrometry, but both methods are approximations, and the solution conditions for the two experiments were dissimilar; the SAXS data that could be best fit by the model structures was from a sample in 600 mM ammonium sulfate and 1% PEG 400. Notably, the consistent conclusion from all the solution methods was that a few geometrically distinct species are present in solution.
Figure 5. SAXS profile of ATC-HL3.
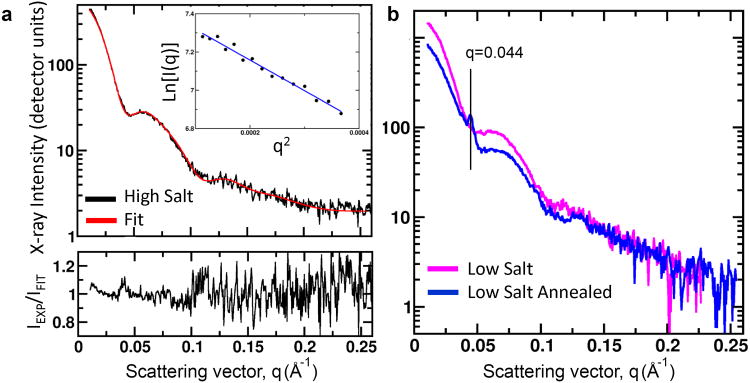
a, The observed small angle X-ray scattering (SAXS) profile is shown (black) for the designed protein (in 600 mM ammonium sulfate), along with a weighted sum of SAXS profiles calculated from atomic models of the designed (24-mer) cube-shaped assembly and the 18-mer and 12-mer assemblies, all in their flexed forms. The Guinier region is shown in the inset; its near-linearity indicating the absence of other aggregated forms of the protein. A residual error plot (bottom), expressed as a ratio of the observed:calculated scattering intensity (with ideal value of 1) is shown at the bottom. The close agreement between the calculated and observed profiles supports the modeling of the assembly in solution as a mixture of the 12-mer, 18-mer, and 24-mer assembly forms. b, Microcrystal peaks observed on the SAXS profile after temperature annealing of a low salt (50 mM ammonium sulfate) condition. The scattering angle of the dominant peak (q=0.044 Å-1) corresponds to the first diffraction peak in the crystal form obtained for the 24-subunit cube (Supplementary Material).
In further SAXS experiments, we probed the effects of solution conditions and thermal annealing on assembly. Samples were heated to 45°C and slowly cooled to 4°C over an 8-hour period. Interestingly, powder rings appeared under solution conditions similar to those that produced single crystals (50 mM MES pH 6.0 and 50 mM ammonium sulfate), indicating the formation of microcrystals. The positions of these peaks in the scattering profile were consistent with strong, low resolution reflections expected for diffraction from the crystal form used for structure determination (Fig. 5b and Supplementary Fig. S16). However, the Rg of the SAXS profile decreased and the overall profile lost sharpness in oscillations, indicating an overall loss of large ordered structures from the solution. Therefore, while nucleation of crystals was promoted by thermal annealing, the overall assembly mixture was not evidently shifted towards the cube-shaped form in solution. In an attempt to drive the protein assembly more fully to the intended cube-shaped structure, we also attempted to partially unfold and then refold and reassemble the protein. We performed denaturation/renaturation experiments using various concentrations of urea, including in the presence of reagents that promote alpha helix formation (e.g. trifluoroethanol and trimethylamine N-oxide), with the view that the helix linker might be the primary source of instability. However, those experiments all produced lesser amounts of large regular assemblies as judged by SAXS and native mass spectrometry; large assemblies could not be recovered in high yield after disassembly.
Discussion
The crystal structure of the 24-subunit cage validated our computational design in atomic detail, thereby supporting the utility of the oligomeric fusion approach for creating large assemblies. The close agreement with the design suggests that the energy landscape for the assembly has a sharp minimum for the intended configuration. However, all of the solution studies provide evidence for a complex energy landscape with minima for other states, while also shedding light on elements of dynamic behavior and routes of assembly.
