

Abstract
Diabetic neuropathy is the most common complication of diabetes, affecting 50% of diabetic patients. Currently, the only treatment for diabetic neuropathy is glucose control and careful foot care. In this review, we discuss the idea that excess glucose overloads the electron transport chain, leading to the production of superoxides and subsequent mitochondrial and cytosolic oxidative stress. Defects in metabolic and vascular pathways intersect with oxidative stress to produce the onset and progression of nerve injury present in diabetic neuropathy. These pathways include the production of advanced glycation end products, alterations in the sorbitol, hexosamine and protein kinase C pathways and activation of Poly-ADP ribose polymerase. New bioinformatics approaches can augment current research and lead to new discoveries to understand the pathogenesis of diabetic neuropathy and to identify more effective molecular therapeutic targets.
Keywords: Diabetes, Neuropathy, Oxidative Stress, Bioinformatics
1. Introduction
Diabetic neuropathy is the most common complication of diabetes. While estimates vary, depending on the methods used to diagnose diabetic neuropathy, it is generally held that at least 50% of all diabetic patients will develop neuropathy in his or her lifetime [1–8]. This high prevalence of neuropathy is likely an underestimate as several recent studies report that patients with impaired fasting glucose and/or impaired glucose tolerance also exhibit neuropathy at the time of diagnosis. Diabetic neuropathy is the most common cause of foot ulcers and non-traumatic amputations in the Western world. Patients with diabetic neuropathy report a poor quality of life secondary to pain, disability and recurrent hospitalizations. It is estimated that in the United States the annual cost of diabetic neuropathy is nearly $11 billion dollars and increasing annually in parallel with the alarming increase in the incidence and prevalence of diabetes (www.diabetes.org).
There are no treatments for diabetic neuropathy other than glycemic control and diligent foot care [1, 3, 7, 9–11]. This is in spite of ongoing research addressing the pathogenesis of the disorder, with the goal to identify mechanism based treatments. In recent years, the idea has emerged that multiple distinct metabolic pathways are impaired leading to a singular end result: enhanced cellular oxidative stress. This review will focus on the relationship between mitochondrial and cytosolic oxidative stress and the biochemical pathways that converge to enhance cellular oxidative stress and the onset and progression of diabetic neuropathy. The reader is referred to the following recent review articles for reviews of the symptoms, staging and treatment of diabetic neuropathy [1–8, 12, 13].
2. Oxidative Stress: Reactive Oxygen/Nitrogen Species (ROS/RNS) Formation in the Mitochondria
The “free radical theory” of aging was proposed by Harman in 1956. Based on his theory of aging, “the reaction of active free radicals, normally produced in organisms, with cellular constituents initiate the changes associated with aging.” Excess generation of free radicals results in upregulation of stress signaling, negatively affecting both life quality and life span. Reactive oxygen (ROS) and reactive nitrogen (RNS) species are linked to multiple disease states [14–18], including the microvascular complications of diabetes [19–23]. Mitochondrial metabolism and the cascade of oxidative phosphorylation are highlighted as key contributors of ROS generation in many diseases.
Mitochondrial oxidative phosphorylation is the major ATP synthetic pathway in eukaryotes. In this process, electrons from reducing substrates are transferred to molecular oxygen (O2) via respiratory chain complexes I–IV. These complexes establish a hydrogen gradient across the inner mitochondrial membrane, and the electrochemical energy of this gradient is then used to drive ATP synthesis by ATP synthase (complex V).
There are four protein complexes associated with the respiratory chain. NADH-ubiquinone oxidoreductase, or complex I, accepts electrons from NADH; these electrons are carried to succinate dehydrogenase, complex II, and used to oxidize succinate to fumarate. Electrons continue to travel down an electrochemical gradient to ubiquinol-cytochrome c oxidoreductase (complex III), and subsequently to cytochrome c oxidase (complex IV), which are finally used to reduce molecular oxygen to water. Even though the majority of molecular oxygen is reduced at complex IV to water via the respiratory chain, 1–4% of the oxygen is incompletely reduced to superoxide (O2•−) [24]. O2•− is the most common ROS and creates other ROS/RNS via various enzymatic or nonenzymatic reactions discussed later in this review.
O2•− generation by mitochondrial electron transport chain is mainly at complexes I and III. It is suggested that O2•− production in complex I is via reverse electron transfer, and is predominately released into the matrix [25]. Autoxidation of the ubisemiquinone radical intermediate (QH•) at complex III is the other source of O2•− generation. Complex III has the capacity to release O2•− to both sides of the mitochondrial inner membrane, however, the Q site closer to the intermembrane space (Qo), is known to be the major site of O2•− production, and the matrix side (Qi) is less likely to form O2•− [26, 27].
As stated above, O2•− is the major ROS produced in the intermembrane space or matrix of mitochondria. As a charged reactive species, O2•− does not readily diffuse across mitochondrial membranes. However, the mitochondrial permeability transition pore might serve as a channel for intermembranous mitochondrial O2•− to pass through the outer mitochondrial membrane and into the cytosol [28]. The conversion of O2•− to hydrogen peroxide (H2O2) by superoxide dismutase (SOD) facilitates permeation through the generation of uncharged ROS, which easily diffuse across the membrane.
There are three isoforms of SOD, SOD 1 or copper zinc SOD (CuZn-SOD), SOD 2 or manganese SOD (Mn-SOD), and SOD 3 or extracellular CuZn-SOD (EC-SOD) [29]. CuZn-SOD (SOD 1) is found in the cytosol, nucleus, and intermembrane space of mitochondria [30], Mn-SOD (SOD 2) is expressed only in the mitochondrial matrix, and SOD 3 is located in the extracellular space. Among all SOD isoforms, SOD 2 is physiologically more important and its genetic elimination, in contrast to other isoforms, is embryonically lethal [31, 32].
H2O2 is reduced enzymatically by catalase and glutathione peroxidase. In the mitochondrial matrix, glutathione peroxidase uses glutathione to convert H2O2 to water. Catalase has higher Km for H2O2 compared to glutathione peroxidase, and can protect against a higher concentration of H2O2 [33]. Other antioxidant enzymes such as gluthathione S-transferase and thioredoxin [34] also help in removal and inactivation of ROS formed in the mitochondria. In the presence of transition metals such as copper and iron, H2O2 generates hydroxyl radical ( OH) via the Fenton reaction or the Haber-Weiss reaction. Hydroxyl radicals are very highly reactive and contribute significantly to local organelle damage through DNA and protein modification.
3. Regulation of Mitochondrial ROS Production
In the process of oxidative phosphorylation, energy carried by electrons is used by complexes I, III, and IV to pump protons out of the matrix. The resulting electrochemical gradient across the mitochondrial inner membrane is used by ATP synthase to drive the synthesis of ATP from ADP. In mitochondria, increased ATP synthesis is regulated by uncoupling proteins. Upon activation of uncoupling proteins (UCP), protons leak across the inner membrane and “uncouple” oxidative metabolism from ATP synthase, resulting in loss of ATP production. Basal and hyperglycemia-induced ROS formation are decreased in dorsal root ganglia sensory neurons that over express UCP [35]. Mitochondrial membrane permeability is increased via activation of UCP by O2•−, resulting in decreased electrochemical potential and further reduction of O2•− generation. Mild mitochondrial depolarization that limits Ca2+ accumulation and reduces reactive species generation (e.g. by limiting nitric oxide synthase, NOS, activity) may explain the protective effect of UPC [36].
Mitochondrial ROS are also regulated by nitric oxide (NO), a diffusible gas produced by NOS. The presence of mitochondrial NOS (Mt NOS) and its activity were reported by Ghafouri and Richter in 1997 [37]. Mt NOS is associated with the matrix face of the mitochondrial inner membrane. The activity of Mt NOS is regulated by intramitochondrial Ca2+ concentration, [Ca2+]m [37]. Elevation of [Ca2+]m increases NO production and leads to reduction in Δψ, while a decrease in Δψ releases Ca2+ from the mitochondria and results in Mt NOS inactivation.
Other ROS/RNS are generated in the mitochondria from interaction of O2•− and other reactive species. Generation of O2•− in the presence of NO results in peroxynitrite (ONOO-) formation. The rate of ONOO− formation is 9.5 × 10−8 M s−1, which exceeds the interaction of NO with cytochrome c oxidase (0.8 × 10−8 M s−1) [38, 39]. Ghafouri et al. (1999) reported that peroxynitrite-induced stress promotes cytochrome c release from the mitochondria and results in apoptosis [40]. Mt NOS is also involved in mitochondrial dysfunction. Nitration of the tyrosine residues [41–43] of proteins and S-nitrosation of protein thiols are very important reactions in the mitochondria [44].
4. ROS/RNS and Diabetic Neuropathy
As discussed above, under normal conditions, neurons have the capacity to neutralize both ROS and RNS [45–47]. Because O2•− and H2O2 are normal products of the mitochondrial electron transport chain, SOD, catalase, and glutathione are normally sufficient to remove these metabolic byproducts (Figure 1) [48]. However, hyperglycemia increases mitochondrial activity and subsequent O2•− production. A surplus production of this primary mitochondrial ROS leads to formation of RNS as outlined in the previous section. Thus, excess mitochondrial activity leads to an overwhelming production of ROS and RNS in a neuron that is already depleted of reducing equivalents and struggling with oxidative stress brought on by other metabolic and inflammatory insults (reviewed below). The buildup of ROS/RNS in the neuron coupled with the inability of the neuron to detoxify the excess ROS and RNS leads to progressive organelle, membrane and nuclear dysfunction.
Figure 1. Oxidative Stress and Mitochondrial Dysfunction [48].
