

Abstract
Emerging data establish dyslipidemia as a significant contributor to the development of diabetic neuropathy. In this review we discuss how separate metabolic imbalances, including hyperglycemia and hyperlipidemia, converge on mechanisms leading to oxidative stress in dorsal root ganglia sensory neurons. We conclude with suggestions for novel therapeutic strategies to prevent or reverse diabetes-induced nerve degeneration.
Significance of the Problem
Diabetes mellitus affects over 20 million people in the United States and the number of diabetic patients is increasing by 5% per year (www.diabetes.org). The most common complication of diabetes is diabetic neuropathy. Depending on the diagnostic criteria used, at least 50% up to 90% of individuals with diabetes will develop diabetic neuropathy. The most common form of diabetic neuropathy is diabetic polyneuropathy, a symmetric loss of nerve function beginning in the toes and progressing in a distal to proximal fashion, yielding what is commonly called a stocking/glove pattern of sensory loss (Edwards, et al., 2008). While all sensory modalities are eventually affected, recent studies show that initially small unmyelinated and thinly myelinated fibers are injured and, especially early in the disease, patients can present with pain. Twenty-five years after the diagnosis of diabetes, the cumulative risk of a lower extremity amputation is 22% and, in the general population, 60% of all lower extremity amputations are secondary to diabetic neuropathy. This represents a cost of over $22 billion per year and a significant loss of quality of life for diabetic patients (Barrett, et al., 2007). Although therapies are available to alleviate the symptoms of diabetic neuropathy, these rarely impact upon the root causes of the disease (Feldman, et al., 2002). The immense physical, psychological, and economic cost of diabetic neuropathy underscores the need for causally targeted therapies (Kles and Vinik, 2006).
Oxidative Stress in Diabetes
There is a growing consensus, driven by both clinical and basic studies, that oxidative stress underlies the development of the microvascular complications of diabetes, including diabetic neuropathy (Low, et al., 1997; Russell, et al., 2008; Vincent, et al., 2008; Ziegler, et al., 2004). In type 1 diabetic patients, the severity of microvascular complications parallels the degree of systemic oxidative stress (Giugliano, et al., 2008; Sullivan and Feldman, 2005). With disease and diabetic neuropathy progression, antioxidant potential decreases while lipid peroxidation products increase. Type 2 diabetic patients have a similar oxidative stress profile which directly relates to the onset and progression of microvascular complications (Greene, et al., 1999; Vincent, et al., 2004b).
In the late 1990s, our group introduced the idea that glucose-mediated oxidative stress injures the peripheral nervous system, leading to eventual death and loss of neurons and supporting Schwann cells (Feldman, et al., 1997; Russell, et al., 2001; Russell, et al., 1999; Russell, et al., 1998). Investigation of the basic mechanisms underlying this process in DRG neurons identified multiple mechanisms by which hyperglycemia mediates DRG neuron injury. Mitochondrial overload is the principal site of reactive oxygen species (ROS) generation in hyperglycemia (Vincent, et al., 2005a). DRG neurons may be preserved in vitro in the face of hyperglycemic insult by uncoupling agents that relieve the mitochondrial overload (Vincent, et al., 2004a) and by lipophilic antioxidants that protect the mitochondria against ROS injury (Vincent, et al., 2005a; Vincent, et al., 2005b).
Additional cellular mechanisms are activated by hyperglycemia to produce ROS. Hyperglycemia leads to the formation of advanced glycation end-products (AGE). DRG neurons express the receptor for AGE (RAGE) and exposure to AGE leads to oxidative stress and injury in DRG neurons that is partially mediated through activation of the nicotinamide adenine dinucleotide phosphate [NAD(P)H] oxidase complex (Vincent, et al., 2007a). In addition, direct exposure to hyperglycemia leads to the activation of NAD(P)H oxidase (Vincent, et al., 2005a). This complex is formed through recruitment of a combination of a p22-phox subunit and 5 NOX subunits (Baumer, et al., 2008). Within neurons, NOX2 (or gp91-phox) and p22-phox are expressed within the cell membrane; p47-phox and p67-phox are recruited from the cytoplasm to the membrane when the neuron is exposed to a NAD(P)H oxidase activating stimulus (Murdoch, et al., 2006). NAD(P)H oxidase activity generates superoxide (O2.-) that promotes mitochondrial dysfunction and apoptosis in the setting of inflammation, neurodegeneration and in atherosclerosis (Silver, et al., 2007; Stamler, 1996). Over time, these mechanisms act in concert with accumulating ROS-induced damage that impairs nerve function and results in the signs and symptoms of diabetic neuropathy (Feldman, et al., 1997; Feldman, et al., 2002; Greene, et al., 1999; Vincent and Feldman, 2004; Vincent, et al., 2004b; Vincent, et al., 2007b).
Pathophysiology of Diabetic Neuropathy: More than Just Glucose
Until recently, we (Feldman, et al., 1997; Feldman, et al., 2002; Greene, et al., 1999; Vincent, et al., 2004b) and other investigators (Brownlee, 2005; Low, et al., 1997; Osawa and Kato, 2005; Tomlinson and Gardiner, 2008) in the field contended that hyperglycemia was the driving force underlying the development of diabetic neuropathy. Our opinions were based originally on results from The Diabetes Control and Complications Trial (DCCT). In the DCCT, type 1 diabetic subjects receiving intensive therapy with an average glycosylated hemoglobin (HbA1c) of 7.2% had a reduced 60% cumulative incidence of diabetic neuropathy when compared to patients receiving conventional treatment (average HbA1c of 9.0%) (The Diabetes Control and Complications Trial Research, 1993). However, the continuing longitudinal study of the DCCT, the Epidemiology of Diabetes Complications and Interventions Cohort (EDIC) yielded unanticipated results 20 years later (Genuth, 2006a; Martin, et al., 2006a; Pop-Busui, et al., 2009). Within one year of discontinuing the DCCT and beginning EDIC, the glycemic control in the two treatment groups equalized to an average HbA1c of 8% (Genuth, 2006a). All 1,300 patients were examined annually for diabetic neuropathy; one decade later patients from the intensive-DCCT cohort had a lower incidence of diabetic neuropathy compared to patients from the conventional-DCCT cohort, despite 10 years of convergent glycemic control (Martin, et al., 2006b). The underlying mechanism(s) of this result is not determined, but one interesting difference is the lipid profiles of the two groups: a subset of the intensive-DCCT cohort has less dyslipidemia than the conventional cohort (1999; Lyons, et al., 2004). This interesting, unanticipated finding is further supported by the Eurodiab Trial, a longitudinal study of over 3,000 individuals with type 1 diabetes (Tesfaye, 2007; Tesfaye, et al., 2005). Of 1,200 subjects who did not have diabetic neuropathy at baseline, hypertension, serum lipids and body mass index were each independently associated with the risk of developing diabetic neuropathy during a 7 year follow-up period. Of these risk factors, dyslipidemia was closely linked with the onset and progression of diabetic neuropathy [reviewed in (Leiter, 2005)]. In support of these findings, we recently evaluated the mechanisms underlying diabetic neuropathy progression using indexes of sural nerve morphometry obtained from two identical randomized, placebo-controlled clinical trials (Wiggin, et al., 2009a). Sural nerve myelinated fiber density, nerve conduction velocities, vibration perception thresholds, clinical symptom scores, and a visual analog scale for pain were analyzed in participants with mild to moderate diabetic neuropathy. A loss of ≥ 500 fibers/mm2 in sural nerve myelinated fiber density over 52 weeks was defined as progressing diabetic neuropathy, and a myelinated fiber density loss of ≤ 100 fibers/mm2 during the same time interval as nonprogressing diabetic neuropathy. In this cohort of participants elevated triglycerides was the only clinical parameter that correlated with a loss of myelinated fiber density, independent of disease duration, age, diabetes control, or other variables (Wiggin, et al., 2009a). The nerve fiber densities, triglycerides, and motor nerve conduction velocities for the two groups are presented in Fig. 1.