The native mass spectrometry data show that trimers are present in the sample at a small but detectable concentration. This points to a likely hierarchical assembly process in which monomeric ATC-HL3 subunits, once produced in E. coli, assemble first into trimers, which then serve as building blocks for forming the higher-order assemblies: the 12-mer, 18-mer and 24-mer. By virtue of our design, the 24-mer should have a lower energy than the 18-mer or 12-mer. Yet the 12-mer and 18-mer are well-populated in solution. If the system represents an equilibrium situation, then the formation of alternate species reflects a balance between the cost of slightly higher helix bending energies and the favorable entropy associated with partitioning into different assembly forms. This would of course be affected by overall concentration. The samples showed a tendency to form – presumably by rearrangement – much larger aggregates when concentrated (data not shown), suggesting the ability to re-equilibrate between assembly states. On the other hand, SAXS studies did not show a strong dependence on concentration, though different solution conditions did indicate changes in average radius of gyration and mass (Supplementary Table S4). Our inability to identify conditions that might give the intended 24-mer cube as a single dominant species suggests the possibility that the assembly outcome might be limited by kinetic effects. By necessity the route to the 24-mer must proceed through smaller species, some of which we show here are capable (owing to flexibility) to form structures in which all the natural protein interfaces are satisfied. Further studies will be required to analyze whether different assembly forms are able to equilibrate or whether the alternate (geometrically non-ideal) assembly forms represent kinetically trapped states.
Regardless of the thermodynamic or kinetic basis, the formation of alternate structures raises an important challenge in the design strategy advanced here. As noted earlier, the oligomer fusion approach makes it possible to realize sophisticated assembly architectures without having to redesign new interfaces into proteins, but this comes at the cost of structural flexibility, which permits alternate assembly states. In the present case our modeling shows that only 15° or 19° bending motions are needed to allow the designed subunit to form the 18-mer or 12-mer structures instead of the intended cube (Supplementary Material). The occurrence here of polymorphic assemblies due to small angular differences recalls among natural proteins the case of clathrin coats. In vitro, clathrin can assemble into several regular shapes, including mini-coats (78-mer), hexagonal barrels (108-mer) and icosahedrally symmetric soccer balls (180-mer). There it was found that a bending of only 8° can lead to different assemblies39. Thus synthetic protein assemblies of the type described here could serve as surrogate systems to further our understanding of natural heterogeneous assemblies.
Looking forward, various strategic variations might allow for greater rigidity and control over assembly states. Linkers that are shorter or stiffer – like an alpha helical coiled-coil for example – might offer an advantage. In addition, it may be possible to combine the oligomer fusion strategy with a more limited application of protein interface design to gain the benefits of both approaches. Finally, some other combinations of symmetries (other than dimer and trimer) allow for fewer alternative outcomes. Less geometrically permissive symmetry combinations would likely make it easier to realize a single intended outcome in future design studies.
Conclusions
A crystal structure of our designed 24-subunit protein cube, the largest and most open assembly created to date from protein subunits, confirmed the symmetry and atomic level accuracy of this novel design. This successful result shows that protein assemblies that are highly porous, yet geometrically regular, can be created using the oligomer fusion strategy. The designed cube has an extremely open structure, and the resulting crystal form generates an extraordinarily porous protein lattice, which could open up new biomaterials applications. Not with standing the successful characterization of this designed protein cube, various solution experiments emphasize that flexibility is intrinsic to assemblies created using the oligomer fusion strategy. In the present study this gives rise to alternate structures in addition to the 24 subunit cube – a 12-subunit tetrahedron and an 18-subunit triangular prism. The flexibility also gives rise to dynamic distortions in solution. These results underscore the importance of monitoring and controlling flexibility to address the challenges of robustly creating highly porous, protein-based assemblies with predetermined structures such as the cube presented here.
Data Deposition
Atomic coordinates of the crystal structure of the protein cage have been deposited in the PDB (ID 4QCC).