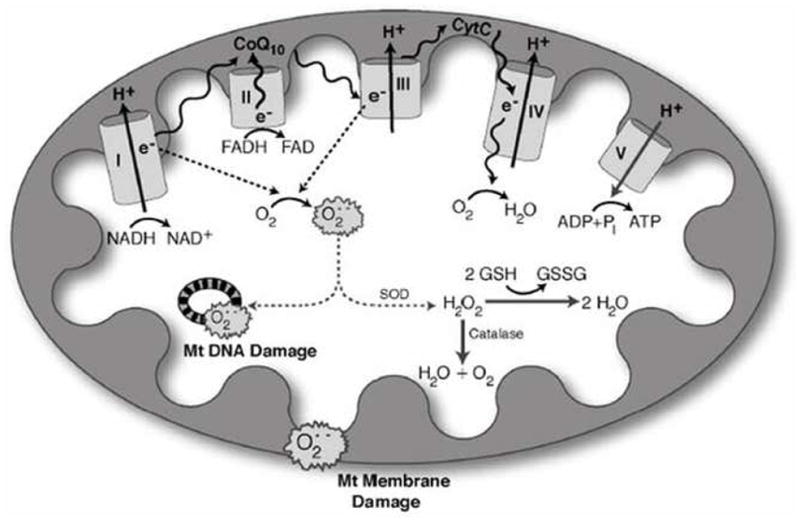
Hyperglycemia increases production of reactive oxygen species (ROS) in mitochondria. NADH and FADH2 produced from the tricarboxylic acid cycle transfer to the mitochondria, where they serve as electron donors to the mitochondrial membrane-associated redox enzyme complexes. The electrons (e−) are shuttled through oxidoreductase complexes I, II, III and IV (cytochrome c), until they are donated to molecular oxygen, forming water. The electron transfer into complexes I, III and IV by NADH (and FADH2 via complex II to complex III) produces a proton gradient at the outer mitochondrial membrane, generating a potential between the inner mitochondrial membrane and outer mitochondrial membrane. This potential drives ATP synthesis, and is crucial for mitochondrial viability, function, and normal metabolism. As electrons are passed from complex II to complex III, however, ROS are produced as by-products. The levels of ROS produced during normal oxidative phosphorylation are minimal, and they are detoxified by cellular antioxidants such as glutathione, catalase and superoxide dismutase. The hyperglycemic cell, on the other hand, shuttles more glucose through the glycolytic and tricarboxylic acid cycles, providing the cell with an over-abundance of NADH and FADH2 electron donors. This produces a high proton gradient across the inner mitochondrial membrane, which increases the turnover of the initial complexes, and thereby produces increased levels of radicals. Accumulation of these radicals, or ROS, is severely detrimental to mitochondrial DNA, mitochondrial membranes and the whole cell. Abbreviations: Cyto-c, cytochrome c; CoQ10, coenzyme Q10; e−, electrons; GSH, glutathione; GSSG, oxidized glutathione; H2O2, hydrogen peroxide; O2•−, superoxide; Pi, phosphate; SOD, superoxide dismutase.
Of note, mitochondria are both the source of ROS/RNS generation and also the first structures to be damaged, putting the neuron at even greater risk. Given the typical distal-proximal length dependent progression of diabetic neuropathy, axons are particularly susceptible to the metabolic and vascular imbalances that lead to diabetic neuropathy [48]. Axons are susceptible to hyperglycemia not only because of their direct access to nerve blood supply, but also because of their large population of mitochondria. Mounting evidence suggest that axons are as, if not more, susceptible to ROS and RNS mediated damage, in part because of their dependence on local mitochondria for energy. As these mitochondria become progressively dysfunctional, axons undergo energy failure which in turn precipitates axonal degeneration [48–50].
Mitochondria are also critical regulators of cell survival signaling pathways, and not surprisingly, oxidative damage to mitochondrial DNA, proteins, and membranes initiates signaling pathways that subsequently lead to apoptosis. Prior to the onset of frank apoptosis, mitochondria damaged by oxidative stress are destroyed via a localized process called mitoptosis. Mitoptosis is regulated, in part, by shifting the balance in the normal mitochondrial fission/fusion equilibrium. The fission of mitochondria is initiated by dynamin related protein 1 (Drp1), which translocates from the cytosol to the mitochondria during times of stress [51]. Excess mitochondrial fission leads to mitoptosis, which then may progress to apoptosis. Drp1 is elevated in in vitro and in vivo models of diabetic neuropathy [48], further promoting mitochondrial dysfunction, energy failure and axonal degeneration.
While numerous studies document the presence of axonal dystrophy and apoptosis in diabetic sensory neurons [52], some studies have failed to detect apoptosis in high glucose treated sensory neurons in culture [53, 54]. A hypothesis that accounts for this is that neurons, with support from trophic factors and antioxidants provided by surrounding glia, are initially able to undergo successful repair. Eventually, though, the cycle of glucose mediated ROS/RNS accumulation results in mitochondrial damage and the injury cascade outlined above: energy failure and axonal degeneration [55, 56].
5. The Intersection of ROS/RNS and Other Metabolic Pathways
Excess production of mitochondrial ROS/RNS is a pivotal step in hyperglycemia-induced nerve damage in diabetic neuropathy. Several other metabolic pathways become perturbed in the nervous system due to continued hyperglycemia. Each of these pathways contributes to the neuronal and axonal injury present in diabetic neuropathy, and also to enhanced levels of oxidative stress present in the diabetic nervous system. In the following sections, each of these pathways will be discussed, and the reader is provided with one or more references for thorough reviews on the individual pathways. Figure 2 is a schematic of the interaction of the pathways in the generation of diabetic neuropathy; the pathways are discussed in the order they are presented in Figure 2, from left to right.
Figure 2. Schematic of Hyperglycemic Effects on Biochemical Pathways in Diabetic Neuropathy [49].

Excessive glucose metabolism generates excess NADH and leads to overload of the electron transport chain causing oxidative stress, damage to Mt, activation of PARP. PARP activation by ROS acts in conjunction with the hexosamine and PKC pathway to induce inflammation and neuronal dysfunction. A combination of oxidative stress and hyperglycemia activate the detrimental pathways of AGE, polyol, hexosamine and PKC pathways which lead to redox imbalance, gene expression disturbances, and further oxidative stress. These pathways also induce inflammation and neuronal dysfunction. Abbreviations: NF-κB, Nuclear factor kappa B; PARP, Poly(ADP-ribose) polymerase; PKC, Protein kinase C; AGE, Advanced glycation endproducts; RNS, Reactive nitrogen species; ROS, Reactive oxygen species, GSH, glutathione; GSSG, oxidized glutathione; UDPGlcNAc, UDP-N-Acetylglucosamine; VEGF, Vascular endothelial growth factor. (Reprinted from Pharmacol Ther. 2008 Jun 13. Diabetic neuropathy: Mechanisms to management, Edwards JL, Vincent AM, Cheng HL, Feldman EL Copy right 2008 with permission form Elsevier).
As the reader reviews the other metabolic pathways believed to produce diabetic neuropathy, it is critical to remember that diabetic neuropathy results from both hyperglycemia-induced damage to nerve cells and axons per se and from neuronal ischemia caused by hyperglycemia-induced decreases in neurovascular flow. We have summarized recent animal models of experimental diabetes and neuropathy with evidence of peripheral nervous system oxidative stress in Table 1.
Table 1.
Rodent Models of Diabetic Neuropathy and Oxidative Stress.
| Rodent Model of Diabetes | Duration of Diabetes | Neuropathy Assessment | OX Stress Measurements | Reference |
|---|---|---|---|---|
| STZ-induced | ||||
| DBA/2J mice | 24 wk | NCV Behavior |
NT HODEs |
Wiggin 2008 (57) |
| Wistar rats | 6 and 12 wk | MNCV | Hexokinase | Gardiner 2007 (58) |
| Sprague–Dawley rats | 8 wk 2 or 8 wk endaravone |
MNC NBF Behavior |
LPO SOD Catalase |
Saini 2007 (59) |
| Sprague–Dawley rats | 6 wk untreated 2 wk reseratrol |
MNCV NBF Behavior |
MDA Peroxynitrite Catalase |
Kumar 2007 (60) |
| Sprague–Dawley rats | 4–12 wk untreated 12 wk enalapril/L-158809 |
MNCV NBF |
O2•− | Coopey 2006 (61) |
| Male Wistar rats | 2 wk untreated 2 wk ISO |
Behavior | NT PAR O2•− |
Ilnytska 2006 (62) |
| Sprague–Dawley rats | 2 wk untreated 10 wk fidarestat |
MNCV NBF |
GSH 8-OHdG Sorbitol |
Kuzumoto 2006 (63) |
| Sprague–Dawley rats | 6 wk untreated 2 wk U83836E |
MNCV NBF Behavior |
SOD Catalase MDA |
Sayyed 2006 (64) |
| Sprague–Dawley rats | 6 wk untreated 2 wk troxol |
MNCV NBF Behavior |
SOD Catalase LPO NOS |
Sharma and Sayyed 2006 (65) |
| Sprague–Dawley rats | 4, 12, 52 wk | MNCV SNCV |
8-OHdG | Schmeichel 2003 (66) |
| C57BL6 mice | 8 wk untreated 1 wk FP15 |
MNCV SNCV |
NT PCr/Cr Sorbitol, Glucose, Fructose |
Obrosova 2005 (67) |
| COX-2 KO mice | 24 wk | MNCV SNCV INFD |
MDA O2•− GSH PG |
Kellogg 2007 (68) |
| AR KO mice | 4, 8, 12 wk | MNCV SNCV |
JNK activation GSH O2•− DNA damage Sorbitol 8-OHdG |
Ho 2006 (69) |
| Galactose-induced Wistar rats PARP KO mice |
4 wk untreated 2 wk 3-aminobenzamide 13 wk |
MNCV SNCV NBF |
PAR PCr/Cr Nerve energy failure |
Obrosova 2004 (70) |
| Spontaneous | ||||
| ZDF rats | 8–40 wk old | MNCV NBF |
NT O2•− |
Oltman 2005 (71) |
| BKS.Cg-m+/+Leprdb/J (BKS-db/db) C57BL/6J STZ-induced |
24 wk old | MNCV SNCV Behavior INFD |
NT | Sullivan 2007 (55) |
| B6.V-Lepob/J | 8 wk old untreated 3 wk FP15/FeTMPS |
MNCV SNCV Behavior INFD |
NT PAR |
Vareniuk 2007 (72) |
| B6.V-Lepob/J | 5 wk old untreated 6 wk fidarestat |
MNCV SNCV Behavior INFD |
NT PAR |
Drel 2006 (73) |
| C57BL6/J High-fat diet | 16 wk old High-fat diet | MNCV SNCV Behavior INFD |
NT 4-HNE PAR 12/15-lipoxygenase Sorbitol |
Obrosova 2007 (74) |
4-HNE, Aminoacid-(4)-hydrosynonenal adducts; 8-OHdG, 8-hydroxy-2′-deoxyguanosine; AR, Aldose reductase; FeTMPS, Fe(III) tetra-mesitylpophyrin octasulfonate; FP15, Fe(III) tetrakis-2-(N-trethylene glycol monomethyl ether)-pyridyl porphyrin; ISO, 1,5-isoquinolinediol; LPO, lipid peroxidation; MDA, malondialdehyde; MNCN, Motor nerve conduction velocities; NBF, Nerve blood flow; NT, Nitrotyrosine; O2•−, Superoxide; PAR, poly(ADP)-ribose; PCr/Cr, phosphocreatinine/creatinine ratio; PG, Prostaglandin content; SNCV, Sensory nerve conduction velocities; SOD, Superoxide dismutase; STZ, Streptozotocin; ZDF, Zucker diabetic fatty.