Fig. 1. Myelinated fiber density (MFD) of the rapidly progressing and nonprogressing diabetic patients.
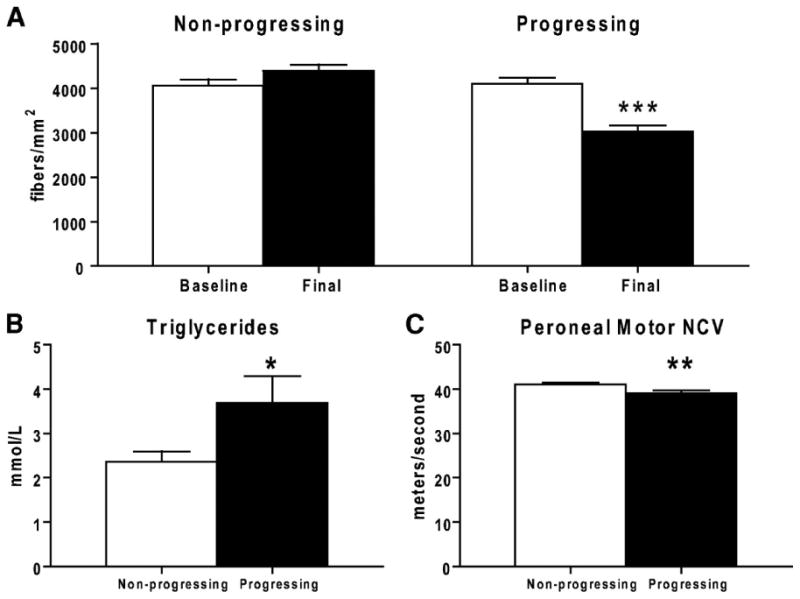
(A): The nonprogressing dataset shows a no change in MFD (fibers/mm2) over 52 weeks, while the progressing dataset shows a highly significant decrease in MFD. Baseline measurements of triglyceride levels (B) and peroneal motor nerve conduction velocity (C) are significantly different between the progressing and nonprogressing participants. *P < 0.05; ** P < 0.01; *** P < 0.0001. (reproduced from (Wiggin, et al., 2009b)).
Dyslipidemia and Neuropathy- an Expanding Problem
The emerging idea that dyslipidemia contributes to the development of diabetic neuropathy may explain the earlier incidence of diabetic neuropathy in individuals with type 2 compared to type 1 diabetes. Lipid profiles are commonly abnormal early in the course of type 2 diabetes in a temporal pattern that correlates with the presence of diabetic neuropathy (2001; Clemens, et al., 2004). In contrast, lipid profiles are nearly always normal in type 1 patients at the time of diabetes diagnosis (Leiter, 2005). Dyslipidemia develops later in the course of type 1 diabetes, and these abnormal lipid profiles coincide with the delayed onset and progression of diabetic neuropathy (Kempler, et al., 2002; Young, et al., 1993). Accumulating data from several large scale trials of patients with type 2 diabetes also point to early dyslipidemia as a major independent risk factor for the development of diabetic neuropathy [reviewed in (Cameron, et al., 2003; Gordon and Robinson, 2006; Leiter, 2005)]. In the United Kingdom Prospective Diabetes Study (UKPDS), 3,867 newly diagnosed type 2 patients were randomized into either intensive treatment with an oral hypoglycemic agent or insulin or conventional treatment with diet. After 10 years, intensive treatment resulted in approximately 1% lower HbA1c versus conventional treatment but there was no difference in the development of diabetic neuropathy between the two groups, which had similar lipid and blood pressure profiles (1998). This finding, at first unexpected in light of the earlier DCCT data, was supported by the VA Cooperative Study, which demonstrated no difference in the prevalence of diabetic neuropathy in type 2 patients over a 2 year period comparing standard and intensive glycemic control (Azad, et al., 1999). These results suggested that independent factors other than glycemic control are critical to the development of diabetic neuropathy (Leiter, 2005).
As with EDIC and Eurodiab, analysis of the UKPDS and VA cooperative data points to dyslipidemia as a critical independent factor for the development of diabetic neuropathy (Leiter, 2005). Type 2 diabetes clusters with risk factors for coronary heart disease including obesity, hypertension, and dyslipidemia; individuals with 2 or more of these factors are diagnosed with the metabolic syndrome (Bonora, 2006; Fonseca, 2005; Grundy, 2005; Zimmet and Alberti, 2008). In a cross sectional study of 548 type 2 diabetic subjects, those with Metabolic Syndrome were twice as likely to have diabetic neuropathy (Isomaa, et al., 2001), and the driving factor was dyslipidemia. In a European study of 85 type 2 diabetic patients with at least two additional metabolic syndrome parameters, the prevalence of microvascular complications, including diabetic neuropathy, increased with each additional parameter present (Isomaa, et al., 2001); abnormalities in LDL profiles were more closely related to diabetic neuropathy than hyperglycemia. Finally, prospective studies of patients with idiopathic neuropathy, including our own recently published work, confirm a higher prevalence of hyperlipidemia than impaired glucose tolerance or hypertension, suggesting that dyslipidemia is an essential factor underlying nerve injury (Gordon and Robinson, 2006; Wiggin, et al., 2009a). Collectively, this evolving and exciting literature links dyslipidemia to the development and progression of diabetic neuropathy. Fig. 2 outlines our current understanding of the factors that contribute to the development of diabetic neuropathy.
Fig. 2. Hyperglycemia and hyperlipidemia contribute to the pathogenesis of diabetic neuropathy.

Adapted from (Brownlee, 2005).
Lipid Modification in Diabetes
We have now employed cell culture and mouse models of diabetic neuropathy and suggest oxLDLs are one notable “lipid factor” responsible for nervous system injury (Vincent, et al., 2009a). LDL is the primary carrier of cholesterol (Hammer, et al., 1995) and vitamin E (Heinecke, 1987) within the plasma. Systemic oxidative stress results in the modification of these lipoproteins, which is well characterized in atherosclerosis (Tsuzura, et al., 2004a; Willems, et al., 1998a). LDLs spontaneously oxidize in the presence of reactive oxygen species such as superoxide (O2.-) to form oxLDLs (Hammer, et al., 1995). Cholesterol carrying LDLs are more prone to oxidation than smaller, high density particles (Krentz, 2003).
OxLDLs are critically involved in endothelial cell dysfunction, evident from the large body of literature implicating OxLDLs in atherosclerotic lesion formation (Ceriello, 2006; Genuth, 2006b; Li and Mehta, 2005). OxLDL is strongly cytotoxic, which may explain the areas of necrosis detected within atherosclerotic lesions (Li and Mehta, 2005; Pennathur and Heinecke, 2007; Thum and Borlak, 2008; Tsuzura, et al., 2004a). In man, oxLDL is a highly analytic marker for macrovascular disease, including stroke and myocardial infarction (Tsimikas, et al., 2005). In patients with types 1 and 2 diabetes, serum levels of oxLDL in proportion to total LDL particles are associated with diabetic neuropathy (Tsuzura, et al., 2004b; Willems, et al., 1998b). Fig. 3 illustrates our findings that oxLDL increase significantly in mice on a high fat diet. These oxLDL can be found in the dorsal root ganglia and the mice develop early signs and symptoms of diabetic neuropathy (Vincent, et al., 2009a).
Fig. 3. Mice on a High Fat Diet Increase oxLDL and Develop Neuropathy.

(A) Pooled plasma samples (2 pools/group, each pool analyzed 3 times) were subjected to fractionation by FPLC, then cholesterol (A) were measured in each fraction. The graph shows the mean and SEM for n=2 pools/group. (B) Oxidative stress measures in the LDL fraction by reverse phase HPLC. HODE, dityrosine, and nitrotyrosine were all significantly increased (p<0.05). (C) Representative IENFD images from one control and one high fat fed mouse footpad. Bar = 50 μm; d=dermis, e=epidermis. White dots indicate nerve fibers counted. (D) In a different mouse study, using obese db/db mice, we immunostained the DRG for ApoB and MDA-oxidized LDL. We observe co-localization of MDA-LDL in green and ApoB in red (yielding a yellow signal) around the neurons (arrows). (A-C reproduced from (Vincent, et al., 2009b)).