Methods
X-ray Crystallography
Purified ATC-HL3 protein in buffer containing 20 mM Tris (pH 8.0) and 100 mM NaCl was concentrated by dialysis in the same buffer condition supplemented with 5% PEG 20,000 (only added to the outside of the dialysis bag) until the concentration reached ∼10 mg/mL. Large cubic crystals grew in 0.1 M MES (pH 6.0), 0.6 M ammonium sulfate and 1% PEG 3350. The hanging-drop technique was used to grow the crystals and the ratio between the protein and reservoir was 2 uL to 1 uL, with a reservoir volume of 500 uL. 5 M lithium acetate was found to be the only suitable cryo-protectant, giving diffraction up to 7 Å on a home x-ray source (Rigaku FRE+/Quantum4 CCD); severe radiation damage was observed in an experiment using a synchrotron x-ray source. The diffraction data set obtained was processed using the XDS package40. A theoretical fusion protein monomer was used as the search model in the molecular replacement program Phaser41, where the space group was determined to be either F23 or F432. Rigid body refinement by Refmac42 led to significantly lower R-factors in the F23 space group. Five cycles of highly constrained jelly-body refinement (sigma=0.01) was carried out and resulted in Rwork and Rfree values of 0.283 and 0.317 respectively. Additional validation of the X-ray structure is described in the Supplementary Materials.
Native mass spectrometry
A purified ATC-HL3 protein solution was buffered exchanged into 200 mM ammonium acetate, pH 7.3 using a centrifugal buffer exchange device (Micro Bio-Spin 6, Bio-Rad) prior to native mass spectrometry analysis. Experiments were conducted using a Q-ToF 2 instrument (Micromass UK Ltd) with a Z-spray source and modifications for high-mass operation as previously described43. Typically, 2 μL of protein solution was electrosprayed from gold-plated borosilicate capillaries prepared in-house. Spectra were externally calibrated by Cesium Iodide solution (200 mg/mL) and processed using MassLynx software (Waters MassLynx V4.1). Stoichiometric analysis of the native macromolecular assemblies in solution was performed by using both mass spectrometry (MS) and tandem mass spectrometry (MS/MS). Tandem mass spectrometry employs collision-induced dissociation on specific macromolecular species, selected via their mass to charge (m/z) ratios. Instrument parameters for MS were 1.50 kV capillary voltage, 150 V sample cone, 20 V extractor cone, 5 V accelerating collision voltage, 1.01 e-2 mbar pirani pressure, 5.05 e-4 mbar penning pressure, 2.57 e-6 To F penning pressure and argon for collisional gas at pressure of 0.2-0.3 MPa. For MS/MS experiments 8959, 12592 and 13278 m/z were selected for the 12, 18 and 24-mer respectively, where the quadrupole resolution was adjusted to encompass the entire charge state of interest. MS/MS instruments settings were identical to those used for MS analysis with the exception of an 150, 200 and 200 accelerating collision voltage for the 12, 18 and 24-mer respectively. Population ratios were estimated using an in-house program, and were corrected for detector efficiency44, 45.
Electron microscopy and image processing
The ATC-HL3 fusion protein was diluted to ∼30 μg/mL in buffer containing 20 mM Tris pH8.0 and 100 mM NaCl. A carbon-coated Maxtaform, 400-mesh Cu/Rh EM specimen grid (Ted Pella, Inc.) was glow-discharged for 30 s. About 3 μl of protein solution was applied to a grid and preserved with 0.75% (w/v) uranyl formate. Images were recorded at a magnification of 52,000× on a 4K × 4K Teitz F416 detector, and ∼ 2 μm under focus, with a Tecnai Spirit electron microscope (FEI), equipped with an LaB6 filament and operating at an acceleration voltage of 120 kV. A total of 157 detector images were automatically acquired using Leginon46. Particle images (∼15,000) were picked using DoG picker47. Two-fold pixel binning of the original particle images resulted in a final pixel size of 4.1 Å. Image alignment and classification were carried out using the Iterative Stable Alignment and Clustering (ISAC) program in the SPARX software package48.
SAXS experiments
ATC-HL3 protein samples were analyzed by small-angle x-ray scattering in eight different buffer conditions (see Supplementary Methods) at the 12.3.1 beamline of the Advanced Light Source at the Berkeley National Laboratory49. The beamline provides 1012 photons/second of 12 keV X-rays focused to a 100 μm spot at the MAR165 detector placed 1.5 m away from the sample. Samples of 20 μL volumes were collected as previously described37. Each protein sample was collected in association with two blank buffers collected before and after the protein sample. Buffers were subtracted from the sample and checked for agreement. Detector images were integrated to display X-ray intensity vs q in Å-1 where q = 4π(sinθ/2)/λ, q is the scattering angle and λ is the X-ray wavelength. The acquired data were analyzed by the program Scatter (available at sibyls.als.lbl.gov), which was used to merge data from each sample, calculate the Rg and mass. Three concentrations were collected at low salt showing no concentration dependence. One concentration was collected for the remaining samples. Each sample was exposed for 0.5, 1, 2 and 5 seconds; the 5 second exposure routinely showed radiation damage and was thus not used for merging profiles. The FoXS program was used to calculate SAXS profiles from atomic models38. Fitting of calculated model SAXS profiles to observed SAXS data is described in Supplementary Material.