5.1. Advanced Glycation Endproducts (AGE) Pathway
Advanced glycation endproducts (AGEs) are non-enzymatically created adducts between reducing sugars or oxaldehydes and proteins, DNA, or lipids [75, 76]. AGEs are thus heterogenous, and are found both inside and outside the cell, where their formation interferes with multiple aspects of cell function. Reactive dicarbonyls are the precursor molecules to AGEs that spontaneously form covalent bonds with proteins or lipids, and are synthesized through three pathways: glucose oxidation, which forms glyoxal; degradation of fructose-lysine adducts (Amadori products); and formation of methylglyoxal through the abnormal metabolism of glycolytic intermediates. Methylglyoxal is highly reactive and causes vascular endothelial cells to become more sensitive to damage [77]. Extracellular formation of protein AGEs not only disrupt cellular adhesion (through interference with cell surface protein/extracellular matrix interactions), but also activate a specific cell-surface receptor for the AGEs, known as RAGE [78].
Activation of RAGE by extracellular AGEs leads to activation of the transcription factor nuclear factor kappa B (NF-κB), which regulates gene expression, apoptosis and inflammation (Figure 2). RAGE activation in diabetic animal models contributes to the onset and progression of diabetic neuropathy. When RAGE knockout mice are made diabetic with streptozotocin (STZ), there is a significant improvement in both electrophysiological and anatomical markers of diabetic neuropathy, compared to the STZ control animals. The diabetic RAGE knockout mice also have decreased expression of NF-κB and protein kinase C in peripheral nerves, and particularly in Schwann cells [76]. RAGE activation in neurons also induces NADPH oxidase activity, which further promotes mitochondrial oxidative stress and dysfunction [79]. The confluence of data strongly suggest that RAGE is a therapeutic target for the treatment of diabetic neuropathy [76, 79–84]. For a detailed discussion on the role of AGE and RAGE in the pathogenesis of diabetic neuropathy and the potential therapeutic efficacy of blocking RAGE in the treatment of diabetic neuropathy, the reader is referred to an excellent 2008 review by Sugimoto and colleagues [85].
5.2. Polyol Pathway
The polyol pathway converts glucose to fructose through a two-step reduction/oxidation: First, aldose reductase reduces glucose to sorbitol, and then sorbitol dehydrogenase oxidizes sorbitol to fructose (Figure 2). Both aldose reductase and sorbitol dehydrogenase are prevalent in tissues prone to diabetic complications. The aldose reductase pathway is susceptible to over activation by a mass-action effect of hyperglycemia, which results in imbalances of two of the pathways metabolites, NADPH and sorbitol. Excess glucose flow through the pathway causes consumption of NADPH, which is required for regeneration of reduced glutathione [86, 87]. The depletion of glutathione secondary to excess aldose reductase activity thus renders the cell susceptible to oxidative stress, as discussed above. Increased production of sorbitol causes the intracellular environment to become hypertonic, and leads to compensatory efflux of other osmolytes such as myo-inositol (MI, important in signal transduction) and taurine (an antioxidant) [22, 88]. Intracellular reducing potential is further diminished by the second step in the polyol pathway, the production of fructose [89]. Hyperglycemia-driven production of excess fructose promotes glycation and further depletion of NADPH. Finally, activation of aldose reductase may also increase formation of diacylglycerol, which activates the deleterious protein kinase C pathway (discussed below) [90, 91]. Several studies of the human aldose reductase gene revealed polymorphisms associated with susceptibility to diabetic complications. Patients with a “high aldose reductase expression” genotype are commonly found to have early diabetic neuropathy while patients with a “low aldose reductase expression” genotype are less susceptible to neuropathy [92–94].
The polyol pathway has and continues to be a target of drug intervention in the treatment of diabetic neuropathy. Aldose reductase inhibitors block the formation of sorbitol preventing NADPH depletion. This leaves sufficient NADPH for glutathione production allowing neurons to mount a cellular defense against ROS/RNS mediated damage. It is now generally believed that it is this mechanism of action that underlies the salutary effects of aldose reductase inhibitors. Peter Oates recently published a thorough review of the polyol pathway and the past and current aldose reductase inhibitor trials in man [95]. As of yet, none of these compounds have shown efficacy in a Phase 3 trial of diabetic neuropathy.
5.3. Hexosamine Pathway
As with the polyol pathway, excess available glucose causes a mass action increase in flux through the hexosamine pathway. Under normal circumstances, a small amount of the glycolytic intermediate fructose-6 phosphate is shunted from glycolysis to the hexosamine pathway. The hexosamine pathway converts fructose-6 phosphate to glucosamine-6 phosphate by glutamine fructose-6 phosphate amidotransferase [96]. Glucosamine-6 phosphate is then converted to uridine diphosphate-N-acetyl glucosamine (UDP-GlcNAc), which is the obligatory substrate for O-GlcNAc transferase, attaching O-GlcNAC to the serine and threonine residues of transcription factors and altering gene expression [19]. Thus, a hyperglycemia-driven increase in flux through this pathway results in abnormalities in gene expression [19, 97, 98]. An increased understanding of O-GlcNAC biology also suggests that O-GlcNACcylation regulates the nutrient sensing role of the hexosamine pathway and has a role in insulin resistance and macrovascular complications [99, 100].
Sp1 is one transcription factor implicated in diabetic complications that is subject to modification by UDP-GlcNAc. Sp1 regulates the expression of many glucose-induced “housekeeping” genes, including tissue type plasminogen activator inhibitor-1 (PAI-1) and transforming growth factor-β1 (TGF-β1) [19, 101]. Interest in plasminogen activator and PAI-1 is based on the premise that impaired fibrinolysis in small neural blood vessels promotes nerve ischemia, leading to oxidative stress and the signs and symptoms of diabetic neuropathy. Data to support this idea come primarily from studies in man. Plasminogen activator expression is lower by 4 to 6 fold in the epineurial and endoneurial microvessels in sural nerves from patients with diabetic neuropathy compared to control nerve biopsies. This lower expression would promote thrombosis and nerve ischemia [102]. This idea is further supported by data from men with type 1 diabetes in the Epidemiology of Diabetes Interventions and Complications Study (EDIC); patients with diabetes neuropathy had higher serum levels of plasminogen activator/PAI-I complexes than those men without neuropathy [103]. Type 2 patients who are obese have higher PAI-I levels which may contribute to the high incidence of diabetic neuropathy in this population [104, 105]. In experimental animals, PAI-I blocks nerve regeneration [106]. More work is needed on experimental models of diabetic neuropathy to fully understand the role of plasminogen activator/PAI-I complexes.
Over expression of TGF-β1 is associated with diabetic nephropathy and contributes to microvascular damage by stimulating collagen matrix production and suppressing mitogenesis of mesangial cells [107, 108]. A recent study by the Russell laboratory identifies a role for TGF-β and other TGF isoforms in experimental diabetic neuropathy. After twelve weeks of STZ diabetes, TGF-β isoforms are increased in the dorsal root ganglia and sciatic nerves of rodents with neuropathy. In parallel, TGF-β isoforms applied directly to dorsal root ganglia cultures in vitro block neurite outgrowth [109]. Collectively, these new findings suggest TGF-β may be a potential new target for diabetic neuropathy, similar to its role in diabetic nephropathy.
5.4. Protein Kinase C (PKC) Pathway
Hyperglycemia stimulates over-activation of the protein kinase C (PKC) pathway by increasing synthesis of diacylglycerol (DAG), which activates PKC. The PKC β-isoform in particular has been linked to the development of retinopathy, nephropathy, and cardiovascular disease [110–112]. Hyperstimulation of PKC causes the overexpression of the angiogenic protein vascular endothelial growth factor (VEGF), PAI-1, NF-κB, and TGF-β, supporting a role for PKC activation in the pathogenesis of diabetic neuropathy (Figure 2). Studies in STZ diabetic rats report that inhibitors of PKC-β improve measures of diabetic neuropathy, including sciatic blood flow and nerve conduction velocity, [88, 113]. While the exact mechanisms by which PKC-β contributes to diabetic neuropathy require further study, PKC-induced vasoconstriction, altered capillary permeability, hypoxia, and nerve basement membrane thickening are all thought to be involved [110, 111]. Overexpression of PKC isoforms also directly induces insulin resistance which can further contribute to the onset of diabetic neuropathy [114, 115]. Treatment of patients with symptomatic diabetic neuropathy with a PKC inhibitor, ruboxistaurin, did not result in clinical improvement [116, 117], which could be due to the fact the drug can not penetrate the blood nerve barrier [111]. A recent review completely discusses the role of PKC and diabetic micro- and macrovascular complications and the therapeutic efficacy of PKC inhibitors [111].
5.5. Poly-ADP Ribose Polymerase (PARP) Pathway
PARP, a nuclear enzyme closely associated with oxidative-nitrosative stress, is expressed in sensory neurons, Schwann cells, and endothelial cells. While hyperglycemia, free radicals, and oxidants stimulate PARP activation, PARP also causes oxidative stress (Figure 2) [118]. PARP cleaves nicotinamide adenine dinucleotide (NAD+) to nicotinamide, and also removes ADP-ribose residues attached to nuclear proteins [119]. PARP’s catalytic activity causes a number of deleterious effects, including changes in gene expression, increases in free radical and oxidant concentration, NAD+ depletion, and shunting of glycolytic intermediates to other pathogenic pathways that can lead to PKC activation and AGE formation [67, 120–122]. In experimental diabetes, these varied effects result in neurovascular abnormalities, neuropathy, decreased nerve conduction velocity thermal and mechanical hyperalgesia, and tactile allodynia [47, 62, 70, 123–125]. Several recent reviews outline the role of PARP activation in diabetic neuropathy and discuss emerging new PARP targeted therapies [126, 127].
5.6. Inflammation
Elevated blood levels of inflammatory proteins, including C-reactive protein and TNF-α are associated with neuropathy [128, 129]. Hsp 27, part of the TNF-α signaling pathway that leads to release of the inflammatory mediators cyclooxygenase-2 (Cox-2), IL-6, and IL-8, was recently found by the Eurodiab study to be elevated in the blood of diabetic patients with neuropathy [130]. As discussed in the previous sections of this review, some inflammatory mediators like TNF-α and TGF-β are regulated by hyperglycemia-driven abnormalities in metabolism and signaling [20, 23]. Excess glucose-mediated activity in the hexokinase and PKC pathways results in activation of signaling intermediates and modified transcription factors, ultimately increasing TGF-β and NF-κB [19]. Similarly, formation of the AGE methylglyoxal results in covalently modifies transcription factors that can lead to aberrant expression of inflammatory proteins, particularly a repressor of angiotensin II called Sp3 [77]. The resulting increase in available angiotensin II activates vascular endothelial cells [77]. Activated endothelial cells in the endoneurium recruit inflammatory cells, leading to local cytokine production, reduced blood flow, and generation of reactive oxygen species [61].
RAGE activation by extracellular AGEs also affect inflammation by causing the upregulation of NF-κB [76], which in turn upregulates Cox-2 [131]. Cox-2 activation results in a feed forward loop: Cox-2 stimulates production of prostaglandin E2 and reactive oxygen species, which go on to further activate NF-κB. NF-κ-B/Cox-2 upregulation is present in the vasculature and peripheral nerves of animal models of diabetes [132]. Either blocking Cox-2 upregulation with a drug or genetic knock-down prevents multiple aspects of diabetic neuropathy, including blood flow and nerve conduction deficits, glutathione depletion, and TNFα upregulation [68, 133].