OxLDLs Mediate Cellular Injury via the Scavenger Receptor, LOX-1
OxLDLs cause apoptotic injury and death in both endothelial cells (Dimmeler, et al., 1997) and neurons (Draczynska-Lusiak, et al., 1998a; Draczynska-Lusiak, et al., 1998b; Keller, et al., 2000; Keller, et al., 1999; Papassotiropoulos, et al., 1996; Schroeter, et al., 2000) In endothelial cells, oxLDL induce multiple events associated with apoptotic injury, including Bid degradation, cytochrome C release, and caspase-3 activation (Vindis, et al., 2005). OxLDL are associated with increased pro-apoptotic Bax and decreased levels of the anti-apoptotic type I IGF receptor (IGF-IR) in smooth muscle cells (Higashi, et al., 2005). Both death receptor and mitochondrial pathways are involved in atherosclerotic-plaque associated apoptosis induced by oxLDL (Napoli, 2003). In neurons, oxLDL induce DNA fragmentation characteristic of apoptosis in DRG (Papassotiropoulos, et al., 1996; Vincent, et al., 2009a), striatal neurons (Draczynska-Lusiak, et al., 1998b; Schroeter, et al., 2000), and PC-12 cells (Draczynska-Lusiak, et al., 1998a). In motor neurons, oxLDL increase reactive oxygen species and activate a caspase-3-dependent death mechanism (Keller, et al., 2000). Specific effects of oxLDL on mitochondria and mitochondrial-mediated apoptotic events in neurons remain unknown.
OxLDL exert effects on cells through two primary cell surface receptors, lectin-like oxidized LDL receptor-1 (LOX-1) on endothelial cells (Chen, et al., 2006) and CD36 on macrophages (Yamashita, et al., 2006). Upon receptor-mediated uptake of oxLDL into endothelial cells, oxLDL is transported through the endothelial cell and extruded into the subendothelial region within tissues (Li and Mehta, 2005). A schematic showing the expression of LOX-1 and the potential effects of oxLDL binding is shown in Fig. 4. LOX-1 expression is upregulated by oxLDL, which in turn increases intracellular O2.- production (Hu, et al., 2003). Shear stress, tumor necrosis factor-α, and free radicals all increase LOX-1 expression levels (Hu, et al., 2003), while lipid lowering drugs decrease expression (Draude, et al., 1999). Hyperglycemia, advanced glycation endproducts, and C-reactive protein also increase LOX-1 expression (Iwashima, et al., 2000; Rudijanto, 2007; Schalkwijk and Stehouwer, 2005). Indeed, glucose stimulates LOX-1 expression in both endothelial cells and macrophages, prevented by antioxidants and inhibitors of MAPK and NF-κB (Dandapat, et al., 2007; Li, et al., 2003; Li, et al., 2004). LOX-1 is upregulated in renal tubules in obese, diabetic rats and its activation leads to inflammation and nephropathy (Dominguez, et al., 2008; Kelly, et al., 2008; Ueno, et al., 2003). Finally, LOX-1 is also expressed on neurons, and polymorphisms in the LOX-1 gene are associated with neurodegenerative disease in humans (Papassotiropoulos, et al., 2005). We examined LOX-1 expression in DRG neurons, and found basal expression that was further increased by exposure to oxLDL (Vincent, et al., 2009a). Selected data are presented in Fig. 5. Subsequent to LOX-1 activation, the DRG neurons rapidly activated NAD(P)H oxidase, increased superoxide generation, and activated a programmed cell death mechanism. DRG neuron injury in the presence of oxLDL was prevented by a LOX-1 blocking antibody, the NAD(P)H oxidase inhibitor apocyanin, or the antioxidant α-lipoic acid (Vincent, et al., 2009a).
Fig. 4. Schematic of Effects of oxLDL Binding to LOX-1.
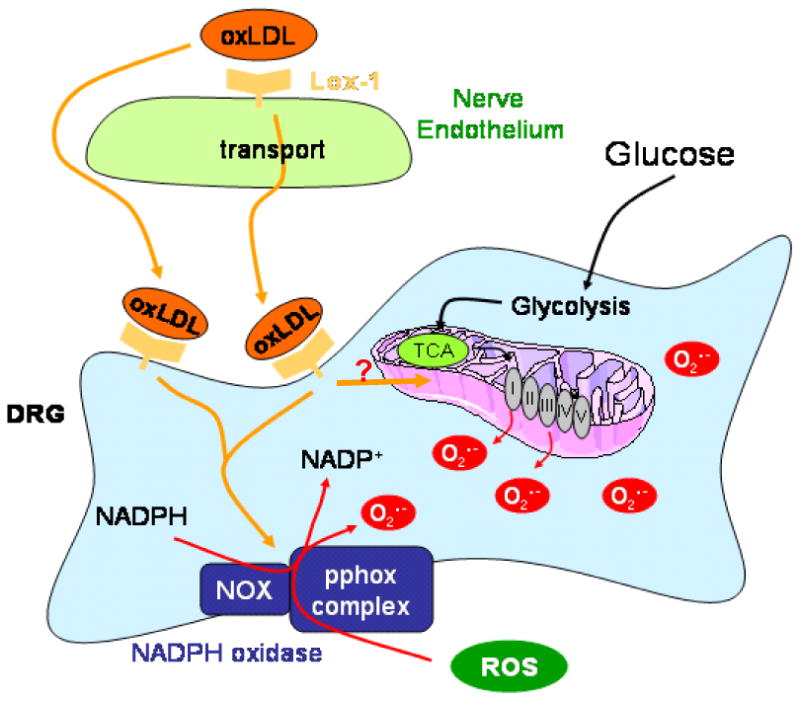
LOX-1 on both vascular endothelial cells and DRG neurons will bind oxLDL. Subsequently, the oxLDL may be endocytosed or transcytosed. Receptor binding initiates a signaling pathway leading to the activation of NAD(P)H oxidase and also may alter mitochondrial generation of reactive oxygen species. Glucose independently affects these same cellular targets.
Fig. 5. High glucose and oxLDL cause cell death in DRG neurons via NAD(P)H Oxidase.

Adult DRG neurons were exposed to high glucose (25.7 mM) or increasing concentrations of oxLDL and then cell death was quantitated by caspase 3 activation after 5 h. DRG neurons were additionally pre-treated with LOX-1 neutralizing antibody (Anti-LOX-1, 100 mg/ml), apocyanin (Apo, 1 μM), or α-lipoic acid (LA, 100 μM). n=9, *p<0.01 compared to untreated control, +p<0.01 compared to no pre-treatment (None). (B) Adult DRG neurons were exposed to 30 μg/ml oxLDL and then immunolabeled for NAD(P)H oxidase subunits p47 or gp91. In (B), adult DRG neurons were exposed to high glucose (25.7 mM) or oxLDL (30 μg/ml) for 1 h or 3 h, then lysed for biochemical assays of NAD(P)H oxidase. *p<0.01 compared to untreated control, +p<0.05 compared to untreated control. (reproduced from (Vincent, et al., 2009b)).
These data suggest that, in diabetes, neurons are exposed to both glucose and oxLDL which independently increase ROS, and glucose may sensitize neurons to oxLDL-mediated damage via upregulation of LOX-1. Interestingly, oxLDL decreases native LDL-receptor expression in a LOX-1-dependent manner (Hu, et al., 2003). Given the critical role for native LDL-receptors in neuronal functioning, synapse maintenance, and myelination following injury (Herz and Bock, 2002), oxLDL could also predispose neurons to glucose-mediated injury by decreasing native LDL-receptor. These ideas await further exploration.