Supplementary Material
Acknowledgments
This work was supported by NSF grant CHE-1332907 (T.O.Y.) and NIH grant R01GM067167 (F.J.A.). We thank Michael Sawaya, Duilio Cascio, Dan McNamara and David Leibly for X-ray data collection at the Advanced Photon Source, the staff at APS beamline 24-ID-C and the National Resource for Automated Macromolecular Microscopy (NRAMM) for support. We thank Dek Woolfson, Neil King, and members of the David Baker laboratory for helpful discussions, Thomas Goddard for advice on modeling in UCSF Chimera. SAXS data collection and analysis at BL12.3.1 at the Advanced Light Source (ALS) was supported by the Integrated Diffraction Analysis Technologies (IDAT) program (DOE/BER), by DOE contract DE-AC02-05CH11231, and by NIH MINOS (R01GM105404).
Footnotes
Author Contributions: Y-T.L. and T.O.Y. conceived the project. Y-T.L., E.R., G.H., K-L.T., A.L., J.A.T., and T.O.Y. performed the experiments and analyzed the data. Y-T.L. and T.O.Y. drafted the manuscript. All other authors contributed to the writing and editing of individual sections of the complete manuscript.
References
- 1.Whitesides GM, Grzybowski B. Self-assembly at all scales. Science. 2002;295:2418–2421. doi: 10.1126/science.1070821. [DOI] [PubMed] [Google Scholar]
- 2.Goodsell DS, Olson AJ. Structural symmetry and protein function. Annu Rev Biophys Biomol Struct. 2000;29:105–153. doi: 10.1146/annurev.biophys.29.1.105. [DOI] [PubMed] [Google Scholar]
- 3.Lucon J, Qazi S, Uchida M, Bedwell GJ, La France B, Prevelige PE, et al. Use of the interior cavity of the P22 capsid for site-specific initiation of atom-transfer radical polymerization with high-density cargo loading. Nat Chem. 2012;4:781–788. doi: 10.1038/nchem.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Douglas T, Young M. Viruses: Making friends with old foes. Science. 2006;312:873–875. doi: 10.1126/science.1123223. [DOI] [PubMed] [Google Scholar]
- 5.Huard DJ, Kane KM, Tezcan FA. Re-engineering protein interfaces yields copper-inducible ferritin cage assembly. Nat Chem Biol. 2013;9:169–176. doi: 10.1038/nchembio.1163. [DOI] [PubMed] [Google Scholar]
- 6.Zhang S. Fabrication of novel biomaterials through molecular self-assembly. Nat Biotechnol. 2003;21:1171–1178. doi: 10.1038/nbt874. [DOI] [PubMed] [Google Scholar]
- 7.King NP, Lai YT. Practical approaches to designing novel protein assemblies. Curr Opin Struct Biol. 2013;23:632–638. doi: 10.1016/j.sbi.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 8.Lai YT, King NP, Yeates TO. Principles for designing ordered protein assemblies. Trends Cell Biol. 2012;22:653–661. doi: 10.1016/j.tcb.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 9.Gradisar H, Jerala R. Self-assembled bionanostructures: proteins following the lead of DNA nanostructures. J Nanobiotechnology. 2014;12:4. doi: 10.1186/1477-3155-12-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gradisar H, Bozic S, Doles T, Vengust D, Hafner-Bratkovic I, Mertelj A, et al. Design of a single-chain polypeptide tetrahedron assembled from coiled-coil segments. Nat Chem Biol. 2013;9:362–366. doi: 10.1038/nchembio.1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fletcher JM, Harniman RL, Barnes FR, Boyle AL, Collins A, Mantell J, et al. Self-assembling cages from coiled-coil peptide modules. Science. 2013;340:595–599. doi: 10.1126/science.1233936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lanci CJ, MacDermaid CM, Kang SG, Acharya R, North B, Yang X, et al. Computational design of a protein crystal. Proc Natl Acad Sci U S A. 2012;109:7304–7309. doi: 10.1073/pnas.1112595109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Padilla JE, Colovos C, Yeates TO. Nanohedra: using symmetry to design self assembling protein cages, layers, crystals, and filaments. Proc Natl Acad Sci U S A. 2001;98:2217–2221. doi: 10.1073/pnas.041614998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sinclair JC, Davies KM, Venien-Bryan C, Noble ME. Generation of protein lattices by fusing proteins with matching rotational symmetry. Nat Nanotechnol. 2011;6:558–562. doi: 10.1038/nnano.2011.122. [DOI] [PubMed] [Google Scholar]