NF-κB participates in a second vicious cycle of inflammation, in which it both induces and is induced by inducible nitric oxide synthase (iNOS) [134, 135]. NO produced by the excess of iNOS contributes to microvascular damage by diminishing the blood supply to nerves [136, 137]. Moreover, NO contributes to axon and myelin degeneration following injury, damages growth cones, and is involved in the development of neuropathic pain [136, 138].
NF-κB appears to be the keystone of the inflammatory pathways that participate in the development of diabetic neuropathy. Chronic NF-κB activation appears to render neurons and blood vessels more susceptible to ischemia-reperfusion injury [139]. The subsequent extensive infiltration of macrophages is further intensified by NF-κB-stimulated release of cytokines from endothelial cells, Schwann cells and neurons [140]. The activation of macrophages leads to further production of cytokines, as well as proteases and reactive oxygen species that lead to myelin breakdown, cellular oxidative damage, and impairment of nerve regeneration [141–143]. Cameron and Cotter (2008) have recently reviewed the role of NF-κB in diabetic neuropathy and the new therapies targeted at decreasing inflammation to halt progression of diabetic neuropathy.
6. The Search for Novel Therapeutic Targets
Glucose control remains the only disease-modifying therapy for diabetic neuropathy [7, 21, 50]. We propose a bioinformatics approach as the next important paradigm in examining the causes and potential treatments of diabetic neuropathy. This novel paradigm will provide insight into disease pathogenesis and identify viable targets for disease-modifying treatments. Analysis of genomic and proteomic data from patients and animal models of diabetic neuropathy will not only validate or refute current hypotheses but will also lead to new ideas to further enhance our understanding of disease onset and progression.
To date, only two animal studies (and no human studies) have addressed alterations in gene expression within the peripheral nervous system under hyperglycemic stress and/or a common treatment for diabetes [57, 144]. Price et al. (2006) performed microarray analyses on Wistar rat dorsal root ganglion neurons, 1, 4, and 8 weeks post-STZ-induced diabetes. Diabetic neuropathy was confirmed by slowed nerve conduction velocities at weeks 4 and 8 [144]. The induction of diabetes, prior to the onset of neuropathy correlated with the upregulation of genes involved in glucose metabolism [144]. By week 4, glutathione transferase was upregulated secondary to conditions of oxidative stress [144]. We recently reported (2008) that genes functionally relevant to metabolism, mitochondria, metal ion binding and general cellular regulation are significantly differentially expressed in the sciatic nerves of type 1 diabetic and healthy mice [57]. The changes in mitochondrial gene expression were linked to the presence of both NF-κB and AP1 binding sites in the proximal promoter, a configuration that was 10-fold over-represented in the regulated mitochondrial genes compared to the overall distribution of transcription factor binding sites in mouse promoter regions. The diabetic mice were treated with Rosiglitazone, which reduced the development of neuropathy, and decreased oxidative stress in the nerve [57]. Genes that were significantly regulated by diabetes then returned to normal levels by Rosiglitazone treatment were analyzed for common transcription factor binding sites, and the results were cross checked in healthy mice treated with Rosiglitazone to determine the direction of causation. Two site combinations, SP1F and ZBPF, and a configuration of two EGRF sites were found to be significant by both approaches [57]. We and others are now making a comprehensive effort to establish the molecular signatures of neural tissues including peripheral nerve and sensory and sympathetic ganglia, from genome wide screening of RNA from human patients and animal models with and without diabetic neuropathy [55, 145]. Our approach for diabetic neuropathy is presented in Figure 3.
Figure 3. Methodologies in Biomarker Research [49].
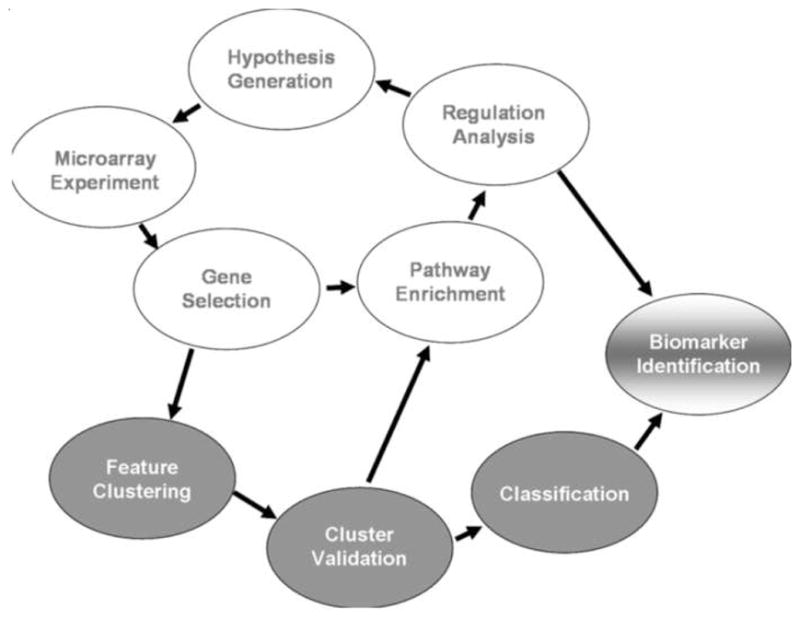
Activities in white are hypothesis driven and attempt to identify biomarkers based on the disequilibrium of identified targets in diabetic neuropathy, leading to an abnormal accumulation of products, such as modified proteins or small molecules. Activities in grey are discovery oriented and seek to identify features of the data set that are predictive of diabetic neuropathy without necessarily corresponding to a single target. (Reprinted from Pharmacol Ther. 2008 Jun 13. Diabetic neuropathy: Mechanisms to management, Edwards JL, Vincent AM, Cheng HL, Feldman EL Copy right 2008 with permission form Elsevier).
The feasibility, power, and utility of using a discovery/bioinformatics approach to uncover disease mechanisms is described for chronic kidney disease by Kretzler and colleagues [146–149]. Human renal biopsies examined by Affymetrix™ microarray analyses and real-time RT-PCR revealed differentially expressed genes between healthy and diseased tissue. These genes were mapped onto known cellular pathways that predicted regulatory elements controlling the observed changes. The regulatory elements were then used to predict the downstream effects of gene expression, including the potential biomarkers of chronic renal disease. We propose a similar comprehensive approach to be applied to diabetic neuropathy to advance our understanding of disease pathogenesis and development of disease-modifying therapies. Of special interest is how this approach, in parallel, leads to biomarker discovery. As outlined in Figure 4, the first step is to employ microarray analyses and confirmatory Q-PCR to analyze gene expression of relevant neuronal and Schwann cell markers followed by validation techniques to detect enriched pathways. These data provide the information needed to predict proteins and macromolecules influenced by gene expression. The use of clustering and classification analysis, while maintaining high standards of mathematical validation, is a valuable tool in discovering the most useful target genes, proteins and macromolecules.
Figure 4. Discovery Approach for Novel Targets in Diabetic Neuropathy (DN) [49].

Genome wide expression profiling of neural tissues from animal models with diabetic neuropathy (DN) will yield differentially regulated transcripts. Analyses of these data using Gene Ontology (GO) will provide the data needed to define categories of genes that are functionally related providing a molecular signature for diabetic neuropathy. Further analyses of these data can define relevant pathways related to functional gene categories and shared promoter modules among members of different gene categories, providing one or more specific targets for disease regulation. These targets can be verified at the mRNA level, confirming the identification of a novel disease target. (Reprinted from Pharmacol Ther. 2008 Jun 13. Diabetic neuropathy: Mechanisms to management, Edwards JL, Vincent AM, Cheng HL, Feldman EL Copy right 2008 with permission form Elsevier).
7. Summary
Ongoing research suggests that multiple metabolic and vascular pathways intersect to produce systemic and neural oxidative stress that underlies the onset and progression of diabetic neuropathy. Therapies based on the mechanisms discussed in the current review have not yet been successful in ameliorating disease progression. We suggest that a new bioinformatics approach to diabetic neuropathy provides promise for the future identification of more promising molecular targets.
Acknowledgments
We thank Kelli Sullivan and Judith Boldt for excellent technical and secretarial support. This work was supported by the A. Alfred Taubman Medical Research Institute, the Program for Neurology Research and Discovery and by NIH T32 NS07222 and NIH UO1 DK076160.