Lipids and Diabetic Neuropathy
If the idea that dyslipidemia contributes to the development of diabetic neuropathy is true (McManis, et al., 1994), lipid lowering drugs may be beneficial in the treatment of diabetic neuropathy. Fenofibrate is a PPARα agonist that lowers plasma lipids by improving their removal by the liver and improving fatty acid metabolism (Aasum, et al., 2008; Harano, et al., 2006). In genetic dyslipidemia in mice, including ApoE knockout, leptin deficient, and LDL receptor knockout mice, fenofibrate improves the lipid profile and increases HDL (Kooistra, et al., 2006; Lie, et al., 2005; Srivastava, et al., 2006). These lipid improvements correlate with prevention of insulin resistance and atherosclerosis (Aasum, et al., 2008; Calkin, et al., 2007; Xie, et al., 2007). Interest in this drug treatment has expanded with the demonstration that fenofibrate dramatically improves hyperglycemia, insulin resistance, albuminuria, and glomerular lesions in db/db mice (Park, et al., 2006). The FIELD trial demonstrated that fenofibrate improves signs and progression of retinopathy and nephropathy (2007; Davis, et al., 2008; Firth, 2008; Keech, et al., 2007; Simo and Hernandez, 2007). The Fremantle Diabetes Study was an observational investigation of 1,237 patients with type 2 diabetes. The data suggest that therapy with a statin or fibrate protects against diabetic peripheral sensory neuropathy, but calls for confirmatory evidence via a randomized clinical trial (Davis, et al., 2008). Fenofibrate also may provide neuroprotection against stroke (Deplanque, et al., 2003). We recently demonstrated potent ability of fenofibrate to prevent hyperglycemia-induced DRG neuron injury in vitro by decreasing mitochondrial O2.- generation (Vincent and Feldman, 2008). We are following this study with an intervention in type 1 diabetic mice and have demonstrated that 0.1% w/w fenofibrate chow significantly decreases total cholesterol and LDL triglycerides (unpublished data).
Future Directions
We maintain our stand that hyperglycemia also is a key mediator of DRG neuron injury particularly in poorly-controlled diabetes. Therefore, compounds that improve glycemic control will assist in the prevention of complications. Metformin, a biguanide compound, improves insulin resistance by reducing gluconeogenesis and enhancing peripheral glucose uptake, promoting reduction of the plasma glucose level (Yoon, et al., 2007). Metformin remains one of the most used glucose regulating drugs in type 2 diabetes (Saenz, et al., 2005) and is used in preclinical trials in mice (Algire, et al., 2008; Yoon, et al., 2007). Interestingly, dyslipidemia increases in adolescent type 1 diabetic patients with poor glycemic control, again highlighting the complex interplay between glycemia and dyslipidemia (Shamir, et al., 2008). Taken together, the data indicate that strategic use of NAD(P)H oxidase inhibition, antioxidants, anti-LOX-1 therapy, anti-hyperglycemia, and lipid lowering therapies will prevent diabetic neuropathy. Each of these components has been tested in rodents with positive results (Cotter and Cameron, 2003; Dominguez, et al., 2008; Park, et al., 2006). Individually, these strategies have not produced significant results in clinical trials, with the exception of α-lipoic acid (Ziegler, et al., 2006). Current investigations are focusing in on metabolic deficits in the axon, particularly at the mitochondria (Edwards, et al., 2009; Figueroa-Romero, et al., 2008; Wiggin, et al., 2008). Further drug refinement and subcellular targeting may be the key to improved efficacy against neuronal injury.
Acknowledgments
The Feldman Laboratory is supported by, Juvenile Diabetes Research Foundation (AMV, ELF), the American Diabetes Association, (AMV, ELF), the Animal Models of Diabetes Complications Consortium (AMDCC; NIH UO1 DK076160, ELF), the Program for Neurology Research and Discovery, and the A. Alfred Taubman Medical Institute.
Literature Cited
- UK Prospective Diabetes Study (UKPDS) Group. Lancet. Vol. 352. UK Prospective Diabetes Study (UKPDS) Group; 1998. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33) pp. 837–853. [PubMed] [Google Scholar]
- Epidemiology of Diabetes Interventions and Complications (EDIC). Design, implementation, and preliminary results of a long-term follow-up of the Diabetes Control and Complications Trial cohort. Diabetes care. 1999;22:99–111. doi: 10.2337/diacare.22.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The effect of aggressive versus standard lipid lowering by atorvastatin on diabetic dyslipidemia: the DALI study: a double-blind, randomized, placebo-controlled trial in patients with type 2 diabetes and diabetic dyslipidemia. Diabetes care. 2001;24:1335–1341. doi: 10.2337/diacare.24.8.1335. [DOI] [PubMed] [Google Scholar]
- Reducing ocular damage in type 2 diabetes: the FIELD study shows fenofibrate benefits. Cardiovascular journal of Africa. 2007;18:400. [PubMed] [Google Scholar]
- Aasum E, Khalid AM, Gudbrandsen OA, How OJ, Berge RK, Larsen TS. Fenofibrate modulates cardiac and hepatic metabolism and increases ischemic tolerance in diet-induced obese mice. Journal of molecular and cellular cardiology. 2008;44:201–209. doi: 10.1016/j.yjmcc.2007.08.020. [DOI] [PubMed] [Google Scholar]
- Algire C, Zakikhani M, Blouin MJ, Shuai JH, Pollak M. Metformin Attenuates the Stimulatory Effect of a High Energy Diet on in vivo H59 Carcinoma Growth. Endocrine-related cancer. 2008 doi: 10.1677/ERC-08-0038. [DOI] [PubMed] [Google Scholar]
- Azad N, Emanuele NV, Abraira C, Henderson WG, Colwell J, Levin SR, Nuttall FQ, Comstock JP, Sawin CT, Silbert C, Rubino FA. The effects of intensive glycemic control on neuropathy in the VA cooperative study on type II diabetes mellitus (VA CSDM) Journal of diabetes and its complications. 1999;13:307–313. doi: 10.1016/s1056-8727(99)00062-8. [DOI] [PubMed] [Google Scholar]
- Barrett AM, Lucero MA, Le T, Robinson RL, Dworkin RH, Chappell AS. Epidemiology, public health burden, and treatment of diabetic peripheral neuropathic pain: a review. Pain medicine (Malden, Mass) 2007;8(Suppl 2):S50–62. doi: 10.1111/j.1526-4637.2006.00179.x. [DOI] [PubMed] [Google Scholar]
- Baumer AT, Ten Freyhaus H, Sauer H, Wartenberg M, Kappert K, Schnabel P, Konkol C, Hescheler J, Vantler M, Rosenkranz S. Phosphatidylinositol 3-kinase-dependent membrane recruitment of Rac-1 and p47phox is critical for alpha-platelet-derived growth factor receptor-induced production of reactive oxygen species. The Journal of biological chemistry. 2008;283:7864–7876. doi: 10.1074/jbc.M704997200. [DOI] [PubMed] [Google Scholar]
- Bonora E. The metabolic syndrome and cardiovascular disease. Ann Med. 2006;38:64–80. doi: 10.1080/07853890500401234. [DOI] [PubMed] [Google Scholar]
- Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- Calkin AC, Jandeleit-Dahm KA, Sebekova E, Allen TJ, Mizrahi J, Cooper ME, Tikellis C. PPARs and diabetes-associated atherosclerosis. Current pharmaceutical design. 2007;13:2736–2741. doi: 10.2174/138161207781662902. [DOI] [PubMed] [Google Scholar]
- Cameron N, Cotter M, Inkster M, Nangle M. Looking to the future: diabetic neuropathy and effects of rosuvastatin on neurovascular function in diabetes models. Diabetes research and clinical practice. 2003;61(Suppl 1):S35–S39. doi: 10.1016/s0168-8227(03)00123-2. [DOI] [PubMed] [Google Scholar]
- Ceriello A. Oxidative stress and diabetes-associated complications. EndocrPract. 2006;12(Suppl 1):60–62. doi: 10.4158/EP.12.S1.60. [DOI] [PubMed] [Google Scholar]
- Chen J, Liu Y, Liu H, Hermonat PL, Mehta JL. Lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) transcriptional regulation by Oct-1 in human endothelial cells: implications for atherosclerosis. BiochemJ. 2006;393:255–265. doi: 10.1042/BJ20050845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens A, Siegel E, Gallwitz B. Global risk management in type 2 diabetes: blood glucose, blood pressure, and lipids--update on the background of the current guidelines. ExpClinEndocrinolDiabetes. 2004;112:493–503. doi: 10.1055/s-2004-821306. [DOI] [PubMed] [Google Scholar]
- Cotter MA, Cameron NE. Effect of the NAD(P)H oxidase inhibitor, apocynin, on peripheral nerve perfusion and function in diabetic rats. Life sciences. 2003;73:1813–1824. doi: 10.1016/s0024-3205(03)00508-3. [DOI] [PubMed] [Google Scholar]
- Dandapat A, Hu C, Sun L, Mehta JL. Small concentrations of oxLDL induce capillary tube formation from endothelial cells via LOX-1-dependent redox-sensitive pathway. Arteriosclerosis, thrombosis, and vascular biology. 2007;27:2435–2442. doi: 10.1161/ATVBAHA.107.152272. [DOI] [PubMed] [Google Scholar]
- Davis TM, Yeap BB, Davis WA, Bruce DG. Lipid-lowering therapy and peripheral sensory neuropathy in type 2 diabetes: the Fremantle Diabetes Study. Diabetologia. 2008;51:562–566. doi: 10.1007/s00125-007-0919-2. [DOI] [PubMed] [Google Scholar]
- Deplanque D, Gele P, Petrault O, Six I, Furman C, Bouly M, Nion S, Dupuis B, Leys D, Fruchart JC, Cecchelli R, Staels B, Duriez P, Bordet R. Peroxisome proliferator-activated receptor-alpha activation as a mechanism of preventive neuroprotection induced by chronic fenofibrate treatment. J Neurosci. 2003;23:6264–6271. doi: 10.1523/JNEUROSCI.23-15-06264.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmeler S, Haendeler J, Galle J, Zeiher AM. Oxidized low-density lipoprotein induces apoptosis of human endothelial cells by activation of CPP32-like proteases. A mechanistic clue to the ‘response to injury’ hypothesis. Circulation. 1997;95:1760–1763. doi: 10.1161/01.cir.95.7.1760. [DOI] [PubMed] [Google Scholar]
- Dominguez JH, Mehta JL, Li D, Wu P, Kelly KJ, Packer CS, Temm C, Goss E, Cheng L, Zhang S, Patterson CE, Hawes JW, Peterson R. Anti-LOX-1 therapy in rats with diabetes and dyslipidemia: ablation of renal vascular and epithelial manifestations. Am J Physiol Renal Physiol. 2008;294:F110–119. doi: 10.1152/ajprenal.00013.2007. [DOI] [PubMed] [Google Scholar]
- Draczynska-Lusiak B, Chen YM, Sun AY. Oxidized lipoproteins activate NF-kappaB binding activity and apoptosis in PC12 cells. Neuroreport. 1998a;9:527–532. doi: 10.1097/00001756-199802160-00028. [DOI] [PubMed] [Google Scholar]
- Draczynska-Lusiak B, Doung A, Sun AY. Oxidized lipoproteins may play a role in neuronal cell death in Alzheimer disease. MolChem Neuropathol. 1998b;33:139–148. doi: 10.1007/BF02870187. [DOI] [PubMed] [Google Scholar]
- Draude G, Hrboticky N, Lorenz RL. The expression of the lectin-like oxidized low-density lipoprotein receptor (LOX-1) on human vascular smooth muscle cells and monocytes and its down-regulation by lovastatin. Biochemical Pharmacology. 1999;57:383–386. doi: 10.1016/s0006-2952(98)00313-x. [DOI] [PubMed] [Google Scholar]
- Edwards JL, Quattrini A, Lentz SI, Figueroa C, Cerri F, Backus C, Hong Y, Feldman EL. Diabetes Regulates Mitochondrial Biogenesis and Fission in Neurons. The Journal of biological chemistry. 2009 doi: 10.1007/s00125-009-1553-y. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards JL, Vincent AM, Cheng HT, Feldman EL. Diabetic neuropathy: mechanisms to management. Pharmacology & therapeutics. 2008;120:1–34. doi: 10.1016/j.pharmthera.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman EL, Stevens MJ, Greene DA. Pathogenesis of diabetic neuropathy. Clinical Neuroscience. 1997;4:365–370. [PubMed] [Google Scholar]
- Feldman EL, Stevens MJ, Russell JW, Sperling MA. Contemporary Endocrinology. Humana Press; 2002. Diabetic peripheral and autonomic neuropathy. [Google Scholar]
- Figueroa-Romero C, Sadidi M, Feldman EL. Mechanisms of disease: the oxidative stress theory of diabetic neuropathy. Reviews in endocrine & metabolic disorders. 2008;9:301–314. doi: 10.1007/s11154-008-9104-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth J. Fenofibrate and diabetic retinopathy. Lancet. 2008;371:722. doi: 10.1016/S0140-6736(08)60334-7. author reply 722. [DOI] [PubMed] [Google Scholar]
- Fonseca VA. The metabolic syndrome, hyperlipidemia, and insulin resistance. ClinCornerstone. 2005;7:61–72. doi: 10.1016/s1098-3597(05)80069-9. [DOI] [PubMed] [Google Scholar]
- Genuth S. Insights from the diabetes control and complications trial/epidemiology of diabetes interventions and complications study on the use of intensive glycemic treatment to reduce the risk of complications of type 1 diabetes. Endocr Pract. 2006a;12(Suppl 1):34–41. doi: 10.4158/EP.12.S1.34. [DOI] [PubMed] [Google Scholar]
- Genuth S. Insights from the diabetes control and complications trial/epidemiology of diabetes interventions and complications study on the use of intensive glycemic treatment to reduce the risk of complications of type 1 diabetes. EndocrPract. 2006b;12(Suppl 1):34–41. doi: 10.4158/EP.12.S1.34. [DOI] [PubMed] [Google Scholar]
- Giugliano D, Ceriello A, Esposito K. Glucose metabolism and hyperglycemia. Am J Clin Nutr. 2008;87:217S–222S. doi: 10.1093/ajcn/87.1.217S. [DOI] [PubMed] [Google Scholar]
- Gordon SA, Robinson SJ. Idiopathic neuropathy, prediabetes and the metabolic syndrome. J NeurolSci. 2006;242:9–14. doi: 10.1016/j.jns.2005.11.020. [DOI] [PubMed] [Google Scholar]
- Greene DA, Stevens MJ, Obrosova I, Feldman EL. Glucose-induced oxidative stress and programmed cell death in diabetic neuropathy. European journal of pharmacology. 1999;375:217–223. doi: 10.1016/s0014-2999(99)00356-8. [DOI] [PubMed] [Google Scholar]
- Grundy SM. A constellation of complications: the metabolic syndrome. ClinCornerstone. 2005;7:36–45. doi: 10.1016/s1098-3597(05)80066-3. [DOI] [PubMed] [Google Scholar]
- Hammer A, Kager G, Dohr G, Rabl H, Ghassempur I, Jurgens G. Generation, characterization, and histochemical application of monoclonal antibodies selectively recognizing oxidatively modified apoB-containing serum lipoproteins. ArteriosclerThrombVascBiol. 1995;15:704–713. doi: 10.1161/01.atv.15.5.704. [DOI] [PubMed] [Google Scholar]