- 15.Lai YT, Cascio D, Yeates TO. Structure of a 16-nm cage designed by using protein oligomers. Science. 2012;336:1129. doi: 10.1126/science.1219351. [DOI] [PubMed] [Google Scholar]
- 16.Lai YT, Tsai KL, Sawaya MR, Asturias FJ, Yeates TO. Structure and flexibility of nanoscale protein cages designed by symmetric self-assembly. J Am Chem Soc. 2013;135:7738–7743. doi: 10.1021/ja402277f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.King NP, Sheffler W, Sawaya MR, Vollmar BS, Sumida JP, Andre I, et al. Computational design of self-assembling protein nanomaterials with atomic level accuracy. Science. 2012;336:1171–1174. doi: 10.1126/science.1219364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.King NP, Bale JB, Sheffler W, McNamara DE, Gonen S, Gonen T, et al. Accurate design of co-assembling multi-component protein nanomaterials. Nature. 2014;510:103–108. doi: 10.1038/nature13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tozawa T, Jones JT, Swamy SI, Jiang S, Adams DJ, Shakespeare S, et al. Porous organic cages. Nat Mater. 2009;8:973–978. doi: 10.1038/nmat2545. [DOI] [PubMed] [Google Scholar]
- 20.Furukawa H, Cordova KE, O'Keeffe M, Yaghi OM. The chemistry and applications of metal-organic frameworks. Science. 2013;341:1230444. doi: 10.1126/science.1230444. [DOI] [PubMed] [Google Scholar]
- 21.Holst JR, Trewin A, Cooper AI. Porous organic molecules. Nat Chem. 2010;2:915–920. doi: 10.1038/nchem.873. [DOI] [PubMed] [Google Scholar]
- 22.Chen JH, Seeman NC. Synthesis from DNA of a molecule with the connectivity of a cube. Nature. 1991;350:631–633. doi: 10.1038/350631a0. [DOI] [PubMed] [Google Scholar]
- 23.Pinheiro AV, Han D, Shih WM, Yan H. Challenges and opportunities for structural DNA nanotechnology. Nat Nanotechnol. 2011;6:763–772. doi: 10.1038/nnano.2011.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shih WM, Quispe JD, Joyce GF. A 1.7-kilobase single-stranded DNA that folds into a nanoscale octahedron. Nature. 2004;427:618–621. doi: 10.1038/nature02307. [DOI] [PubMed] [Google Scholar]
- 25.Iinuma R, Ke Y, Jungmann R, Schlichthaerle T, Woehrstein JB, Yin P. Polyhedra self-assembled from DNA tripods and characterized with 3D DNA-PAINT. Science. 2014;344:65–69. doi: 10.1126/science.1250944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Afonin KA, Bindewald E, Yaghoubian AJ, Voss N, Jacovetty E, Shapiro BA, et al. In vitro assembly of cubic RNA-based scaffolds designed in silico. Nat Nanotechnol. 2010;5:676–682. doi: 10.1038/nnano.2010.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pavone V, Zhang SQ, Merlino A, Lombardi A, Wu Y, Degrado WF. Crystal structure of an amphiphilic foldamer reveals a 48-mer assembly comprising a hollow truncated octahedron. Nat Commun. 2014;5:3581. doi: 10.1038/ncomms4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ueno T. Porous Protein Crystals as Reaction Vessels. Chemistry-a European Journal. 2013;19:9096–9102. doi: 10.1002/chem.201300250. [DOI] [PubMed] [Google Scholar]
- 29.Molino Nicholas M, Wang SW. Caged protein nanoparticles for drug delivery. Curr Opin Biotechnol. 2014;28:75–82. doi: 10.1016/j.copbio.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peer D, Karp JM, Hong S, FaroKHzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 31.Walters MJ, Srikannathasan V, McEwan AR, Naismith JH, Fierke CA, Toone EJ. Characterization and crystal structure of Escherichia coli KDPGal aldolase. Bioorg Med Chem. 2008;16:710–720. doi: 10.1016/j.bmc.2007.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saul FA, Arie JP, Vulliez-le Normand B, Kahn R, Betton JM, Bentley GA. Structural and functional studies of FkpA from Escherichia coli, a cis/trans peptidyl-prolyl isomerase with chaperone activity. J Mol Biol. 2004;335:595–608. doi: 10.1016/j.jmb.2003.10.056. [DOI] [PubMed] [Google Scholar]