References
- 1.Boulton AJ, Vinik AI, Arezzo JC, Bril V, Feldman EL, Freeman R, Malik RA, Maser RE, Sosenko JM, Ziegler D. Diabetic neuropathies: a statement by the American Diabetes Association. Diabetes care. 2005;28:956–962. doi: 10.2337/diacare.28.4.956. [DOI] [PubMed] [Google Scholar]
- 2.Martin CL, Albers J, Herman WH, Cleary P, Waberski B, Greene DA, Stevens MJ, Feldman EL. Neuropathy among the diabetes control and complications trial cohort 8 years after trial completion. Diabetes care. 2006;29:340–344. doi: 10.2337/diacare.29.02.06.dc05-1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feldman EL, Stevens MJ, Russell JW, Peltier A, Inzucchi S, Porte JD, Sherwin RS, Baron A. The Diabetes Mellitus Manual. McGraw-Hill; 2005. Somatosensory neuropathy; pp. 366–384. [Google Scholar]
- 4.Singleton JR, Smith AG, Russell J, Feldman EL. Polyneuropathy with Impaired Glucose Tolerance: Implications for Diagnosis and Therapy. Current treatment options in neurology. 2005;7:33–42. doi: 10.1007/s11940-005-0004-4. [DOI] [PubMed] [Google Scholar]
- 5.Feldman EL, Stevens MJ, Russell JW, Greene DA. Somatosensory neuropathy. In: Porte D Jr, SRS S, Baron A, editors. Ellenberg and Rifkin’s Diabetes Mellitus. Philadelphia: McGraw Hill; 2002. pp. 771–788. [Google Scholar]
- 6.Feldman EL, Stevens MJ, Russell JW, Sperling MA. Contemporary Endocrinology. Humana Press; 2002. Diabetic peripheral and autonomic neuropathy. [Google Scholar]
- 7.Little AA, Edwards JL, Feldman EL. Diabetic neuropathies. Practical neurology. 2007;7:82–92. [PubMed] [Google Scholar]
- 8.Martin CL, Albers J, Herman WH, Cleary P, Waberski B, Greene DA, Stevens MJ, Feldman EL. Neuropathy among the diabetes control and complications trial cohort 8 years after trial completion. Diabetes care. 2006;29:340–344. doi: 10.2337/diacare.29.02.06.dc05-1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feldman EL, Stevens MJ, Russell JW, Greene DA, Porte D, Jr, Sherwin RS, Baron A. Ellenberg and Rifkin’s Diabetes Mellitus. McGraw Hill; 2003. Somatosensory neuropathy; pp. 771–788. [Google Scholar]
- 10.Vinik AI, Maser RE, Mitchell BD, Freeman R. Diabetic autonomic neuropathy. Diabetes care. 2003;26:1553–1579. doi: 10.2337/diacare.26.5.1553. [DOI] [PubMed] [Google Scholar]
- 11.Vinik AI, Mehrabyan A. Diabetic neuropathies. The Medical clinics of North America. 2004;88:947–999. xi. doi: 10.1016/j.mcna.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 12.Feldman EL, Stevens MJ, Thomas PK, Brown MB, Canal N, Greene DA. A practical two-step quantitative clinical and electrophysiological assessment for the diagnosis and staging of diabetic neuropathy. Diabetes care. 1994;17:1281–1289. doi: 10.2337/diacare.17.11.1281. [DOI] [PubMed] [Google Scholar]
- 13.Boulton AJ, Vinik AI, Arezzo JC, Bril V, Feldman EL, Freeman R, Malik RA, Maser RE, Sosenko JM, Ziegler D. Diabetic neuropathies: a statement by the American Diabetes Association. Diabetes care. 2005;28:956–962. doi: 10.2337/diacare.28.4.956. [DOI] [PubMed] [Google Scholar]
- 14.Hoye AT, Davoren JE, Wipf P, Fink MP, Kagan VE. Targeting mitochondria. Accounts of chemical research. 2008;41:87–97. doi: 10.1021/ar700135m. [DOI] [PubMed] [Google Scholar]
- 15.Lee MY, Griendling KK. Redox signaling, vascular function, and hypertension. Antioxidants & redox signaling. 2008;10:1045–1059. doi: 10.1089/ars.2007.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu T, Finkel T. Free radicals and senescence. Experimental cell research. 2008;314:1918–1922. doi: 10.1016/j.yexcr.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paravicini TM, Touyz RM. NADPH oxidases, reactive oxygen species, and hypertension: clinical implications and therapeutic possibilities. Diabetes care. 2008;31(Suppl 2):S170–180. doi: 10.2337/dc08-s247. [DOI] [PubMed] [Google Scholar]
- 18.Skulachev VP. A biochemical approach to the problem of aging: “megaproject” on membrane-penetrating ions. The first results and prospects. Biochemistry. 2007;72:1385–1396. doi: 10.1134/s0006297907120139. [DOI] [PubMed] [Google Scholar]
- 19.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 20.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 21.Vincent AM, Edwards JL, Sadidi M, Feldman EL. The antioxidant response as a drug target in diabetic neuropathy. Current drug targets. 2008;9:94–100. doi: 10.2174/138945008783431754. [DOI] [PubMed] [Google Scholar]
- 22.Vincent AM, Feldman EL. New insights into the mechanisms of diabetic neuropathy. Reviews in endocrine & metabolic disorders. 2004;5:227–236. doi: 10.1023/B:REMD.0000032411.11422.e0. [DOI] [PubMed] [Google Scholar]
- 23.Vincent AM, Russell JW, Low P, Feldman EL. Oxidative stress in the pathogenesis of diabetic neuropathy. Endocrine reviews. 2004;25:612–628. doi: 10.1210/er.2003-0019. [DOI] [PubMed] [Google Scholar]
- 24.Erusalimsky JD, Moncada S. Nitric oxide and mitochondrial signaling: from physiology to pathophysiology. Arterioscler Thromb Vasc Biol. 2007;27:2524–2531. doi: 10.1161/ATVBAHA.107.151167. [DOI] [PubMed] [Google Scholar]
- 25.Lambert AJ, Brand MD. Inhibitors of the Quinone-binding Site Allow Rapid Superoxide Production from Mitochondrial NADH:Ubiquinone Oxidoreductase (Complex I) J Biol Chem. 2004;279:39414–39420. doi: 10.1074/jbc.M406576200. [DOI] [PubMed] [Google Scholar]
- 26.Han D, Williams E, Cadenas E. Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem J. 2001;353:411–416. doi: 10.1042/0264-6021:3530411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muller FL, Liu Y, Van Remmen H. Complex III Releases Superoxide to Both Sides of the Inner Mitochondrial Membrane. J Biol Chem. 2004;279:49064–49073. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- 28.Han D, Antunes F, Canali R, Rettori D, Cadenas E. Voltage-dependent Anion Channels Control the Release of the Superoxide Anion from Mitochondria to Cytosol. J Biol Chem. 2003;278:5557–5563. doi: 10.1074/jbc.M210269200. [DOI] [PubMed] [Google Scholar]
- 29.Faraci FM, Didion SP. Vascular Protection: Superoxide Dismutase Isoforms in the Vessel Wall. Arterioscler Thromb Vasc Biol. 2004;24:1367–1373. doi: 10.1161/01.ATV.0000133604.20182.cf. [DOI] [PubMed] [Google Scholar]
- 30.Okado-Matsumoto A, Fridovich I. Subcellular Distribution of Superoxide Dismutases (SOD) in Rat Liver. Cu,Zn-SOD IN MITOCHONDRIA. J Biol Chem. 2001;276:38388–38393. doi: 10.1074/jbc.M105395200. [DOI] [PubMed] [Google Scholar]
- 31.Lebovitz RM, Zhang H, Vogel H, Cartwright J, Jr, Dionne L, Lu N, Huang S, Matzuk MM. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proceedings of the National Academy of Sciences. 1996;93:9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, Huang T-T, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nature genetics. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 33.Suttorp N, Toepfer W, Roka L. Antioxidant defense mechanisms of endothelial cells: glutathione redox cycle versus catalase. Am J Physiol Cell Physiol. 1986;251:C671–680. doi: 10.1152/ajpcell.1986.251.5.C671. [DOI] [PubMed] [Google Scholar]
- 34.Zhang R, Al-Lamki R, Bai L, Streb JW, Miano JM, Bradley J, Min W. Thioredoxin-2 Inhibits Mitochondria-Located ASK1-Mediated Apoptosis in a JNK-Independent Manner. Circulation research. 2004;94:1483–1491. doi: 10.1161/01.RES.0000130525.37646.a7. [DOI] [PubMed] [Google Scholar]
- 35.Vincent AM, Gong C, Brownlee M, Russell JW. Glucose induced neuronal programmed cell death is regulated by manganese superoxide dismutase and uncoupling protein-1. Endocrine Society Abstracts P1-289. 2001:210. [Google Scholar]
- 36.Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science (New York, NY. 2005;307:384–387. doi: 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]
- 37.Ghafourifar P, Richter C. Nitric oxide synthase activity in mitochondria. FEBS letters. 1997;418:291–296. doi: 10.1016/s0014-5793(97)01397-5. [DOI] [PubMed] [Google Scholar]
- 38.Poderoso JJ, Carreras MC, Schopfer F, Lisdero CL, Riobo NA, Giulivi C, Boveris AD, Boveris A, Cadenas E. The reaction of nitric oxide with ubiquinol: kinetic properties and biological significance. Free radical biology & medicine. 1999;26:925–935. doi: 10.1016/s0891-5849(98)00277-9. [DOI] [PubMed] [Google Scholar]
- 39.Poderoso JJ, Lisdero C, Schopfer F, Riobo N, Carreras MC, Cadenas E, Boveris A. The regulation of mitochondrial oxygen uptake by redox reactions involving nitric oxide and ubiquinol. The Journal of biological chemistry. 1999;274:37709–37716. doi: 10.1074/jbc.274.53.37709. [DOI] [PubMed] [Google Scholar]
- 40.Ghafourifar P, Schenk U, Klein SD, Richter C. Mitochondrial nitric-oxide synthase stimulation causes cytochrome c release from isolated mitochondria. Evidence for intramitochondrial peroxynitrite formation. The Journal of biological chemistry. 1999;274:31185–31188. doi: 10.1074/jbc.274.44.31185. [DOI] [PubMed] [Google Scholar]
- 41.Marcondes S, Turko IV, Murad F. Nitration of succinyl-CoA:3-oxoacid CoA-transferase in rats after endotoxin administration. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:7146–7151. doi: 10.1073/pnas.141222598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aulak KS, Koeck T, Crabb JW, Stuehr DJ. Proteomic method for identification of tyrosine-nitrated proteins. Methods in molecular biology (Clifton, NJ. 2004;279:151–165. doi: 10.1385/1-59259-807-2:151. [DOI] [PubMed] [Google Scholar]
- 43.Turko IV, Marcondes S, Murad F. Diabetes-associated nitration of tyrosine and inactivation of succinyl-CoA:3-oxoacid CoA-transferase. Am J Physiol Heart Circ Physiol. 2001;281:H2289–2294. doi: 10.1152/ajpheart.2001.281.6.H2289. [DOI] [PubMed] [Google Scholar]