- Harano Y, Yasui K, Toyama T, Nakajima T, Mitsuyoshi H, Mimani M, Hirasawa T, Itoh Y, Okanoue T. Fenofibrate, a peroxisome proliferator-activated receptor alpha agonist, reduces hepatic steatosis and lipid peroxidation in fatty liver Shionogi mice with hereditary fatty liver. Liver Int. 2006;26:613–620. doi: 10.1111/j.1478-3231.2006.01265.x. [DOI] [PubMed] [Google Scholar]
- Heinecke JW. Free radical modification of low-density lipoprotein: mechanisms and biological consequences. Free Radic Biol Med. 1987;3:65–73. doi: 10.1016/0891-5849(87)90040-2. [DOI] [PubMed] [Google Scholar]
- Herz J, Bock HH. Lipoprotein receptors in the nervous system. Annual Review of Biochemistry. 2002;71:405–434. doi: 10.1146/annurev.biochem.71.110601.135342. [DOI] [PubMed] [Google Scholar]
- Higashi Y, Peng T, Du J, Sukhanov S, Li Y, Itabe H, Parthasarathy S, Delafontaine P. A redox-sensitive pathway mediates oxidized LDL-induced downregulation of insulin-like growth factor-1 receptor. J Lipid Res. 2005;46:1266–1277. doi: 10.1194/jlr.M400478-JLR200. [DOI] [PubMed] [Google Scholar]
- Hu B, Li D, Sawamura T, Mehta JL. Oxidized LDL through LOX-1 modulates LDL-receptor expression in human coronary artery endothelial cells. BiochemBiophys ResCommun. 2003;307:1008–1012. doi: 10.1016/s0006-291x(03)01295-6. [DOI] [PubMed] [Google Scholar]
- Isomaa B, Henricsson M, Almgren P, Tuomi T, Taskinen MR, Groop L. The metabolic syndrome influences the risk of chronic complications in patients with type II diabetes. Diabetologia. 2001;44:1148–1154. doi: 10.1007/s001250100615. [DOI] [PubMed] [Google Scholar]
- Iwashima Y, Eto M, Hata A, Kaku K, Horiuchi S, Ushikubi F, Sano H. Advanced glycation end products-induced gene expression of scavenger receptors in cultured human monocyte-derived macrophages. BiochemBiophys ResCommun. 2000;277:368–380. doi: 10.1006/bbrc.2000.3685. [DOI] [PubMed] [Google Scholar]
- Keech AC, Mitchell P, Summanen PA, O'Day J, Davis TM, Moffitt MS, Taskinen MR, Simes RJ, Tse D, Williamson E, Merrifield A, Laatikainen LT, d'Emden MC, Crimet DC, O'Connell RL, Colman PG. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): a randomised controlled trial. Lancet. 2007;370:1687–1697. doi: 10.1016/S0140-6736(07)61607-9. [DOI] [PubMed] [Google Scholar]
- Keller JN, Hanni KB, Kindy MS. Oxidized high-density lipoprotein induces neuron death. Experimental neurology. 2000;161:621–630. doi: 10.1006/exnr.1999.7276. [DOI] [PubMed] [Google Scholar]
- Keller JN, Hanni KB, Pedersen WA, Cashman NR, Mattson MP, Gabbita SP, Friebe V, Markesbery WR. Opposing actions of native and oxidized lipoprotein on motor neuron-like cells. Experimental neurology. 1999;157:202–210. doi: 10.1006/exnr.1999.7043. [DOI] [PubMed] [Google Scholar]
- Kelly KJ, Wu P, Patterson CE, Temm C, Dominguez JH. LOX-1 and inflammation: a new mechanism for renal injury in obesity and diabetes. Am J Physiol Renal Physiol. 2008;294:F1136–1145. doi: 10.1152/ajprenal.00396.2007. [DOI] [PubMed] [Google Scholar]
- Kempler P, Tesfaye S, Chaturvedi N, Stevens LK, Webb DJ, Eaton S, Kerenyi Z, Tamas G, Ward JD, Fuller JH. Autonomic neuropathy is associated with increased cardiovascular risk factors: the EURODIAB IDDM Complications Study. DiabetMed. 2002;19:900–909. doi: 10.1046/j.1464-5491.2002.00821.x. [DOI] [PubMed] [Google Scholar]
- Kles KA, Vinik AI. Pathophysiology and treatment of diabetic peripheral neuropathy: the case for diabetic neurovascular function as an essential component. Current diabetes reviews. 2006;2:131–145. doi: 10.2174/157339906776818569. [DOI] [PubMed] [Google Scholar]
- Kooistra T, Verschuren L, de Vries-van der Weij J, Koenig W, Toet K, Princen HM, Kleemann R. Fenofibrate reduces atherogenesis in ApoE*3Leiden mice: evidence for multiple antiatherogenic effects besides lowering plasma cholesterol. Arteriosclerosis, thrombosis, and vascular biology. 2006;26:2322–2330. doi: 10.1161/01.ATV.0000238348.05028.14. [DOI] [PubMed] [Google Scholar]
- Krentz AJ. Lipoprotein abnormalities and their consequences for patients with type 2 diabetes. Diabetes ObesMetab. 2003;5(Suppl 1):S19–S27. doi: 10.1046/j.1462-8902.2003.0310.x. [DOI] [PubMed] [Google Scholar]
- Leiter LA. The prevention of diabetic microvascular complications of diabetes: is there a role for lipid lowering? Diabetes research and clinical practice. 2005;68(Suppl 2):S3–14. doi: 10.1016/j.diabres.2005.03.015. [DOI] [PubMed] [Google Scholar]
- Li D, Mehta JL. Oxidized LDL, a critical factor in atherogenesis. Cardiovascular Research. 2005;68:353–354. doi: 10.1016/j.cardiores.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Li L, Sawamura T, Renier G. Glucose enhances endothelial LOX-1 expression: role for LOX-1 in glucose-induced human monocyte adhesion to endothelium. Diabetes. 2003;52:1843–1850. doi: 10.2337/diabetes.52.7.1843. [DOI] [PubMed] [Google Scholar]
- Li L, Sawamura T, Renier G. Glucose enhances human macrophage LOX-1 expression: role for LOX-1 in glucose-induced macrophage foam cell formation. Circulation Research. 2004;94:892–901. doi: 10.1161/01.RES.0000124920.09738.26. [DOI] [PubMed] [Google Scholar]
- Lie J, Lankhuizen IM, Gross B, van Gent T, van Haperen R, Scheek L, Staels B, de Crom R, van Tol A. Fenofibrate reverses the decline in HDL cholesterol in mice overexpressing human phospholipid transfer protein. Biochimica et biophysica acta. 2005;1738:48–53. doi: 10.1016/j.bbalip.2005.10.002. [DOI] [PubMed] [Google Scholar]
- Low PA, Nickander KK, Tritschler HJ. The roles of oxidative stress and antioxidant treatment in experimental diabetic neuropathy. Diabetes. 1997;46(Suppl 2):S38–42. doi: 10.2337/diab.46.2.s38. [DOI] [PubMed] [Google Scholar]
- Lyons TJ, Jenkins AJ, Zheng D, Lackland DT, McGee D, Garvey WT, Klein RL. Diabetic retinopathy and serum lipoprotein subclasses in the DCCT/EDIC cohort. Invest OphthalmolVisSci. 2004;45:910–918. doi: 10.1167/iovs.02-0648. [DOI] [PubMed] [Google Scholar]
- Martin CL, Albers J, Herman WH, Cleary P, Waberski B, Greene DA, Stevens MJ, Feldman EL. Neuropathy among the diabetes control and complications trial cohort 8 years after trial completion. Diabetes care. 2006a;29:340–344. doi: 10.2337/diacare.29.02.06.dc05-1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin CL, Albers J, Herman WH, Cleary P, Waberski B, Greene DA, Stevens MJ, Feldman EL. Neuropathy among the diabetes control and complications trial cohort 8 years after trial completion. Diabetes care. 2006b;29:340–344. doi: 10.2337/diacare.29.02.06.dc05-1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManis PG, Windebank AJ, Kiziltan M. Neuropathy associated with hyperlipidemia. Neurology. 1994;44:2185–2186. doi: 10.1212/wnl.44.11.2185. [DOI] [PubMed] [Google Scholar]
- Murdoch CE, Grieve DJ, Cave AC, Looi YH, Shah AM. NADPH oxidase and heart failure. CurrOpinPharmacol. 2006;6:148–153. doi: 10.1016/j.coph.2005.10.008. [DOI] [PubMed] [Google Scholar]