- 33.Kantardjieff KA, Rupp B. Matthews coefficient probabilities: Improved estimates for unit cell contents of proteins, DNA, and protein-nucleic acid complex crystals. Protein Sci. 2003;12:1865–1871. doi: 10.1110/ps.0350503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rose PW, Bi CX, Bluhm WF, Christie CH, Dimitropoulos D, Dutta S, et al. The RCSB Protein Data Bank: new resources for research and education. Nucleic Acids Res. 2013;41:D475–D482. doi: 10.1093/nar/gks1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Inokuma Y, Yoshioka S, Ariyoshi J, Arai T, Hitora Y, Takada K, et al. X-ray analysis on the nanogram to microgram scale using porous complexes. Nature. 2013;495:461–466. doi: 10.1038/nature11990. [DOI] [PubMed] [Google Scholar]
- 36.Hernandez H, Robinson CV. Determining the stoichiometry and interactions of macromolecular assemblies from mass spectrometry. Nat Protoc. 2007;2:715–726. doi: 10.1038/nprot.2007.73. [DOI] [PubMed] [Google Scholar]
- 37.Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL, 2nd, Tsutakawa SE, et al. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nat Methods. 2009;6:606–612. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schneidman-Duhovny D, Hammel M, Sali A. FoXS: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res. 2010;38:W540–544. doi: 10.1093/nar/gkq461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fotin A, Cheng Y, Sliz P, Grigorieff N, Harrison SC, Kirchhausen T, et al. Molecular model for a complete clathrin lattice from electron cryomicroscopy. Nature. 2004;432:573–579. doi: 10.1038/nature03079. [DOI] [PubMed] [Google Scholar]
- 40.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 43.Sobott F, Hernandez H, McCammon MG, Tito MA, Robinson CV. A tandem mass spectrometer for improved transmission and analysis of large macromolecular assemblies. Anal Chem. 2002;74:1402–1407. doi: 10.1021/ac0110552. [DOI] [PubMed] [Google Scholar]
- 44.Brunton AN, Fraser GW, Lees JE, Turcu IC. Metrology and modeling of microchannel plate x-ray optics. Appl Opt. 1997;36:5461–5470. doi: 10.1364/ao.36.005461. [DOI] [PubMed] [Google Scholar]
- 45.Ebong IO, Morgner N, Zhou M, Saraiva MA, Daturpalli S, Jackson SE, et al. Heterogeneity and dynamics in the assembly of the heat shock protein 90 chaperone complexes. Proc Natl Acad Sci U S A. 2011;108:17939–17944. doi: 10.1073/pnas.1106261108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, et al. Automated molecular microscopy: The new Leginon system. J Struct Biol. 2005;151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 47.Voss NR, Yoshioka CK, Radermacher M, Potter CS, Carragher B. DoG Picker and TiltPicker: Software tools to facilitate particle selection in single particle electron microscopy. J Struct Biol. 2009;166:205–213. doi: 10.1016/j.jsb.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang Z, Fang J, Chittuluru J, Asturias FJ, Penczek PA. Iterative stable alignment and clustering of 2D transmission electron microscope images. Structure. 2012;20:237–247. doi: 10.1016/j.str.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dyer KN, Hammel M, Rambo RP, Tsutakawa SE, Rodic I, Classen S, et al. High-Throughput SAXS for the Characterization of Biomolecules in Solution: A Practical Approach. Structural Genomics: General Applications. 2014;1091:245–258. doi: 10.1007/978-1-62703-691-7_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.