- 44.Mannick JB, Schonhoff C, Papeta N, Ghafourifar P, Szibor M, Fang K, Gaston B. S-Nitrosylation of mitochondrial caspases. The Journal of cell biology. 2001;154:1111–1116. doi: 10.1083/jcb.200104008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 46.Obrosova IG, Drel VR, Oltman CL, Mashtalir N, Tibrewala J, Groves JT, Yorek MA. Role of nitrosative stress in early neuropathy and vascular dysfunction in streptozotocin-diabetic rats. American journal of physiology. 2007;293:E1645–1655. doi: 10.1152/ajpendo.00479.2007. [DOI] [PubMed] [Google Scholar]
- 47.Obrosova IG, Drel VR, Pacher P, Ilnytska O, Wang ZQ, Stevens MJ, Yorek MA. Oxidative-nitrosative stress and poly(ADP-ribose) polymerase (PARP) activation in experimental diabetic neuropathy: the relation is revisited. Diabetes. 2005;54:3435–3441. doi: 10.2337/diabetes.54.12.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leinninger GM, Edwards JL, Lipshaw MJ, Feldman EL. Mechanisms of disease: mitochondria as new therapeutic targets in diabetic neuropathy. Nature clinical practice. 2006;2:620–628. doi: 10.1038/ncpneuro0320. [DOI] [PubMed] [Google Scholar]
- 49.Edwards JL, Vincent AM, Cheng HL, Feldman EL. Pharmacology & therapeutics. 2008. Diabetic neuropathy: Mechanisms to management. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leinninger GM, Backus C, Sastry AM, Yi YB, Wang CW, Feldman EL. Mitochondria in DRG neurons undergo hyperglycemic mediated injury through Bim, Bax and the fission protein Drp1. Neurobiology of disease. 2006;23:11–22. doi: 10.1016/j.nbd.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 51.Arnoult D, Rismanchi N, Grodet A, Roberts RG, Seeburg DP, Estaquier J, Sheng M, Blackstone C. Bax/Bak-dependent release of DDP/TIMM8a promotes Drp1-mediated mitochondrial fission and mitoptosis during programmed cell death. Curr Biol. 2005;15:2112–2118. doi: 10.1016/j.cub.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 52.Brussee V, Guo G, Dong Y, Cheng C, Martinez JA, Smith D, Glazner GW, Fernyhough P, Zochodne DW. Distal degenerative sensory neuropathy in a long-term type 2 diabetes rat model. Diabetes. 2008;57:1664–1673. doi: 10.2337/db07-1737. [DOI] [PubMed] [Google Scholar]
- 53.Cheng C, Zochodne DW. Sensory neurons with activated caspase-3 survive long-term experimental diabetes. Diabetes. 2003;52:2363–2371. doi: 10.2337/diabetes.52.9.2363. [DOI] [PubMed] [Google Scholar]
- 54.Gumy LF, Bampton ET, Tolkovsky AM. Hyperglycaemia inhibits Schwann cell proliferation and migration and restricts regeneration of axons and Schwann cells from adult murine DRG. Molecular and cellular neurosciences. 2008;37:298–311. doi: 10.1016/j.mcn.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 55.Sullivan KA, Hayes JM, Wiggin TD, Backus C, Su Oh S, Lentz SI, Brosius F, 3rd, Feldman EL. Mouse models of diabetic neuropathy. Neurobiology of disease. 2007;28:276–285. doi: 10.1016/j.nbd.2007.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vincent AM, Russell JW, Sullivan KA, Backus C, Hayes JM, McLean LL, Feldman EL. SOD2 protects neurons from injury in cell culture and animal models of diabetic neuropathy. Experimental neurology. 2007;208:216–227. doi: 10.1016/j.expneurol.2007.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wiggin TD, Kretzler M, Pennathur S, Sullivan KA, Brosius FC, Feldman EL. Rosiglitazone Treatment Reduces Diabetic Neuropathy in STZ Treated DBA/2J Mice. Endocrinology. 2008 doi: 10.1210/en.2008-0869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gardiner NJ, Wang Z, Luke C, Gott A, Price SA, Fernyhough P. Expression of hexokinase isoforms in the dorsal root ganglion of the adult rat and effect of experimental diabetes. Brain Res. 2007;1175:143–154. doi: 10.1016/j.brainres.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 59.Saini AK, Kumar HSA, Sharma SS. Preventive and curative effect of edaravone on nerve functions and oxidative stress in experimental diabetic neuropathy. European journal of pharmacology. 2007;568:164–172. doi: 10.1016/j.ejphar.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 60.Kumar A, Kaundal RK, Iyer S, Sharma SS. Effects of resveratrol on nerve functions, oxidative stress and DNA fragmentation in experimental diabetic neuropathy. Life sciences. 2007;80:1236–1244. doi: 10.1016/j.lfs.2006.12.036. [DOI] [PubMed] [Google Scholar]
- 61.Coppey LJ, Davidson EP, Rinehart TW, Gellett JS, Oltman CL, Lund DD, Yorek MA. ACE inhibitor or angiotensin II receptor antagonist attenuates diabetic neuropathy in streptozotocin-induced diabetic rats. Diabetes. 2006;55:341–348. doi: 10.2337/diabetes.55.02.06.db05-0885. [DOI] [PubMed] [Google Scholar]
- 62.Ilnytska O, Lyzogubov VV, Stevens MJ, Drel VR, Mashtalir N, Pacher P, Yorek MA, Obrosova IG. Poly(ADP-ribose) polymerase inhibition alleviates experimental diabetic sensory neuropathy. Diabetes. 2006;55:1686–1694. doi: 10.2337/db06-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kuzumoto Y, Kusunoki S, Kato N, Kihara M, Low PA. Effect of the aldose reductase inhibitor fidarestat on experimental diabetic neuropathy in the rat. Diabetologia. 2006;49:3085–3093. doi: 10.1007/s00125-006-0400-7. [DOI] [PubMed] [Google Scholar]
- 64.Sayyed SG, Kumar A, Sharma SS. Effects of U83836E on nerve functions, hyperalgesia and oxidative stress in experimental diabetic neuropathy. Life sciences. 2006;79:777–783. doi: 10.1016/j.lfs.2006.02.033. [DOI] [PubMed] [Google Scholar]
- 65.Sharma SS, Sayyed SG. Effects of trolox on nerve dysfunction, thermal hyperalgesia and oxidative stress in experimental diabetic neuropathy. Clinical and experimental pharmacology & physiology. 2006;33:1022–1028. doi: 10.1111/j.1440-1681.2006.04481.x. [DOI] [PubMed] [Google Scholar]
- 66.Schmeichel AM, Schmelzer JD, Low PA. Oxidative injury and apoptosis of dorsal root ganglion neurons in chronic experimental diabetic neuropathy. Diabetes. 2003;52:165–171. doi: 10.2337/diabetes.52.1.165. [DOI] [PubMed] [Google Scholar]
- 67.Obrosova IG, Mabley JG, Zsengeller Z, Charniauskaya T, Abatan OI, Groves JT, Szabo C. Role for nitrosative stress in diabetic neuropathy: evidence from studies with a peroxynitrite decomposition catalyst. Faseb J. 2005;19:401–403. doi: 10.1096/fj.04-1913fje. [DOI] [PubMed] [Google Scholar]
- 68.Kellogg AP, Wiggin TD, Larkin DD, Hayes JM, Stevens MJ, Pop-Busui R. Protective effects of cyclooxygenase-2 gene inactivation against peripheral nerve dysfunction and intraepidermal nerve fiber loss in experimental diabetes. Diabetes. 2007;56:2997–3005. doi: 10.2337/db07-0740. [DOI] [PubMed] [Google Scholar]
- 69.Ho EC, Lam KS, Chen YS, Yip JC, Arvindakshan M, Yamagishi S, Yagihashi S, Oates PJ, Ellery CA, Chung SS, Chung SK. Aldose reductase-deficient mice are protected from delayed motor nerve conduction velocity, increased c-Jun NH2-terminal kinase activation, depletion of reduced glutathione, increased superoxide accumulation, and DNA damage. Diabetes. 2006;55:1946–1953. doi: 10.2337/db05-1497. [DOI] [PubMed] [Google Scholar]
- 70.Obrosova IG, Li F, Abatan OI, Forsell MA, Komjati K, Pacher P, Szabo C, Stevens MJ. Role of poly(ADP-ribose) polymerase activation in diabetic neuropathy. Diabetes. 2004;53:711–720. doi: 10.2337/diabetes.53.3.711. [DOI] [PubMed] [Google Scholar]
- 71.Oltman CL, Coppey LJ, Gellett JS, Davidson EP, Lund DD, Yorek MA. Progression of vascular and neural dysfunction in sciatic nerves of Zucker diabetic fatty and Zucker rats. American journal of physiology. 2005;289:E113–122. doi: 10.1152/ajpendo.00594.2004. [DOI] [PubMed] [Google Scholar]
- 72.Vareniuk I, Pavlov IA, Drel VR, Lyzogubov VV, Ilnytska O, Bell SR, Tibrewala J, Groves JT, Obrosova IG. Nitrosative stress and peripheral diabetic neuropathy in leptin-deficient (ob/ob) mice. Experimental neurology. 2007;205:425–436. doi: 10.1016/j.expneurol.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 73.Drel VR, Mashtalir N, Ilnytska O, Shin J, Li F, Lyzogubov VV, Obrosova IG. The leptin-deficient (ob/ob) mouse: a new animal model of peripheral neuropathy of type 2 diabetes and obesity. Diabetes. 2006;55:3335–3343. doi: 10.2337/db06-0885. [DOI] [PubMed] [Google Scholar]
- 74.Obrosova IG, Ilnytska O, Lyzogubov VV, Pavlov IA, Mashtalir N, Nadler JL, Drel VR. High-fat diet induced neuropathy of pre-diabetes and obesity: effects of “healthy” diet and aldose reductase inhibition. Diabetes. 2007;56:2598–2608. doi: 10.2337/db06-1176. [DOI] [PubMed] [Google Scholar]
- 75.Ahmed N. Advanced glycation endproducts--role in pathology of diabetic complications. Diabetes research and clinical practice. 2005;67:3–21. doi: 10.1016/j.diabres.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 76.Toth C, Rong LL, Yang C, Martinez J, Song F, Ramji N, Brussee V, Liu W, Durand J, Nguyen MD, Schmidt AM, Zochodne DW. RAGE and Experimental Diabetic Neuropathy. Diabetes. 2007 doi: 10.2337/db07-0339. [DOI] [PubMed] [Google Scholar]
- 77.Yao D, Taguchi T, Matsumura T, Pestell R, Edelstein D, Giardino I, Suske G, Rabbani N, Thornalley PJ, Sarthy VP, Hammes HP, Brownlee M. High glucose increases angiopoietin-2 transcription in microvascular endothelial cells through methylglyoxal modification of mSin3A. The Journal of biological chemistry. 2007;282:31038–31045. doi: 10.1074/jbc.M704703200. [DOI] [PubMed] [Google Scholar]
- 78.Ramasamy R, Yan SF, Schmidt AM. Arguing for the motion: yes, RAGE is a receptor for advanced glycation endproducts. Molecular nutrition & food research. 2007;51:1111–1115. doi: 10.1002/mnfr.200700008. [DOI] [PubMed] [Google Scholar]
- 79.Vincent AM, Perrone L, Sullivan KA, Backus C, Sastry AM, Lastoskie C, Feldman EL. Receptor for advanced glycation end products activation injures primary sensory neurons via oxidative stress. Endocrinology. 2007;148:548–558. doi: 10.1210/en.2006-0073. [DOI] [PubMed] [Google Scholar]
- 80.Ramasamy R, Vannucci SJ, Yan SS, Herold K, Yan SF, Schmidt AM. Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology. 2005;15:16R–28R. doi: 10.1093/glycob/cwi053. [DOI] [PubMed] [Google Scholar]