- Napoli C. Oxidation of LDL, atherogenesis, and apoptosis. Annals of the New York Academy of Sciences. 2003;1010:698–709. doi: 10.1196/annals.1299.127. [DOI] [PubMed] [Google Scholar]
- Osawa T, Kato Y. Protective role of antioxidative food factors in oxidative stress caused by hyperglycemia. Annals of the New York Academy of Sciences. 2005;1043:440–451. doi: 10.1196/annals.1333.050. [DOI] [PubMed] [Google Scholar]
- Papassotiropoulos A, Ludwig M, Naib-Majani W, Rao GS. Induction of apoptosis and secondary necrosis in rat dorsal root ganglion cell cultures by oxidized low density lipoprotein. Neuroscience Letters. 1996;209:33–36. doi: 10.1016/0304-3940(96)12595-7. [DOI] [PubMed] [Google Scholar]
- Papassotiropoulos A, Wollmer MA, Tsolaki M, Brunner F, Molyva D, Lutjohann D, Nitsch RM, Hock C. A cluster of cholesterol-related genes confers susceptibility for Alzheimer's disease. J ClinPsychiatry. 2005;66:940–947. [PubMed] [Google Scholar]
- Park CW, Zhang Y, Zhang X, Wu J, Chen L, Cha DR, Su D, Hwang MT, Fan X, Davis L, Striker G, Zheng F, Breyer M, Guan Y. PPARalpha agonist fenofibrate improves diabetic nephropathy in db/db mice. Kidney international. 2006;69:1511–1517. doi: 10.1038/sj.ki.5000209. [DOI] [PubMed] [Google Scholar]
- Pennathur S, Heinecke JW. Oxidative stress and endothelial dysfunction in vascular disease. Current diabetes reports. 2007;7:257–264. doi: 10.1007/s11892-007-0041-3. [DOI] [PubMed] [Google Scholar]
- Pop-Busui R, Low PA, Waberski BH, Martin CL, Albers JW, Feldman EL, Sommer C, Cleary PA, Lachin JM, Herman WH. Effects of prior intensive insulin therapy on cardiac autonomic nervous system function in type 1 diabetes mellitus: the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications study (DCCT/EDIC) Circulation. 2009;119:2886–2893. doi: 10.1161/CIRCULATIONAHA.108.837369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudijanto A. The expression and down stream effect of lectin like-oxidized low density lipoprotein 1 (LOX-1) in hyperglycemic state. Acta medica Indonesiana. 2007;39:36–43. [PubMed] [Google Scholar]
- Russell JW, Berent-Spillson A, Vincent AM, Freimann CL, Sullivan KA, Feldman EL. Oxidative injury and neuropathy in diabetes and impaired glucose tolerance. Neurobiology of disease. 2008 doi: 10.1016/j.nbd.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell JW, Gong C, Vincent A, Berent AR, Mentzer LE, Brownlee M. Nitric oxide (NO) and mitochondrial manganese superoxide dismutase (MnSOD) regulate glucose-induced oxidative stress and programmed cell death in neurons. Neurology. 2001;56(Suppl 3):A394. [Google Scholar]
- Russell JW, Sullivan KA, Windebank AJ, Herrmann DN, Feldman EL. Neurons undergo apoptosis in animal and cell culture models of diabetes. Neurobiology of disease. 1999;6:347–363. doi: 10.1006/nbdi.1999.0254. [DOI] [PubMed] [Google Scholar]
- Russell JW, van Golen C, Feldman EL. IGF-I prevents glucose mediated loss of mitochondrial membrane integrity and programmed cell death in models of diabetic neuropathy. Annals of Neurology. 1998;44:439. [Google Scholar]
- Saenz A, Fernandez-Esteban I, Mataix A, Ausejo M, Roque M, Moher D. Metformin monotherapy for type 2 diabetes mellitus. Cochrane database of systematic reviews. 2005:CD002966. doi: 10.1002/14651858.CD002966.pub3. Online. [DOI] [PubMed] [Google Scholar]
- Schalkwijk CG, Stehouwer CD. Vascular complications in diabetes mellitus: the role of endothelial dysfunction. ClinSci (Lond) 2005;109:143–159. doi: 10.1042/CS20050025. [DOI] [PubMed] [Google Scholar]
- Schroeter H, Williams RJ, Matin R, Iversen L, Rice-Evans CA. Phenolic antioxidants attenuate neuronal cell death following uptake of oxidized low-density lipoprotein. Free Radic Biol Med. 2000;29:1222–1233. doi: 10.1016/s0891-5849(00)00415-9. [DOI] [PubMed] [Google Scholar]
- Shamir R, Kassis H, Kaplan M, Naveh T, Shehadeh N. Glycemic control in adolescents with type 1 diabetes mellitus improves lipid serum levels and oxidative stress. Pediatric diabetes. 2008;9:104–109. doi: 10.1111/j.1399-5448.2007.00313.x. [DOI] [PubMed] [Google Scholar]
- Silver AE, Beske SD, Christou DD, Donato AJ, Moreau KL, Eskurza I, Gates PE, Seals DR. Overweight and obese humans demonstrate increased vascular endothelial NAD(P)H oxidase-p47(phox) expression and evidence of endothelial oxidative stress. Circulation. 2007;115:627–637. doi: 10.1161/CIRCULATIONAHA.106.657486. [DOI] [PubMed] [Google Scholar]
- Simo R, Hernandez C. Fenofibrate for diabetic retinopathy. Lancet. 2007;370:1667–1668. doi: 10.1016/S0140-6736(07)61608-0. [DOI] [PubMed] [Google Scholar]
- Srivastava RA, Jahagirdar R, Azhar S, Sharma S, Bisgaier CL. Peroxisome proliferator-activated receptor-alpha selective ligand reduces adiposity, improves insulin sensitivity and inhibits atherosclerosis in LDL receptor-deficient mice. Molecular and cellular biochemistry. 2006;285:35–50. doi: 10.1007/s11010-005-9053-y. [DOI] [PubMed] [Google Scholar]
- Stamler JS. Alzheimer's disease. A radical vascular connection. Nature. 1996;380:108–111. [PubMed] [Google Scholar]
- Sullivan KA, Feldman EL. New developments in diabetic neuropathy. Curr Opin Neurol. 2005;18:586–590. doi: 10.1097/01.wco.0000178825.56414.52. [DOI] [PubMed] [Google Scholar]
- Tesfaye S. Advances in the management of painful diabetic neuropathy. Clinical medicine (London, England) 2007;7:113–114. doi: 10.7861/clinmedicine.7-2-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesfaye S, Chaturvedi N, Eaton SE, Ward JD, Manes C, Ionescu-Tirgoviste C, Witte DR, Fuller JH. Vascular risk factors and diabetic neuropathy. The New England journal of medicine. 2005;352:341–350. doi: 10.1056/NEJMoa032782. [DOI] [PubMed] [Google Scholar]
- The Diabetes Control and Complications Trial Research G. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. New England Journal of Medicine. 1993;329:977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- Thum T, Borlak J. Lox-1 receptor blockade abrogates OXLDL induced oxidative DNA damage and prevents activation of the transcriptional repressor OCT-1 in human coronary arterial endothelium. The Journal of biological chemistry. 2008 doi: 10.1074/jbc.M708309200. [DOI] [PubMed] [Google Scholar]
- Tomlinson DR, Gardiner NJ. Glucose neurotoxicity. Nature reviews. 2008;9:36–45. doi: 10.1038/nrn2294. [DOI] [PubMed] [Google Scholar]
- Tsimikas S, Brilakis ES, Miller ER, McConnell JP, Lennon RJ, Kornman KS, Witztum JL, Berger PB. Oxidized phospholipids, Lp(a) lipoprotein, and coronary artery disease. N EnglJ Med. 2005;353:46–57. doi: 10.1056/NEJMoa043175. [DOI] [PubMed] [Google Scholar]