- 81.Tanji N, Markowitz GS, Fu C, Kislinger T, Taguchi A, Pischetsrieder M, Stern D, Schmidt AM, D’Agati VD. Expression of advanced glycation end products and their cellular receptor RAGE in diabetic nephropathy and nondiabetic renal disease. J Am Soc Nephrol. 2000;11:1656–1666. doi: 10.1681/ASN.V1191656. [DOI] [PubMed] [Google Scholar]
- 82.Coughlan MT, Forbes JM, Cooper ME. Role of the AGE crosslink breaker, alagebrium, as a renoprotective agent in diabetes. Kidney Int Suppl. 2007:S54–60. doi: 10.1038/sj.ki.5002387. [DOI] [PubMed] [Google Scholar]
- 83.Hudson BI, Schmidt AM. RAGE: a novel target for drug intervention in diabetic vascular disease. Pharmaceutical research. 2004;21:1079–1086. doi: 10.1023/b:pham.0000032992.75423.9b. [DOI] [PubMed] [Google Scholar]
- 84.Bucciarelli LG, Wendt T, Qu W, Lu Y, Lalla E, Rong LL, Goova MT, Moser B, Kislinger T, Lee DC, Kashyap Y, Stern DM, Schmidt AM. RAGE blockade stabilizes established atherosclerosis in diabetic apolipoprotein E-null mice. Circulation. 2002;106:2827–2835. doi: 10.1161/01.cir.0000039325.03698.36. [DOI] [PubMed] [Google Scholar]
- 85.Sugimoto K, Yasujima M, Yagihashi S. Role of advanced glycation end products in diabetic neuropathy. Current pharmaceutical design. 2008;14:953–961. doi: 10.2174/138161208784139774. [DOI] [PubMed] [Google Scholar]
- 86.Djordjevic VB. Free radicals in cell biology. International review of cytology. 2004;237:57–89. doi: 10.1016/S0074-7696(04)37002-6. [DOI] [PubMed] [Google Scholar]
- 87.Mathers J, Fraser JA, McMahon M, Saunders RD, Hayes JD, McLellan LI. Antioxidant and cytoprotective responses to redox stress. Biochemical Society symposium. 2004:157–176. doi: 10.1042/bss0710157. [DOI] [PubMed] [Google Scholar]
- 88.Nakamura J, Kato K, Hamada Y, Nakayama M, Chaya S, Nakashima E, Naruse K, Kasuya Y, Mizubayashi R, Miwa K, Yasuda Y, Kamiya H, Ienaga K, Sakakibara F, Koh N, Hotta N. A protein kinase C-beta-selective inhibitor ameliorates neural dysfunction in streptozotocin-induced diabetic rats. Diabetes. 1999;48:2090–2095. doi: 10.2337/diabetes.48.10.2090. [DOI] [PubMed] [Google Scholar]
- 89.Feldman EL, Stevens MJ, Greene DA. Pathogenesis of diabetic neuropathy. Clinical neuroscience (New York, NY. 1997;4:365–370. [PubMed] [Google Scholar]
- 90.Uehara K, Yamagishi S, Otsuki S, Chin S, Yagihashi S. Effects of polyol pathway hyperactivity on protein kinase C activity, nociceptive peptide expression, and neuronal structure in dorsal root ganglia in diabetic mice. Diabetes. 2004;53:3239–3247. doi: 10.2337/diabetes.53.12.3239. [DOI] [PubMed] [Google Scholar]
- 91.Yamagishi S, Uehara K, Otsuki S, Yagihashi S. Differential influence of increased polyol pathway on protein kinase C expressions between endoneurial and epineurial tissues in diabetic mice. Journal of neurochemistry. 2003;87:497–507. doi: 10.1046/j.1471-4159.2003.02011.x. [DOI] [PubMed] [Google Scholar]
- 92.Demaine AG. Polymorphisms of the aldose reductase gene and susceptibility to diabetic microvascular complications. Current medicinal chemistry. 2003;10:1389–1398. doi: 10.2174/0929867033457359. [DOI] [PubMed] [Google Scholar]
- 93.Donaghue KC, Margan SH, Chan AK, Holloway B, Silink M, Rangel T, Bennetts B. The association of aldose reductase gene (AKR1B1) polymorphisms with diabetic neuropathy in adolescents. Diabet Med. 2005;22:1315–1320. doi: 10.1111/j.1464-5491.2005.01631.x. [DOI] [PubMed] [Google Scholar]
- 94.Thamotharampillai K, Chan AK, Bennetts B, Craig ME, Cusumano J, Silink M, Oates PJ, Donaghue KC. Decline in neurophysiological function after 7 years in an adolescent diabetic cohort and the role of aldose reductase gene polymorphisms. Diabetes care. 2006;29:2053–2057. doi: 10.2337/dc06-0678. [DOI] [PubMed] [Google Scholar]
- 95.Oates PJ. Aldose Reductase, Still a Compelling Target for Diabetic Neuropathy. Current drug targets. 2008 doi: 10.2174/138945008783431781. in press. [DOI] [PubMed] [Google Scholar]
- 96.Thornalley PJ. The potential role of thiamine (vitamin B(1)) in diabetic complications. Current diabetes reviews. 2005;1:287–298. doi: 10.2174/157339905774574383. [DOI] [PubMed] [Google Scholar]
- 97.Kolm-Litty V, Sauer U, Nerlich A, Lehmann R, Schleicher ED. High glucose-induced transforming growth factor beta1 production is mediated by the hexosamine pathway in porcine glomerular mesangial cells. The Journal of clinical investigation. 1998;101:160–169. doi: 10.1172/JCI119875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sayeski PP, Kudlow JE. Glucose metabolism to glucosamine is necessary for glucose stimulation of transforming growth factor-alpha gene transcription. The Journal of biological chemistry. 1996;271:15237–15243. doi: 10.1074/jbc.271.25.15237. [DOI] [PubMed] [Google Scholar]
- 99.Dias WB, Hart GW. O-GlcNAc modification in diabetes and Alzheimer’s disease. Molecular bioSystems. 2007;3:766–772. doi: 10.1039/b704905f. [DOI] [PubMed] [Google Scholar]
- 100.Love DC, Hanover JA. The hexosamine signaling pathway: deciphering the “O-GlcNAc code”. Sci STKE. 2005;2005:re13. doi: 10.1126/stke.3122005re13. [DOI] [PubMed] [Google Scholar]
- 101.Du XL, Edelstein D, Rossetti L, Fantus IG, Goldberg H, Ziyadeh F, Wu J, Brownlee M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:12222–12226. doi: 10.1073/pnas.97.22.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hafer-Macko CE, Ivey FM, Sorkin JD, Macko RF. Microvascular tissue plasminogen activator is reduced in diabetic neuropathy. Neurology. 2007;69:268–274. doi: 10.1212/01.wnl.0000266391.20707.83. [DOI] [PubMed] [Google Scholar]
- 103.Maser RE, Ellis D, Erbey JR, Orchard TJ. Do tissue plasminogen activator-plasminogen activator inhibitor-1 complexes relate to the complications of insulin-dependent diabetes mellitus? Pittsburgh Epidemiology of Diabetes Complications Study. Journal of diabetes and its complications. 1997;11:243–249. doi: 10.1016/s1056-8727(96)00040-2. [DOI] [PubMed] [Google Scholar]
- 104.Aso Y, Matsumoto S, Fujiwara Y, Tayama K, Inukai T, Takemura Y. Impaired fibrinolytic compensation for hypercoagulability in obese patients with type 2 diabetes: association with increased plasminogen activator inhibitor-1. Metabolism: clinical and experimental. 2002;51:471–476. doi: 10.1053/meta.2002.31334. [DOI] [PubMed] [Google Scholar]
- 105.Erem C, Hacihasanoglu A, Celik S, Ovali E, Ersoz HO, Ukinc K, Deger O, Telatar M. Coagulation and fibrinolysis parameters in type 2 diabetic patients with and without diabetic vascular complications. Med Princ Pract. 2005;14:22–30. doi: 10.1159/000081919. [DOI] [PubMed] [Google Scholar]
- 106.Nilsson A, Moller K, Dahlin L, Lundborg G, Kanje M. Early changes in gene expression in the dorsal root ganglia after transection of the sciatic nerve; effects of amphiregulin and PAI-1 on regeneration. Brain research. 2005;136:65–74. doi: 10.1016/j.molbrainres.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 107.Kanwar YS, Wada J, Sun L, Xie P, Wallner EI, Chen S, Chugh S, Danesh FR. Diabetic nephropathy: mechanisms of renal disease progression. Experimental biology and medicine (Maywood, NJ. 2008;233:4–11. doi: 10.3181/0705-MR-134. [DOI] [PubMed] [Google Scholar]
- 108.Zhu Y, Usui HK, Sharma K. Regulation of transforming growth factor beta in diabetic nephropathy: implications for treatment. Seminars in nephrology. 2007;27:153–160. doi: 10.1016/j.semnephrol.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Anjaneyulu M, Berent-Spillson A, Inoue T, Choi J, Cherian K, Russell JW. Transforming growth factor-beta induces cellular injury in experimental diabetic neuropathy. Experimental neurology. 2008;211:469–479. doi: 10.1016/j.expneurol.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Arikawa E, Ma RC, Isshiki K, Luptak I, He Z, Yasuda Y, Maeno Y, Patti ME, Weir GC, Harris RA, Zammit VA, Tian R, King GL. Effects of insulin replacements, inhibitors of angiotensin, and PKCbeta’s actions to normalize cardiac gene expression and fuel metabolism in diabetic rats. Diabetes. 2007;56:1410–1420. doi: 10.2337/db06-0655. [DOI] [PubMed] [Google Scholar]
- 111.Das Evcimen N, King GL. The role of protein kinase C activation and the vascular complications of diabetes. Pharmacol Res. 2007;55:498–510. doi: 10.1016/j.phrs.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 112.Veves A, King GL. Can VEGF reverse diabetic neuropathy in human subjects? The Journal of clinical investigation. 2001;107:1215–1218. doi: 10.1172/JCI13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cameron NE, Cotter MA. Effects of protein kinase Cbeta inhibition on neurovascular dysfunction in diabetic rats: interaction with oxidative stress and essential fatty acid dysmetabolism. Diabetes/metabolism research and reviews. 2002;18:315–323. doi: 10.1002/dmrr.307. [DOI] [PubMed] [Google Scholar]
- 114.Cortright RN, Azevedo JL, Jr, Zhou Q, Sinha M, Pories WJ, Itani SI, Dohm GL. Protein kinase C modulates insulin action in human skeletal muscle. American journal of physiology. 2000;278:E553–562. doi: 10.1152/ajpendo.2000.278.3.E553. [DOI] [PubMed] [Google Scholar]
- 115.Naruse K, Rask-Madsen C, Takahara N, Ha SW, Suzuma K, Way KJ, Jacobs JR, Clermont AC, Ueki K, Ohshiro Y, Zhang J, Goldfine AB, King GL. Activation of vascular protein kinase C-beta inhibits Akt-dependent endothelial nitric oxide synthase function in obesity-associated insulin resistance. Diabetes. 2006;55:691–698. doi: 10.2337/diabetes.55.03.06.db05-0771. [DOI] [PubMed] [Google Scholar]
- 116.Casellini CM, Barlow PM, Rice AL, Casey M, Simmons K, Pittenger G, Bastyr EJ, 3rd, Wolka AM, Vinik AI. A 6-month, randomized, double-masked, placebo-controlled study evaluating the effects of the protein kinase C-beta inhibitor ruboxistaurin on skin microvascular blood flow and other measures of diabetic peripheral neuropathy. Diabetes care. 2007;30:896–902. doi: 10.2337/dc06-1699. [DOI] [PubMed] [Google Scholar]
- 117.Vinik AI, Bril V, Kempler P, Litchy WJ, Tesfaye S, Price KL, Bastyr EJ., 3rd Treatment of symptomatic diabetic peripheral neuropathy with the protein kinase C beta-inhibitor ruboxistaurin mesylate during a 1-year, randomized, placebo-controlled, double-blind clinical trial. Clinical therapeutics. 2005;27:1164–1180. doi: 10.1016/j.clinthera.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 118.Obrosova IG, Julius UA. Role for poly(ADP-ribose) polymerase activation in diabetic nephropathy, neuropathy and retinopathy. Current vascular pharmacology. 2005;3:267–283. doi: 10.2174/1570161054368634. [DOI] [PubMed] [Google Scholar]
- 119.Southan GJ, Szabo C. Poly(ADP-ribose) polymerase inhibitors. Current medicinal chemistry. 2003;10:321–340. doi: 10.2174/0929867033368376. [DOI] [PubMed] [Google Scholar]
- 120.Du X, Matsumura T, Edelstein D, Rossetti L, Zsengeller Z, Szabo C, Brownlee M. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. The Journal of clinical investigation. 2003;112:1049–1057. doi: 10.1172/JCI18127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Garcia Soriano F, Virag L, Jagtap P, Szabo E, Mabley JG, Liaudet L, Marton A, Hoyt DG, Murthy KG, Salzman AL, Southan GJ, Szabo C. Diabetic endothelial dysfunction: the role of poly(ADP-ribose) polymerase activation. Nature medicine. 2001;7:108–113. doi: 10.1038/83241. [DOI] [PubMed] [Google Scholar]
- 122.Apfel SC. Nerve growth factor for the treatment of diabetic neuropathy: what went wrong, what went right, and what does the future hold? International review of neurobiology. 2002;50:393–413. doi: 10.1016/s0074-7742(02)50083-0. [DOI] [PubMed] [Google Scholar]
- 123.Li F, Drel VR, Szabo C, Stevens MJ, Obrosova IG. Low-dose poly(ADP-ribose) polymerase inhibitor-containing combination therapies reverse early peripheral diabetic neuropathy. Diabetes. 2005;54:1514–1522. doi: 10.2337/diabetes.54.5.1514. [DOI] [PubMed] [Google Scholar]
- 124.Pacher P, Liaudet L, Soriano FG, Mabley JG, Szabo E, Szabo C. The role of poly(ADP-ribose) polymerase activation in the development of myocardial and endothelial dysfunction in diabetes. Diabetes. 2002;51:514–521. doi: 10.2337/diabetes.51.2.514. [DOI] [PubMed] [Google Scholar]
- 125.Zheng L, Szabo C, Kern TS. Poly(ADP-ribose) polymerase is involved in the development of diabetic retinopathy via regulation of nuclear factor-kappaB. Diabetes. 2004;53:2960–2967. doi: 10.2337/diabetes.53.11.2960. [DOI] [PubMed] [Google Scholar]
- 126.Pacher P. Poly(ADP-ribose) polymerase inhibition as a novel therapeutic approach against intraepidermal nerve fiber loss and neuropathic pain associated with advanced diabetic neuropathy: a commentary on “PARP Inhibition or gene deficiency counteracts intraepidermal nerve fiber loss and neuropathic pain in advanced diabetic neuropathy”. Free radical biology & medicine. 2008;44:969–971. doi: 10.1016/j.freeradbiomed.2007.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pacher P, Szabo C. Role of the peroxynitrite-poly(ADP-ribose) polymerase pathway in human disease. The American journal of pathology. 2008;173:2–13. doi: 10.2353/ajpath.2008.080019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gonzalez-Clemente JM, Mauricio D, Richart C, Broch M, Caixas A, Megia A, Gimenez-Palop O, Simon I, Martinez-Riquelme A, Gimenez-Perez G, Vendrell J. Diabetic neuropathy is associated with activation of the TNF-alpha system in subjects with type 1 diabetes mellitus. Clinical endocrinology. 2005;63:525–529. doi: 10.1111/j.1365-2265.2005.02376.x. [DOI] [PubMed] [Google Scholar]
- 129.Gomes MB, Piccirillo LJ, Nogueira VG, Matos HJ. Acute-phase proteins among patients with type 1 diabetes. Diabetes & metabolism. 2003;29:405–411. doi: 10.1016/s1262-3636(07)70051-4. [DOI] [PubMed] [Google Scholar]
- 130.Gruden G, Bruno G, Chaturvedi N, Burt D, Schalkwijk C, Pinach S, Stehouwer CD, Witte DR, Fuller JH, Cavallo Perin P. Serum Hsp27 and Diabetic Complications in the Eurodiab Prospective Complications Study: a Novel Circulating Marker for Diabetic Neuropathy. Diabetes. 2008 doi: 10.2337/db08-0009. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lee KM, Kang BS, Lee HL, Son SJ, Hwang SH, Kim DS, Park JS, Cho HJ. Spinal NF-kB activation induces COX-2 upregulation and contributes to inflammatory pain hypersensitivity. The European journal of neuroscience. 2004;19:3375–3381. doi: 10.1111/j.0953-816X.2004.03441.x. [DOI] [PubMed] [Google Scholar]
- 132.Kellogg AP, Pop-Busui R. Peripheral nerve dysfunction in experimental diabetes is mediated by cyclooxygenase-2 and oxidative stress. Antioxidants & redox signaling. 2005;7:1521–1529. doi: 10.1089/ars.2005.7.1521. [DOI] [PubMed] [Google Scholar]
- 133.Matsunaga A, Kawamoto M, Shiraishi S, Yasuda T, Kajiyama S, Kurita S, Yuge O. Intrathecally administered COX-2 but not COX-1 or COX-3 inhibitors attenuate streptozotocin-induced mechanical hyperalgesia in rats. European journal of pharmacology. 2007;554:12–17. doi: 10.1016/j.ejphar.2006.09.072. [DOI] [PubMed] [Google Scholar]
- 134.Hasnis E, Bar-Shai M, Burbea Z, Reznick AZ. Mechanisms underlying cigarette smoke-induced NF-kappaB activation in human lymphocytes: the role of reactive nitrogen species. J Physiol Pharmacol. 2007;58(Suppl 5):275–287. [PubMed] [Google Scholar]
- 135.Kim YW, Zhao RJ, Park SJ, Lee JR, Cho IJ, Yang CH, Kim SG, Kim SC. Anti-inflammatory effects of liquiritigenin as a consequence of the inhibition of NF-kappaB-dependent iNOS and proinflammatory cytokines production. British journal of pharmacology. 2008 doi: 10.1038/bjp.2008.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Levy D, Zochodne DW. NO pain: potential roles of nitric oxide in neuropathic pain. Pain Pract. 2004;4:11–18. doi: 10.1111/j.1533-2500.2004.04002.x. [DOI] [PubMed] [Google Scholar]
- 137.Zochodne DW, Levy D. Nitric oxide in damage, disease and repair of the peripheral nervous system. Cellular and molecular biology (Noisy-le-Grand, France) 2005;51:255–267. [PubMed] [Google Scholar]
- 138.McDonald DS, Cheng C, Martinez JA, Zochodne DW. Regenerative arrest of inflamed peripheral nerves: role of nitric oxide. Neuroreport. 2007;18:1635–1640. doi: 10.1097/WNR.0b013e3282f03fff. [DOI] [PubMed] [Google Scholar]
- 139.Wang Y, Schmeichel AM, Iida H, Schmelzer JD, Low PA. Enhanced inflammatory response via activation of NF-kappaB in acute experimental diabetic neuropathy subjected to ischemia-reperfusion injury. Journal of the neurological sciences. 2006;247:47–52. doi: 10.1016/j.jns.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 140.Yamagishi S, Ogasawara S, Mizukami H, Yajima N, Wada R, Sugawara A, Yagihashi S. Correction of protein kinase C activity and macrophage migration in peripheral nerve by pioglitazone, peroxisome proliferator activated-gamma-ligand, in insulin-deficient diabetic rats. Journal of neurochemistry. 2008;104:491–499. doi: 10.1111/j.1471-4159.2007.05050.x. [DOI] [PubMed] [Google Scholar]
- 141.Kawamura N, Dyck PJ, Schmeichel AM, Engelstad JK, Low PA, Dyck PJ. Inflammatory mediators in diabetic and non-diabetic lumbosacral radiculoplexus neuropathy. Acta neuropathologica. 2008;115:231–239. doi: 10.1007/s00401-007-0326-2. [DOI] [PubMed] [Google Scholar]
- 142.Tesch GH. Role of macrophages in complications of type 2 diabetes. Clinical and experimental pharmacology & physiology. 2007;34:1016–1019. doi: 10.1111/j.1440-1681.2007.04729.x. [DOI] [PubMed] [Google Scholar]
- 143.Conti G, Scarpini E, Baron P, Livraghi S, Tiriticco M, Bianchi R, Vedeler C, Scarlato G. Macrophage infiltration and death in the nerve during the early phases of experimental diabetic neuropathy: a process concomitant with endoneurial induction of IL-1beta and p75NTR. Journal of the neurological sciences. 2002;195:35–40. doi: 10.1016/s0022-510x(01)00684-0. [DOI] [PubMed] [Google Scholar]
- 144.Price SA, Zeef LA, Wardleworth L, Hayes A, Tomlinson DR. Identification of changes in gene expression in dorsal root ganglia in diabetic neuropathy: correlation with functional deficits. Journal of neuropathology and experimental neurology. 2006;65:722–732. doi: 10.1097/01.jnen.0000228199.89420.90. [DOI] [PubMed] [Google Scholar]
- 145.Sullivan KA, Lentz SI, Roberts JL, Jr, Feldman EL. Criteria for creating and assessing mouse models of diabetic neuropathy. Current drug targets. 2008;9:3–13. doi: 10.2174/138945008783431763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Cohen CD, Klingenhoff A, Boucherot A, Nitsche A, Henger A, Brunner B, Schmid H, Merkle M, Saleem MA, Koller KP, Werner T, Grone HJ, Nelson PJ, Kretzler M. Comparative promoter analysis allows de novo identification of specialized cell junction-associated proteins. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:5682–5687. doi: 10.1073/pnas.0511257103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Schmid H, Cohen CD, Henger A, Irrgang S, Schlondorff D, Kretzler M. Validation of endogenous controls for gene expression analysis in microdissected human renal biopsies. Kidney international. 2003;64:356–360. doi: 10.1046/j.1523-1755.2003.00074.x. [DOI] [PubMed] [Google Scholar]
- 148.Schmid H, Henger A, Cohen CD, Frach K, Grone HJ, Schlondorff D, Kretzler M. Gene expression profiles of podocyte-associated molecules as diagnostic markers in acquired proteinuric diseases. J Am Soc Nephrol. 2003;14:2958–2966. doi: 10.1097/01.asn.0000090745.85482.06. [DOI] [PubMed] [Google Scholar]
- 149.Schmid H, Henger A, Kretzler M. Molecular approaches to chronic kidney disease. Current opinion in nephrology and hypertension. 2006;15:123–129. doi: 10.1097/01.mnh.0000214770.11609.fb. [DOI] [PubMed] [Google Scholar]