- Tsuzura S, Ikeda Y, Suehiro T, Ota K, Osaki F, Arii K, Kumon Y, Hashimoto K. Correlation of plasma oxidized low-density lipoprotein levels to vascular complications and human serum paraoxonase in patients with type 2 diabetes. Metabolism: clinical and experimental. 2004a;53:297–302. doi: 10.1016/j.metabol.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Tsuzura S, Ikeda Y, Suehiro T, Ota K, Osaki F, Arii K, Kumon Y, Hashimoto K. Correlation of plasma oxidized low-density lipoprotein levels to vascular complications and human serum paraoxonase in patients with type 2 diabetes. Metabolism: clinical and experimental. 2004b;53:297–302. doi: 10.1016/j.metabol.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Ueno T, Kaname S, Takaichi K, Nagase M, Tojo A, Onozato ML, Fujita T. LOX-1, an oxidized low-density lipoprotein receptor, was upregulated in the kidneys of chronic renal failure rats. Hypertens Res. 2003;26:117–122. doi: 10.1291/hypres.26.117. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Edwards JL, Sadidi M, Feldman EL. The antioxidant response as a drug target in diabetic neuropathy. Current drug targets. 2008;9:94–100. doi: 10.2174/138945008783431754. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Feldman EL. New insights into the mechanisms of diabetic neuropathy. Reviews in endocrine & metabolic disorders. 2004;5:227–236. doi: 10.1023/B:REMD.0000032411.11422.e0. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Feldman EL. Can drug screening lead to candidate therapies for testing in diabetic neuropathy? Antioxidants & redox signaling. 2008;10:387–393. doi: 10.1089/ars.2007.1815. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Hayes JM, McLean LL, Vivekanandan-Giri A, Pennathur S, Feldman EL. Dyslipidemia-Induced Neuropathy in Mice: the Role of oxLDL/LOX-1. Diabetes. 2009a doi: 10.2337/db09-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent AM, Hayes JM, McLean LL, Vivekanandan-Giri A, Pennathur S, Feldman EL. Dyslipidemia-induced neuropathy in mice: the role of oxLDL/LOX-1. 2009b doi: 10.2337/db09-0047. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent AM, McLean LL, Backus C, Feldman EL. Short-term hyperglycemia produces oxidative damage and apoptosis in neurons. Faseb J. 2005a;19:638–640. doi: 10.1096/fj.04-2513fje. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Olzmann JA, Brownlee M, Sivitz WI, Russell JW. Uncoupling proteins prevent glucose-induced neuronal oxidative stress and programmed cell death. Diabetes. 2004a;53:726–734. doi: 10.2337/diabetes.53.3.726. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Perrone L, Sullivan KA, Backus C, Sastry AM, Lastoskie C, Feldman EL. Receptor for advanced glycation end products activation injures primary sensory neurons via oxidative stress. Endocrinology. 2007a;148:548–558. doi: 10.1210/en.2006-0073. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Russell JW, Low P, Feldman EL. Oxidative stress in the pathogenesis of diabetic neuropathy. Endocrine reviews. 2004b;25:612–628. doi: 10.1210/er.2003-0019. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Russell JW, Sullivan KA, Backus C, Hayes JM, McLean LL, Feldman EL. SOD2 protects neurons from injury in cell culture and animal models of diabetic neuropathy. Experimental neurology. 2007b;208:216–227. doi: 10.1016/j.expneurol.2007.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent AM, Stevens MJ, Backus C, McLean LL, Feldman EL. Cell culture modeling to test therapies against hyperglycemia-mediated oxidative stress and injury. Antioxidants & redox signaling. 2005b;7:1494–1506. doi: 10.1089/ars.2005.7.1494. [DOI] [PubMed] [Google Scholar]
- Vindis C, Elbaz M, Escargueil-Blanc I, Auge N, Heniquez A, Thiers JC, Negre-Salvayre A, Salvayre R. Two distinct calcium-dependent mitochondrial pathways are involved in oxidized LDL-induced apoptosis. ArteriosclerThrombVascBiol. 2005;25:639–645. doi: 10.1161/01.ATV.0000154359.60886.33. [DOI] [PubMed] [Google Scholar]
- Wiggin TD, Kretzler M, Pennathur S, Sullivan KA, Brosius FC, Feldman EL. Rosiglitazone Treatment Reduces Diabetic Neuropathy in STZ Treated DBA/2J Mice. Endocrinology. 2008 doi: 10.1210/en.2008-0869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggin TD, Sullivan KA, Pop-Busui R, Amato A, Sima AA, Feldman EL. Elevated triglycerides correlate with progression of diabetic neuropathy. Diabetes. 2009a;58:1634–1640. doi: 10.2337/db08-1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggin TD, Sullivan KA, Pop-Busui R, Amato A, Sima AAF, Feldman EL. Elevated Triglycerides Correlate with Progression of Diabetic Neuropathy. Diabetes. 2009b doi: 10.2337/db08-1771. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems D, Dorchy H, Dufrasne D. Serum antioxidant status and oxidized LDL in well-controlled young type 1 diabetic patients with and without subclinical complications. Atherosclerosis. 1998a;137(Suppl):S61–64. doi: 10.1016/s0021-9150(97)00320-1. [DOI] [PubMed] [Google Scholar]
- Willems D, Dorchy H, Dufrasne D. Serum antioxidant status and oxidized LDL in well-controlled young type 1 diabetic patients with and without subclinical complications. Atherosclerosis. 1998b;137(Suppl):S61–S64. doi: 10.1016/s0021-9150(97)00320-1. [DOI] [PubMed] [Google Scholar]
- Xie W, Nie Y, Du L, Zhang Y, Cai G. Preventive effects of fenofibrate on insulin resistance, hyperglycaemia, visceral fat accumulation in NIH mice induced by small-dose streptozotocin and lard. Pharmacol Res. 2007;55:392–399. doi: 10.1016/j.phrs.2007.01.014. [DOI] [PubMed] [Google Scholar]
- Yamashita S, Hirano KI, Kuwasako T, Janabi M, Toyama Y, Ishigami M, Sakai N. Physiological and pathological roles of a multi-ligand receptor CD36 in atherogenesis; insights from CD36-deficient patients. MolCell Biochem. 2006 doi: 10.1007/s11010-005-9031-4. [DOI] [PubMed] [Google Scholar]
- Yoon SH, Han EJ, Sung JH, Chung SH. Anti-diabetic effects of compound K versus metformin versus compound K-metformin combination therapy in diabetic db/db mice. Biological & pharmaceutical bulletin. 2007;30:2196–2200. doi: 10.1248/bpb.30.2196. [DOI] [PubMed] [Google Scholar]
- Young MJ, Boulton AJM, Macleod AF, Williams DRR, Sonksen PH. A multicentre study of the prevalence of diabetic peripheral neuropathy in the United Kingdom hospital clinic population. Diabetologia. 1993;36:150–154. doi: 10.1007/BF00400697. [DOI] [PubMed] [Google Scholar]
- Ziegler D, Ametov A, Barinov A, Dyck PJ, Gurieva I, Low PA, Munzel U, Yakhno N, Raz I, Novosadova M, Maus J, Samigullin R. Oral treatment with alpha-lipoic acid improves symptomatic diabetic polyneuropathy: the SYDNEY 2 trial. Diabetes care. 2006;29:2365–2370. doi: 10.2337/dc06-1216. [DOI] [PubMed] [Google Scholar]
- Ziegler D, Sohr CG, Nourooz-Zadeh J. Oxidative stress and antioxidant defense in relation to the severity of diabetic polyneuropathy and cardiovascular autonomic neuropathy. Diabetes care. 2004;27:2178–2183. doi: 10.2337/diacare.27.9.2178. [DOI] [PubMed] [Google Scholar]
- Zimmet P, Alberti G. The metabolic syndrome: progress towards one definition for an epidemic of our time. Nature clinical practice. 2008;4:239. doi: 10.1038/ncpendmet0834. [DOI] [PubMed] [Google Scholar]